

Abstract
Molecular motors such as kinesin and dynein use the energy derived from ATP hydrolysis to walk processively along microtubule tracks and transport various cargoes inside the cell. Recent advancements in fluorescent protein (FP) research enable motors to be fluorescently labeled such that single molecules can be visualized inside cells in multiple colors. The performance of these fluorescent tags can vary depending on their spectral properties and a natural tendency for oligomerization. Here we present a survey of different fluorescent tags fused to kinesin-1 and studied by single-molecule motility assays of mammalian cell lysates. We tested eight different FP tags and found that seven of them display sufficient fluorescence intensity and photostability to visualize motility events. Although none of the FP tags interfere with the enzymatic properties of the motor, four of the tags (EGFP, monomeric EGFP, tagRFPt, and mApple) cause aberrantly long motor run lengths. This behavior is unlikely to be due to electrostatic interactions and is probably caused by tag-dependent oligomerization events that appear to be facilitated by fusion to the dimeric kinesin-1. We also compared the single-molecule performance of various fluorescent SNAP and HALO ligands. We found that although both green and red SNAP ligands provide sufficient fluorescent signal, only the tetramethyl rhodamine (TMR) HALO ligand provides sufficient signal for detection in these assays. This study will serve as a valuable reference for choosing fluorescent labels for single-molecule motility assays.
Introduction
Cytoskeletal molecular motors are enzymes that catalyze the hydrolysis of ATP, converting the released energy into mechanical work inside the cell (1). Some kinesin, dynein, and myosin motors are dimeric, processive motors that transport specific cargoes along cytoskeletal tracks. Kinesin-1, for instance, is a dimeric motor that walks hand-over-hand in 8 nm steps along microtubules (2). After the discovery of kinesin-1 in the 1980s (3,4), investigators studied the motility properties of this protein by attaching purified motors to large polystyrene beads that simulated cellular cargoes (5). The subsequent development of total internal reflection fluorescence (TIRF) microscopy allowed scientists to visualize the motility of single kinesin motors labeled by small organic fluorophores such as Cy3 and Cy5 (6). The identification and optimization of fluorescent proteins (FPs) (7,8) provided a powerful technique for genetically labeling proteins, and allowed the single-molecule properties of kinesin motors in cells to be directly compared with their properties in vitro (9–11). Thus, there is a growing demand for bright FPs of various output colors that are applicable for single-molecule studies both in cells and in vitro.
A large number of FPs are now available that are derived from a variety of different organisms and display variable spectral properties and biostability (8). Most FPs consist of either a green FP (GFP)-like fold (typically green emission) or DsRed-like fold (typically red emission) structure consisting of an interior tripeptide chromophore that is protected by an exterior β-barrel, where the fluorescent properties are defined by the chromophore structure and local environment of the barrel interior (8). Although direct comparisons of the spectral properties of various FPs have been made, these properties can vary widely depending on the excitation method (e.g., arc lamp versus laser excitation) and experimental environment (12). Additionally, alternative methods for fluorescently labeling proteins have been developed, such as SNAP (13) and HALO (14) tags, which are engineered enzymes that link covalently to small fluorescent ligands. However, relatively few studies have compared and tested FPs and enzyme-based fluorophores under the same experimental conditions, especially for single-molecule applications.
Here, we tested 10 different fluorescent tags for single-molecule imaging of a truncated dimeric form of kinesin-1. We first confirmed the fluorescence intensities and photostability in single-molecule motility assays, and then compared the motility properties of each fusion protein as obtained by kymograph analysis. We found that the fluorescent tags do not affect the velocity of the motor, but several FPs (EGFP, mEGFP, tagRFPt, and mApple) lead to aberrantly long run lengths. We determined that this effect is not due to electrostatic interactions, but rather reflects the tendency of these FPs to oligomerize. In addition, we found that the utility of enzyme-based tags such as SNAP and HALO depends on the fluorescent ligand, and these tags do not outperform the FPs. Overall, this study provides a valuable survey of fluorophores for single-molecule, TIRF-based imaging.
Materials and Methods
Plasmids
A truncated, constitutively active version of the kinesin-1 motor rat KIF5C (aa 1-560; KHC(1-560) (9,10)) was used in this work. Different fluorescent tags were genetically fused to the C-terminus via restriction digestion and ligation. Most constructs were generated using AgeI and BsrGI enzymes: KHC(1-560)-HALO was generated using AgeI and MfeI, and KHC(1-560)-mNeGr was generated using Acc65I and BsrGI. Short linker sequences situated between KHC(1-560) and the tag protein were from a multiple cloning site: LVPGGGGGGGGGPVAT for EGFP-, mEGFP-, 2xmCh-, mApple-, and tagRFPt-tagged motors; LVPRARDPPVAT for SNAP-, HALO-, tdTom-, and mCit-tagged motors; and LVPRARDPLE for the mNeGr-tagged motor. Plasmids encoding the HALO tag, SNAP tag, and mNeonGreen were purchased from Promega (Madison, WI), New England Biolabs (Ipswich, MA), and Allele Biotechnology (San Diego, CA) respectively. mApple was obtained from Addgene (#54567; Cambridge, MA) and the plasmid was provided by the laboratory of M. Davidson (Florida State University) (15). A tandem dimer of mCherry was synthesized by DNA 2.0 (Menlo Park, CA). Plasmids encoding tagRFPt and tdTomato were gifts from D. Cai (University of Michigan). A plasmid encoding EGFP was a gift from J. Swanson (University of Michigan). Monomeric GFP (A206K) was generated by QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) of EGFP. All plasmids were verified by DNA sequencing.
Cell culture, transfection, lysis, and normalization of motor concentration
COS7 cells were cultured, transfected, and lysed as described previously (9,16). Briefly, COS7 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) + 10% (vol/vol), Fetal Clone III (Thermo Fisher, Waltham, MA), and 2 mM L-glutamine at 37°C with 5% (vol/vol) CO2. Cells were transfected with 1 μg of plasmid DNA using Expressfect (Denville Scientific, Metuchen, NJ). After overnight expression (16 h), the cells were trypsinized and harvested by low-speed centrifugation at 1,500 × g at 4°C. The pellet was washed once in DMEM and resuspended in 25 μL lysis buffer (25 mM HEPES/KOH, 115 mM potassium acetate, 5 mM sodium acetate, 5 mM MgCl2, 0.5 mM EGTA, and 1% Triton X-100, pH 7.4) freshly supplemented with 1 mM ATP, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitors (10 μg/mL leupeptin, 5 μg/mL chymostatin, 3 μg/mL elastatinal, and 1 mg/mL pepstatin). After the lysate was clarified by centrifugation at 16,000 × g at 4°C, aliquots were snap-frozen in liquid nitrogen and stored at −80°C. The amount of motor in the COS7 lysates was normalized across constructs by a dot-blot in which increasing volumes of COS7 lysates were spotted onto a nitrocellulose membrane that was air-dried and immunoblotted with a monoclonal antibody to kinesin-1 (MAb1614; Millipore, Billerica, MA). The spots within the linear regime were quantified to normalize the motor concentration across lysates.
