

Abstract
Analyses of genome-wide association study (GWAS) data have revealed that detectable genetic mosaicism involving large (>2 Mb) structural autosomal alterations occurs in a fraction of individuals. We present results for a set of 24,849 genotyped individuals (total GWAS set II [TGSII]) in whom 341 large autosomal abnormalities were observed in 168 (0.68%) individuals. Merging data from the new TGSII set with data from two prior reports (the Gene-Environment Association Studies and the total GWAS set I) generated a large dataset of 127,179 individuals; we then conducted a meta-analysis to investigate the patterns of detectable autosomal mosaicism (n = 1,315 events in 925 [0.73%] individuals). Restricting to events >2 Mb in size, we observed an increase in event frequency as event size decreased. The combined results underscore that the rate of detectable mosaicism increases with age (p value = 5.5 × 10−31) and is higher in men (p value = 0.002) but lower in participants of African ancestry (p value = 0.003). In a subset of 47 individuals from whom serial samples were collected up to 6 years apart, complex changes were noted over time and showed an overall increase in the proportion of mosaic cells as age increased. Our large combined sample allowed for a unique ability to characterize detectable genetic mosaicism involving large structural events and strengthens the emerging evidence of non-random erosion of the genome in the aging population.
Main Text
Detectable mosaicism is the presence of two or more genetically distinct populations of cells in an individual who has developed from a single zygote.1 The clonal expansion of acquired post-zygotic mutations, such as large-scale gains, losses, and copy-neutral uniparental disomy, can result in the co-existence of aberrant cellular populations with normal germline DNA.2 Clonal mosaicism can also contribute to diverse phenotypes depending on developmental timing, the tissue involved, the genomic location of the mutation, and the percentage of cellular populations affected.3–5 Compared to constitutional defects in the same regions, mosaic abnormalities can result in milder phenotypes, as observed for neurofibromatosis type 1 (MIM 162200) and trisomy 21 (MIM 190685); interestingly, these same mutations have been observed in apparently healthy individuals.6–8 A spectrum of clinical phenotypes, including Maffucci syndrome (MIM 614569),9,10 McCune-Albright syndrome (MIM 174800),11 nevus sebaceus (MIM 162900),12 Ollier disease (MIM 166000),9,10 Proteus syndrome (MIM 176920),13 and mosaic RASopathies,14 have been associated with mosaicism.
Until recently, estimates of the rates of human mosaicism involving large structural events were unavailable.15 Early evidence demonstrated somatic mosaicism in monozygotic twins16 and differentiated human tissues17 but provided no estimates of rates in human populations. The combination of large datasets and improved methodology for analysis of genome-wide SNP microarray data has enabled genome-wide surveys of large structural mosaic events in blood and buccal DNA.18,19 An initial population-based case-control genome-wide association study (GWAS) of 1,991 individuals with bladder cancer reported autosomal mosaic abnormalities (e.g., structural events >2 Mb) in blood or buccal DNA from 1.7% of the overall study sample.18 Subsequent analyses of a larger set of GWASs involving 57,853 individuals, described as the total GWAS set I (TGSI), presented evidence that clonal mosaicism was strongly associated with greater age and weakly associated with male gender and overall solid-tumor risk, in particular lung and kidney cancer.20 A concurrently published study, involving 50,222 individuals, from the Gene-Environment Association Studies (GENEVA) Consortium observed an association between mosaicism and age, although no significant associations were observed with gender or solid tumors.21 An additional study by Forsberg and colleagues detected age-related structural changes in leukocyte DNA from paired monozygotic twins and single-born subjects in 3.4% of individuals aged 60 years or older but failed to detect mosaic events in individuals aged 55 years or younger.22 Other studies have subsequently confirmed the presence of detectable autosomal mosaicism in older populations,23,24 as well as demonstrated an age-specific relationship with mosaicism on the Y chromosome.25 Furthermore, recent evidence indicates that somatic mosaicism might be an important contributor to unexpected familial recurrences of genomic disorders.26
We confirmed the presence of clonal mosaic events greater than 2 Mb in an independent sample set of cancer-affected individuals and control individuals and conducted a meta-analysis of the events from our sample set and the two prior investigations to refine our understanding of the landscape of events. Study subjects from our new sample set, hereafter referred to as the total GWAS set II (TGSII), were drawn from published GWASs investigating cancer-susceptibility risk and were analyzed in the Cancer Genomics Research Laboratory of the National Cancer Institute. TGSII includes 24,849 participants drawn from 46 studies on populations of European, Asian, and African descent. Approval by the institutional review board for each study was confirmed, and written informed consent was obtained.
