

Abstract
Aims
Sudden death resulting from cardiac arrhythmias is the most common consequence of cardiac disease. Certain arrhythmias caused by abnormal impulse formation including catecholaminergic polymorphic ventricular tachycardia (CPVT) are associated with delayed afterdepolarizations resulting from diastolic Ca2+ release (DCR) from the sarcoplasmic reticulum (SR). Despite high response of CPVT to agents directly affecting Ca2+ cycling, the incidence of refractory cases is still significant. Surprisingly, these patients often respond to treatment with Na+ channel blockers. However, the relationship between Na+ influx and disturbances in Ca2+ handling immediately preceding arrhythmias in CPVT remains poorly understood and is the object of this study.
Methods and results
We performed optical Ca2+ and membrane potential imaging in ventricular myocytes and intact cardiac muscles as well as surface ECGs on a CPVT mouse model with a mutation in cardiac calsequestrin. We demonstrate that a subpopulation of Na+ channels (neuronal Na+ channels; nNav) colocalize with ryanodine receptor Ca2+ release channels (RyR2). Disruption of the crosstalk between nNav and RyR2 by nNav blockade with riluzole reduced and also desynchronized DCR in isolated cardiomyocytes and in intact cardiac tissue. Such desynchronization of DCR on cellular and tissue level translated into decreased arrhythmias in CPVT mice.
Conclusions
Thus, our study offers the first evidence that nNav contribute to arrhythmogenic DCR, thereby providing a conceptual basis for mechanism-based antiarrhythmic therapy.
Keywords: Ventricular arrhythmias, Neuronal Na+ channels, Diastolic Ca2+ release
1. Introduction
Sudden arrhythmic death is a leading cause of mortality in the USA.1 Arrhythmias caused by abnormal impulse generation occur both in patients with structural heart disease,2 as well as in seemingly healthy individuals.3 In the latter, fatal arrhythmias are often associated with aberrant diastolic Ca2+ release (DCR) during exercise or stress-induced catecholamine surge.3 These are called Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT).4 Recent evidence suggests that CPVT is related to mutations in proteins comprising the ryanodine receptor Ca2+ release channel (RyR2) complex, including the RyR2 itself and the sarcoplasmic reticulum (SR) Ca2+-binding protein calsequestrin (CASQ2).5,6 These mutations appear to alter the ability of RyR2 to become refractory following systolic Ca2+ release, allowing it to remain open during diastole and leak SR Ca2+.7–10 This can result in DCR, which in turn can be translated into membrane depolarization via electrogenic Na+/Ca2+ exchange (NCX) resulting in triggered ventricular arrhythmia.8,10–12
Despite high response of CPVT to combination of Ca2+ channel- and β-blockers, the incidence of refractory cases is still significant.13 Surprisingly, based on the Ca2+-dependent mechanisms, such patients often respond to treatment with Na+ channel blockade.13 However, the precise contribution of Na+ influx to aberrant DCR and arrhythmogenesis is unclear. Data presented herein strongly suggest that a subpopulation of Na+ channels, neuronal Na+ channels (nNav), colocalize with RyR2. The electrogenic NCX couples Na+ fluxes in these compartments to Ca2+ influx and efflux as well as membrane potential. Disruption of coupling between nNav and RyR2 by nNav blockade reduced and desynchronized DCR in isolated cardiomyocytes as well as in tissue and decreased arrhythmias in CPVT mice. Thus, our study offers the first support for the concept that nNav contribute to the genesis and synchronization of arrhythmogenic DCR and identifies nNav as a target for mechanism-based antiarrhythmic therapy.
2. Methods
All animal procedures were approved by The Ohio State University Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 2011).
2.1. Myocyte isolation, confocal Ca2+ imaging, Na+ current, and membrane potential recordings
Ventricular myocytes were obtained by enzymatic isolation from 3- to 5-month-old R33Q (in C57BL/6 background) and age-matched C57BL/6 WT (Jackson Jax Lab) male mice. Mice were anaesthetized with isoflurane, and once a deep level of anaesthesia was reached, confirmed by lack of response to noxious stimuli, the heart was rapidly removed and perfused via a Langendorff as previously described.8 Single ventricular myocytes were isolated using Liberase Blendzymes (Roche, Applied Science, IN, USA). Whole-cell patch clamp recordings of action potentials were performed with an Axopatch 200B amplifier using external solution that contained (in mM): 140 NaCl, 5.4 KCl, 2.0 CaCl2, 0.5 MgCl2, 10 HEPES, and 5.6 glucose (pH 7.4). Patch pipettes were filled with a solution that contained (in mM): 90 K-aspartate, 50 KCl, 5 MgATP, 5 NaCl, 1 MgCl2, 0.1 Tris GTP, 10 HEPES, and 0.1 Fluo-4 K-salt (Molecular Probes, Eugene, OR, USA) (pH 7.2). Sodium currents (INa) were recorded using internal solution containing (in mM): 10 NaCl, 20 TEACl, 123 CsCl, 1 MgCl2, 0.1 Tris GTP, 5 MgATP, 10 HEPES, and 10 BAPTA (pH 7.2). The extracellular bathing solution contained (in mM): 10 NaCl, 130 TEACl, 4 CsCl, 0.4 CaCl2, 2 MgCl2, 0.05 CdCl2, 10 HEPES, and 10 glucose; pH was maintained at 7.4 with CsOH. Whole-cell capacitance and series resistance compensation (≥60%) was applied along with leak subtraction. Electrical field stimulation experiments were performed using the following external solution (in mM): 140 NaCl, 5.4 KCl, 2.0 CaCl2, 0.5 MgCl2, 10 HEPES, and 5.6 glucose (pH 7.4). For Ca2+ spark recordings in permeabilized myocytes, the cardiac myocytes were permeabilized with saponin (0.01% for 20–30 s). The internal solution contained (in mM): 120 K-aspartate, 20 KCl, 0.81 MgCl2, 1 KH2PO4, 0.5 EGTA (free [Ca2+] ∼50 nm), 3 MgATP, 10 phosphocreatine, 0.03 Fluo-4 K-salt, 20 HEPES (pH 7.2), and 5 U/mL creatine phosphokinase. To assess the SR Ca2+ load, 20 mM caffeine was applied at the end of the experiments. Intracellular Ca2+ cycling was monitored by a Nikon A1 laser scanning confocal microscope equipped with a ×60 1.4 NA oil objective. For intact myocytes, we used the cytosolic Ca2+-sensitive indicators Fluo-4 AM (Ca2+ sparks) and Fluo-4FF AM (Ca2+ waves). The fluorescent probes were excited with the 488 nm line of an argon laser, and emission was collected at 500–600 nm. Fluo-4/Fluo-4FF fluorescence was recorded in the line scan mode of the confocal microscope (0.207 µm per pixel, 1–5 ms per line). For Ca2+ wave recordings, myocytes were paced at 0.3 Hz using extracellular platinum electrodes to obtain a comprehensive distribution of the first DCR latencies. Any DCR event (i.e. wave, wavelet) that increased cell-wide fluorescence intensity above 10% of the signal generated by the preceding stimulated Ca2+ transient was included in the analysis. The fluorescence emitted was expressed as F/F0, where F is the fluorescence at time t and F0 represents the background signal. All experiments were performed at room temperature (26°C).