SNAP and HALO ligand labeling
COS7 cells expressing KHC(1-560)-SNAP or KHC(1-560)-HALO were labeled with cell-permeable SNAP or HALO ligands before lysis. The indicated ligand (SNAP-Cell Oregon Green, NEB #S9104S; SNAP-Cell 505-STAR, NEB #S9103S; SNAP-Cell TMR-STAR, NEB #S9105S; HALOTag Oregon Green ligand, Promega #G2801; HALOTag diAcFAM ligand, Promega #G8272; or HALOTag TMR ligand, Promega #G8251) was added to 1 mL of DMEM in a six-well, 35-mm plate per manufacturer’s suggestion (5 μM for SNAP-Cell Oregon Green, 5 μM for SNAP-Cell 505-STAR, 3 μM for SNAP-Cell TMR-STAR, 1 μM for HALOTag Oregon Green, 1 μM for HALOTag diAcFAM ligand, and 5 μM for HALOTag TMR ligand). After a 30 min incubation, the cells were washed three times in DMEM and incubated in DMEM for 30 min before lysis to remove unbound ligand.
Single-molecule motility assays
All assays were performed as described previously (16) at room temperature in a narrow flow-cell (∼10 μL volume) prepared by attaching a clean #1.5 coverslip to a glass slide with double-sided tape. HiLyte-647-labeled microtubules were polymerized from purified tubulin (Cytoskeleton, Denver, CO) in BRB80 buffer (80 mM PIPES/KOH, 1 mM EGTA, and 1 mM MgCl2, pH 6.8) supplemented with 1 mM GTP at 37°C for 15 min. Polymerized microtubules were stored at room temperature after addition of five volumes of prewarmed BRB80 containing 20 μM taxol and an additional 15 min incubation at 37°C. Polymerized microtubules were diluted in P12 buffer (12 mM PIPES/KOH, 1 mM EGTA, and 2 mM MgCl2, pH 6.8) containing 20 μM taxol and then infused into a flow-cell and incubated for 5 min at room temperature to adsorb onto the coverslip. Subsequently, 50 μL of blocking buffer (10 mg/mL bovine serum albumin (BSA) in P12 buffer with 10 μM taxol) was introduced and incubated for 10 min to prevent nonspecific binding of kinesin motors onto the coverslip surface. Finally, lysates containing equal amounts of motor proteins (typically 0.1–1.0 μL) were added to the flow chambers in a motility mixture in either P12 buffer or physiological-salt buffer (25 mM HEPES/KOH, 115 mM potassium acetate, 5 mM sodium acetate, 5 mM MgCl2, and 0.5 mM EGTA, pH 7.4). The ionic strength (IS) of each buffer was calculated based on the molar concentration of each ion and its counterion based on BioMol.net (http://www.biomol.net). Each motility mixture also contained 2 mM ATP, 10 mg/mL BSA, 10 μM taxol, and oxygen-scavenging components to reduce photobleaching (1 mM dithiothreitol, 1 mM MgCl2, 10 mM glucose, 0.1 mg/mL glucose oxidase, and 0.08 mg/mL catalase). The motility data for each construct were obtained from at least two independent protein preparations.
Image acquisition
Images for single-molecule motility assays were acquired using a Nikon TiE/B microscope with a 100× 1.49 NA oil immersion TIRF objective (Nikon, Melville, NY) equipped with three 20 mW diode lasers (488 nm, 561 nm, and 640 nm) combined into a single fiber and controlled via AOTF (Agilent, Santa Clara, CA). Images were collected via an EMCCD detector (iXon X3 DU897, 512 × 512, 16 μm array; Andor, Belfast, UK). For imaging in green and/or red, the microscope used a dual-band laser polychroic mirror (ZT488/561rpc; Chroma Technology, Rockingham, VT), a dual-band sputtered emission filter (ZET488/561m; Chroma), and a dual-band sputtered cleanup filter (ZET488/561×; Chroma), and either a 488 nm (2 mW power, used for EGFP, mEGFP, mNeGr, mCit, SNAP-OreGr488, SNAP-505-STAR, HALO-OreGr488, and HALO-diAcFAM) or 561 nm (4 mW power, used for tdTom, tagRFPt, mApple, 2xmCh, SNAP-TMR, and HALO-TMR) laser was used for TIR-based illumination. Images were acquired continuously with 100 ms exposures, and image acquisition was controlled by Nikon Elements software.
Kymograph analysis
To avoid complications arising from variation in the signal/noise ratio or point spread function for different fluorescent tags, motility events were analyzed by kymograph analysis. Maximum intensity projections were generated to determine the location of microtubules, and kymographs were generated (width = 5 pixels) along these tracks using Elements (Nikon). Only constant velocity events of at least five frames (500 ms) were considered, and stalled events were ignored. Kymographs were generated with distance on the y axis and time on the x axis. Run length was defined as the vertical component of the kymograph, which is the distance traveled along the microtubule, in micrometers. Dwell time was defined as the horizontal component of the kymograph, which is the time spent in a constant velocity segment, in seconds. Velocity was defined as the run length divided by dwell time, in micrometers per second. The significance of the run length and velocity differences between motor populations was determined by Mann-Whitney-Wilcoxon tests in MATLAB (The MathWorks, Natick, MA) (see Tables S2–S5 in the Supporting Material).
Cumulative distribution functions
Cumulative distribution functions (CDFs) of velocities and run lengths were generated via MATLAB and mean values were obtained as described previously (17). CDFs were used for statistical analysis because they are continuous and do not introduce subjective binning. Using MATLAB, run-length CDFs above a minimum t0 = 0.5 μm were fit to the hypothetical CDF for an exponential distribution using a nonlinear least-squares fit with the free parameter t.
