

Abstract
Co-expression of wild-type human superoxide dismutase 1 (WT-hSOD1) with ALS mutant hSOD1 accelerates disease onset relative to mice expressing only mutant protein. Here, we analyzed the effect of co-expressed WT-hSOD1 in two established mutant mouse models (L126Z and G37R), and a new model that expresses the first 102 amino acids of SOD1 with mutations at histidines 46, 48 and 63 to eliminate Cu binding (Cu-V103Z). A subset of Cu-V103Z mice developed paralysis between 500 and 730 days. Similar to mice expressing L126Z-SOD1, the spinal cords of this new model showed SOD1 immunoreactive fibrillar inclusions. Co-expression of WT-hSOD1 with Cu-V103Z SOD1 moderately accelerated the age to paralysis, similar in magnitude to WT/L126Z mice. In either combination of these bigenic mice, the severity of fibrillar inclusion pathology was diminished and unreactive to antibodies specific for the C terminus of WT protein. Co-expression of WT-hSOD1 fused to yellow fluorescent protein (WT-hSOD1:YFP) with G37R-hSOD1 produced earlier disease, and spinal cords of paralyzed bigenic mice showed YFP fluorescent inclusion-like structures. In bigenic L126Z/WT-hSOD1:YFP mice, disease was not accelerated and WT-hSOD1:YFP remained diffusely distributed. A combination of split luciferase complementation assays and affinity capture-binding assays demonstrated that soluble G37R-hSOD1 efficiently and tightly bound soluble WT-hSOD1, whereas soluble forms of the Cu-V103Z and L126Z variants demonstrated low affinity. These data indicate that WT-hSOD1 may indirectly augment the toxicity of mutant protein by competing for protective factors, but disease onset seems to be most accelerated when WT-hSOD1 interacts with mutant SOD1 and becomes misfolded.
INTRODUCTION
Mutations in the gene encoding superoxide dismutase 1 (hSOD1) account for ∼20% of the cases of familial amyotrophic lateral sclerosis (fALS) (www.alsod.iop.kcl.uk). The vast majority of SOD1-linked ALS cases are associated with dominant inheritance patterns in which the affected individuals harbor one wild-type allele and one allele carrying a mutation (1). Initial work to characterize the impact of disease causing mutations on the biology of SOD1 demonstrated that interactions between the normal protein and the mutant protein occurred (2), but the role of such interactions in disease pathogenesis was uncertain. SOD1 is a relatively small enzyme comprised of 153 amino acids; in its active state the protein homodimerizes to form the mature enzyme with each subunit binding 1 atom of Zn and 1 atom of Cu (3). To date, >165 mutations in >75 positions in the enzyme have been identified in patients diagnosed with ALS (www.alsod.iop.kcl.uk). The impact of these mutations on enzymatic activity varies greatly and it has not been possible to define a single mechanism by which these mutations cause disease (4,5). One common feature of mutant SOD1 proteins is that they exhibit a high tendency to aberrantly aggregate into high-molecular-weight structures that are insoluble in non-ionic detergents (6,7).
Multiple studies in transgenic mouse models have provided insight into the role of interactions between WT and mutant SOD1 in the pathogenesis of fALS. An initial effort to lower the expression of WT mouse SOD1 in the presence of G85R-hSOD1 was designed to test whether changes in the level of baseline SOD1 enzymatic function would impact disease onset or progression (8). This study included an experiment in which mice that overexpress WT-hSOD1 were mated with the G85R mice as a means to raise enzymatic levels. Although this initial study reported no evidence that raising or lowering the levels of enzymatic activity or WT-hSOD1 levels had any effect on any aspect of disease (8), many subsequent studies have now firmly established that overexpression of WT-hSOD1 in the presence of mutant SOD1 can dramatically accelerate the onset of disease (9–13). The most dramatic effects have been observed in studies that use a line of mice created by Gurney et al. (GurWT) (14) that are recognized as the line with the highest level of expression (10). Mating of the GurWT mice to mice that express low levels of the A4V variant of hSOD1 (which do not develop disease) induces paralysis at 10–12 months; mating of the GurWT mice to mice that express low levels of the G37R variant of hSOD1 [PrP-G37R-SOD1, which develop disease >20 months as heterozygotes (15)], induces paralysis at 5–6 months of age (10). Mating the Gur-WT mice to mice that express low levels of G93A-hSOD1 [Thy1-G93A-SOD1, which also develop paralysis at >20 months (11)] induces disease by 12 months of age. Finally, the age to paralysis in mice that express the G85R variant can be accelerated by a factor of 2 by mating to the Gur-WT line (13). Similarly, the age to paralysis in mice that express the L126Z variant can be reduced by 20–40% by mating to mice expressing high levels of WT-hSOD1 (9,10). Notably, however, when L126Z mice were crossed to WT-Line 76 mice, which express at lower levels than the GurWT (16), no acceleration in disease was observed (10). Importantly, the lower-expressing WT-line 76 mice were also used in the initial Bruijn study that reported no change in disease course in bigenic WT/G85R mice (8). In mice that co-express the WT and truncation mutants of SOD1, it has been possible to demonstrate that WT proteins misfold and acquire insolubility in non-ionic detergents as mice develop paralysis (9,10). Collectively, these mouse modeling studies have demonstrated that the most potent enhancer of mutant SOD1 toxicity that has been identified to date is the introduction of multiple copies of human genomic DNA encoding WT-hSOD1.
As noted above, >160 mutations in SOD1 have been described in patients with ALS (www.alsod.iop.kcl.uk). The vast majority of these mutations are missense point mutations. There are two examples of SOD1 mutations in individuals with ALS that would cause frame-shift mutations to produce an extremely truncated 35 amino acid protein (mutations at positions 27 and 29). Although these would be predicted to be loss of allelic expression mutations due to nonsense-mediated decay of the aberrant mRNAs (for review 17), whether these mutations are truly disease causing has not be confirmed. There are many more examples of mutations that either shift the reading frame or introduce early termination codons, in the last coding exon of the gene (www.alsod.iop.kcl.uk). These truncation variants are presumed to be expressed and produce truncated mutant proteins (18,19). To determine whether truncation variants of SOD1 that terminate earlier than the last coding exon could produce disease, Deng et al. created mice in which a termination codon was engineered into a modified human genomic DNA fragment such that the resulting mRNA would escape nonsense-mediated decay and a 116 amino acid variant of SOD1 would be produced (12). Although mice heterozygous for transgenes encoding this mutant did not develop disease, the authors described one potentially homozygous animal that became paralyzed at 10 months of age (12). Following a previously described strategy to induce disease by raising overall SOD1 levels (9), the authors produced mice that co-expressed SOD1-116x with WT-hSOD1 yielding mice that developed paralysis by 13–18 months (12). Importantly, the authors reported that the co-expressed WT protein appeared to be present, to an unspecified degree, in inclusions present in the spinal cords of paralyzed mice (12).
Whether these findings indicate that the addition of high levels of WT-hSOD1 heightens the toxicity of mutant SOD1 or whether the WT protein acquires the toxicity embodied in the mutant protein has not been resolved. The level of WT-hSOD1 expression clearly influences the degree to which the co-expression of the protein modulates disease onset in mice expressing mutant hSOD1. In general, however, the effect of co-expressed WT-hSOD1 on the toxicity of truncated mutant SOD1 is more modest. We suspected that these truncation mutants may not interact with WT protein very efficiently because they lack portions of the normal dimer interface. In the present study, we have used multiple approaches to determine the relative ability of WT-hSOD1 to interact with mutant hSOD1, examining several fALS variants that have been previously studied in mice and introducing a new model based on the expression of an extremely truncated experimental mutant. Our findings suggest that the magnitude of the effect of WT-hSOD1 on disease onset, when co-expressed with mutant SOD1, correlates with the strength of heterodimeric interactions between soluble WT and soluble mutant protein.
RESULTS
To further refine the minimal elements in SOD1 that are required for a misfolded protein to induce motor neuron disease, we created a variant of human SOD1 that encodes a termination at codon position 103, which lies just C terminal to the 6th beta strand domain of the protein. This position for inserting the stop mutation was chosen based on cell culture experiments in which a range of SOD1 early truncation mutants were examined for stability. In a prior study, we observed that an SOD1 mutant terminating at residue 75 was very unstable (20). In unpublished work, we observed a similar instability for mutants terminating at residue 90, which represents the C terminus of the 5th β-strand. Through an empirical assessment of truncated SOD1 constructs, we determined that a protein terminating at residue 103 showed steady-state accumulation levels similar to natural truncation mutations (data not show). Because of the risk involved in generating new lines of mice, we chose to test the 103 termination variant because we reasoned that the instability of mutants any shorter might confound interpretation of data if the mice failed to develop paralysis. Thus, the 103 truncation mutant may represent the most truncated mutant of SOD1 that is feasible to test in transgenic mice.
The 103 termination mutation was introduced into a previously described fragment of human SOD1 genomic DNA in which exons 3, 4 and 5 had been fused into a single cDNA exon (18) (Fig. 1A). To limit aberrant Cu binding by a severely misfolded SOD1 polypeptide, we mutated the Cu-binding histidines at positions 46, 48 and 63 to R, Q and G [which represent fALS associated (R and Q) and an experimental mutation]. We termed this novel experimental variant as Cu-V103Z. Two founder lines, designated E-17 and D-14, were identified that expressed the transgene mRNA at levels comparable with pre-existing lines of mice that are known to develop motor neuron disease (Fig. 1B). The most relevant comparisons are between L126Z mice (Line 171), which develop paralysis by 12–14 months of age (18), and H46R/H48Q mice (Line 139), which develop paralysis by 7–9 months of age (21). Only one animal from the E-17 line of Cu-V103Z mice developed paralysis before 24 months of age. However, a much larger number of mice from the D-14 line of Cu-V103Z mice developed obvious hindlimb paralysis between 16 and 24 months of age (Fig. 1C). Because of the late onset, many D-14 animals were found dead with no record of prolonged paralysis and a large number of animals from both lines reached the maximum age of 24 months without any obvious signs of motor neuron disease (Fig. 1C). To determine whether co-expression of WT-hSOD1 would accelerate disease in this model, we crossed the GurWT mice (14) to Cu-V103Z (D-14) mice. In a cohort of 10 bigenic mice, all developed paralysis by 12–20 months of age (Fig. 1C). The presence of typical motor neuron disease in this new model further refines the minimal SOD1 polypeptide necessary to produce paralysis, and to interact with WT-hSOD1 to accelerate disease, to the first 103 amino acids.
Figure 1.