TGSII genotyping was carried out on commercially available Illumina Infinium BeadArray human assays (HumanHap610, HumanHap660W, HumanHap1M, OmniExpress, Omni1, Omni2.5, and Omni5). Assays, specimens, and participants met the following criteria for inclusion: (1) information was available on the first cancer site, or individuals were determined to be cancer free, (2) the minimum genotype completion rate was 88%, (3) SDs were less than 0.33 and 0.05 for the final corrected log2 R ratio (LRR) and B allele frequency (BAF), respectively, and (4) genetic identity was consistent across duplicate samples.
To assess copy-number changes and allelic imbalances, we estimated LRR and BAF. LRR provides a metric for assessing copy-number change via the calculation of log2 of the ratio of observed total signal intensities to expected signal intensities for a SNP. LRR values greater than 0 indicate mosaic copy gain, whereas values less than 0 indicate loss. The BAF, a measure of allelic imbalance, is calculated as the ratio of signal intensity between two alleles at each SNP in relation to estimated genotype clusters; it is thus a calculation of the frequency of the B allele for a biallelic SNP with alleles A and B. BAF values for heterozygous SNPs that deviate from 0.5 are indicative of mosaic copy-number changes or copy-neutral changes associated with acquired uniparental disomy.
To improve accuracy, we used a quantile normalization approach similar to that used by Staaf et al.27 and Diskin et al.28 because LRR and BAF estimates from Illumina GenomeStudio software suffer biases from assay chemistry and DNA concentration. For TGSI and TGSII, a mosaic-alteration-detection algorithm incorporated into the software package R-GADA (based on genome alteration detection analysis) was used to detect clonal mosaic events greater than 2 Mb in size from corrected LRR and BAF values.19,29 The GADA segmentation algorithm detected clonal mosaic regions by identifying breakpoints in Bdev values with the use of sparse Bayesian learning and backward elimination. The assigned event type was based on the mean LRR value, and the mosaic proportion of abnormal cells was estimated from the BAF values. The detection algorithm has been previously validated with laboratory techniques (e.g., single tandem repeat, multiplex ligation-dependent probe amplification, and fluorescent in situ hybridization)18 and is described in greater detail in the methods and supplementary material of the original Jacobs et al. analysis.20 Investigators blind to study outcomes conducted a manual review to confirm events in TGSI and TGSII. Common modifications included adjusting boundaries of mosaic events or merging or separating adjacent events.
To improve our understanding of the landscape of clonal mosaic events, we first combined TGSI20 and TGSII, both of which were analyzed with the identical pipeline described above. Subsequently, we extracted events >2 Mb from the Laurie et al. study (GENEVA).21 GENEVA used a circular-binary-segmentation algorithm with the Bioconductor packages DNACopy and GWASTools. The overall reproducibility between the GENEVA algorithm and TGSI algorithm in detecting events >2 Mb was assessed in 5,510 lung cancer samples included in both GENEVA and TGSI. The comparability in called events >2 Mb was 75%; 83 of the total 111 events were detected by both algorithms, 20 were detected by TGSI only, and 8 were detected by GENEVA only. The 5,510 replicated lung cancer samples and 235 individuals with inadequate consent were removed from the GENEVA sample in the analyses described herein. For more details on the detection methods, please see the methods and supplementary materials in the previously published TGSI20 and GENEVA21 reports, which include laboratory confirmation of select samples in TGSI.
In the 24,849 individuals in TGSII, our method detected 341 clonal mosaic events across 255 autosomes in 168 individuals (Table S1). We detected 69 events (20%) with mosaic copy gain, 90 events (26%) with mosaic copy loss, and 163 events (48%) with copy-neutral acquired uniparental disomy. 19 events (6%) were complex in nature and not amenable to distinct classification.