The mean run length was then determined by adding the minimum run length t0 to t. Errors were estimated by the bootstrap technique (17). Each distribution was resampled 200 times and fit to the appropriate equation. The standard deviation of the fitted parameter over the resampled data sets was taken as the error for each fitted quantity.
Velocity CDFs were fit to the hypothetical CDF for a normal distribution using a nonlinear least-squares fit with the free parameters μ (mean) and σ (standard deviation).
Photobleaching assays
Quantitative photobleaching assays were performed as described previously (16). Small volumes of COS7 lysates (∼1:20 dilution from motility conditions) containing equal amounts of FP-tagged motors were diluted in blocking solution (15 mg/mL BSA in P12 buffer) and flowed into an empty chamber. The motors were allowed to incubate for 2 min to nonspecifically adsorb to the glass surface. Then, 50 μL of blocking solution was introduced to remove nonadsorbed motors. The surface-bound motors were imaged in TIRF with increased laser power (10 mW for 488 nm; 6 mW for 561 nm). The fluorescence intensity profile of ∼200 motors from two independent protein preparations for each construct was then plotted as a function of time, and the number of discrete photobleaching steps was counted and displayed as a histogram for the population.
Calculation of landing rates
The landing rate [Events/(μm × s × nM)] for each construct was determined from the single-molecule motility data by counting the number of motility events and dividing by observation time, microtubule length, and motor concentration as described previously (18,19). Time was defined as the recording time (in seconds) and microtubule length was determined by measuring the length of the HiLyte-647-labeled microtubule in the 640 nm channel (in micrometers). Motor concentration (in nanomolars) was determined by comparing the amount of KHC in each lysate with a known amount of purified KHC-FLAG via quantitative western blotting with a monoclonal antibody to kinesin-1 (MAb1614; Millipore, Billerica, MA). The number of motility events was determined for least four different microtubules for each construct from two independent experiments.
Estimating the fraction of run lengths above a certain threshold
The probability distribution function (PDF) for a run-length distribution is calculated as follows, where the mean of the distribution is defined as .
The probability of x falling between 3 μm and ∞ is the definite integral of this function, where λ = 1 to reflect a mean run length of 1 μm:
Results
The fluorescent tag can influence the kinesin-1 run length in single-molecule motility assays
To determine whether different fluorescent tags can affect kinesin motility properties, we fused a variety of fluorescent tags to the C-terminus of a constitutively active version of the kinesin-1 motor KIF5C(1-560) (Fig. 1 A). We tested 10 fluorescent markers: four green FPs, four red FPs, and two enzyme-based tags that can be labeled with cell-permeable small organic fluorophores (Table 1). These fluorophores represent a wide range of potential tags in the green-red spectrum, were derived from different organisms, and are well-characterized in terms of oligomeric state (20,21).
Figure 1.
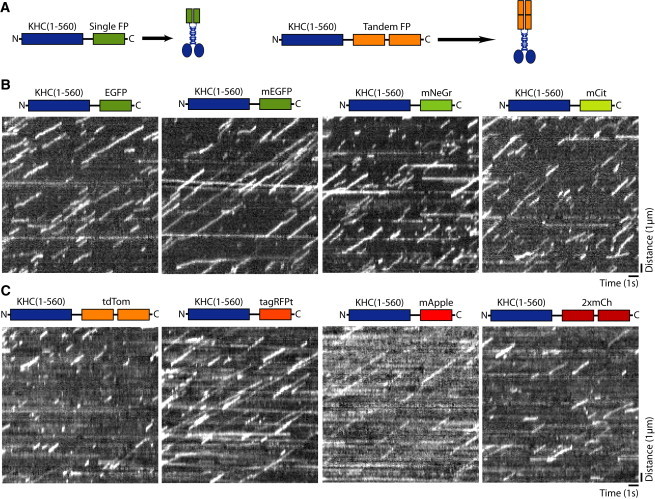
A survey of FPs for labeling kinesin-1 in single-molecule motility assays. (A) Schematic. A dimeric, constitutively active kinesin-1 motor (KHC(1-560), blue) was tagged at the C-terminus with single or tandem FPs. (B and C) Lysates of COS7 cells expressing the indicated KHC(1-560)-FP motors were analyzed in single-molecule motility assays via TIRF microscopy. Representative kymographs were generated from the movies for KHC(1-560) tagged with the indicated (B) green FPs or (C) red FPs. Time is on the x axis (scale bar, 1 s) and distance is on the y axis (scale bar, 1 μm). To see this figure in color, go online.
Table 1.
Properties of the fluorescent tags used in this study
| Fluorescent protein | Excitationa (nm) | Emissionb (nm) | Brightnessc | Photostabilityd | Oligomeric state | Source organism (original protein) | Reference |
|---|---|---|---|---|---|---|---|
| Enhanced GFP (EGFP) | 488 | 507 | 34 | 174 | weak dimer | A. victoria (GFP) | (35,36) |
| Monomeric EGFP (mEGFP) | 488 | 507 | 34 | 150 | monomer | A. victoria (GFP) | (23) |
| Monomeric NeonGreen (mNeGr) | 506 | 517 | 94 | 158 | monomer | B. lanceolatum (LanYFP) | (26) |
| Monomeric Citrine (mCit) | 516 | 529 | 59 | 49 | monomer | A. victoria (GFP) | (23,30) |
| Tandem Tomato (tdTom) | 554 | 581 | 95 | 98 | tandem dimer | Discosoma sp.(DsRed) | (37) |
| Tag RFP-t (tagRFPt) | 555 | 584 | 33 | 337 | monomer (12) or weak dimer (29) | E. quadricolor (eqFP578) | (12,38) |
| Monomeric Apple (mApple) | 568 | 592 | 37 | 4.8e | monomer | Discosoma sp.(DsRed) | (12) |
| Tandem monomeric Cherry (2xmCh) | 587 | 610 | 32f | unknown | tandem dimer | Discosoma sp.(DsRed) | (37) |
| SNAP tag | ligand dependent | monomer | H. sapiens (O6-alkylguanine-DNA alkyltransferase) | (13) | |||
| HALO tag | ligand dependent | monomer | Rhodococcus sp. (haloalkane dehalogenase) | (14) |
Major excitation peak.
Major emission peak.
Product of extinction coefficient and quantum yield as reported in reference cited for each fluorophore, in (mM × cm)−1. Brightness values originally summarized in Shaner et al. (21).
Defined as the time for bleaching from an initial emission rate of 1,000 photons/s down to 500 photons/s under arc lamp illumination.
mApple shows much higher photostability under confocal illumination; see Shaner et al. (12).