2.2. Cardiac muscles confocal Ca2+ and membrane potential imaging
Muscles for confocal Ca2+ and membrane potential imaging were prepared as previously described.8 Briefly, the hearts were removed and placed in a modified Krebs–Henseleit (KH) buffer, containing (in mmol/L) 120 NaCl, 5 KCl, 2MgSO4, 1.2 NaH2PO4, 20 NaHCO3, 0.25 CaCl2, and 10 glucose (pH 7.4), equilibrated with 95% O2–5% CO2. Twenty millimolar 2,3-butanedione monoxime (BDM) was also added to the dissection buffer to minimize cutting injury. The recording chamber was placed on the stage of an Olympus FluoView 1000 laser scanning confocal microscope, equipped with a 5 mW He–Ne laser and a UPLSAPO 60X water objective (NA 1.20, working distance 0.28 mm). Muscles were loaded with the cytosolic Ca2+ indicator Rhod-2 AM for 45 min and di-4-ANBDQBS (Richard D. Berlin Center for Cell Analysis and Modeling, University of Connecticut Health Center, USA) for 10 min. The muscles were superfused with KH buffer containing 5 mM BDM and 10–20 µM blebbistatin to stop cell contraction. The Ca2+ concentration was also raised in the KH buffer to 1.5 mM. Line-scan images were acquired at the rate of 8 µs/pixel. The fluorescence emitted was expressed as F/F0, where F is the fluorescence at time t and F0 represents the background signal. All experiments were performed at room temperature.
2.3. Confocal microscopy of immunolabelled myocytes and image analysis
Isolated ventricular myocytes from R33Q hearts were plated on laminin-coated glass coverslips, fixed with 4% paraformaldehyde (5 min), permeabilized with 0.1% Triton X-100, and washed with PBS. Endogenous immunoglobulin was blocked using a mouse-on-mouse blocking reagent (M.O.M. kit; Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature and subsequently incubated with primary antibodies overnight at 4°C. After washing, goat secondary antibodies (anti-mouse and anti-rabbit) conjugated to Alexa Fluor (488, 594; Life Technologies, Grand Island, NY, USA) were added for 1 h. Coverslips were mounted to using ProLong Gold Anti-Fade Mounting Kit (Molecular Probes).
Immunolabelled myocytes were imaged on a TCS SP8 laser scanning confocal microscope equipped with a ×63/1.4 numerical aperture oil objective (Leica, Buffalo Grove, IL, USA). Custom software written in Matlab (Mathworks, Natick, MA) was used to analyse images for colocalization and cross-correlation as well as to perform fast Fourier transform (FFT) morphological analysis. Briefly, colocalization was assessed from plots of red vs. green intensity as previously described.14 Additionally, the cross-correlation coefficient between the intensities of red and green immunofluorescent signals was calculated both row-wise as well as for the averaged intensity profile over a whole myocyte. Further morphological analysis was performed by calculating the two-dimensional (2D) FFT for the red and green channels of each image. The amplitude of the 2D FFT for the red and green channels, each itself in the form of a 2D image with the same dimensions as the original image, was then overlaid. Periodicity of immunofluorescent signal was assessed from the location and amplitude of peaks on the FFTs and the relative periodicities of the red and green immunofluorescent signals assessed from the overlaid FFTs of the respective channels. The antibodies used were rabbit anti-NaV1.1, 1.3, 1.6 (Alomone, Jerusalem, Israel), 1.5 (generous gift from Dr Peter Mohler), and mouse monoclonal anti-RyR2 (Pierce Antibodies, Rockford, IL, USA).
2.4. ECG recordings
ECG recordings were performed before and after epinephrine and caffeine challenge. Continuous ECG recordings were obtained from mice anaesthetized with isoflurane, at minimum effective concentration (1–1.5%). Subcutaneous needle electrodes were applied to the left, right upper limb, and right lower limb for ECG recording (PL3504 PowerLab 4/35, ADInstruments). After baseline recording (5 min.), each mouse received intraperitoneally vehicle (DMSO), riluzole (10 mg/kg), or SN-6 (40 mg/kg). After 5–10 min from the initial intervention, animals were exposed to an intraperitoneally epinephrine (Epi, 1.5 mg/kg) and caffeine (Caff, 120 mg/kg) challenge, and ECG recording continued for 10 min. ECG recordings were analysed using the LabChart 7.3 program (ADInstruments). Ventricular tachycardia (VT) was defined as three or more premature ectopies, while arrhythmia was defined as frequent ectopies, bigeminies, and/or VT.
2.5. Reagents
Unless otherwise stated, all chemicals were purchased from Sigma (St Louis, MO, USA), Torcis (Bristol, UK), or Alomone. Fluorescent dyes were purchased from either Molecular Probes or Richard D. Berlin Center for Cell Analysis and Modeling (University of Connecticut Health Center, USA).
2.6. Data analysis
Membrane potential and INa analysis were performed using pCLAMP9 software (Molecular Devices, Sunnyvale, CA, USA). Line scanning images of Ca2+ and membrane potential were normalized for baseline fluorescence.8 Ca2+ and membrane potential imaging data were processed using ImageJ and Origin software. Statistical analysis of the data was performed using a two-tailed Student's t-test for paired and unpaired data or a single factor ANOVA. The Šidák correction was applied to adjust for multiple comparisons. A Fisher's exact and a Mantel–Haenszel test were used to test differences in nominal data. All values are reported as means ± SEM unless otherwise noted. A value of P < 0.05 was considered statistically significant.