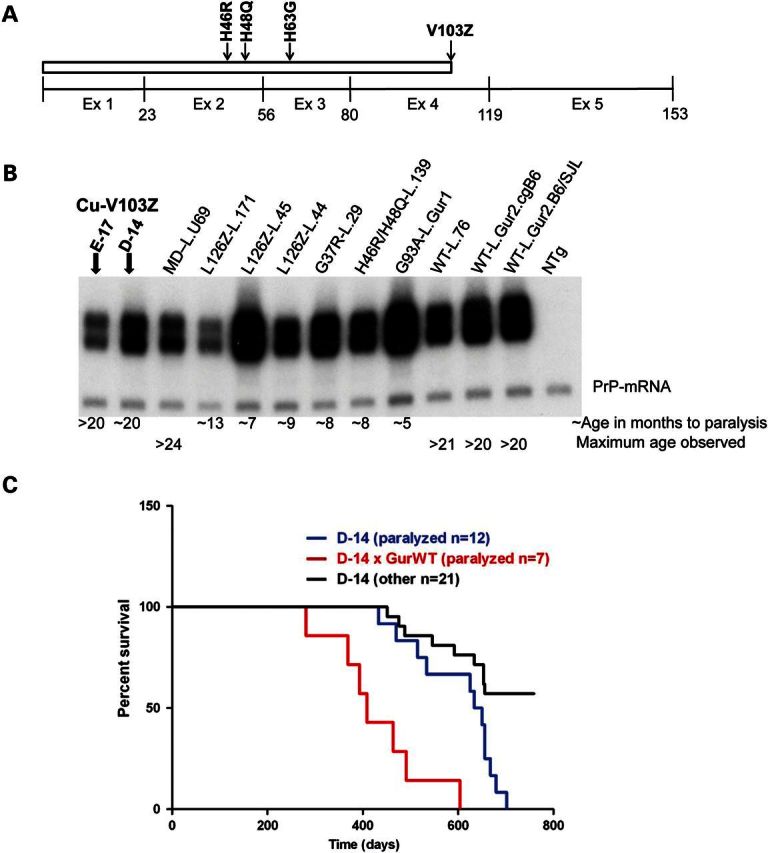
Characteristics of Cu-V103Z mice. Diagrammatic representation of Cu-V103Z construct (A). Representative northern blot of transgene mRNA from newly developed and pre-existing lines of SOD1 mice (B). Transgenic mice designations (mutant name and line designation): Cu-V103Z Line E-17, V103Z Line D-14, MD Line U69, L126Z Line 171, L126Z Line 45, L126Z Line 44A, G37R Line 29, 46/48 Line 139, G93A Gur1, WT Line76, WT Gur Congenic (C57BL/6J), GurWT-SJL, NTg). Note that only one animal from line E-17 developed obvious paralysis (confirmed by autopsy to have motor neuron disease). Survival plot for line D-14 Cu-V013Z mice and mice that are bigenic for line D-14 Cu-V103Z and Gur2 WT-hSOD1 transgenes (C). D-14 mice that were observed to develop end-stage paralysis are identified on the graph. D-14 (other) refers to mice that were found dead with no record of prolonged paralysis. Because of advanced age, some of these mice may have had multiple disease states that hastened death before paralytic symptoms were obvious. In our colony, most hybrid B6/C3 mice over 20 months of age will have developed tumors in one or more vital organs.
Pathologically, the paralyzed Cu-V103Z mice showed motor neuron loss typical of the mutant SOD1 models (not shown). The most striking new feature of this model was that within surviving motor neurons, we observed wispy fibrillar immunoreactivity to ubiquitin antibodies (Fig. 2A) that was also recognized by an SOD1 antibody raised against the whole human protein (22) (Fig. 2B), and visualized by Thioflavin S staining (Fig. 2C; Supplementary Material, Fig. S1). To determine whether WT-hSOD1 may be co-localized with these fibrillar structures in bigenic WT/ Cu-V103Z mice, we immunostained tissues with two different SOD1 antibodies, termed C4F6 and SEDI that recognize defined sequences in the protein. The C4F6 antibody (23) recognizes an epitope that includes residues 90–93 and can show some preference for mutant over WT-hSOD1 in reactivity (24). The SEDI antibody recognizes residues 145–151 at the C terminus of the protein (25) and thus cannot bind the Cu-V103Z variant. In paralyzed mice expressing only the Cu-V103Z variant, the C4F6 antibody recognized the fibrillar structures described above (Fig. 3A–C, Supplementary Material, Figs S2 and S3). In paralyzed mice co-expressing the Cu-V103Z variant with WT-hSOD1, the frequency of C4F6 reactive structures was lower and there appeared a reactivity to round punctate structures in the neuropil, including structures that resembled vacuoles (Fig. 3D–F). Importantly, immunostaining of bigenic WT/Cu-V103Z mice with the SEDI antibody identified only the punctate neuropil structures (Fig. 3G–I) and these structures were also detected in mice expressing only the WT protein (Fig. 3J–L). Although there may be some co-localization of these proteins in the C4F6-reactive neuropil structures, such a conclusion cannot be easily drawn because the C4F6 antibody can cross react with overexpressed WT-hSOD1 in fixed mouse tissues (24). The most informative finding was that in this model, we were not able to detect co-localization of WT-hSOD1 in fibrillar structures formed by the Cu-V103Z mutant.
Figure 2.

Fibrillar inclusion pathology in paralyzed D-14 Cu-V103Z mice. Representative images of the ventral horn of the spinal cord after ubiquitin immunostaining (A), SOD1 immunostaining (B) and Thioflavin S staining (C) comparing with nontransgenic mice (D–F) and WT-hSOD1 mice (GurWT) (G–I). Scale bar = 10 µm.
Figure 3.
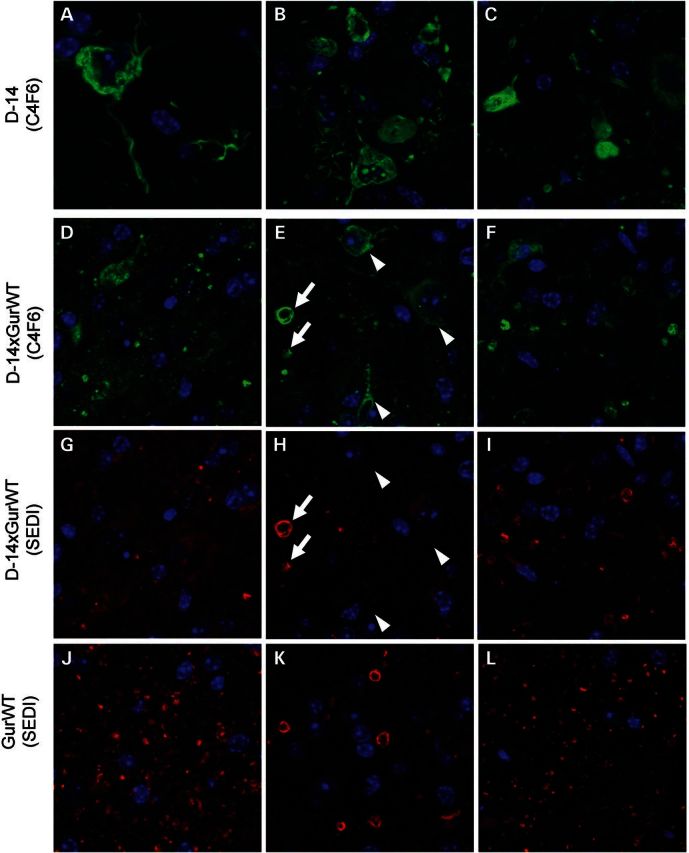
WT-hSOD1 does not co-localize to fibrillar inclusions formed by the Cu-V103Z SOD1 mutant. Representative images of the ventral horn of the spinal cord from three different animals (at least six sections per animal), genotype indicated to left, immunostained with C4F6 (A–F, green) or the SEDI (G–L, red) antibodies. (A)–(C) are images from three different animals that express only Cu-V103Z SOD1. (D) and (G), (E) and (H), (F) and (I) are paired images from three different WT/Cu-V103Z bigenic animals co-stained with both antibodies, showing each color channel individually. (J)–(L) are images from three different Gur WT animals. Arrowheads indicate the location of fibrillary structures stained by C4F6. Arrows indicate the vacuole structures stained by both C4F6 and SEDI.
Less severe fibrillar inclusions in bigenic mice expressing WT and truncated mutant SOD1
Bigenic mice that co-express WT-hSOD1 with a more natural truncation mutant, L126Z, have been previously described (10). To examine interactions between WT-hSOD1 and L126Z-hSOD1 in paralyzed transgenic mice that co-express these proteins, we used immunofluorescence with the C4F6 and SEDI antibodies. In tissue sections from mice expressing only the L126Z mutant, using antigen retrieval techniques that involve steaming in citrate buffers (24), the C4F6 antibody recognized fibrillar inclusions that ring the nucleus (Fig. 4A). In contrast, using the same antigen retrieval conditions, the SEDI antibody did not react with these fibrillar structures and instead produced astrocyte staining (Fig. 4B). For the C4F6 antibody, there was no reactivity in age-matched nontransgenic littermates. (Fig. 4C), but for the SEDI antibody, there was less abundant but clear reactivity to astrocytes (Fig. 4D). The SEDI reactivity to astrocytes may be nonspecific and elaborated by antigen retrieval, possibly further augmented in activated astrocytes. However, because of shared epitopes between human and mouse SOD1 (25), such reactivity could be recognition of endogenous mouse SOD1 that is induced by antigen retrieval.
Figure 4.
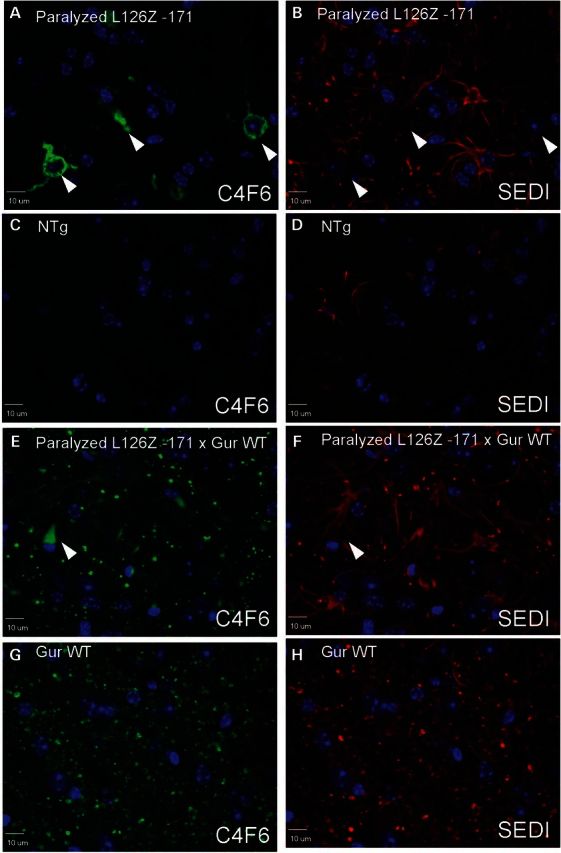
Co-expression of WT-hSOD1 diminishes the frequency of fibrillar inclusions formed by the L126Z-SOD1 mutant. Representative images of immunostaining of spinal cord sections (n = at least 3, with at least 6 sections analyzed per animal) from paralyzed L126Z Line −171 (A and B, age ∼12 months) and bigenic Gur WT/L126Z Line −171 (E and F; age 7–8 months) with C4F6 antibody (green) and SEDI antibody (red). Images from age-matched NTg littermates (C and D) and age-matched mice expressing only WT-hSOD1 (G and H) are shown. Scale bar = 10 µm. (A) and (B), (C) and (D), (E) and (F), and (G) and (H) are paired images from the same sections, showing each color channel individually. Arrowheads indicate the location of fibrillary structures stained by C4F6.