Defined as twice the reported value of single mCherry in Shaner et al. (12).
To examine the motility properties of each fluorophore-tagged KHC(1-560), we transfected COS7 cells with the construct of interest, harvested cell lysates, and performed single-molecule motility assays using TIRF microscopy (16) (Movies S1, S2, S3, and S4). The motility of each construct can be analyzed in a kymograph where run distance is displayed vertically and time is displayed horizontally (Fig. 1, B and C). An important parameter for imaging at the single-molecule level and determining motility properties via kymographs or automated tracking software is the brightness of the fluorophore. In our hands, mApple was barely detectable above the background fluorescence, mCitrine and tagRFPt provided relatively weak fluorescent signals, and EGFP, mEGFP, mNeGr, tdTom, and 2xmCh provided significantly brighter signals (Fig. 1, B and C; Movies S1 and S2). For all of the FP-tagged motors tested, the majority of the processive events terminated abruptly rather than being preceded by a loss in signal, indicating that the rate of photobleaching was significantly slower than the typical off-rate of a motile event (Fig. 1, B and C).
For the enzyme-based tags, KHC(1-560) was labeled with either SNAP or HALO proteins and the indicated fluorescent ligand was added before cell lysis (Fig. 2 A; Movies S3 and S4). For the SNAP tag, the TMR ligand provided the strongest signal, whereas the green fluorescent ligands (OreGr-488 and 505-STAR) were significantly weaker (Fig. 2 B). For the HALO tag, the TMR ligand provided sufficient signal for tracking at the single-molecule level, whereas the OreGr488 signal was much weaker and no labeling was detected for the diAcFAM ligand (Fig. 2 C). Similar to what was observed for FP-tagged motors (Fig. 1, B and C), SNAP-505-STAR, SNAP-TMR, and HALO-TMR showed minimal photobleaching under our experimental conditions (Fig. 2, B and C).
Figure 2.

Survey of enzyme tags and fluorescent ligands for labeling kinesin-1 in single-molecule motility assays. (A) Schematic. A dimeric, constitutively active kinesin-1 motor (KHC(1-560), blue) was tagged at the C-terminus with an enzyme (SNAP or HALO) tag that covalently links to a fluorescent dye. (B and C) Enzyme-tagged KHC(1-560) motors expressed in COS7 cells were labeled with the indicated dyes before cell lysis and analysis by TIRF microscopy. (B and C) Representative kymographs were generated from the movies of (B) KHC(1-560)-SNAP and (C) KHC(1-560)-HALO motors in cell lysates. Time is on the x axis (scale bar, 1 s) and distance is on the y axis (scale bar, 1 μm). To see this figure in color, go online.
For each fluorescently tagged KHC(1-560) construct, the run lengths and velocities of individual motility events were determined from the kymographs and plotted as a histogram for the population (Figs. 3, A–C, and S3). The mean run lengths and velocities were then determined by fitting each distribution to its corresponding cumulative distribution function (CDF) (Figs. S1 and S2). The mean velocities were similar across the different fluorescently tagged KHC(1-560) constructs (Figs. 3 E and S3; Table S1), although the slightly decreased velocities of tagRFPt- and mApple-tagged motors were found to be statistically different (Table S2). In contrast, the mean run lengths were highly variable between the different fluorescently tagged constructs, ranging from 0.76 ± 0.02 μm for KHC(1-560)-tdTom to 1.81 ± 0.09 μm for KHC(1-560)-mApple (Figs. 3 D and S3; Table S1). These variations in run length were statistically significant (Table S3) and did not correlate with the photostability, published oligomeric state, or source organism of the fluorescent tag (Table 1), suggesting that these factors did not contribute to the observed FP-dependent run lengths.
Figure 3.
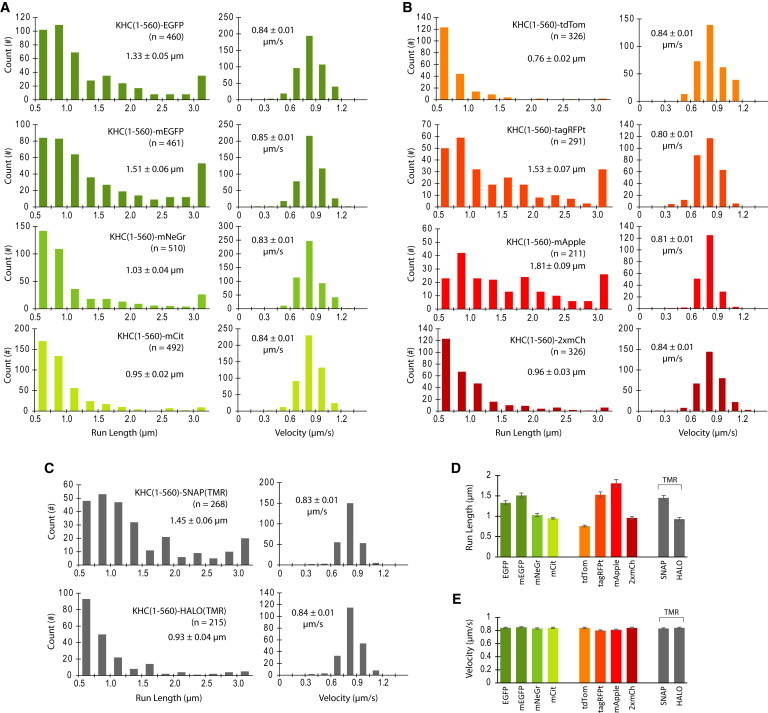
Motility properties of fluorescently tagged KHC(1-560) motors in P12 motility buffer. (A–C) The run lengths and velocities for individual KHC(1-560) motors tagged with (A) green FPs, (B) red FPs, or (C) enzyme tags were determined via kymograph analysis and then plotted as histograms for the population. The mean run length and velocity values (insets) were obtained by fit to the CDF (see Figs. S1 and S2). Error is reported as the standard deviation from bootstrapping. (D and E) Comparison of the mean (D) run length and (E) velocity values for each motor as determined by CDF fit. Data are presented as the mean ± SD from bootstrapping. To see this figure in color, go online.