Detected events from our TGSII were combined with TGSI, and then a meta-analysis with GENEVA was performed on a sample set of a total of 127,179 individuals, in whom 1,315 mosaic events were detected in 925 participants; the overall rate of individuals with detected mosaicism was 0.73% (95% confidence interval [CI] = 0.68–0.78%). Of the 925 participants with detected events, 797 (86%) had only one event, and 128 (14%) harbored multiple mosaic events. Compared to the Poisson expectation, which is that only seven individuals have multiple events, a highly significant excess of individuals with multiple events was observed (p value = 6.5 × 10−30). Although complex rearrangements affecting multiple regions of the genome could partially account for the excess, differences in mosaic proportion of events in some individuals suggest more complex mechanisms over long periods of time as well.
Approximately half of the detected events were mosaic copy-neutral uniparental disomy (48%), followed by mosaic losses (34%) and mosaic gains (17%). To characterize genomic location and potential recurring events, we generated Circos plots.30 The majority of mosaic gains were observed on chromosomes 8, 12, and 15, the majority of mosaic losses were observed on chromosomes 13 and 20, and mosaic copy-neutral events were primarily observed on chromosomes 9 and 14 (Figure 1). Detected mosaic events clustered regionally on chromosomal arms on the basis of their copy-number state (Table 1). Mosaic copy-neutral events occurred primarily on the telomeric ends of chromosomes; 33% included the p telomere, and 54% included the q telomere. Mosaic losses were commonly observed in interstitial chromosomal regions that did not involve telomeres or centromeres. Of all of the copy-number states, mosaic gains most commonly involved whole chromosomes. Only 1.7% of events were interstitial and spanned the centromere, suggesting that for interstitial mosaic events, involvement of a centromere could be uncommon.
Figure 1.
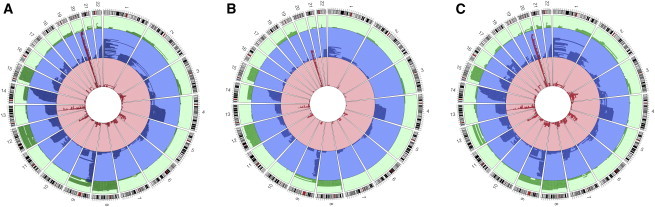
Genomic Locations of the Combined 1,334 Events Overall and by Cancer Status
Green indicates mosaic copy gains, blue represents mosaic copy-neutral events, and red represents mosaic losses.
(A) All 1,334 events from the combined GENEVA, TGSI, and TGSII analysis.
(B) Events in cancer-free control individuals.
(C) Events in individuals with solid tumors.
Table 1.
Distribution of Mosaic Copy-Number State by Chromosomal-Arm Location
| Gain | Neutral | Loss | Total | |||||
|---|---|---|---|---|---|---|---|---|
| Whole chromosome | 60 | (65%, 27%) | 25 | (27%, 4%) | 8 | (9%, 2%) | 93 | (100%, 7%) |
| Telomeric p | 18 | (7%, 8%) | 205 | (78%, 33%) | 40 | (15%, 9%) | 263 | (100%, 20%) |
| Telomeric q | 67 | (15%, 30%) | 338 | (75%, 54%) | 44 | (10%, 10%) | 449 | (100%, 35%) |
| Interstitial | 73 | (16%, 33%) | 51 | (11%, 8%) | 344 | (74%, 77%) | 468 | (100%, 36%) |
| Spans centromere | 5 | (22%, 2%) | 10 | (43%, 2%) | 8 | (35%, 2%) | 23 | (100%, 2%) |
| Total | 223 | (17%, 100%) | 629 | (49%, 100%) | 444 | (34%, 100%) | 1,296 | (100%, 100%) |
Events classified as “whole chromosome” from the combined dataset span an entire chromosome, “telomeric p” and “telomeric q” are events that include the telomere on the p or q arm, respectively, “interstitial” events do not include a telomere or centromere, and “spans centromere” indicates interstitial events that overlap a chromosome’s centromere. Event counts are indicated in parentheses (row percent, column percent).