To determine whether WT-hSOD1 in bigenic WT/L126Z mice might be co-localized with fibrillar inclusions, sections were co-stained with the SEDI antibody, which does not recognize the L126Z variant. In previous work, we had determined that spinal cords of paralyzed mice that co-express WT and L126Z-hSOD1 contained detergent-insoluble complexes of WT-hSOD1 that co-sedimented with complexes formed by L126Z-hSOD1 (10), and thus we expected to observe inclusions containing WT protein. However, in paralyzed mice that co-express L126Z and WT-hSOD1, we observed a greatly diminished frequency of the fibrillar inclusions recognized by C4F6 (Fig. 4E). Most of the C4F6 immunoreactivity was limited to punctate neuropil staining. These neuropil structures also reacted with the C-terminal SEDI antibody (Fig. 4F), but the same C4F6 and SEDI immunoreactive punctate structures were also seen in age-matched mice that express only WT-hSOD1 (Fig. 4G and H). The SEDI antibody also reacted to astrocytes in these transgenics, but these same structures were observed in nontransgenic mice. Thus, we could not demonstrate definitively that WT-hSOD1 was recruited into the fibrillar inclusions formed by L126Z-hSOD1. Moreover, at end-stage, the overall frequency of fibrillar inclusions was less in WT/L126Z bigenic mice than mice expressing L126Z alone.
Bigenic mice for the Gur WT-hSOD1 and PrP-SOD1-G37R-Line 110 transgene arrays develop motor neuron disease by 6 months of age whereas mice harboring the PrP-SOD1-G37R transgenes alone develop symptoms after 20 months of age (10). Determining whether the G37R and WT-hSOD1 proteins co-aggregate in motor neurons in these mice has been difficult because we lack antibodies that definitively distinguish the two proteins. One antibody that we tested for specific reactivity to misfolded G37R-hSOD1 was the C4F6 antibody. Recognition of mutants such as the G37R variant of SOD1 by C4F6 occurs because the mutation produces structural changes that expose an epitope that minimally includes amino acids 90–96 (24,26). Natively folded WT-hSOD1 is unreactive, but presumably WT protein induced to misfold could acquire the reactivity to this antibody. Indeed, older GurWT mice develop a less severe form of the vacuolar pathology than what is seen in paralyzed mice expressing the G37R variant, but these vacuoles can be stained with C4F6 antibody (24). Despite attempts with multiple SOD1 antibodies [hSOD1 peptide antibody (27), whole protein antibody (22) and C4F6 (23)], we could not determine whether WT-hSOD1 was induced to form inclusions in bigenic G37R/WT SOD1 mice (not shown).
Because we lacked antibodies that could distinguish the G37R variant from WT-hSOD1, we turned to recently developed mice that express WT-hSOD1 fused to yellow fluorescent protein (YFP) (28). Mice expressing WT-hSOD1:YFP do not develop ALS or accumulate YFP-labeled inclusion-like structures in spinal motor neurons (28). To visualize how the presence of mutant hSOD1 impacts WT-hSOD1, we crossed the PrP-SOD1-G37R mice to the WT-hSOD1:YFP mice. Similar to native WT-hSOD1, the co-expression of WT-SOD1:YFP with G37R-SOD1 in PrP-G37R-SOD1 mice produced paralysis at earlier ages (13–15 months; Fig. 5A). For comparison, we crossed the L126Z mice with WT-SOD1:YFP mice. In this latter case, the age to paralysis was not obviously accelerated (Fig. 5B); a finding that is consistent with our previous studies of G37R and L126Z mice crossed to different strains of WT-hSOD1 mice (10).
Figure 5.
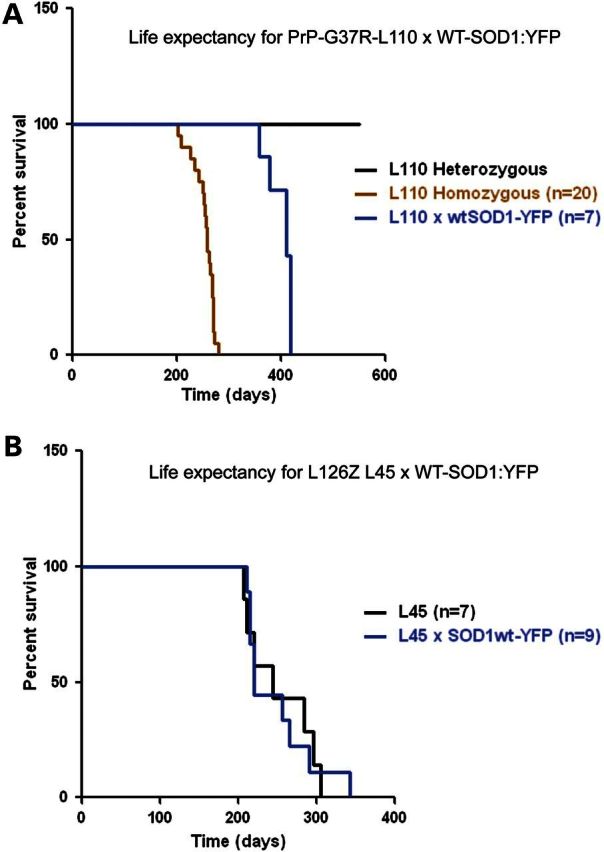
Survival plots for bigenic G37R/WT-SOD1:YFP and L126Z/WT-SOD1:YFP mice. The age at which PrP-SOD1-G37R-L110 mice reach a humane paralytic endpoint is shortened by either co-expressing WT-SOD1:YFP (n = 7) or intercrossing to produce mice homozygous for the transgene (n = 20) (A). Life expectancy of L126Z (L45) (n = 7) is not shortened by co-expressing WT-SOD1:YFP (n = 9) (B).
Using these models, we examined the impact of misfolded mutant SOD1 on the morphological distribution of WT-SOD1:YFP. In the spinal cords of paralyzed bigenic G37R/WT-YFP mice, we observed a complete re-organization of the WT-hSOD1:YFP protein into inclusion-like structures (Fig. 6A). In contrast, in littermates expressing only the WT-hSOD1:YFP protein, the fluorescence remained in a diffuse pattern of distribution (Fig. 6B). Nontransgenic animals show no fluorescence as expected (Fig. 6C). In the spinal cords of paralyzed bigenic L126Z/WT-YFP mice, we observed that the YFP fluorescence was diffusely distributed throughout the cytosol of motor neurons with no obvious inclusions detected (Fig. 6D). A similar pattern of YFP fluorescence was observed in littermate control expressing the WT-SOD1:YFP alone (Fig. 6E), and again no fluorescence was observed in nontransgenic littermates (Fig. 6F). Thioflavin-S staining of tissues from L126Z/WT-SOD1:YFP mice demonstrated the presence of fibrillar inclusions indicating that the co-expression of WT-SOD1:YFP had not abrogated aggregation of the L126Z mutant protein (Supplementary Material, Figs S5A and B; C-nontransgenic). Similarly, immunostaining of spinal cord sections from paralyzed bigenic L126Z/WT-SOD1:YFP mice with C4F6 antibody demonstrated fibrillar inclusions (Supplementary Material, Fig. S5D–F). Thus, the earlier appearance of disease in the bigenic G37R/WT-SOD1:YFP mice was accompanied by what appears to reorganization of WT-SOD1:YFP into inclusion-like structures, whereas in the bigenic L126Z/WT-SOD1:YFP disease was not accelerated and WT-SOD1:YFP remained diffusely distributed.
Figure 6.
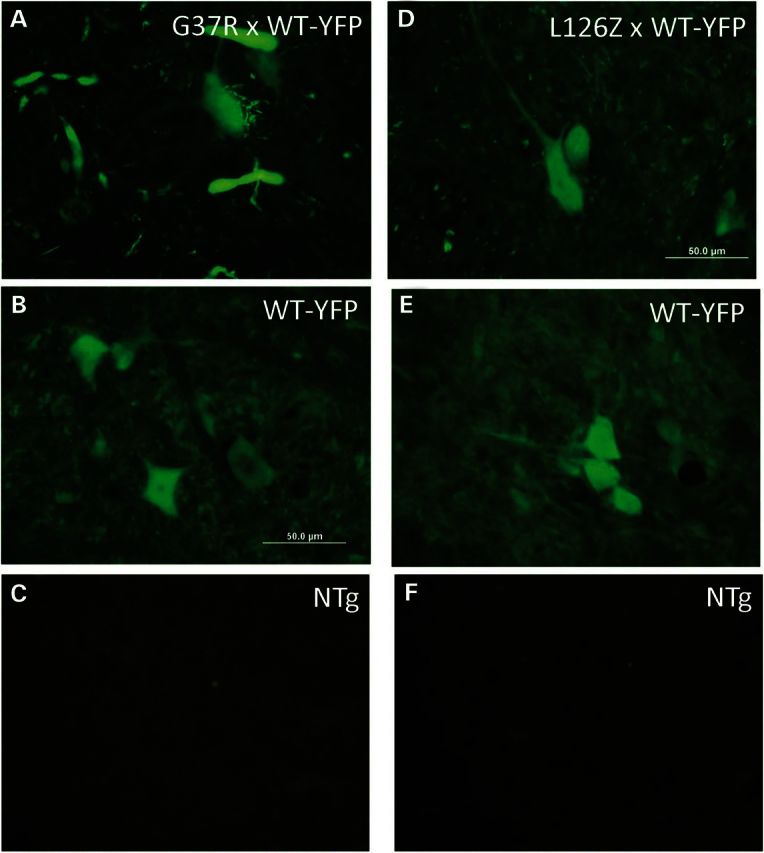
Analysis of WT-SOD1:YFP distribution in mice co-expressing G37R or L126Z mutant SOD1. Direct YFP fluorescence was imaged from freshly cut cryostat sections (20 μm thickness). Bigenic G37R/WT-SOD1:YFP (A) with littermate controls expressing WT-hSOD1 (B) and NTg controls (C). Bigenic L126Z/WT-SOD1:YFP (D) with littermates expressing WT-hSOD1:YFP and NTg controls (E and F).