FP-specific oligomerization influences the kinesin-1 run length independently of electrostatics
Analysis of the run-length histograms indicated that the kinesin-1 constructs with the longest mean run lengths displayed a significant fraction of events with a run length > 3 μm (rightmost bins in Fig. 3, A–C). For events characterized by an exponential run-length distribution with a mean of 1 μm, the probability of a motile event with a run length > 3 μm is <5% (see Materials and Methods). However, the percentage of motile events > 3 μm was 7.6% for KHC(1-560)-EGFP, 11.5% for KHC(1-560)-mEGFP, 11.0% for KHC(1-560)-tagRFPt, 12.3% for KHC(1-560)-mApple, and 7.5% for KHC(1-560)-SNAP-TMR. Thus, the increased number of run lengths > 3 μm appears to be the defining feature that distinguishes fluorophores that influence kinesin-1 from those that do not.
We speculated that differences in surface charge between FPs may influence the motor’s run length due to interactions of the FP with the negatively charged microtubule surface. That is, charge-based interactions between specific FPs and the microtubule could anchor the motor and increase the run length without affecting the velocity under no-load conditions (22). To investigate this possibility, we carried out single-molecule motility assays for each fluorescently tagged KHC(1-560) motor under physiological IS and pH conditions (IS = 145 mM, pH 7.4; Fig. 4). Similar to what was observed for motility in standard P12 motility buffer (IS = 28 mM, pH 6.8; Fig. 3), the fluorescent tag had minimal effects on the mean velocity values (Fig. 4 E; Table S4) but caused significant changes in the motor’s run length in a fluorophore-dependent manner (Fig. 4 D; Table S5). Importantly, KHC(1-560) motors tagged with EGFP, mEGFP, tagRFPt, mApple and SNAP-TMR still displayed an increased number of run lengths > 3 μm in the physiological buffer (rightmost bins in the run-length histograms in Fig. 4, A–C). These results indicate that the influence of the fluorescent tag on the motor’s run length is likely not due to electrostatic interactions between the fluorescent marker and the microtubule.
Figure 4.
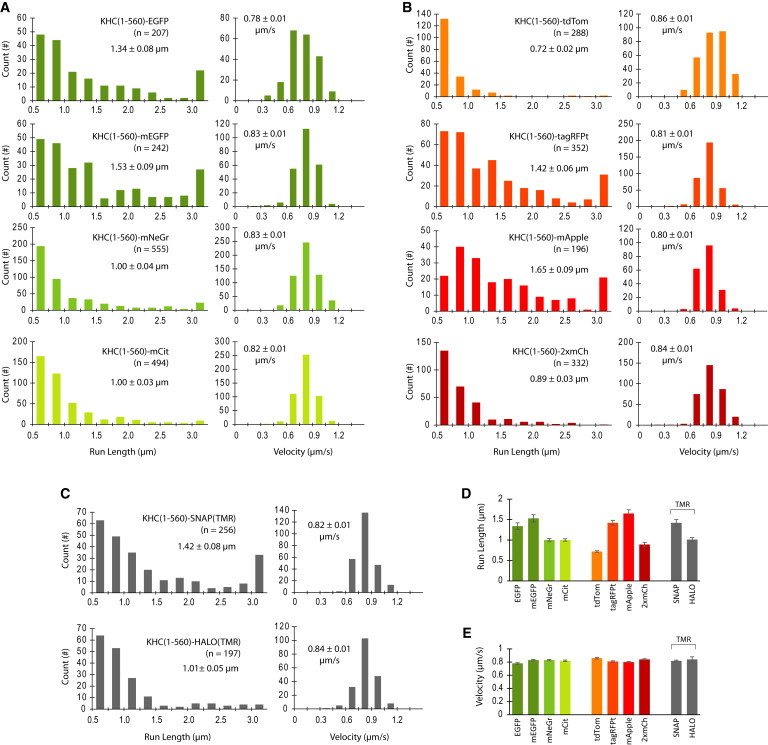
Motility properties of fluorescently tagged KHC(1-560) motors in physiological salt conditions. (A–C) The run lengths and velocities for individual KHC(1-560) motors tagged with (A) green FPs, (B) red FPs, or (C) enzyme tags were determined via kymograph analysis and then plotted as histograms for the population. The mean run length and velocity values (insets) were obtained by CDF fit (see Figs. S1 and S2). Error is reported as the standard deviation from bootstrapping. (D and E) Comparison of the mean (D) run length and (E) velocity values for each motor as determined by CDF fit. Data are presented as the mean ± SD from bootstrapping. To see this figure in color, go online.
We then speculated that oligomerization of the fluorescent tags might play a role in aberrant kinesin-1 run lengths. To examine this possibility, we compared the run-length values for KHC(1-560) motors tagged with EGFP, which is susceptible to weak dimerization, with the run-length values of motors tagged with mEGFP, whose A206K mutation abolishes FP dimerization (23). Surprisingly, fusion of mEGFP resulted in a higher percentage of KHC(1-560) motile events > 3 μm (11.0% for mEGFP versus 7.6% for EGFP; Fig. 3 A) and led to a statistically significant run-length increase under physiological buffer conditions (1.53 ± 0.09 μm for mEGFP versus 1.34 ± 0.08 μm for EGFP; Fig. 4 A; Table S5). These results suggest that simple dimerization of FPs is not the underlying cause of kinesin-1’s increased run length.
Since fusion of the FPs to dimeric proteins such as KHC(1-560) could cause proximity-induced interactions that were not observed in previous ensemble assays of FP dimerization (24), we directly measured the oligomeric state of seven of the fluorescently tagged motors using single-molecule photobleaching assays. We were unable to perform these assays on KHC(1-560)-mApple because the signal was too weak, or on the SNAP- and HALO-tagged motors due to complications from incomplete labeling. Importantly, the KHC(1-560)-FP constructs that showed aberrantly long run lengths (EGFP, mEGFP, and tagRFPt; Figs. 3 and 4) also showed a significant portion of molecules with more photobleaching steps than the expected value for a dimeric kinesin motor (Fig. 5). This was particularly striking for KHC(1-560)-mEGFP and KHC(1-560)-tagRFPt, where three or four photobleaching steps were frequently observed (Fig. 5), although just two steps are expected for a dimeric motor, suggesting that this oligomerization was tag dependent. Overall, the strong correlation between KHC(1-560)-FP oligomerization and run length suggests that a number of FPs can undergo unanticipated homo-interactions when brought into close proximity upon fusion to a dimeric protein.
Figure 5.