3. Results
3.1. Neuronal Na+ channel modulation regulates DCR
To determine whether nNav possess a unique action on intracellular Ca2+ handling that may serve as a substrate for triggered arrhythmias, we assessed the impact of nNav blockade on Ca2+ sparks and DCR in the form of Ca2+ waves in cardiomyocytes isolated from hearts expressing a CPVT-associated CASQ2 mutation (R33Q) and exposed to isoproterenol (Iso).15 In accord with previous observations,16 R33Q cardiomyocytes evidenced a high rate of SR Ca2+ leak that corresponded to high Ca2+ spark frequency and prolonged duration of release. Tetrodotoxin (TTX) at nanomolar concentration is known to selectively inhibit nNav without significantly affecting cardiac-type Na+ channels (Nav1.5) or directly inhibiting RyR2.17–20 Line-scan confocal Ca2+ imaging showed that 100 nM TTX significantly decreased the frequency of Iso-promoted Ca2+ sparks and DCR in resting and paced cardiomyocytes, respectively (Figure 1A–E). We also assessed DCR synchrony, a marker of Ca2+ release refractoriness.8,10 TTX significantly reduced DCR synchrony, as demonstrated by a broadened distribution of DCR first latencies and an increase in the S.D. of latency (Figure 1F). Notably, TTX's antiarrhythmic effects were not accompanied by alterations in the SR Ca2+ content or Ca2+ transient amplitude (see Supplementary material online, Figure S1A–C), which is consistent with a previous report that utilized TTX concentrations of up to 5 µM.21 The sensitivity of DCR in R33Q ventricular cardiomyocytes to 100 nM TTX might have resulted from an adaptive up-regulation of nNav. To address such possibility, we assessed peak Na+ current (INa) in R33Q and WT myocytes. There was no significant difference in INa density between the two study cohorts (Figure 2A and B).
Figure 1.
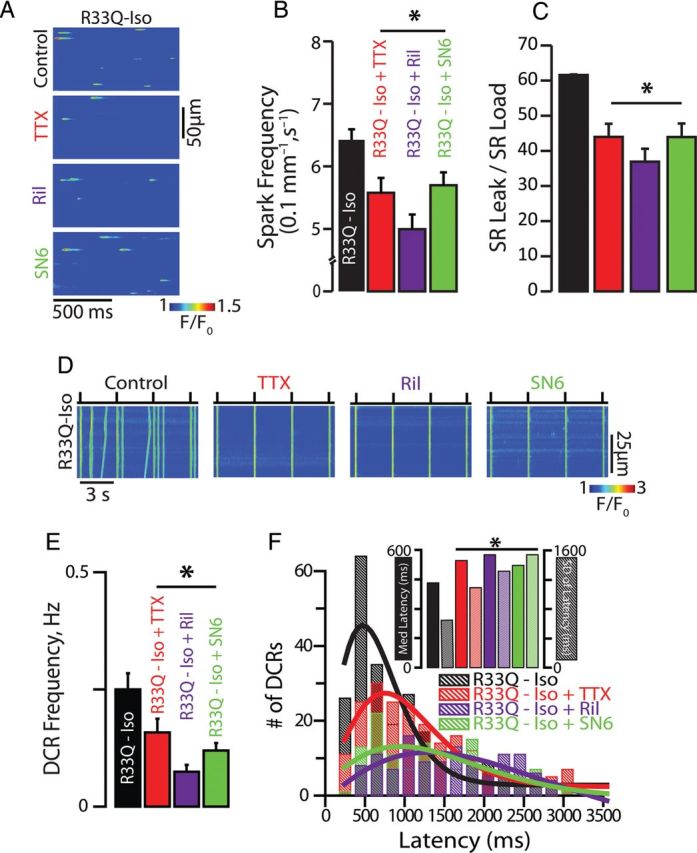
Neuronal Na+ channel blockade disrupts Na+/Ca2+ signalling and suppresses DCR. (A) Representative examples of the line-scan images of Ca2+ sparks recorded in R33Q ventricular cardiomyocytes. Cells were treated with TTX (100 nM), riluzole (Ril, 10 µM), and SN6 (5 µM) (B) TTX (n = 1054 events from 47 cells isolated from four mice), Ril (n = 1675 events from 59 cells isolated from three mice), and SN6 (n = 2811 events from 84 cells isolated from three mice) significantly decreased Ca2+ spark frequency in Iso-treated R33Q myocytes (n = 2442 events from 90 cells isolated from seven mice) (C) resulting in a reduction of SR Ca2+ load normalized spark-mediated SR Ca2+ leak (=spark mass × spark frequency/SR Ca2+ content) (n = 38, 19, 22, and cells isolated from 3–4 mice for TTX, Ril, and SN6, respectively) compared with Iso alone (n = 41 cells isolated from seven mice, *P < 0.05). (D) Representative examples of the line-scan images of Iso-stimulated R33Q and WT ventricular cardiomyocytes paced at 0.3 Hz. (E) TTX (n = 66 cells from four mice), Ril (n = 67 cells from two mice), and SN6 (n = 50 cells from three mice) significantly decreased DCR events in the form of Ca2+ waves in Iso-treated R33Q myocytes (n = 93 cells isolated from eight mice), (F) delayed the latency to the first DCR, as well as reduced DCR synchronicity as demonstrated by a broadened distribution of DCR first latencies and (inset) an increase in the median latency and S.D. of the latency compared with Iso alone (TTX, n = 208 events; Ril, n = 118 events; SN6, n = 129 events vs. Iso alone, n = 237 events, *P < 0.05).
Figure 2.

Effect of neuronal Na+ channel blockade with riluzole on peak Na+ current. (A) Inward Na+ currents obtained by 250 ms depolarization steps from holding potential −120 to 0 mV in 5 mV increments at 3 s intervals. (B) Corresponding peak I/V relationship (n = 9 cells from five mice for WT and n = 6 cells from two mice for R33Q). (C) Reduction of Na+ current by 100 nM TTX and 10 µM Ril (n = 5 cells from two mice for TTX, n = 7 cells from two mice for Ril in WT, and n = 4 cells from two mice for Ril in R33Q, *P < 0.05). (D) The decaying phase of Na+ current traces was fitted to 2-exponential function yielding two time constants, one slow (τis) and the other to fast component (τif). The effect of 10 µM Ril in R33Q cardiomyocytes (n = 4 cells from two mice, *P < 0.05) on the decaying phase of Na+ current with two components, slow (τis) and a fast (τif). (E) Reduction of the slow (τis) component of decaying phases of Na+ current by 100 nM TTX and 10 µM Ril in WT cardiomyocytes (n = 4 cells from two mice and n = 6 cells from two mice, respectively, *P < 0.05).