Analysis of interaction between WT and mutant SOD1
To assess and quantify the strength of interactions between WT-hSOD1 and the G37R and L126Z variants, we developed an ex vivo split luciferase assay (29). We produced a series of fusion proteins in which WT-hSOD1, G37R-hSOD1, L126-hSOD1 and Cu-V103Z were fused to the N- (gLuc1) or C-terminal (gLuc2) domains of Gaussia luciferase. As controls, constructs expressing only the gLuc1 or gLuc2 domain of gLuc were generated as well as constructs in which the SOD1 component of the fusion protein was mutated at residues 50 and 51 to monomerize SOD1 (Mono) (30,31). Each construct was then independently transfected into CHO cells, lysates of each transfected culture were prepared, and the level of SOD1-gLuc fusion protein was quantified by immunoblot. These data were then used to prepare ex vivo reactions in which lysates from the cells were mixed to produce equimolar mixtures of the fusion proteins in all possible combinations. Lysates used in generating the luciferase activity data were subjected to SDS–PAGE and immunoblotting to verify that the levels of the two fusion proteins were similar (Supplementary Material, Figs S6 and S7). We expected that each fusion protein in the respective lysates would be homodimerized. We further anticipated that the homodimers would exchange subunits to produce new heterodimeric species that bring the two halves of gLuc together to produce activity. This assumption was based upon a prior study that had demonstrated that addition of pure homodimers of hSOD1 to lysates of CHO cells produced heterodimers of human/CHO SOD1 that reach equilibrium with pure homodimers of each species in 2 h at 37°C (2).
To validate the assay, we first compared the level of luciferase activity generated when WT-hSOD1-gLuc1 and WT-hSOD1-gLuc2 were co-expressed to the activity when these fusions were co-expressed with gLuc fusions to variants of WT SOD1 (Mono). The luciferase activity of all combinations of WT-hSOD1-gLuc with Mono-gLuc was significantly less than that of WT-hSOD1-gLuc1 and WT-hSOD1-gLuc2 (P < 0.0002–0.0005; Supplementary Material, Fig. S8A). Indeed, the level of luciferase activity for the various combinations of Mono-gLuc was not statistically different from what was detected when gLuc1 and gLuc2, alone, were mixed. To allow for comparisons across different experiments, luciferase activity values for all combinations run on any given day were normalized to values obtained when WT-hSOD1-gLuc1 and WT-hSOD1-gLuc2 were co-expressed (Table 1; scatter plots of raw data are provide in Supplementary Material, Fig. S8A and B). Compared with mixtures of lysates from cells transfected with WT-hSOD1-gLuc1 and WT-hSOD1-gLuc2, mixtures of lysates from cells expressing WT-hSOD1-gLuc1 with G37R-hSOD1-gLuc2 and mixtures of G37R-hSOD1-gLuc1 with WT-hSOD1-gLuc2, produced significantly less luciferase activity (P < 0.01–0.0008, Supplementary Material, Fig. S8B) but more robust luciferase activity than any pair of fusions of truncation mutations (P < 0.0015–0.0007, Supplementary Material, Fig. S8B) (Table 1). Similarly, compared with interactions between WT-hSOD1 subunits, interactions of L126Z-gLuc fusions, or Cu-V103Z fusions, with WT-hSOD1-gLuc fusions or with themselves were significantly lower (P < 0.0008) (Supplementary Material, Fig. S8B, Table 1). Indeed, in comparing normalized data values, the level of interactions between the L126Z or Cu-V103Z mutants and WT-hSOD1 was lower than interactions between WT-hSOD1 and the monomerized variants of WT-hSOD1-mono-gLuc. Note that the co-expression of the gLuc1 and gLuc2 domains of luciferase alone produced a weak signal and the level of signal from co-expression of WT with L126Z, after normalization, was below the level of this control (Table 1). Overall, these data indicate that very weak interactions occur between the L126Z or Cu-V103Z mutant and WT-hSOD1 subunits with much stronger interactions between the G37R-hSOD1 mutant and WT-hSOD1.
Table 1.
Summary of luciferase activity for all combinations of SOD1-gLuc split luciferase fusions tested
| Combination | WT-gLuc1 | G37R-gLuc1 | L126Z-gLuc1 | CuV103Z-gLuc1 | Mono-gLuc1 | gLuc1 | PBS |
|---|---|---|---|---|---|---|---|
| WT-L2 | 100a | 42 ± 6 | 5.5 ± 1 | 9 ± 0.6 | 11 ± 2 | 11 ± 3 | |
| G37R-L2 | 62 ± 15 | 24 ± 4 | |||||
| L126Z-L2 | 1.2 ± 0.2 | 1.7 ± 0.3 | |||||
| CuV103Z-L2 | 3 ± 0.6 | 7 ± 1.9 | |||||
| WTmono-L2 | 11 ± 6 | 20 ± 5 | |||||
| L2 | 15 ± 3 | 19 ± 9 | |||||
| PBS | 0 |
The luciferase activity of every combination was statistically lower than that of WT-L1 + WT-L2, P < 0.05 t-test, two-tailed, unequal variance (see Supplementary Material, Figs S8 and S9 for statistical analyses).
To demonstrate and quantify heterodimeric interactions between WT and mutant SOD1 subunits, we developed a version of WT-hSOD1 that encoded a C-terminal AviTag (32). The small peptide AviTag creates a substrate for a bacterial enzyme that biotinylates the peptide and facilitates affinity purification. These WT-SOD1-AviTag constructs were co-transfected with untagged versions of WT, A4V, G37R, G85R and L126Z-hSOD1 to assess the strength of interactions. As expected WT-hSOD1 easily bound the SOD1-AviTag protein (Fig. 7A and B; note SOD1-AviTag migrates at a higher relative molecular weight than WT-hSOD1 in standard SDS–PAGE). Strong interactions were also noted for G37R variant with WT-hSOD1-AviTtag (Fig. 7A and B). For comparison, we examined the binding of A4V and G85R variants to WT-hSOD1, finding weaker interactions than observed for WT or the G37R variant (Supplementary Material, Fig. S8), but still much stronger than the L126Z mutant, which is missing elements of the dimer interface. This mutant generally failed to bind SOD1-Avitag (Fig. 7A; Supplementary Material, Fig. S9). For the L126Z variant, we observed some nonspecific binding to the avidin column (Fig. 7A, arrowheads) and this background binding was subtracted when immunoblots shown in Figure 1 (and replicates, n at least 3) were quantified. Quantification of the data suggested that the G37R variant binds very avidly and efficiently to WT-hSOD1, whereas the L126z variant binds very weakly (Supplementary Material, Fig. S9).
Figure 7.

Analysis of heterodimeric interactions between WT and mutant SOD1. A version of WT-hSOD1 that encoded a C-terminal AviTag was co-expressed with untagged versions of WT, G37R and L126Z-hSOD1 to assess the strength of interactions. As expected WT-hSOD1 easily bound the SOD1-AviTag protein (labeled SOD1-AviTag); note SOD1-AviTag migrates at a higher relative Mr to WT-hSOD1 in standard SDS–PAGE). To no great surprise, the L126Z variant, which is truncated and missing elements of the dimer interface, generally failed to bind SOD1-AviTag (see labels in figure). For the L126Z variant, we observed some nonspecific binding to the avidin column (arrowheads).
Co-expressed WT-SOD1:YFP does not increase the abundance of G37R-SOD1
Although we observed induced misfolding of WT-SOD1:YFP when co-expressed with G37R-SOD1, the misfolding of the WT protein could be a secondary event that is unrelated to toxicity. One plausible explanation for the accelerated disease in the bigenic G37R/WT-SOD1:YFP mice could be that the heterodimerization of the mutant protein with the WT protein protects the mutant from degradation. In studies involving cultured cells that co-express G85R-hSOD1 with WT-hSOD1 such stabilization was not observed (2), but the dimerization of G85R-hSOD1 with WT-hSOD1 could be weaker than what occurs for G37R-hSOD1 and WT-hSOD1. Additionally, recent studies have demonstrated that engineered obligate (fused) heterodimers of WT and mutant SOD1 have a longer half-life than similarly engineered obligate homodimers of mutant SOD1 (33). To directly test whether G37R-SOD1 levels were raised by co-expression of WT-hSOD1, we examined spinal cords of presymptomatic G37R/WT-SOD1:YFP bigenic. The altered molecular weight of YFP fusion protein allows unambiguous detection of G37R-SOD1. When compared with mice expressing G37R-SOD1 alone, the levels of G37R protein in bigenic mice were not significantly elevated (Supplementary Material, Fig. S10).
WT-hSOD1 does not appear to oligomerize with mutant SOD1
Although the degree to which co-expressed WT-hSOD1 accelerates disease when co-expressed with L126Z mutant SOD1 is less than when WT-hSOD1 is co-expressed with G37R, the effect is measurable (10). A statistically significant acceleration in age to paralysis has been observed when GurWT mice were crossed with a line of mice that express the L126Z variant at low levels (10). Although we did not detect WT-hSOD1 as co-depositing in fibrillar inclusions in L126Z/WT bigenic mice, it is possible that WT-hSOD1 could be interacting with L126Z mutant SOD1 to form assemblies of misfolded protein that are not large conglomerates that produce inclusions. Soluble oligomeric forms of mutant SOD1 have been detected in the spinal cords of multiple lines of mutant SOD1 mice including mice that express the G127X truncation mutant (34).
To initially test whether we could produce mixed assemblies of L126Z and WT-hSOD1, we used a cell culture overexpression system. To separate large insoluble assemblies of mutant SOD1 from smaller soluble assemblies, cells were treated with digitonin to gently disrupt the cell membrane and release soluble SOD1 into the aqueous buffer (6). These soluble preparations were loaded directly onto blue-native gels (35), avoiding freeze–thaw, and the gels were run on ice to limit any dissociation that could occur as the gel heated during electrophoresis. Cells were transfected with vectors encoding WT-hSOD1 and two ALS variants A4V and L126Z. For comparison, we included samples of WT-hSOD1 purified from yeast in a native condition (as isolated) and WT-hSOD1 that had been treated to remove Cu (apo-SOD1). The gels were prepared in duplicate and immunoblotted with two antibodies. Two important technical points were that the gels were transferred to PVDF and the membrane was immersed in 8% acetic acid for 15 min to fix the proteins to the membrane (omitting this step greatly reduced immunostaining presumably because the proteins washed off the membrane). After rinsing the membrane in water several times, it was soaked in methanol to remove most of the Coomassie blue dye (component of the BN-gel) and then re-equilibrated in water before proceeding with immunostaining. One antibody used in immunostaining was an antisera that recognizes amino acids 124–136 [termed m/hSOD1 antibody (27)] that would be found in WT and A4V variants, but not the L126Z variant of hSOD1. The second antibody used was raised against a peptide sequence corresponding to amino acids 24–36 [termed hSOD1 antibody (27)] of hSOD1, recognizing WT, A4V and L126Z hSOD1. Thus, we envisioned that in cells co-expressing L126Z and WT-hSOD1 we would be able to observe whether the presence of the mutant SOD1 induced the WT protein to form higher molecular-weight species by reactivity of the WT protein to the m/hSOD1 antibody.