It is notable that losses at certain regions, such as 20q (chr20: 40,425,000–42,155,000; UCSC Human Genome Browser hg18) and 13q14 (chr13: 49,590,000–49,983,100; UCSC hg18), are observed in leukemias (e.g., myelogenous and lymphoblastic leukemia, respectively).31–33 Clustering of losses in these regions suggests that these events are non-random. Moreover, aggregation of mosaic-event locations on chromosomal arms by copy-number state suggests common mechanisms. For example, copy-neutral telomeric events could be due to mitotic recombination followed by clonal expansion. Breakpoint analyses of regions surrounding mosaic events might aid in understanding mechanisms responsible for event initiation, but the current resolution of event boundaries in SNP microarrays is limited as a result of insufficient probe density. Further work is required to investigate the different types of events that could lead to large structural mosaicism.
To identify characteristics associated with increased risk of large clonal mosaic events, we evaluated age, gender, ancestry, and cancer status. Age at time of DNA collection was available for all GENEVA and TGSI participants, but for TGSII participants, the date of diagnosis (for cancer subjects) or the age at the time of participation (for control subjects) was substituted when age at time of DNA collection was missing. Categorical variables were constructed for the 5-year age groups of 50–54, 55–59, 60–64, 65–69, and 70–74 and for 75 years or older, whereas individuals under 50 years of age were considered the reference group. We used reference populations from the HapMap project34 and the GLU (Genotype Library and Utilities) software package to estimate continental-ancestry proportions for each individual. Terms were fit for percentage of African and Asian ancestry, whereas European ancestry served as the referent. Indicator variables were used for adjusting analyses for effects related to the individual contributing studies. Sensitivity analyses using mixed models, case-control matching, and pooled analyses were also used to investigate the robustness of statistically significant findings. All statistical analyses were performed in R version 3.0.1 on a 64-bit Unix platform.35
Increasing age is the variable most strongly associated with clonal mosaicism (Figure 2). In logistic regression models adjusted for gender, ancestry, cancer subtype, and contributing study, highly significant age effects were observed in both a meta-analysis and a pooled analysis of GENEVA, TGSI, and TGSII (Figure 3A). The effect of the 5-year age groups was significantly associated with detectable clonal mosaicism overall (p value = 5.5 × 10−31), showed no evidence of heterogeneity across the study (p value = 0.71), and remained significant when the analysis was restricted to a subset of cancer-free control individuals (p value = 8.92 × 10−12). Compared to individuals under 50 years old, individuals aged 75 years or older had an approximate 6-fold increase in detection of large-scale mosaic events (p value = 2.22 × 10−16, 95% CI = 4.16–10.09).
Figure 2.
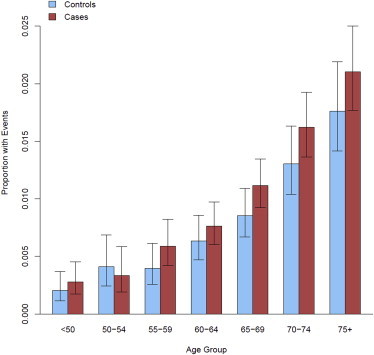
Proportion of Mosaic Individuals across 5-Year Age Groups in the Combined GENEVA, TGSI, and TGSII Dataset by Cancer Status
Affected individuals are in red, and cancer-free control individuals are in blue. Error bars represent 95% CIs. An overall significant relationship in the proportion of individuals with mosaic events was observed with age (p value = 1.1×10−30).
Figure 3.
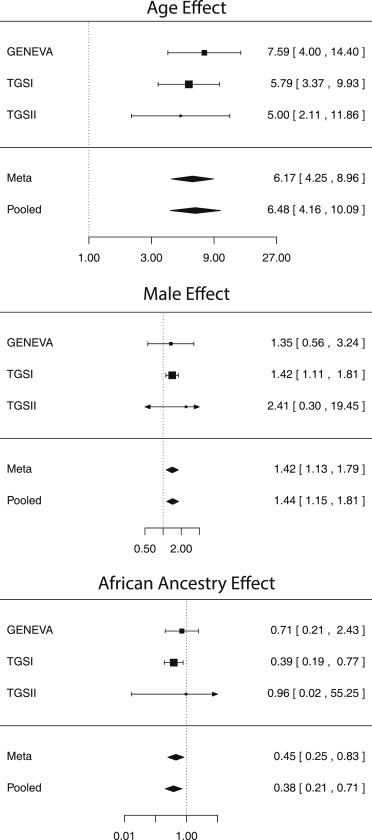
Forest Plots of Associations with Clonal Mosaicism
Associations between clonal mosaicism and (A) age-group, (B) gender, and (C) ancestry.