Next, we investigated the antiarrhythmic effects of nNav inhibition using riluzole,22 an agent with an established clinical efficacy in management of amyotrophic lateral sclerosis (ALS).23 First, we assessed whether riluzole inhibits the TTX-sensitive component of INa. To this end, we compared INa reduction between 100 nM TTX and 10 µM riluzole. In WT cardiomyocytes, we observed no significant difference in the reduction of peak INa (32.5 ± 7.7 vs. 35.1 ± 4.2%) between the two pharmacological interventions (Figure 2C). Furthermore, both riluzole and TTX facilitated INa inactivation as evidenced by a reduction in the slow component of the decaying phase of INa (Figure 2D and E). Importantly, effects of 10 µM riluzole on excitability (Supplementary material online, Figure S2A and B), Ca2+ release (Figure 1A–E; Supplementary material online, Figure S1A–C), and Ca2+ release synchrony (Figure 1E) were similar to those observed with TTX.20,24 Lastly, riluzole did not affect Ca2+ spark frequency in permeabilized cardiomyocytes (8.73 ± 0.49 vs. 7.69 ± 0.36 Sparks/100 µm/s, P = ns, n = 634–700 events), suggesting that riluzole lacks a direct effect on RyR2.
To assess whether modulation of nNav inactivation25 (Figure 2D and E) can in part contribute to the generation of DCR in quiescent (non-stimulated) myocytes, we facilitated as well as slowed nNav inactivation with riluzole (10 µM) and β-pompilidotoxin (β-PMTX, 50 µM), respectively.26 In catecholamine-free wild-type (WT) myocytes, we observed riluzole-mediated decrease in Ca2+ spark frequency (Figure 3A–C). On the other hand, β-PMTX increased frequency of Ca2+ sparks in WT myocytes despite the omission of catecholamines (Figure 3A–C). Furthermore, the enhanced Na+ flux at rest translated into increased rate of DCR frequency in stimulated, catecholamine-exposed WT cardiomyocytes (Figure 3D and E). Additionally, such rise in DCR was coupled to abbreviated latency to the first DCR and reduced SR Ca2+ content (Figure 3F; Supplementary material online, Figure S1E and F). This reduction in SR Ca2+ content can perhaps be ascribed to a large reverse mode NCX (Na+-out and Ca2+-in) in the junctional cleft prompted by enhanced Na+ influx via the incompletely inactivated nNav. Of note, the effects of β-PMTX on Ca2+ cycling were reversed with riluzole (Figure 3A–E; Supplementary material online, Figure S1D–F). Importantly, the effects of nNav modulation were independent of global phosphorylation state of the cardiomyocyte. Neither Ca2+/calmodulin-dependent protein kinase II inhibition with KN-93 (5 µM) in the presence of Iso nor the complete omission of Iso from the superfusate prevented nNav modulation of aberrant Ca2+ release (Figure 3A–C; Supplementary material online, Figure S2C and D). Taken together these results strongly suggest that a specific subpopulation of Na+ channels (i.e. nNav) exert a modulatory effect on DCR, which likely involves NCX.
Figure 3.
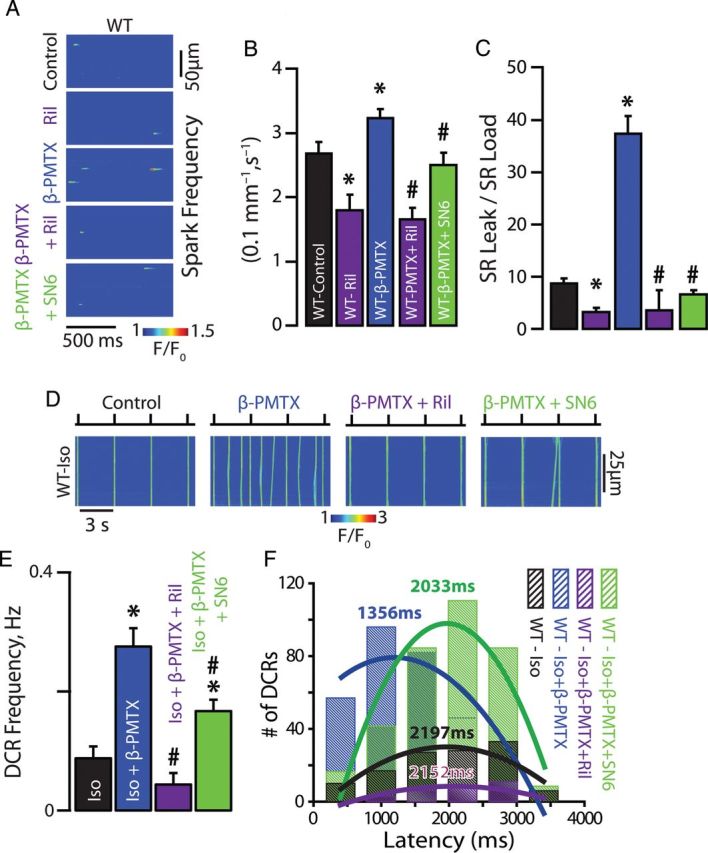
Neuronal Na+ channel contribute to Na+/Ca2+ signalling and DCR. (A) Representative examples of the line-scan images of Ca2+ sparks recorded in WT ventricular cardiomyocytes. nNav blockade with Ril in WT myocytes reduced Ca2+ spark frequency, while augmentation of the current passed by nNav with β-PMTX increased Ca2+ spark frequency. (B) Ril decreased (n = 199 events from 49 cells isolated from two mice) while β-PMTX increased Ca2+ spark frequency (n = 2413 events from 159 cells isolated from six mice) relative to catecholamine-free control (n = 1363 events from 115 cells isolated from eight mice). (C) Ril (n = 27 cells isolated from two mice) decreased while β-PMTX (n = 36 cells isolated from six mice) increased SR Ca2+ load normalized spark-mediated SR Ca2+ leak relative to catecholamine-free control (n = 29 cells isolated from eight mice, *P < 0.05). Ril and SN6 significantly reversed the effects of β-PMTX on Ca2+ spark properties (n = 293 and 753 events from 51 and 65 cells isolated from three mice for Ca2+ spark, respectively, and n = 27 and 24 cells isolated from three mice for SR Ca2+ load normalized spark-mediated SR Ca2+ leak, respectively, #P < 0.05). (D) Representative examples of the line-scan images of Iso-stimulated WT ventricular cardiomyocytes paced at 0.3 Hz. (E) Augmentation of the current passed by nNav with β-PMTX in WT myocytes (n = 95 cells isolated from seven mice for β-PMTX and n = 38 cells isolated from five mice for Iso alone, *P < 0.05) increased DCR frequency and (F) shortened the latency to the first DCR relative to Iso alone (n = 309 events for PMTX and n = 121 for Iso alone, *P < 0.05). Ril and SN6 significantly reversed the effects of β-PMTX on DCR frequency (n = 30 and 86 cells isolated from three mice, respectively, # P < 0.05, * P < 0.05 vs. Iso alone) and latency (n = 97 and 343 events, respectively).