In cells expressing WT-hSOD1, binding to m/hSOD1(124–136) antibody was generally weak, but a faint pair of bands could be detected that co-migrated with apoSOD1 purified from yeast (Fig. 8A, compare lanes 2 and 7). In these gels, as-isolated WT-hSOD1 migrated to a position near the 66 kDa marker. The predicted size of a homodimer of hSOD1 is 32 kDa, but migration of the protein in nondenaturing gels is not solely based on size (36). Blots of cell lysates with the anti-hSOD1(24–36) antibody generally produced stronger reactivity, and again the WT protein from these cells showed a banding pattern similar to apoSOD1. In cells expressing the A4V variant, a smear of reactivity was routinely detected that was spread across the length of the gel with either antibody (Fig. 8A and B, lane 3). Immunoblots of cell lysates containing the L126Z variant produced no reactive band with the m/hSOD1(124–136) antibody (Fig. 8A, lane 4) as expected, whereas blots with the hSOD1 (24–26) antibody produced two reactive species, one of which migrated faster than apoSOD1 and one that migrated at a position consistent with a larger assembly (Fig. 8B, lane 4). Immunoblots of lysates co-expressing WT and L126Z hSOD1 with the hSOD1(124–136) antibody detected only the WT protein and its relative migration position was unchanged from that of cells expressing only the WT protein. Thus, when co-expressed in cultured cells, we do not observe L126Z hSOD1 to induce WT-hSOD1 to form high-molecular-weight species.
Figure 8.

Immunoblots of Blue-native gels to detect oligomeric SOD1 in cell models. HEK293 cells were transiently transfected with vectors expressing the variants of SOD1 noted on the figure. UTf denotes untransfected cells. Cell lysates were prepared by incubation in digitonin and the lysates were then analyzed by BNGE as described in Materials and Methods. After transfer to PDVF membrane and fixation as described in Materials and Methods, the membranes were probed with m/hSOD1 antibody (27) (A), which will detect only the full-length WT-hSOD1 protein, or the hSOD1 antibody (27) (B), which will detect both the full-length WT-hSOD1 protein and the L126Z-hSOD1 protein.
Using the same BN-gel strategy, we examined spinal cords of mice that express the L126Z mutant, WT-hSOD1 and bigenic mice to determine whether we could detect evidence that the L126Z mutant induced WT-hSOD1 to form high-molecular-weight species in vivo. For this study, we focused on presymptomatic mice so as to avoid the influence of large inclusion-like structures that form late in disease; and to look for early changes in WT-hSOD1 that could be associated with earlier disease onset. The same pair of antibodies was used to detect either WT-hSOD1 specifically or WT and L126Z-hSOD1 on immunoblots of BN-gels (Fig. 9). As was the case in the cell culture experiments, immunoblots of BN-gels with the m/hSOD1(124–136) antibody detected only the WT-hSOD1 protein (Fig. 9A, arrowhead). The relative migration of the WT-hSOD1 protein in bigenic mice was no different from that of mice expressing WT-hSOD1 alone (Fig. 9A, arrowhead). In animals of both genotypes, most of the WT-hSOD1 migrated as a broad band at 66 kDa similar to what was seen in cell models and for isolated pure hSOD1 (see Fig. 8). A minor band of immunoreactivity migrated much faster than predicted for even monomeric hSOD1, and which was not observed in the cell culture model (see Fig. 8), may represent a species that has been modified by a negatively charged moiety (Fig. 9A, asterisk). Note that the fast migrating species of WT-hSOD1 seen with the m/hSOD1(124–136) antibody was also detected when the hSOD1(24–36) antibody was used to immunoblot BN-gels (Fig. 9B, asterisk). The anti-hSOD1(24–36) antibody also revealed the electrophoretic migration of the L126Z mutant (Fig. 9B, arrow). Most of the protein migrated to a single position that was similar in size to what was observed in the cell model (see Fig. 8), although there was a faint smear of higher molecular-weight reactivity seen in the 66–146 kDa range in mice expressing only L126Z (Fig. 9B). There was also a faint smear of reactivity in the 1048 kDa range in preparations from mice expressing L126Z and the bigenic mice (Fig. 9B). We interpret these findings as evidence that in spinal cord preparations from presymptomatic mice a fraction (difficult to estimate actual percentage) of the L126Z protein migrates to a size consistent with an oligomeric assembly of the protein. We do not see evidence that WT-hSOD1 co-expressed with L126Z-SOD1 migrates aberrantly.
Figure 9.
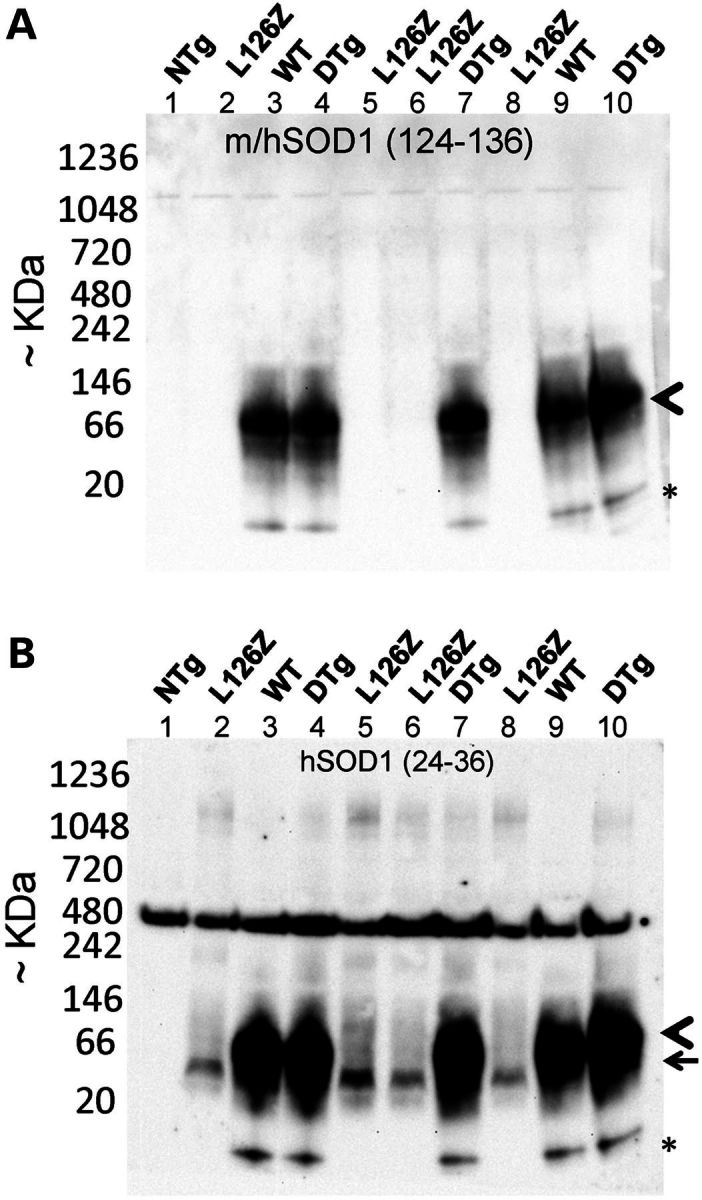
Immunoblots of Blue-native gels to detect oligomeric SOD1 in spinal cords of transgenic mice. Spinal cords from mice expressing L126Z-hSOD1 Line 45, WT-hSOD1 GurWT or bigenic mice (DTG) were prepared as described in Materials and Methods and analyzed by BNGE and immunoblotting with the m/hSOD1 (A) and the hSOD1 (B) antibodies. The arrowhead marks the position of WT-hSOD1 homodimer, the arrow marks the position of L126Z protein, and the asterisk marks the position of an unknown low-molecular-weight derivative of WT-hSOD1—potentially modified by a negative charge since it seems to react to antibodies recognizing sequences near the N- and C terminus.
DISCUSSION
In the present study, we provide the first description of a novel variant of hSOD1 that is truncated at residue 103 and encodes mutations at histidines 46, 48 and 63 to eliminate Cu binding, termed the Cu-V103Z. Two lines of mice were identified that expressed transgene mRNA at levels similar to previously characterized lines of mutant SOD1 mice that develop paralysis. Both of these lines were found to develop motor neuron disease and paralysis, but disease was incompletely penetrant. This mutant produced an interesting pathology that consisted of fibrillar inclusion structures. We observed that we could accelerate the onset, and increase the penetrance, of disease produced by the Cu-V103Z mutant by co-expressing high levels of WT-hSOD1. In bigenic Cu-V103Z/WT mice, the frequency of fibrillar inclusions was diminished and we observed no evidence that WT-hSOD1 co-deposited with the mutant protein. These findings prompted us to re-examine pathologic interactions between WT and mutant SOD1 using previously characterized lines of mice expressing the G37R and L126Z mutants, and using mice that express WT-hSOD1 fused to YFP. Co-expression of WT-hSOD1 with the G37R mutant produces a very robust acceleration in the age that paralysis develops whereas the effect of WT-hSOD1 on age to paralysis in mice that express the L126Z and Cu-V103Z mutants is less robust. In both of these latter cases, disease can be accelerated, but not to the degree seen when WT-hSOD1 is co-expressed with the G37R variant. Using antibodies that recognize specific sequences in SOD1, we demonstrate that mice that co-express WT-hSOD1 and L126Z-hSOD1 do not produce inclusions that contain both proteins. This finding is somewhat surprising because we had previously demonstrated that in paralyzed mice that co-express these proteins, WT-hSOD1 co-sedimented with detergent-insoluble L126Z-hSOD1 (10). To directly visualize binding of WT-hSOD1 to mutant SOD1 in inclusions in mice, we generated bigenic mice that co-express mutant SOD1 with a WT-hSOD1:YFP fusion protein to demonstrate that co-expression of G37R-hSOD1 with WT-SOD1:YFP induces the latter protein to redistribute to inclusion-like structures. Co-expression of the L126Z variant with WT-SOD1:YFP did not induce these inclusion-like structures. Using a split luciferase assay, we demonstrate WT-hSOD1 interacts much more strongly with G37R-hSOD1 than does either the L126Z mutant or the Cu-V103Z mutant. Using WT-hSOD1 engineered to encode a small peptide substrate for biotinylation, we provide additional evidence that G37R variant hSOD1 strongly interacts with WT-hSOD1, whereas the L126Z mutant interacted very weakly. Finally, we attempt to detect aberrant interactions between WT and L126Z-SOD1 using blue-native gel electrophoresis to detect oligomeric assemblies of mutant SOD1. The L126Z mutant produced a distinct electrophoretic entity that migrated to a size consistent with an oligomeric assembly. We could find no evidence that WT-hSOD1 was capable of co-assembling with these putative oligomers. Thus, the weaker effect of WT-hSOD1 on the toxicity of L126Z-hSOD1 seems to be based on poor interaction between these two proteins. While it is difficult to distill all of these findings into a single summary statement, our data indicate that the strength of interactions between WT and mutant SOD1 seems to be one factor that influences the age to paralysis in co-expressing bigenic mice.