Our meta-analysis strengthens the robust age association previously observed in GENEVA and TGSI. Although the inclusion of age at diagnosis as a substitute for age at the time of DNA collection could have introduced measurement error in individual TGSII participants, the TGSII and overall pooled age association agree with the original age estimates from the GENEVA and TGSI studies. Although an association with age was observed, it is important to note that our analysis does not provide insight into whether the events were generated early in life and later positively selected by rapid expansion of a second clonal population or generated later in life as a result of decreased cellular diversity and senescence.
The effect of gender was also significant. Mosaic events were more frequently observed in males than in females (0.98% versus 0.56%, respectively; Figure 3B). Removing sex-specific cancers (e.g., endometrial and prostate cancers) and adjusting for ancestry, 5-year age group, cancer subtype, and contributing study, we found a significant association with male gender (odds ratio [OR] = 1.44, 95% CI = 1.15–1.81, p value = 0.002) and no evidence of heterogeneity of effect across the study (p value = 0.88). In the 37,942 cancer-free control individuals, the gender association was of similar magnitude but marginally not significant (OR = 1.39, p value = 0.06), most likely as a result of the reduced number of individuals with events (n = 303). The elevated rates of mosaic events observed in males could be partially attributable to higher male-specific rates of hematologic malignancies, a set of malignancies previously found to be associated with clonal mosaicism.20,21,23 Further work will be required for understanding the scope and implications of this observation.
An association between genotype-inferred ancestry (defined as the percentage of continental origin from Africa, Asia, or Europe) and clonal mosaic events was also evident (Figure 3C). Logistic regression analyses adjusted for gender, 5-year age group, cancer subtype, and study indicated that relative to individuals of European ancestry, individuals of African ancestry were at a reduced risk (OR = 0.38, 95% CI = 0.21–0.71, p value = 0.003). Heterogeneity testing detected no heterogeneity (p value = 0.66), and the effect retained significance when the analysis was restricted to cancer-free control individuals (OR = 0.23, 95% CI = 0.09–0.60, p value = 0.003). No significant difference was observed between Asian and European ancestry. Mechanisms relating to ancestry-specific differences in rates of clonal mosaicism are poorly understood, and further work is needed for better understanding this relationship.
We further investigated the associations between clonal mosaicism and risk of solid (non-hematological) tumors overall and of tumor subtypes as per the previous TGSI finding.20 Analyses adjusted for gender, ancestry, 5-year age group, and contributing study indicated that solid tumors were associated with clonal mosaicism in blood or buccal tissue (OR = 1.29, 95% CI = 1.11–1.50, p value = 8.1 × 10−4). Additional cancer-specific analyses were performed for all solid-tumor subtypes present in our study (Table 2). Endometrial, kidney, liver, lung, and prostate cancers suggested preliminary evidence of significant associations with clonal mosaicism; however, only lung cancer maintained statistical significance after correction for multiple testing. Interestingly, all solid-tumor subtypes showing preliminary evidence of an association with mosaicism had ORs greater than 1, but the sample sizes per tumor were small, and the estimates were unstable for the risk effects measured. Circos plots showing mosaic-event location for cancer-free control individuals and for individuals with solid tumors are displayed in Figures 1B and 1C. No differences in event clustering or copy-number state were observed among the individuals with solid tumors. Lung cancer was the primary contributor to the overall cancer association, and it is notable that lung cancer is a smoking-related cancer; however, previous data suggest that smoking is not significantly associated with autosomal mosaicism in blood tissue.20,21 Potential mechanisms linking clonal mosaicism in blood to solid-tumor risk might include poor overall genome maintenance unable to repair genomic alterations or immunologic dysfunction in mosaic immune cells and the subsequent poor clearance of pre-cancerous cells from solid tissues. Further studies are required to determine whether detectable clonal mosaicism could be a useful biomarker in screening individuals for increased risk of developing solid tumors. This is in contrast to hematological malignancies, in which detectable mosaicism in blood could be an early indicator of leukemic and pre-leukemic clones.20,21,23
Table 2.