To assess the role of NCX in the interaction between nNav and RyR2, we inhibited reverse mode NCX (Na+-out and Ca2+-in) with SN6 (5 µM).27,28 Effects on DCR similar to those observed with nNav blockade were obtained with SN6 (Figures 1 and 3; Supplementary material online, Figure S1). Furthermore, no significant effect was observed on forward mode NCX (Ca2+-out and Na+-in) as assessed by the decay of the caffeine-induced Ca2+ transient (Supplementary material online, Figure S1C, inset). These data suggest that NCX can regulate Ca2+ release29 and is an integral part of the pro-arrhythmic interaction between nNav and RyR2 within R33Q cardiomyocytes.
3.2. Neuronal Na+ channels and RyR2 colocalize
Experiments in isolated myocytes suggest that localization of nNav in close proximity to SR Ca2+-release machinery is consistent with their role in precipitating DCR. Therefore, we performed confocal immunolocalization experiments in ventricular myocytes isolated from R33Q hearts. Over 80% of the nNav (Nav1.1, 1.3, 1.6) immunofluorescent signal coincided with RyR2 labelling (Figure 4A–C and E–G) and exhibited a periodic pattern consistent with T-tubular localization (Supplementary material online, Figure S3A–C, E–G, S4, and S5A).30,31 On the other hand, very little spatial cross-correlation was observed between regions labelled with cardiac-type Na+ channels (Nav1.5) and RyR2 (Figure 4D and H; Supplementary material online, Figure S3D, H, and S5B). These results strongly suggest that nNav colocalize with RyR2 in the same subcellular regions in the R33Q mouse ventricle and in part may explain nNav contribution to the regulation of DCR in a setting of leaky RyR2.
Figure 4.
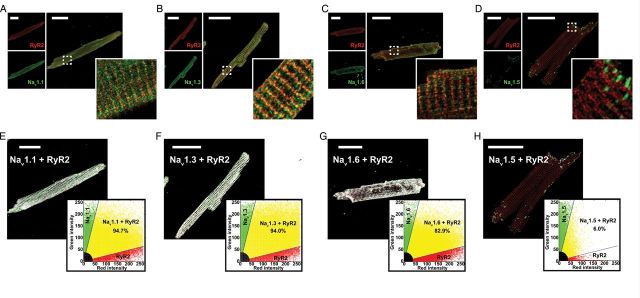
Neuronal Na+ channels and RyR2 colocalize to the same discrete subcellular regions. Confocal micrographs of myocytes from R33Q mutant mice with red labelling for RyR2s (top left) and green labelling (bottom left) for (A) Nav1.1, (B) Nav1.3, (C) Nav1.6, and (D) Nav1.5 and the overlay of the two signals (right). Scale bars represent 30 µm. The dashed white box marks the region displayed in high magnification in the insets. Results of colocalization analysis for RyR2 (red) and (E) Nav1.1, (F) Nav1.3, (G) Nav1.6, and (H) Nav1.5. Images show colocalizing pixels in white, pixels with only RyR2 signal in red, and only Nav1.x signal in green. Inset plots show green (Nav1.x) signal intensity plotted against red (RyR2) signal intensity. Pixels with colocalizing signals are marked in yellow, only red signal in red, only green signal in green, and weak signal intensity (background fluorescence) in black.
3.3. Neuronal Na+ channel blockade desynchronizes DCR in tissue
We examined the role of nNav in DCR and focal excitation in tissue. We hypothesized that in a setting of leaky RyR2,32,33 nNav could facilitate temporal alignment or synchronization of DCRs, and the resultant membrane depolarization in neighbouring cells accounting for the generation of ventricular extrasystole across the myocardium. Therefore, we performed simultaneous confocal imaging of membrane potential and cytosolic Ca2+ in cardiac muscle preparations from R33Q mice during nNav blockade. Both riluzole (10 µM) and TTX (100 nM) significantly reduced frequency and synchronicity of Iso-promoted DCR (Figure 5A–E). Correspondingly, both drugs reduced the frequency of extrasystolic action potentials (Figure 5E). Thus, nNav blockade with riluzole can reduce DCR, its synchronicity, and the resultant triggered arrhythmias in cardiac muscle.
Figure 5.

Neuronal Na+ channel blockade desynchronizes DCR in tissue. Representative Ca2+ (top) and voltage (bottom) line-scan images of Iso-treated R33Q muscle preparations paced at 1 Hz before (A) and after (B) exposure to riluzole (Ril, 10 µM), along with the corresponding Rhod-2 fluorescence profiles from each myocyte and average di-4-ANBDQBS signal. Dashed white lines outline myocyte borders. Red asterisks indicate tissue-wide extrasystolic Ca2+ release (DCR) that resulted in triggered activity (TA). (C) Temporal distribution of DCR in cardiac muscles (n = 264 events for Iso alone, n = 355 for Ril and n = 372 for 100 nM TTX). (D) Median latencies to first DCR (solid bars) and their S.D. (hashed bars) were increased by Ril (n = 355 events) and TTX (n = 372 events) relative to Iso alone (n = 264 events). (E) DCRs and TA frequency per second (Hz) before and after treatment with Ril (10 µM; n = 26 cells from four preparations isolated from three mice, *P < 0.05) or TTX (100 nM; n = 29 cells from three preparations isolated from three mice, *P < 0.05).