Mechanisms by which co-expression of WT-hSOD1 hastens the onset of paralysis in mouse models
In synthesizing our data, we can begin to define correlates that implicate potential mechanisms by which the presence of WT-hSOD1 accelerates motor neuron degeneration. From our study of interactions between WT and mutant SOD1 as soluble entities, we determined that the relative affinity of WT for the set of mutants we examined is G37R>>>L126Z or Cu-V103Z. Relating these data to the magnitude of the effect of WT-hSOD1 on disease course in mutant mice is difficult because the levels of mutant SOD1 in different lines are not identical and the level of mutant SOD1 clearly impacts the degree to which added WT-hSOD1 expression impacts disease course in mutant mice (10). However, a few direct comparisons can be made in looking at previous studies. First, paralysis can be induced in PrP-G37R-SOD1 mice after crossing in the WT transgene array from either the Gur-WT mice or the lower expressing WT-Line 76 mice (10). Second, bigenic mice of Gur-WT with a line of L126Z mice that develop disease at 7–9 months developed paralysis 3 months earlier than the parental L126Z mice; but bigenic of WT-Line 76 with the same L126Z parental line developed paralysis no earlier (10). In the present study, we extend the dataset by adding in our findings of crosses between GurWT mice and our new Cu-V103Z mutant, and crosses between G37R and L126Z mice with WT-SOD1:YFP mice. For the Cu-V103Z mutant, the added expression of WT-hSOD1 hastened the age to paralysis and increased the penetrance of motor neuron disease, but the age to paralysis was accelerated only ∼25%. A similar degree of acceleration was observed when L126Z mice were made bigenic with GurWT mice (10). Crosses of WT-SOD1:YFP mice with PrP-SOD1-G37R mice produced bigenic offspring that developed paralysis at less than half the age of PrP-SOD1-G37R mice alone (current colony develops symptoms at 20–24 months), whereas bigenic mice created by crosses of L126Z mice and WT-SOD1:YFP mice develop paralysis no earlier. From this set of data, we conclude that the effects of WT-hSOD1 on disease course in mice expressing the L126Z, and Cu-V103Z, mutant are of a lower magnitude than the effects on the G37R mutant.
From a previous study, we knew that the G37R variant could form active heterodimers with WT-hSOD1 (36). Presumably, the interactions we have quantified here are reflective of normal dimeric interactions, which could have two potential effects. First, such interactions could stabilize the G37R mutant and increase its half-life, thereby effectively increasing the dose of mutant protein. Second, we could envision that a misfolded species of mutant SOD1 can interact with the WT protein and template misfolding of WT-hSOD1, causing the WT protein to potentially acquire the toxic property embodied in mutant protein. In regard to the first scenario, recent studies have suggested that artificially joining mutant and WT-hSOD1 in obligate heterodimers produces a stabilizing effect on the mutant protein to increase resistance to protease digestion (33). However, previous study of natural heterodimers in cell models indicated that the rate of mutant SOD1 degradation is not influenced by the presence of WT-hSOD1 and that the subunits of SOD1 dimers freely exchange (2). Our analysis of G37R-SOD1 levels in bigenic G37R/WT-SOD1:YFP mice finds no evidence to substantiate a mechanism of action in which the half-life of the mutant protein is extended, which would presumably elevate steady-state levels, by interaction with WT SOD1. Additionally, although the effect of WT-hSOD1 on the toxicity of the L126Z and Cu-V103Z mutants is less than the effect on the G37R variant, it remains that co-expressed WT SOD1 accelerates the course of disease caused by the two truncation mutants. Our analysis of interactions between WT-hSOD1 and these truncation mutants indicate that there would be little opportunity for WT to influence the stability of these mutants through stable interactions. Thus, we do not favor a model that explains the data based solely on some type of interaction between mutant and WT that extends the half-life of the mutant and thus increases the effective dose of mutant protein. In regard to the second scenario, evidence that mutant SOD1 can template a propagating misfolding of WT-hSOD1 has recently been demonstrated in cell culture models (37). Our data from the study of mice co-expressing WT-SOD1:YFP and G37R-hSOD1 are consistent with templated misfolding as we observe a re-organization of the WT-SOD1:YFP protein into inclusion-like structures. However, whether the two proteins actually co-aggregate to form inclusions is uncertain because we lack antibodies that can distinguish between G37R and WT-hSOD1 (or WT-SOD1:YFP) (Supplementary Material, Fig. S11).
Since the appearance of the inclusion-like structures is viewed as a hallmark of misfolded protein, it seems impossible to dissociate misfolded SOD1 from toxicity because we only observe misfolding of the mutant protein when mice exhibit motor neuron disease (15,38). How the formation of the large inclusions plays into toxicity is unclear given that large detergent-insoluble conglomerates of mutant protein appear late in the disease in fALS SOD1 mice (39). Indeed, in our bigenic WT/L126Z and WT/Cu-V103Z mice, the burden of fibrillar inclusions at end stage was lower than mice that developed paralysis from the expression of only mutant SOD1. This finding could be viewed as additional evidence that larger inclusion structures are not the critical toxic entities. Still, it is clear that the accumulation of detergent-insoluble conglomerates of mutant protein and the appearance of pathologic inclusions provides what amounts to a biomarker of SOD1 protein misfolding. Thus in our interactions between WT-SOD1:YFP and G37R-hSOD1, we clearly observed the biomarker of WT-SOD1:YFP misfolding by its reorganization into inclusion-like structures; data that is consistent with the notion that the misfolded conformation of mutant SOD1 was transferred, or templated, to the WT-hSOD1 component of the fusion protein to increase the overall toxic burden of misfolded SOD1. We cannot conclude that the inclusion structures harbor toxicity, only that they are good indicators of a history of misfolding and that the misfolding that produces inclusion structures seems to be associated with toxicity.
Given the evidence for a templating mechanism when G37R is co-expressed with WT-hSOD1, we assumed that the same mechanism is involved when WT-hSOD1 is co-expressed with other fALS variants. In the case of the L126Z combined with WT-hSOD1, we could not provide evidence of co-assembly of the L126Z and WT proteins in the fibrillar inclusions that are characteristic of the L126Z mice. In prior work, we had observed that WT-hSOD1 in paralyzed L126Z/WT SOD1 bigenic mice acquires detergent insolubility that is a hallmark of aggregation (10). However, in this prior study, we did not investigate whether we could detect WT-hSOD1 in aggregates pathologically. In the interval since this study, we became familiar with using the C4F6 and SEDI antibodies to detect pathologic aggregates of SOD1 (6,24). Additionally, in studies in cell models, we observed that acquisition of detergent-insoluble conformations by SOD1 does not necessarily equate to inclusion formation (6). Thus, our prior observation that WT-hSOD1 acquires detergent insolubility in paralyzed L126Z mice is further refined here by demonstrating that the WT protein does not appear to be co-depositing in inclusions. Importantly, in our prior study by Prudencio et al., we observed that L126Z mice bigenic for WT-hSOD1 from the low expressing line 76 mice accumulated detergent insoluble WT-hSOD1 but did not develop disease any earlier (10). We interpreted this finding as evidence that detergent-insoluble WT-hSOD1 might not be contributing significantly to toxicity.
Our new work reported here reveals additional complexity by observing that the frequency of fibrillar inclusions in L126Z/WT SOD1 bigenic mice was generally less severe than what we observed in end-stage L126Z mice. This outcome parallels what we observed in prior studies of WT and mutant SOD1 aggregation in cell culture models where we observed that the co-expression of WT-hSOD1 generally slowed the rate of mutant SOD1 aggregation (10,40). In split luciferase assays, which we presume could report normal dimeric interactions as well as aberrant multimerization, we found little evidence of interaction between WT-hSOD1 and truncation mutants. Additionally, we could not produce evidence that WT and L126Z proteins interact to form oligomeric assemblies. It is possible that for the L126Z variant the mechanism by which the co-expression of WT-hSOD1 at high levels induces disease involves a process other than direct templating of WT-hSOD1 misfolding. We can envision an alternate mechanism in which the excess WT protein competes for factors that prevent the misfolding of, or enhance the clearance of, the L126Z or Cu-V103Z mutant, and indirectly increases the toxicity of the protein. Clearly, the potential exists that if WT competes for some factor, then such competition could also occur in mice bigenic for WT and G37R-hSOD1. Hence, we would argue that there may be multiple explanations for how WT-hSOD1 overexpression enhances the toxicity of mutant SOD1.
Implications for human SOD1-linked fALS
The primary effect of co-expressing WT-hSOD1 with mutant SOD1 in mice is to hasten the onset of disease. There are also multiple examples of lines of mutant mice that express mutant SOD1 below the threshold to induce disease on their own, but if bigenic for WT-hSOD1 then they develop the expected phenotype (9–13). In these later cases, the added WT-hSOD1 appears to directly contribute to breaching the expression threshold and presumably the WT protein acquires the “toxic” conformations held by mutant hSOD1 when the two proteins are co-expressed. When expressed at very high levels, WT-hSOD1 appears to be able to spontaneously acquire the toxic conformation that produces ALS-like symptoms (41). However, the role that WT-hSOD1 plays in modulating the onset of disease in humans, when present at physiologic levels, is unclear. The age of onset for SOD1-linked fALS in humans varies greatly. For example, humans that inherit the L126Z mutation have a median age of onset of 42 years of age while humans that inherit the G37R mutation have median age of onset of 40 years of age; in both cases, the mode of inheritance is complete penetrance [n = 14 and 8 cases, respectively, see Supplementary Information in (7)]. Our data from mice would have predicted that the age of onset for individuals with the G37R mutation might have been much earlier than for the L126Z mutation if templating of WT-hSOD1 misfolding by mutant SOD1 contributes to disease onset in humans. Thus, although the mouse data clearly indicate the potential for interactions between WT and mutant SOD1 to contribute to pathogenesis of SOD1-linked fALS, it is difficult to discern the manifestations of such interactions in the characteristics of human disease. We note, however, that there is great variability in the age of onset in humans harboring SOD1 mutations and the sources of this variation are presently unknown.
The potential role of SOD1 misfolding in the pathogenesis of nonfamilial ALS remains controversial. Preparations of oxidatively modified WT-hSOD1 can impair axonal transport in model systems and SOD1 modified in this manner reacts with antibodies that selectively react with misfolded mutant SOD1 (42). One of these antibodies, C4F6, produced diffusely distributed reactivity in spinal tissues from a subset of sporadic ALS cases (42). However, we did not observe the C4F6 reactivity seen by Bosco et al. in human ALS in a cohort of 25 cases (24); Brotherton et al. demonstrated that familial cases of SOD1-linked ALS demonstrate C4F6 reactive fibrillar inclusions, whereas sporadic cases lack evidence of similar features of specific reactivity (43). Forsberg et al. reported that SOD1 antibodies, recognizing sequences (amino acids 4–20 or 131–153) that are normally inaccessible in natively folded protein, produced diffusely distributed puncta of immunoreactivity in 22 of 23 spinal cords from nonfamilial ALS patients (44). Recent studies by Grad et al. have used immunoprecipitation with antibodies that recognize a normally inaccessible epitope at the C terminus of SOD1 to discern changes in the conformation of soluble WT-hSOD1 in spinal cord tissues from sporadic ALS (37). The observation that misfolded forms of WT-hSOD1 can propagate between cells provides additional suggestive evidence for a role of WT-hSOD1 in nonfamilial ALS. Although these studies provide evidence of conformational changes in WT-hSOD1, whether WT-hSOD1 plays a causative in familial or sporadic ALS remains controversial and difficult to prove.