Solid-Tumor Associations with Clonal Mosaicism
| n | OR | 95% CI | p Value | |
|---|---|---|---|---|
| Overall | 46,831 | 1.29 | 1.11–1.50 | 0.0008 |
| Bladder | 4,995 | 1.29 | 0.90–1.85 | 0.168 |
| Breast | 2,814 | 1.06 | 0.53–2.10 | 0.869 |
| Endometrium | 872 | 2.66 | 1.16–6.12 | 0.021 |
| Esophagus | 1,910 | 0.89 | 0.34–2.34 | 0.821 |
| Glioma | 1,729 | 0.78 | 0.30–2.06 | 0.622 |
| Kidney | 1,565 | 1.81 | 1.06–3.11 | 0.031 |
| Liver | 13 | 10.40 | 1.32–81.9 | 0.026 |
| Lung | 13,015 | 1.54 | 1.21–1.97 | 0.001 |
| Ovary | 543 | 2.60 | 0.93–7.28 | 0.069 |
| Pancreas | 3,923 | 0.83 | 0.53–1.29 | 0.404 |
| Prostate | 10,456 | 1.27 | 1.00–1.60 | 0.046 |
| Skin | 1,949 | 0.96 | 0.43–2.13 | 0.911 |
| Stomach | 2,278 | 1.52 | 0.70–3.28 | 0.292 |
| Testis | 649 | 1.77 | 0.24–13.0 | 0.573 |
Combined analysis of solid-tumor associations adjusted for gender, ancestry, 5-year age group, and contributing study. “n” denotes total sample size. ORs and 95% CIs are reported for solid tumors overall and by cancer subtype.
An association between rate and event size was noted: smaller events were observed to have higher rates than larger events (Figure 4). An inverse exponential relationship fit the trend well (R2 = 0.81). This association was also present in the substrata of mosaic gains, copy-neutral events, and losses. This suggests that smaller autosomal events are more frequent and that larger autosomal events are relatively more rare. Event size was also investigated across copy-number states (Figure 5A). With a median event size of 60.8 Mb, mosaic copy gains, on average, were largest. Mosaic copy-neutral events and mosaic losses had median sizes of 39.8 and 17.0 Mb, respectively. The larger average size of mosaic gains and copy-neutral events might highlight the detrimental nature of induced monosomy and reduced copy number of mosaic losses and provide insight into understanding which mutational events undergo clonal selection. By restricting our analysis to events larger than 2 Mb in size to reduce the false-positive rate of our detection algorithm, we most likely missed many smaller mosaic events, from mosaic point mutations to events several kilobases in size. Extrapolating our observations to smaller events suggests that the copy-number distribution of these events is most likely skewed toward mosaic losses. This could prove to have great importance in both disease risk and heterogeneity of disease phenotypes. Although the association between event rate and size was present in every strata of copy-number state, the overall association might be skewed by the abundance of small copy losses in likely driver regions of hematologic cancers, such as losses at 13q14 and 20q. Further refinements in genotyping technologies and detection algorithms targeted at detecting smaller mosaic events in next-generation sequencing should refine our understanding of the landscape of detectable clonal mosaicism.
Figure 4.
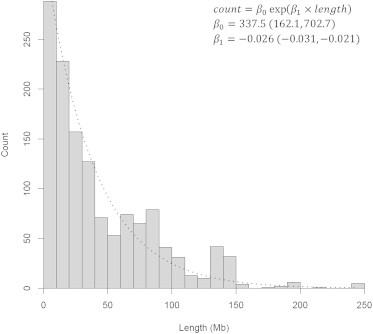
Relationship between Mosaic Event Size and Detected Rate
Counts of detected mosaic events from the combined analysis are plotted in 10-Mb bin sizes. An inverse exponential trend, represented by the dotted line, was fit to the counts (R2 = 0.81). Fitted coefficients (β) and 95% CIs are displayed in the plot.
Figure 5.