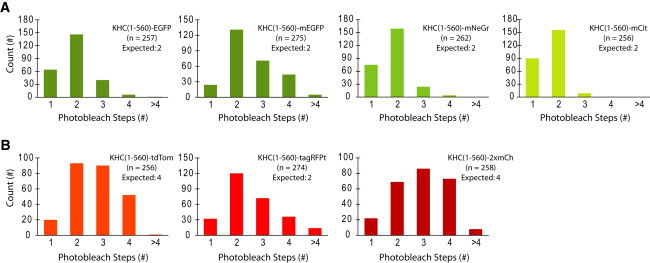
Quantification of oligomeric state by single-molecule photobleaching analysis. (A and B) The amount of (A) green FP-tagged or (B) red FP-tagged KHC(1-560) motors in cell lysates was normalized by western blot. Equal amounts of motor were immobilized on a glass surface and the fluorescence intensity over time was recorded with high-intensity laser excitation. The number of photobleaching steps was quantified for individual molecules and the population data were plotted as histograms. (Expected) denotes the expected number of photobleaching steps for a dimeric motor. To see this figure in color, go online.
FP tags can also influence the motor’s landing rate
While analyzing the motility properties of KHC(1-560) motors tagged with different fluorophores, we noticed that the number of motility events also seemed to vary between constructs. To quantify this, we normalized the amount of each tagged KHC(1-560) motor across constructs and quantified the landing rate of each motor as the number of motility events per unit time per unit microtubule length per nanomolar of motor. As reported previously (6), the landing rate depended only weakly on IS, with most motors showing a slightly decreased affinity for the microtubule under physiological salt conditions (Fig. 6). For the green FPs, mNeGr appeared to positively influence the landing rate of kinesin-1, as more motility events were observed for KHC(1-560)-mNeGr than for other motors at the same concentration (Fig. 6). This construct also appeared stickier in our experiments, typically decorating the imaging surface more than other constructs (horizontal lines in Fig. 1 B and data not shown). For the red FPs, little difference in landing rate was observed between the constructs (Fig. 6), although fusion to mApple caused a decrease in kinesin-1 motility events, perhaps due to detection issues due to weaker signal (Fig. 1 C). Since equal amounts of FP-, SNAP-, and HALO-tagged motors were added to the assay, the landing rate provides an estimate of the labeling efficiency of each SNAP- and HALO-ligand. For both SNAP and HALO tags, the TMR ligand was more efficient at labeling (we estimate that up to 80% of motors were labeled with at least one TMR ligand) than any of the green dyes (Fig. 6). It is interesting to note that all of the FP-labeled motors in these assays displayed higher landing rates than that determined for a purified, bacterially-expressed KHC(1-555)-GFP motor (19), perhaps due to the higher activity of motors expressed in mammalian cells.
Figure 6.

Landing rates of fluorescently tagged KHC(1-560) motors. The amount of fluorescently tagged KHC(1-560) motors in cell lysates was normalized, equal amounts of motor were added to single-molecule motility assays, and the landing rate was quantified as the number of events per micrometer microtubule per second per nanomolar of KHC(1-560). Error is provided as the mean ± SE. At least four different microtubules from two different experiments were quantified for each construct. To see this figure in color, go online.
Discussion
In this study, we compared the performance of 10 different fluorescent tags (eight FPs, SNAP, and HALO) in single-molecule motility assays when fused to dimeric kinesin-1. We found that mNeGr and mCit provide the best green FP tags for imaging of kinesin-1 motors, whereas EGFP and mEGFP have a high tendency to oligomerize. Although mNeGr is preferable for its brightness with 488 nm excitation, this FP appeared to be stickier in our single-molecule assays. We found that, based on its well-behaved motility properties and low oligomerization tendency, mCherry provides the best red FP tag for imaging, and one can augment its low fluorescence intensity by generating a tandem dimer. Importantly, we found that a subset of the fluorophores (EGFP, mEGFP, tagRFPt, mApple, and SNAP) caused aberrant run lengths, presumably due to oligomerization. These results emphasize that FPs are not inert tags and can influence the behavior of their fusion partner. In most single-molecule studies of kinesin motors, one can ignore the aberrant run lengths generated by the FP partner by excluding the longest bin of motile events, as this leads to only relatively small reductions in run length after mean analysis (25). Aberrant run lengths due to FP oligomerization are more likely to influence the interpretation of motor behavior when run length is used as a measure of motor output.
Essentially all FPs exist in nature as either tight dimers or tetramers driven by interactions between their barrel exteriors, and thus most FP-fusion proteins have a tendency to oligomerize (21). For many experimental applications, these oligomeric tendencies can be alleviated by mutagenesis of the dimer interface, at least when examined using biochemical techniques such as size-exclusion chromatography (26) and analytical ultracentrifugation (23). Although most of the FPs used in this study (mEGFP, mNeGr, mCit, tagRFPt, and mApple) were previously found to be monomeric in solution, the FPs may be prone to oligomerization artifacts when a high local concentration is generated by fusion to oligomeric proteins (24). For example, tagRFP and EGFP were susceptible to oligomerization when fused to the cytoplasmic face of a resident endoplasmic reticulum membrane protein (27). Similarly, superfolder GFP, mCherry, and to a lesser extent tagRFPt formed aberrant foci when expressed as fusion proteins to oligomeric Clp protease in E. coli (28). Additionally, many red FPs have been found to cause artificial puncta formation when used to label secretory pathway components (29). This FP oligomerization likely depends on the location of the fluorescent tag. In the case of our KHC(1-560) motors, for instance, fusion of the FPs to the coiled-coil stalk of the truncated motor likely facilitates FP oligomerization, whereas placement of the FPs N-terminal of the motor domains or C-terminal of the tail domains may be less likely to cause aberrant interactions. These results emphasize the need to independently confirm the oligomeric state of each fusion protein, especially as FPs continue to evolve (8).
It is interesting to note that this tendency of FPs to oligomerize when fused to a multimeric protein does not seem to be conserved among FPs derived from the same source protein. We find, for example, that EGFP and mEGFP seem to be prone to oligomerization, but mCit does not, although only a handful of amino acids differ among these FPs. mCit differs from mEGFP by the amino acid changes L64F, T65G, V68L, Q69M, S72A, and T203Y, and all of these mutations are on the interior of the barrel. A previous study showed that GFPmut3, which shares the T65G and S72A mutations with mCit, is substantially less likely to form oligomeric artifacts in E. coli, suggesting that these residues on the interior of the barrel can unexpectedly influence oligomerization (28). Additionally, cysteine residues C48 and C70 of GFP variants have been shown to cause oligomerization via formation of disulphide bonds, and mutations in adjacent residues, such as Q69M, could inhibit this behavior (30,31). Together, these results suggest that small perturbations on either the surface or the interior of the FP can greatly influence these oligomeric tendencies, and a predictive trend is difficult to identify.