Conclusions
Overall, our findings provide an example of a case in which SOD1 encoding a mutation linked to fALS appears to template the misfolding of WT-hSOD1:YFP to produce inclusion-like structures in mice that develop disease at an accelerated rate. In contrast, mice that co-express a truncation mutant of SOD1 failed to template WT-hSOD1 misfolding and failed to accelerate disease. The outcomes of crosses between truncation mutants and untagged WT-hSOD1 suggest that the WT protein may also compete for factors that modulate the folding or degradation of mutant SOD1. Improving our understanding of how co-expression of WT-hSOD1 accelerates disease in these models may reveal the basis for SOD1 toxicity in motor neurons.
MATERIALS AND METHODS
Transgenic mice
To create the transgene constructs for the Cu-V103Z mutant, we started with a genomic fragment of the human SOD1 gene that was similar to a construct used to make SOD1-L126Z transgenic mice (18) in which the introns between exons 3, 4 and 5 had been removed to create a new fused exon. The 5′ and 3′ untranslated portions of the genomic fragment were unaltered. In addition to the previously described SOD1-L126Z construct, we had generated a version of the L126Z mutant that also encoded mutations at histidines that comprise the Cu-binding site of SOD1 (positions 46, 48, 63 and 120) to Arg, Glu, Gly and Gly, respectively. This construct, however, was never used to make transgenic mice. In the present study, we repurposed this Cu deficient version of L126Z to make the Cu-V103Z mutation by replacing the codon for Val at position 103 codon with a stop codon, using PCR approaches. Each coding exon and flanking intronic sequences were verified by sequence analysis before injection into mouse embryos (C3H/HeJ × C57BL/6J F2). Founders were identified by PCR amplification of DNA extracted from tail biopsy using the primers Hu-S: TCA AGC GAT TCT CCT GCC T; Mo-S: TAC ATA TAG GGG TTT ACT TCA T; Mo/Hu-AS: CAC ATT GCC CAR GTC TCC A (R = A/G) as previously described (18).
Other lines of SOD1 expressing mice that were used in this study included mice expressing WT and G93A hSOD1 developed by Gurney et al. (14), lines 45 and 171 of L126Z-SOD1 mice described by Wang et al. (18), line 110 of PrP-SOD1-G37R mice described by Wang et al. (15), and mice expressing fusion proteins of WT and G85R-hSOD1 with YFP described by Wang et al. (28) [graciously provided by Drs. Jiou Wang (Johns Hopkins School of Public Health) and Arthur Horwich (Yale University)]. For all previously described strains of mice, animals were identified by PCR amplification of DNA extracted from tail biopsies as described in prior descriptions of the animals. All lines of mice used in this study were maintained by crossing transgenic males to nontransgenic (C57BL/6J × C3/HeJ F1) females (Jackson Laboratories, ME, USA). Note that both the GurWT and Gur-G93A lines of mice were also bred to B6/C3 F1 females (>10 generations), which switched the background strain of these mice from B6/SJL to B6/C3 so that all lines of mice would be on the same background. All procedures involving animal handling and processing were approved by the University of Florida Institutional Animal Care and Use Committee, following guidelines set forth by the National Institutes of Health.
Neuropathologic studies
All the mice were euthanized by exsanguination and perfusion with phosphate-buffered saline (PBS) under isoflurane anesthesia. After the brain and spinal column were removed, the tissues were immersion fixed in 4% of paraformaldehyde in PBS at 4°C for 48 h. Tissues were moved to PBS and stored at 4°C until processed for sectioning. Spinal cords were carefully dissected from spinal columns. One hemibrain and half of the spinal cord segments were frozen and cut into 20 µm thickness in a cryostat for direct YFP imaging using a fluorescent microscope, the other hemibrain and some spinal cord segments were embedded in paraffin and cut into 6 µm serial sections. Immunofluorescence, immunohistochemical and Thioflavin S stainings of 6 µm paraffin sections were performed according to standard protocols as previously described (21). The antibodies used include the SEDI antibody [rabbit peptide antiserum; generous gift of Janice Robertson at the University of Toronto, Canada (25)] used at 1 : 100 dilution from the provided stock), the C4F6 antibody (23) (1 : 200, MédiMabs, Montréal, Québec, Canada), the wSOD1 [rabbit antiserum raised against the whole protein (22)], ubiquitin antibody (5–25, 1 : 1000, Covance, NJ, USA), YFP antibody (JL-8, 1 : 200, Clontech, CA, USA). Olympus DSU-IX81 Spinning Disc Confocal microscope and an Olympus BX60 epifluorsecence microscope were used to capture the fluorescent images.
Biotinylation and AviTag capture assays
HEK 293 FTs were co-transfected with WT-hSOD-AviTag and untagged SOD plasmids using Lipofectamine 2000 according to the manufacturer's protocol. After 24 h, the cells were rinsed with 1× PBS, then 10 mm Tris, pH 7.4, harvested by scraping and resuspended in 10 mm Tris with Protease Inhibitor Cocktail, (Sigma #P8340). The cells were lysed by repeated freeze–thaw in a dry ice/ethanol bath and a 42°C water bath. The cell lysate was spun at 3000g for 2 min. The cell lysate was incubated with biotin protein ligase (Genecopoeia #BIRA500) for at least 2 h at 30°C and purified through a ProbeQuant50 column (GE Healthcare # 28-9034-08). Monomeric avidin agarose beads (Thermo Scientific #PI20228) were washed with 1× PBS and the biotinylated cell lysate was bound for 1–2 h at RT or overnight at 4°C. The avidin agarose was spun at 3000g for 2 min and washed three times with PBS-T (PBS with 0.05% Tween 20), transferring it to a new tube in the process. The final agarose pellet was resuspended in an equal volume of 2× Laemmli buffer and boiled prior to loading on an 18% Tris–glycine gel. The gel was electrophoresed, transferred to nitrocellulose and blotted with the wSOD-1 antibody.
BN–PAGE and immunoblotting
The protocol utilized was provided by the supplier of BN-gels after the publication by Shagger and von Jagow (34), and similar to a recent analysis of mutant SOD1 expressed in cultured cells (45). HEK 293FT cells were transfected using Lipofectamine 2000 according to the manufacturer's protocol. The total amount of DNA was kept constant regardless of whether there were one or two plasmids. After 24 h, cells were rinsed with 10 mm Tris, pH 7.4, harvested by pipetting using a large bore tip in 50 mm Bis–Tris, 16 mm HCl, 50 mm NaCl and centrifuged at 3000g for 2 min. The cell pellet was resuspended in the above buffer plus 1% digitonin and mixed on the nutator for 30 min at RT. The lysed cells were pelleted at 3000g for 2 min. The supernatant (S1) was centrifuged in an Airfuge for 5 min. This supernatant (S2) was mixed with 4× Sample NativePAGE sample buffer (Life Technologies, BN 2003) and G-250 sample additive (Life Technologies, BN 2004) and run on 3–12% Bis–Tris gel with Dark Blue Cathode buffer at 150 v for 45 min, with Light Blue Cathode Buffer at 150 v for 15 min and with Light Blue Cathode buffer at 250 v for 1 h, according to Life Technologies' NativePAGE gel system. The protein was transferred to PVDF and fixed on the membrane in 8% acetic acid for 15 min. The membrane was rinsed in water, then methanol to destain, then water before immunoblotting with the rabbit anti-peptide antibodies m/hSOD and hSOD (27).
To prepare extracts of spinal cord for BNGE, tissues were dounce homogenized to a 10% (w/v) homogenate in 50 mm Bis–Tris, 16 mm HCl and 50 mm NaCl containing 1 : 100 (v/v) protease inhibitor cocktail (Sigma, St. Louis, MO, USA). Digitonin was then added to 0.5% for each sample, incubated with rocking at room temperature for 30 min, and then centrifuged at 3000g for 2 min. The supernatant (S1) was spun in the Airfuge for 5 min. This supernatant (S2) was mixed with 4× Sample NativePAGE sample buffer (Life Technologies, BN 2003) and G-250 sample additive (Life Technologies, BN 2004) and run on 4–16% Bis–Tris gel with Dark Blue Cathode buffer at 150 v for 45 min, with Light Blue Cathode Buffer at 150 v for 15 min and with Light Blue Cathode buffer at 250 v for 1 h, according to Life Technologies' NativePAGE gel system. The protein was transferred to PVDF and fixed on the PVDF membrane by drying overnight at room temperature. The membrane was then rinsed in methanol to re-wet and destain, then water before immunoblotting with the m/hSOD or hSOD antibodies.
Split luciferase assays
Fusions of SOD1cDNA to cDNA for humanized gLuc1 and gLuc2 (46) were created by PCR-based strategies. CHO cells were transfected with each SOD-gLuc1 or -gLuc2 plasmid individually using Lipofectamine 2000 according to the manufacturer's protocol and allowed to incubate for 24 h. Cells were rinsed with 1× PBS and harvested by pipetting in 1 ml of 1× PBS. Cells were pelleted at 5000g for 2 min and frozen at −80°C overnight. Cells were resuspended in 1× PBS with protease inhibitor cocktail (Sigma #P8340), and lysed by repeated freeze–thaw cycling in a dry ice/ethanol bath and a 42°C water bath. The cell lysate was centrifuged at 5000g for 2 min and an aliquot of the supernatant was boiled in Laemmli buffer and run on an 18% Tris–glycine gel. The proteins were transferred to nitrocellulose before western blotting with the wSOD1 antibody. The resulting image was captured with a FluorChem E imager (Protein Simple, Santa Clara, CA, USA), and quantified using Alpha View SA software provided with the imager. The volumes of lysates used in the reaction were normalized such that an equal amount of each SOD-split luciferase protein was used in subsequent ex vivo reactions.