3.4. Effect of neuronal Na+ channel blockade on CPVT
We next examined the therapeutic efficacy of disrupting the crosstalk between nNav and the leaky RyR2 on the cellular level that desynchronized DCR in intact tissue. To this end, we assessed arrhythmia inducibility in R33Q mice. A caffeine and epinephrine challenge induced frequent ventricular bigeminy (Figure 6A), which degenerated into polymorphic VT. After riluzole treatment (10 mg/kg), half of the R33Q animals remained in sinus rhythm (Figure 6B) despite the caffeine and epinephrine challenge. Overall, riluzole reduced by half ventricular arrhythmias, including frequent ventricular extrasystoles and bigeminies (Figure 6D), while completely suppressing VTs in all R33Q mice tested (Figure 6E). Consistent with observations in isolated cardiomyocytes, riluzole's antiarrhythmic effect in R33Q mice was modulated by perturbations of NCX-dependent Na+/Ca2+ signalling. NCX inhibition with SN-6 (40 mg/kg, Figure 6C–E) resulted in a reduced overall arrhythmia burden. Taken together, these data suggest that perturbing local crosstalk between nNav and RyR2 via nNav blockade exerts an antiarrhythmic effect in vivo by reducing and desynchronizing DCR.
Figure 6.

Effect of neuronal Na+ channel blockade on CPVT. (A) Representative ECG recordings of R33Q mutant mice before (top) and after (bottom) injection (i.p.) of epinephrine (Epi, 1.5 mg/kg) and caffeine (Caff, 120 mg/kg). As illustrated in the ECG trace, Epi + Caff challenge resulted in numerous repetitive ventricular extrasystoles (arrows), bigeminy, and the induction of ventricular tachycardia (VT). (B) ECG of R33Q mice after administration of Ril (10 mg/kg), targeting plasma concentrations comparable to those achieved in experiments conducted in isolated cardiomyocytes,34 before (top) and after (bottom) Epi + Caff challenge. (C) ECG of SN-6 (40 mg/kg) treated mice 10 min before (top) and after (bottom) Epi + Caff challenge. (D) Arrhythmia and (E) ventricular tachycardia (VT) incidence (%) in R33Q mice exposed to Epi + Caff during various interventions (n = 6–11 mice, *P < 0.05). (F) Na+/Ca2+ signalling: colocalization to the same discrete subcellular region allows crosstalk between neuronal Na+ channels (nNav) and leaky Ca2+ release channels (RyR2) through the NCX resulting in aberrant DCR that underlie ventricular extrasystoles, which in turn trigger VT.
4. Discussion
Here we studied CPVT-associated arrhythmogenesis resulting from aberrant DCR.15,35 Previous studies showed that CPVT-linked mutations disrupt RyR2 gating, thereby precipitating synchronous DCR and triggered arrhythmias.5,8,36 In recent years, Na+ channel blockade has emerged as a potential antiarrhythmic strategy in genetic and acquired models of Ca2+-mediated arrhythmias.13,19,21 Previous studies have suggested two mechanisms for the antiarrhythmic effect of Na+ channel blockers such as flecainide: (i) direct inhibition of the RyR237 or (ii) decreased membrane excitability due to cardiac-type Na+ channel (Nav1.5) blockade uncoupling DCR from membrane depolarization.38 Our results demonstrate a novel mechanism for Ca2+-mediated arrhythmogenesis which involves Na+/Ca2+ signalling independent of direct RyR2 block and reduced excitability. Importantly, disruption of the Na+/Ca2+ signalling via inhibition of neuronal Na+ channels (nNav) effectively suppressed arrhythmias in CPVT mice, thus suggesting a new therapeutic approach in management of CPVT.
Initially, the antiarrhythmic effect of Na+ channel blockers, such as flecainide, was attributed to the direct inhibition of the RyR2.37 Yet, riluzole did not reduce spark frequency in permeabilized myocytes in the present study arguing against its interacting directly with RyR2. Subsequent studies have suggested that decreased membrane excitability as a result of Na+ channel blockade uncouples DCR from membrane depolarization.38 However, neither 10 µM riluzole in our study nor 100 nM TTX in previous ones altered membrane excitability.19,20,39 Importantly, our findings point to a distinct microdomain where nNav are localized near RyR2s (Figure 4), forming the functional unit underlying arrhythmogenic DCR in CPVT (Figure 6F). This is in accord with previous studies that have demonstrated the presence of nNav in the T-tubule.31,40–43 We further demonstrate using pharmacological modulation of nNav (i.e. inhibition with TTX and riluzole and augmentation with β-PMTX) as well as NCX activity (SN6) that the unique interplay between Na+ and Ca2+ handling within this domain is key to the pro-arrhythmic process. More precisely, in CPVT where cardiomyocytes exhibit increased baseline pro-arrhythmic RyR2 Ca2+ leak, nNav, and NCX blockade reduced DCR and suppressed arrhythmic activity on cellular level (Figures 1 and 3) and in tissue isolated from CPVT hearts (Figure 5). The results of these functional experiments taken together with colocalization of nNav (but not cardiac isoform of Na+ channel) with RyR2 make a very compelling case for the involvement of nNav in the modulation of the arrhythmic DCR. However, this does not preclude the cardiac isoform of the Na+ channel from participating in the arrhythmogenic process. Future studies will need to address the involvement of this particular subpopulation of Na+ channels in aberrant Ca2+ handling.
The concept of Na+/Ca2+ signalling in the vicinity of RyR2 that our data suggest is supported by previous observations that, reverse mode NCX (Na+-out/Ca2+-in) activated by the TTX-sensitive inward Na+ current early during the action potential, can contribute to stimulated Ca2+ release.17,44–47 These studies demonstrated that, during a normal heart beat, Na+ channels modulate the efficiency of Ca2+ release by supplying the necessary Na+ for reverse NCX, which in turn can facilitate RyR2 function (i.e. priming RyR2). The present study extends this concept to elucidate the mechanism of Ca2+-dependent arrhythmias by providing the first evidence that the crosstalk between nNav along with NCX and RyR2 is critical to aberrant DCR and triggered activity. Specifically, it has been previously demonstrated that nNav inactivate at relatively high potentials48 and carry a substantial residual Na+ current during depolarization.24 nNav have also been shown to exhibit substantial activity at negative membrane potentials,25 where they are prone to activation and do not completely inactivate.48 Thus, nNav activity in both stimulated and quiescent cardiomyocytes could account for the nNav-dependent effects observed herein. Our results further suggest that Na+ influx through the not fully inactivated nNav (Figure 2D)24 in the wake of electrical stimuli (Figures 1D–F and 3D–F) or those that open at rest (Figures 1A–C and 3A–C) may facilitate DCR by enhancing NCX-dependent Ca2+ accumulation in the space between the sarcolemma and the RyR2. This microdomain Ca2+ accumulation in turn promotes Ca2+-induced Ca2+ release via RyR2 that are sensitized due to the presence of a pathogenic mutation in CASQ2 (Figures 1, 3, and 6F).7–10 While a direct measurement of microdomaine Na+ and Ca2+ flux in a physiologically relevant setting might be presently not feasible, our functional and structural assays targeting the key components of the Na+/Ca2+ signalling provide strong evidence for its role in CPVT-associated arrhythmogenesis. Importantly, they reveal Na+/Ca2+ signalling and in particular nNav as a therapeutic target. Future studies will have to, however, address the specific subtypes of nNav involved in this pro-arrhythmic process.