In this study, we also assessed the relative effectiveness of various cell-permeable SNAP and HALO ligands for single-molecule imaging of kinesin-1 motors. We found that for both SNAP and HALO tags, the TMR ligand consistently provided the brightest fluorescence signal in single-molecule motility assays (Fig. 2) and the highest efficiency of labeling as determined by the motor landing rate (Fig. 6). The HALO tag and TMR ligand were recently used to label dynactin subunits expressed in COS7 cells for single-molecule analysis of dynein motility (32). We also found that kinesin-1 motors tagged with SNAP-TMR and HALO-TMR showed different run lengths in single-molecule motility assays (Fig. 3), suggesting that, like the FPs, the SNAP tag can undergo oligomerization. Indeed, long run lengths were also observed for motors labeled with 505-STAR, whereas the dim fluorescence of the OreGr488 ligand led to short detectable runs. For the SNAP tag, one could improve the labeling efficiency by using proteins with improved kinetics, such as the SNAPf tag (33). A recent comprehensive analysis of various cell-impermeable SNAP ligands in single-molecule studies also showed high variability in the effectiveness of the dyes for single-molecule imaging (34). These results emphasize that fluorescent dyes need to be tested under specific experimental conditions.
Conclusions
Here, we compared 10 fluorescent tags used to label kinesin-1 motors in single-molecule motility assays and found that a subset of these tags (EGFP, mEGFP, tagRFPt, mApple, and SNAP-TMR) are susceptible to low-order oligomer formation, which can introduce aberrant behavior of the kinesin-1 motor. Thus, these proteins should be avoided when they need to be fused to an oligomeric partner or in an integral membrane fusion where the local concentration is very high. Additionally, we compared the fluorescent tags in terms of single-molecule imaging quality. Of the green FPs studied, mNeonGreen provides the best imaging quality and has a relatively low susceptibility to oligomerization, but appears to stick to other proteins and the glass surface. mCitrine appears to be the most well-behaved of the green FPs, but is not optimized for 488 nm excitation. Of the red FPs studied, tandem mCherry provides the best imaging quality without compromising native motor properties. SNAP and HALO tags provide great versatility for imaging in multiple colors, but typically are not brighter or more photostable than FPs in single-molecule studies. Overall, this study should serve as a valuable reference for designing single-molecule experiments in multiple colors.
Author Contributions
S.R.N. and K.J.V. designed the research. S.R.N. and M.F.N. performed the research. S.R.N. analyzed the data. S.R.N. and K.J.V. wrote the manuscript with input from all authors.
Acknowledgments
We thank members of the Verhey lab for help and discussions. We also thank D. Cai and J. Swanson for the gift of plasmids.
This work was supported by a grant to K.J.V. (NIH NIGMS RO1070862).
Supporting Material
Lysates of COS7 cells expressing KHC(1-560) labeled with GFPs in standard P12 motility buffer were imaged by TIRF microscopy at room temperature (Ti-E/B; Nikon). Frames were acquired continuously with 100 ms exposure. Top panel: KHC(1-560) labeled with EGFP; second panel: KHC(1-560) labeled with monomeric EGFP; third panel: KHC(1-560) labeled with mNeGr; bottom panel: KHC(1-560) labeled with mCit. Scale bar = 5 μm.
Lysates of COS7 cells expressing KHC(1-560) labeled with RFPs in standard P12 motility buffer were imaged by TIRF microscopy at room temperature (Ti-E/B; Nikon). Frames were acquired continuously with 100 ms exposure. Top panel: KHC(1-560) labeled with tdTomato; second panel: KHC(1-560) labeled with tagRFPt; third panel: KHC(1-560) labeled with mApple; bottom panel: KHC(1-560) labeled with 2xmCh. Scale bar = 5 μm.
Lysates of COS7 cells expressing ligand-labeled KHC(1-560)-SNAP in standard P12 motility buffer were imaged by TIRF microscopy at room temperature (Ti-E/B; Nikon). Frames were acquired continuously with 100 ms exposure. Top panel: KHC(1-560)-SNAP labeled with SNAP-OreGr488; middle panel: KHC(1-560)-SNAP labeled with SNAP-505-STAR; bottom panel: KHC(1-560)-SNAP labeled with SNAP-TMR. Scale bar = 5 μm.
Lysates of COS7 cells expressing ligand-labeled KHC(1-560)-HALO in standard P12 motility buffer were imaged by TIRF microscopy at room temperature (Ti-E/B; Nikon). Frames were acquired continuously with 100 ms exposure. Top panel: KHC(1-560)-HALO labeled with HALO-OreGr488; middle panel: KHC(1-560)-HALO labeled with HALO-diAcFAM; bottom panel: KHC(1-560)-HALO labeled with HALO-TMR. Scale bar = 5 μm.