The total amount of lysate needed to produce a signal in the linear range of a Biotek Synergy HT plate reader was determined empirically. Each lysate was diluted in 1× PBS separately and supplemented with BSA to 0.1%. Mixtures of the two lysates were incubated at 37°C for 1 h. Luciferase activity was measured by injecting 50 µl native coelenterazine (Nanolight Technologies, cat# 303-1) and reading in a Biotek plate reader.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by a grant from the National Institutes of Neurological Disease and Stroke (P01 NS049134—Program Project).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr Pamela McLean (Department of Neuroscience, Mayo Clinic, Jacksonville) for providing the humanized Gaussia split luciferase constructs. We thank Dr Janice Robertson for providing the SEDI anti-SOD1 antibody. We are grateful for helpful advice from Drs David Eisenberg, Julian Whitelegge and Joan S. Valentine.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J.P., Deng H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 2.Borchelt D.R., Guarnieri M., Wong P.C., Lee M.K., Slunt H.S., Xu Z.S., Sisodia S.S., Price D.L., Clevel D.W. Superoxide dismutase 1 subunits with mutations linked to familial amyotrophic lateral sclerosis do not affect wild-type subunit function. J. Biol. Chem. 1995;270:3234–3238. doi: 10.1074/jbc.270.7.3234. [DOI] [PubMed] [Google Scholar]
- 3.Fridovich I. Superoxide dismutases. Adv. Enzymol. Relat. Areas Mol. Biol. 1974;41:35–97. doi: 10.1002/9780470122860.ch2. [DOI] [PubMed] [Google Scholar]
- 4.Williamson T.L., Corson L.B., Huang L., Burlingame A., Liu J., Bruijn L.I., Cleveland D.W. Toxicity of ALS-linked SOD1 mutants. Science. 2000;288:399a–399a. doi: 10.1126/science.288.5465.399a. [DOI] [PubMed] [Google Scholar]
- 5.Bruijn L.I., Miller T.M., Clevel D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 6.Prudencio M., Borchelt D.R. Superoxide dismutase 1 encoding mutations linked to ALS adopts a spectrum of misfolded states. Mol. Neurodegener. 2011;6:77. doi: 10.1186/1750-1326-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prudencio M., Hart P.J., Borchelt D.R., Andersen P.M. Variation in aggregation propensities among ALS-associated variants of SOD1: Correlation to human disease. Hum. Mol. Genet. 2009;18:3217–3226. doi: 10.1093/hmg/ddp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruijn L.I., Houseweart M.K., Kato S., Anderson K.L., Anderson S.D., Ohama E., Reaume A.G., Scott R.W., Clevel D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 9.Deng H.X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G.H., Khan M.S., Hung W.Y., Bigio E.H., et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. USA. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prudencio M., Durazo A., Whitelegge J.P., Borchelt D.R. An examination of wild-type SOD1 in modulating the toxicity and aggregation of ALS-associated mutant SOD1. Hum. Mol. Genet. 2010;19:4774–4789. doi: 10.1093/hmg/ddq408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaarsma D., Teuling E., Haasdijk E.D., De Zeeuw C.I., Hoogenraad C.C. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. 2008;28:2075–2088. doi: 10.1523/JNEUROSCI.5258-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng H.X., Jiang H., Fu R., Zhai H., Shi Y., Liu E., Hirano M., Dal Canto M.C., Siddique T. Molecular dissection of ALS-associated toxicity of SOD1 in transgenic mice using an exon-fusion approach. Hum. Mol. Genet. 2008;17:2310–2319. doi: 10.1093/hmg/ddn131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L., Deng H.X., Grisotti G., Zhai H., Siddique T., Roos R.P. Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum. Mol. Genet. 2009;18:1642–1651. doi: 10.1093/hmg/ddp085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurney M.E., Pu H., Chiu A.Y., Dal Canto M.C., Polchow C.Y., Alexander D.D., Caliendo J., Hentati A., Kwon Y.W., Deng H.X. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 15.Wang J., Xu G., Slunt H.H., Gonzales V., Coonfield M., Fromholt D., Copeland N.G., Jenkins N.A., Borchelt D.R. Coincident thresholds of mutant protein for paralytic disease and protein aggregation caused by restrictively expressed superoxide dismutase cDNA. Neurobiol. Dis. 2005;20:943–952. doi: 10.1016/j.nbd.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Wong P.C., Pardo C.A., Borchelt D.R., Lee M.K., Copeland N.G., Jenkins N.A., Sisodia S.S., Cleveland D.W., Price D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 17.Maquat L.E. When cells stop making sense: Effects of nonsense codons on RNA metabolism in vertebrate cells. RNA. 1995;1:453–465. [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J., Xu G., Li H., Gonzales V., Fromholt D., Karch C., Copeland N.G., Jenkins N.A., Borchelt D.R. Somatodendritic accumulation of misfolded SOD1-L126Z in motor neurons mediates degeneration: {Alpha}B-crystallin modulates aggregation. Hum. Mol. Genet. 2005;14:2335–2347. doi: 10.1093/hmg/ddi236. [DOI] [PubMed] [Google Scholar]
- 19.Jonsson P.A., Ernhill K., Andersen P.M., Bergemalm D., Brannstrom T., Gredal O., Nilsson P., Marklund S.L. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127:73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- 20.Wang J., Xu G., Borchelt D.R. Mapping superoxide dismutase 1 domains of non-native interaction: roles of intra- and intermolecular disulfide bonding in aggregation. J. Neurochem. 2006;96:1277–1288. doi: 10.1111/j.1471-4159.2005.03642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J., Xu G., Gonzales V., Coonfield M., Fromholt D., Copeland N.G., Jenkins N.A., Borchelt D.R. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol. Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- 22.Ratovitski T., Corson L.B., Strain J., Wong P., Cleveland D.W., Culotta V.C., Borchelt D.R. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum. Mol. Genet. 1999;8:1451–1460. doi: 10.1093/hmg/8.8.1451. [DOI] [PubMed] [Google Scholar]
- 23.Urushitani M., Ezzi S.A., Julien J.P. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 2007;104:2495–2500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ayers J.I., Xu G., Pletnikova O., Troncoso J.C., Hart P.J., Borchelt D.R. Conformatonal specificity of the C4F6 SOD1 antibody; low frequency of reactivity in sporadic ALS cases. Acta Neuropathol. Commun. 2014;2:55. doi: 10.1186/2051-5960-2-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rakhit R., Robertson J., Vande V.C., Horne P., Ruth D.M., Griffin J., Cleveland D.W., Cashman N.R., Chakrabartty A. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat. Med. 2007;13:754–759. doi: 10.1038/nm1559. [DOI] [PubMed] [Google Scholar]
- 26.Rotunno M.S., Auclair J.R., Maniatis S., Shaffer S.A., Agar J., Bosco D.A. Identification of a misfolded region in superoxide dismutase 1 that is exposed in amyotrophic lateral sclerosis. J. Biol. Chem. 2014;289:28527–28538. doi: 10.1074/jbc.M114.581801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruijn L.I., Becher M.W., Lee M.K., Anderson K.L., Jenkins N.A., Copeland N.G., Sisodia S.S., Rothstein J.D., Borchelt D.R., Price D.L., et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 28.Wang J., Farr G.W., Zeiss C.J., Rodriguez-Gil D.J., Wilson J.H., Furtak K., Rutkowski D.T., Kaufman R.J., Ruse C.I., Yates J.R., III, et al. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc. Natl. Acad. Sci. USA. 2009;106:1392–1397. doi: 10.1073/pnas.0813045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S.B., Sato M., Tao H. Split gaussia luciferase-based bioluminescence template for tracing protein dynamics in living cells. Anal. Chem. 2009;81:67–74. doi: 10.1021/ac801658y. [DOI] [PubMed] [Google Scholar]
- 30.Bertini I., Piccioli M., Viezzoli M.S., Chiu C.Y., Mullenbach G.T. A spectroscopic characterization of a monomeric analog of copper, zinc superoxide dismutase. Eur. Biophys. J. 1994;23:167–176. doi: 10.1007/BF01007608. [DOI] [PubMed] [Google Scholar]
- 31.Qualls D.A., Crosby K., Brown H., Borchelt D.R. An analysis of interactions between fluorescently-tagged mutant and wild-type SOD1 in intracellular inclusions. PLoS ONE. 2013;8:e83981. doi: 10.1371/journal.pone.0083981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Driegen S., Ferreira R., van Zon A., Strouboulis J., Jaegle M., Grosveld F., Philipsen S., Meijer D. A generic tool for biotinylation of tagged proteins in transgenic mice. Transgenic Res. 2005;14:477–482. doi: 10.1007/s11248-005-7220-2. [DOI] [PubMed] [Google Scholar]
- 33.Weichert A., Besemer A.S., Liebl M., Hellmann N., Koziollek-Drechsler I., Ip P., Decker H., Robertson J., Chakrabartty A., Behl C., et al. Wild-type cu/zn superoxide dismutase 1 stabilizes mutant variants by heterodimerization. Neurobiol. Dis. 2014;62:479–488. doi: 10.1016/j.nbd.2013.10.027. [DOI] [PubMed] [Google Scholar]
- 34.Zetterstrom P., Graffmo K.S., Andersen P.M., Brannstrom T., Marklund S.L. Composition of soluble misfolded superoxide dismutase-1 in murine models of amyotrophic lateral sclerosis. Neuromolecular Med. 2013;15:147–158. doi: 10.1007/s12017-012-8204-z. [DOI] [PubMed] [Google Scholar]
- 35.Schagger H., von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 36.Borchelt D.R., Lee M.K., Slunt H.S., Guarnieri M., Xu Z.S., Wong P.C., Brown R.H., Jr, Price D.L., Sisodia S.S., Cleveland D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grad L.I., Yerbury J.J., Turner B.J., Guest W.C., Pokrishevsky E., O'Neill M.A., Yanai A., Silverman J.M., Zeineddine R., Corcoran L., et al. Intercellular propagated misfolding of wild-type cu/zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA. 2014;111:3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prudencio M., Lelie H., Brown H.H., Whitelegge J.P., Valentine J.S., Borchelt D.R. A novel variant of human SOD1 harboring ALS-associated and experimental mutations in metal-binding residues and free cysteines lacks toxicity in vivo. J. Neurochem. 2012;121:305–314. doi: 10.1111/j.1471-4159.2012.07690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karch C.M., Prudencio M., Winkler D.D., Hart P.J., Borchelt D.R. Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. USA. 2009;106:7774–7779. doi: 10.1073/pnas.0902505106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prudencio M., Durazo A., Whitelegge J.P., Borchelt D.R. Modulation of mutant superoxide dismutase 1 aggregation by co-expression of wild-type enzyme. J. Neurochem. 2009;108:1009–1018. doi: 10.1111/j.1471-4159.2008.05839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graffmo K.S., Forsberg K., Bergh J., Birve A., Zetterstrom P., Andersen P.M., Marklund S.L., Brannstrom T. Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013;22:51–60. doi: 10.1093/hmg/dds399. [DOI] [PubMed] [Google Scholar]
- 42.Bosco D.A., Morfini G., Karabacak N.M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B.A., Lemay N., McKenna-Yasek D., et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brotherton T.E., Li Y., Cooper D., Gearing M., Julien J.P., Rothstein J.D., Boylan K., Glass J.D. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc. Natl. Acad. Sci. USA. 2012;109:5505–5510. doi: 10.1073/pnas.1115009109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forsberg K., Jonsson P.A., Andersen P.M., Bergemalm D., Graffmo K.S., Hultdin M., Jacobsson J., Rosquist R., Marklund S.L., Brannstrom T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE. 2010;5:e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown H.H., Borchelt D.R. Analysis of mutant SOD1 electrophoretic mobility by blue native gel electrophoresis; evidence for soluble multimeric assemblies. PLoS ONE. 2014;9:e104583. doi: 10.1371/journal.pone.0104583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Danzer K.M., Kranich L.R., Ruf W.P., Cagsal-Getkin O., Winslow A.R., Zhu L., Vanderburg C.R., McLean P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.