An additional unanswered question that our study attempts to tackle is how DCR can occur synchronously in sufficient amount of myocytes to elicit a ventricular extrasystole within the entire myocardium. An extrasystolic trigger must provide a depolarizing current source large enough to overcome the electrical load of connected neighbouring myocytes serving as current sinks.49 Our data suggest that in addition to facilitating arrhythmogenic DCR on the cellular level, Na+/Ca2+ signalling synchronizes aberrant DCR across the myocardium which results in triggered activity in intact cardiac tissue (Figure 5). The latter effect enables DCR-induced depolarization to overcome the source-sink mismatch, thereby generating ventricular extrasystole.
Interestingly, over 25 years ago Na+ channel blockade has emerged as a promising strategy in management of Ca2+-mediated arrhythmias due to heart failure.50 However, reductions of electrical excitability have proved to be pro-arrhythmic in these patients, and class Ic antiarrhythmics are contraindicated in the population with structural heart disease due to an increased risk of arrhythmic death.51,52 Importantly, the reduction in electrical excitability in the present as well as our previous work was not observed during selective blockade of nNav,19 which nonetheless proved to be antiarrhythmic. Taken together, these data suggest a novel arrhythmogenic mechanism and provide clues to its structural underpinnings. Importantly, they also form the basis for a novel mechanism-based antiarrhythmic therapy utilizing a clinically available nNav blocker without compromising electrical excitability, thus causing pro-arrhythmic responses.19,51
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by National Institutes of Health (NIH) Grants (HL074045, HL063043) to S.G., (HL098039-03) to P.B.R. and (HL084583, HL083422, HL114383) to P.J.M.; and ACCP Cardiology PRN Investigator Development Research Award to P.B.R.
Acknowledgements
We thank Dr Robert Gourdie for his generous assistance with preparation of confocal micrographs of immunolabeled cardiomyocytes.
Conflict of interest: none declared.
References
- 1.Kong MH, Fonarow GC, Peterson ED, Curtis AB, Hernandez AF, Sanders GD, Thomas KL, Hayes DL, Al-Khatib SM. Systematic review of the incidence of sudden cardiac death in the United States. J Am Coll Cardiol 2011;57:794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation 1998;98:2404–2414. [DOI] [PubMed] [Google Scholar]
- 3.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 1995;91:1512–1519. [DOI] [PubMed] [Google Scholar]
- 4.Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol 2002;190:1–6. [DOI] [PubMed] [Google Scholar]
- 5.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BEC, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 2006;116:2510–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001;103:196–200. [DOI] [PubMed] [Google Scholar]
- 7.Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B, Escobar AL. Calsequestrin 2 deletion shortens the refractoriness of Ca2+ release and reduces rate-dependent Ca2+-alternans in intact mouse hearts. J Mol Cell Cardiol 2012;52:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunello L, Slabaugh JL, Radwanski PB, Ho H-T, Belevych AE, Lou Q, Chen H, Napolitano C, Lodola F, Priori SG, Fedorov VV, Volpe P, Fill M, Janssen PML, Györke S. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc Natl Acad Sci USA 2013;110:10312–10317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loaiza R, Benkusky NA, Powers PP, Hacker T, Noujaim S, Ackerman MJ, Jalife J, Valdivia HH. Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 2013;112:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belevych AE, Terentyev D, Terentyeva R, Ho H-T, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Györke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res 2012;110:569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radwański PB, Belevych AE, Brunello L, Carnes CA, Györke S. Store-dependent deactivation: cooling the chain-reaction of myocardial calcium signaling. J Mol Cell Cardiol 2013;58:77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ter Keurs HEDJ, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev 2007;87:457–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knollmann BC. Power and pitfalls of using transgenic mice to optimize therapy for CPVT – a need for prospective placebo-controlled clinical trials in genetic arrhythmia disorders. Heart Rhythm 2010;7:1683–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu C, Barker RJ, Hunter AW, Zhang Y, Jourdan J, Gourdie RG. Quantitative analysis of ZO-1 colocalization with Cx43 gap junction plaques in cultures of rat neonatal cardiomyocytes. Microsc Microanal 2005;11:244–248. [DOI] [PubMed] [Google Scholar]
- 15.Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D, Spedito A, Scelsi M, Villani L, Esposito G, Boncompagni S, Protasi F, Volpe P, Priori SG. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res 2008;103:298–306. [DOI] [PubMed] [Google Scholar]
- 16.Liu N, Denegri M, Dun W, Boncompagni S, Lodola F, Protasi F, Napolitano C, Boyden PA, Priori SG. Abnormal propagation of calcium waves and ultrastructural remodeling in recessive catecholaminergic polymorphic ventricular tachycardia. Circ Res 2013;113:142–152. [DOI] [PubMed] [Google Scholar]
- 17.Torres NS, Larbig R, Rock A, Goldhaber JI, Bridge JHB. Na+ currents are required for efficient excitation-contraction coupling in rabbit ventricular myocytes: a possible contribution of neuronal Na+ channels. J Physiol 2010;588:4249–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, Yin H, Knollmann BC. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2011;4:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radwański PB, Greer-Short A, Poelzing S. Inhibition of Na+ channels ameliorates arrhythmias in a drug-induced model of Andersen-Tawil syndrome. Heart Rhythm 2013;10:255–263. [DOI] [PubMed] [Google Scholar]
- 20.Buchanan JW, Jr, Saito T, Gettes LS. The effects of antiarrhythmic drugs, stimulation frequency, and potassium-induced resting membrane potential changes on conduction velocity and dV/dtmax in guinea pig myocardium. Circ Res 1985;56:696–703. [DOI] [PubMed] [Google Scholar]