References
- 1.Vale R.D. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 2.Yildiz A., Tomishige M., Selvin P.R. Kinesin walks hand-over-hand. Science. 2004;303:676–678. doi: 10.1126/science.1093753. [DOI] [PubMed] [Google Scholar]
- 3.Brady S.T. A novel brain ATPase with properties expected for the fast axonal transport motor. Nature. 1985;317:73–75. doi: 10.1038/317073a0. [DOI] [PubMed] [Google Scholar]
- 4.Vale R.D., Reese T.S., Sheetz M.P. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Block S.M., Goldstein L.S., Schnapp B.J. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- 6.Vale R.D., Funatsu T., Yanagida T. Direct observation of single kinesin molecules moving along microtubules. Nature. 1996;380:451–453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalfie M., Tu Y., Prasher D.C. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 8.Dedecker P., De Schryver F.C., Hofkens J. Fluorescent proteins: shine on, you crazy diamond. J. Am. Chem. Soc. 2013;135:2387–2402. doi: 10.1021/ja309768d. [DOI] [PubMed] [Google Scholar]
- 9.Cai D., Verhey K.J., Meyhöfer E. Tracking single kinesin molecules in the cytoplasm of mammalian cells. Biophys. J. 2007;92:4137–4144. doi: 10.1529/biophysj.106.100206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai D., McEwen D.P., Verhey K.J. Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLoS Biol. 2009;7:e1000216. doi: 10.1371/journal.pbio.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norris S.R., Soppina V., Verhey K.J. A method for multiprotein assembly in cells reveals independent action of kinesins in complex. J. Cell Biol. 2014;207:393–406. doi: 10.1083/jcb.201407086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaner N.C., Lin M.Z., Tsien R.Y. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juillerat A., Gronemeyer T., Johnsson K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem. Biol. 2003;10:313–317. doi: 10.1016/s1074-5521(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 14.Los G.V., Encell L.P., Wood K.V. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 15.Kremers G.J., Hazelwood K.L., Piston D.W. Photoconversion in orange and red fluorescent proteins. Nat. Methods. 2009;6:355–358. doi: 10.1038/nmeth.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soppina V., Norris S.R., Verhey K.J. Dimerization of mammalian kinesin-3 motors results in superprocessive motion. Proc. Natl. Acad. Sci. USA. 2014;111:5562–5567. doi: 10.1073/pnas.1400759111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorn K.S., Ubersax J.A., Vale R.D. Engineering the processive run length of the kinesin motor. J. Cell Biol. 2000;151:1093–1100. doi: 10.1083/jcb.151.5.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romberg L., Pierce D.W., Vale R.D. Role of the kinesin neck region in processive microtubule-based motility. J. Cell Biol. 1998;140:1407–1416. doi: 10.1083/jcb.140.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seitz A., Kojima H., Mandelkow E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002;21:4896–4905. doi: 10.1093/emboj/cdf503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kremers G.J., Gilbert S.G., Piston D.W. Fluorescent proteins at a glance. J. Cell Sci. 2011;124:157–160. doi: 10.1242/jcs.072744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaner N.C., Steinbach P.A., Tsien R.Y. A guide to choosing fluorescent proteins. Nat. Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 22.Visscher K., Schnitzer M.J., Block S.M. Single kinesin molecules studied with a molecular force clamp. Nature. 1999;400:184–189. doi: 10.1038/22146. [DOI] [PubMed] [Google Scholar]
- 23.Zacharias D.A., Violin J.D., Tsien R.Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 24.Olenych S.G., Claxton N.S., Davidson M.W. The fluorescent protein color palette. Curr. Protoc. Cell Biol. 2007;Chapter 21 doi: 10.1002/0471143030.cb2105s36. Unit 21.5. [DOI] [PubMed] [Google Scholar]
- 25.Zhu C., Dixit R. Single molecule analysis of the Arabidopsis FRA1 kinesin shows that it is a functional motor protein with unusually high processivity. Mol. Plant. 2011;4:879–885. doi: 10.1093/mp/ssr077. [DOI] [PubMed] [Google Scholar]
- 26.Shaner N.C., Lambert G.G., Wang J. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods. 2013;10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costantini L.M., Fossati M., Snapp E.L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 2012;13:643–649. doi: 10.1111/j.1600-0854.2012.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landgraf D., Okumus B., Paulsson J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods. 2012;9:480–482. doi: 10.1038/nmeth.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han L., Zhao Y., Zhang M. RFP tags for labeling secretory pathway proteins. Biochem. Biophys. Res. Commun. 2014;447:508–512. doi: 10.1016/j.bbrc.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Griesbeck O., Baird G.S., Tsien R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 31.Jain R.K., Joyce P.B., Gorr S.U. Oligomerization of green fluorescent protein in the secretory pathway of endocrine cells. Biochem. J. 2001;360:645–649. doi: 10.1042/0264-6021:3600645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayloo S., Lazarus J.E., Holzbaur E.L. Dynactin functions as both a dynamic tether and brake during dynein-driven motility. Nat. Commun. 2014;5:4807. doi: 10.1038/ncomms5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun X., Zhang A., Corrêa I.R., Jr. Development of SNAP-tag fluorogenic probes for wash-free fluorescence imaging. ChemBioChem. 2011;12:2217–2226. doi: 10.1002/cbic.201100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bosch P.J., Corrêa I.R., Jr., Subramaniam V. Evaluation of fluorophores to label SNAP-tag fused proteins for multicolor single-molecule tracking microscopy in live cells. Biophys. J. 2014;107:803–814. doi: 10.1016/j.bpj.2014.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heim R., Cubitt A.B., Tsien R.Y. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- 36.Cormack B.P., Valdivia R.H., Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 37.Shaner N.C., Campbell R.E., Tsien R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 38.Merzlyak E.M., Goedhart J., Chudakov D.M. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods. 2007;4:555–557. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Lysates of COS7 cells expressing KHC(1-560) labeled with GFPs in standard P12 motility buffer were imaged by TIRF microscopy at room temperature (Ti-E/B; Nikon). Frames were acquired continuously with 100 ms exposure. Top panel: KHC(1-560) labeled with EGFP; second panel: KHC(1-560) labeled with monomeric EGFP; third panel: KHC(1-560) labeled with mNeGr; bottom panel: KHC(1-560) labeled with mCit. Scale bar = 5 μm.
Lysates of COS7 cells expressing KHC(1-560) labeled with RFPs in standard P12 motility buffer were imaged by TIRF microscopy at room temperature (Ti-E/B; Nikon). Frames were acquired continuously with 100 ms exposure. Top panel: KHC(1-560) labeled with tdTomato; second panel: KHC(1-560) labeled with tagRFPt; third panel: KHC(1-560) labeled with mApple; bottom panel: KHC(1-560) labeled with 2xmCh. Scale bar = 5 μm.
Lysates of COS7 cells expressing ligand-labeled KHC(1-560)-SNAP in standard P12 motility buffer were imaged by TIRF microscopy at room temperature (Ti-E/B; Nikon). Frames were acquired continuously with 100 ms exposure. Top panel: KHC(1-560)-SNAP labeled with SNAP-OreGr488; middle panel: KHC(1-560)-SNAP labeled with SNAP-505-STAR; bottom panel: KHC(1-560)-SNAP labeled with SNAP-TMR. Scale bar = 5 μm.
Lysates of COS7 cells expressing ligand-labeled KHC(1-560)-HALO in standard P12 motility buffer were imaged by TIRF microscopy at room temperature (Ti-E/B; Nikon). Frames were acquired continuously with 100 ms exposure. Top panel: KHC(1-560)-HALO labeled with HALO-OreGr488; middle panel: KHC(1-560)-HALO labeled with HALO-diAcFAM; bottom panel: KHC(1-560)-HALO labeled with HALO-TMR. Scale bar = 5 μm.