- 21.Sikkel MB, Collins TP, Rowlands C, Shah M, O'Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT. Flecainide reduces Ca(2+) spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc Res 2013;98:286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song JH, Huang CS, Nagata K, Yeh JZ, Narahashi T. Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J Pharmacol Exp Ther 1997;282:707–714. [PubMed] [Google Scholar]
- 23.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994;330:585–591. [DOI] [PubMed] [Google Scholar]
- 24.Conforti L, Tohse N, Sperelakis N. Tetrodotoxin-sensitive sodium current in rat fetal ventricular myocytes--contribution to the plateau phase of action potential. J Mol Cell Cardiol 1993;25:159–173. [DOI] [PubMed] [Google Scholar]
- 25.Duclohier H. Neuronal sodium channels in ventricular heart cells are localized near T-tubules openings. Biochem Biophys Res Commun 2005;334:1135–1140. [DOI] [PubMed] [Google Scholar]
- 26.Schiavon E, Stevens M, Zaharenko AJ, Konno K, Tytgat J, Wanke E. Voltage-gated sodium channel isoform-specific effects of pompilidotoxins. FEBS J 2010;277:918–930. [DOI] [PubMed] [Google Scholar]
- 27.Niu C-F, Watanabe Y, Ono K, Iwamoto T, Yamashita K, Satoh H, Urushida T, Hayashi H, Kimura J. Characterization of SN-6, a novel Na+/Ca2+ exchange inhibitor in guinea pig cardiac ventricular myocytes. Eur J Pharmacol 2007;573:161–169. [DOI] [PubMed] [Google Scholar]
- 28.Iwamoto T, Inoue Y, Ito K, Sakaue T, Kita S, Katsuragi T. The exchanger inhibitory peptide region-dependent inhibition of Na+/Ca2+ exchange by SN-6 [2-[4-(4-nitrobenzyloxy)benzyl]thiazolidine-4-carboxylic acid ethyl ester], a novel benzyloxyphenyl derivative. Mol Pharmacol 2004;66:45–55. [DOI] [PubMed] [Google Scholar]
- 29.Neco P, Rose B, Huynh N, Zhang R, Bridge JHB, Philipson KD, Goldhaber JI. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J 2010;99:755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jayasinghe ID, Cannell MB, Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys J 2009;97:2664–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maier SKG, Westenbroek RE, McCormick KA, Curtis R, Scheuer T, Catterall WA. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 2004;109:1421–1427. [DOI] [PubMed] [Google Scholar]
- 32.Maltsev AV, Maltsev VA, Mikheev M, Maltseva LA, Sirenko SG, Lakatta EG, Stern MD. Synchronization of stochastic Ca2(+) release units creates a rhythmic Ca2(+) clock in cardiac pacemaker cells. Biophys J 2011;100:271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stern MD, Kort AA, Bhatnagar GM, Lakatta EG. Scattered-light intensity fluctuations in diastolic rat cardiac muscle caused by spontaneous Ca++-dependent cellular mechanical oscillations. J Gen Physiol 1983;82:119–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milane A, Tortolano L, Fernandez C, Bensimon G, Meininger V, Farinotti R. Brain and plasma riluzole pharmacokinetics: effect of minocycline combination. J Pharm Pharm Sci 2009;12:209–217. [DOI] [PubMed] [Google Scholar]
- 35.Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, Napolitano C, Williams SC, Volpe P, Priori SG, Gyorke S. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res 2006;98:1151–1158. [DOI] [PubMed] [Google Scholar]
- 36.Terentyev D, Viatchenko-Karpinski S, Györke I, Volpe P, Williams SC, Györke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc Natl Acad Sci USA 2003;100:11759–11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AAM, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 2009;15:380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu N, Denegri M, Ruan Y, Avelino-Cruz JE, Perissi A, Negri S, Napolitano C, Coetzee WA, Boyden PA, Priori SG. Short communication: flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ Res 2011;109:291–295. [DOI] [PubMed] [Google Scholar]
- 39.Weiss S, Benoist D, White E, Teng W, Saint DA. Riluzole protects against cardiac ischaemia and reperfusion damage via block of the persistent sodium current. Br J Pharmacol 2010;160:1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maier SKG, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci USA 2002;99:4073–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westenbroek RE, Bischoff S, Fu Y, Maier SKG, Catterall WA, Scheuer T. Localization of sodium channel subtypes in mouse ventricular myocytes using quantitative immunocytochemistry. J Mol Cell Cardiol 2013;64:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin X, Liu N, Lu J, Zhang J, Anumonwo JMB, Isom LL, Fishman GI, Delmar M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm 2011;8:1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin X, O'Malley H, Chen C, Auerbach D, Foster M, Shekhar A, Zhang M, Coetzee W, Jalife J, Fishman GI, Isom L, Delmar M. Scn1b deletion leads to increased tetrodotoxin-sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J Physiol 2014; doi:10.1113/jphysiol.2014.277699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larbig R, Torres N, Bridge JHB, Goldhaber JI, Philipson KD. Activation of reverse Na+-Ca2+ exchange by the Na+ current augments the cardiac Ca2+ transient: evidence from NCX knockout mice. J Physiol 2010;588:3267–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipp P, Niggli E. Sodium current-induced calcium signals in isolated guinea-pig ventricular myocytes. J Physiol 1994;474:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sipido KR, Carmeliet E, Pappano A. Na+ current and Ca2+ release from the sarcoplasmic reticulum during action potentials in guinea-pig ventricular myocytes. J Physiol 1995;489:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res 2009;104:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ulbricht W. Sodium channel inactivation: molecular determinants and modulation. Physiol Rev 2005;85:1271–1301. [DOI] [PubMed] [Google Scholar]
- 49.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J 2010;99:1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.The Cardiac Arrhythmia Pilot Study. The CAPS investigators. Am J Cardiol 1986;57:91–95. [DOI] [PubMed] [Google Scholar]
- 51.Starmer CF, Lastra AA, Nesterenko VV, Grant AO. Proarrhythmic response to sodium channel blockade. Theoretical model and numerical experiments. Circulation 1991;84:1364–1377. [DOI] [PubMed] [Google Scholar]
- 52.Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991;324:781–788. [DOI] [PubMed] [Google Scholar]