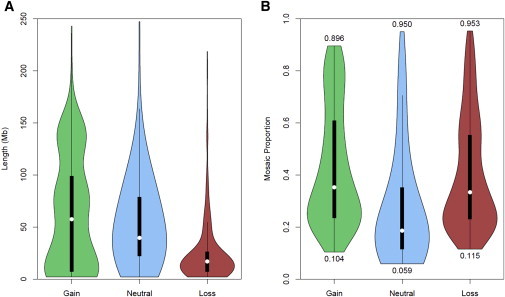
Event Size and Mosaic-Proportion Distribution across Copy-Number State
Violin plots of combined sample event size (A) and mosaic proportion (B) in relationship to mosaic copy gains, copy-neutral uniparental disomies, and copy losses. Boxplots with white circles denoting the median and thick black boxes representing the interquartile range are encapsulated in kernel density plots of the distribution of event length. Numbers below and above the violin plots of mosaic proportion (B) indicate the minimum and maximum detected range observed for each respective event’s copy-number state.
Mosaic proportion, namely, the percentage of cells with large structural events that differ from germline DNA, was distinct across event copy-number state (Figure 5B). Mosaic copy gains and copy losses were observed, on average, to have higher mosaic proportions (medians of 0.35 and 0.33, respectively), whereas mosaic copy-neutral events had a lower median proportion of 0.19. The range of the mosaic proportions provides insight into the detectable range of our methodology. Copy-neutral mosaic events can be detected in the range 0.06–0.95, whereas mosaic copy gains can be detected in the range 0.10–0.90.
The relationship between age and mosaic proportion was also investigated (Figure 6). A significant positive association between age and mosaic proportion was observed for copy-neutral events (p value = 1.2 × 10−4), suggesting that on a population level, mosaic proportion might gradually increase with age. To further assess the evolution of mosaic events over time, we analyzed serial samples from the Prostate, Lung, Colon, and Ovary Prevention Trial (PLCO). Two to four DNA samples collected at least 1 year apart were analyzed for PLCO individuals with detectable clonal mosaicism in TGSI and TGSII. In total, we tracked large structural mosaic events for 58 autosomes (Figure 7) detected in 47 individuals. The set included 24 events with mosaic loss, 27 with copy-neutral loss of heterozygosity, and 7 with gain. Although examples existed where events had stable or decreasing mosaic proportion over time, most events were observed to increase in mosaic proportion. Fitting a linear mixed model with a zero intercept and a random effect for each event, our analysis suggests that with each year increase in age, the overall fraction of mosaic proportion increases on average by approximately 1.44% (p value = 3.3 × 10−7). Significant increases in mean mosaic proportion were seen over time in the strata of mosaic losses and mosaic copy-neutral events, but because of limited sample size (n = 7), the increase was not significant for mosaic gains. Together, these observations suggest that most detectable mosaic events confer some form of selective advantage that enables cellular clones to increase in frequency over time in relation to cells with normal karyotypes.
Figure 6.
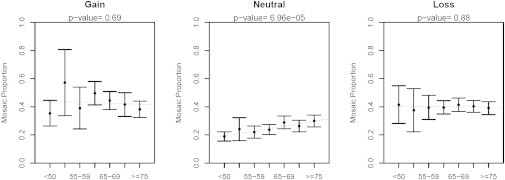
Characterization of Changes in Mosaic Proportion with Age
Mosaic proportion is stratified across event copy-number state (columns) and age category (x axis) from the combined analysis. Points represent estimates of the mean, and error bars indicate 95% CI around the mean. Best-fit regression lines are plotted with dotted lines, and p values are shown for slopes that are significantly different from 0.
Figure 7.
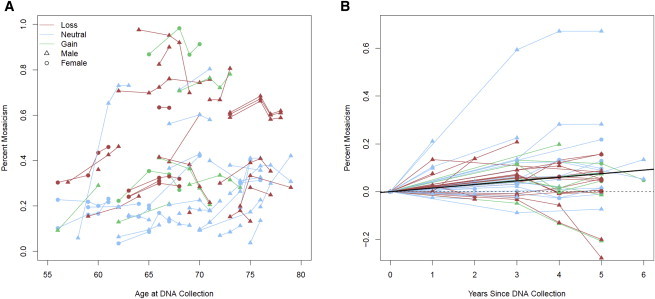
Changes in Percentage of Mosaicism of Serial Samples over Time
(A) Lines (red for copy loss, blue for copy neutral, and green for copy gain) connect DNA-collection time points (triangles represent males, and circles represent females) for each mosaic chromosome (n = 58) to track changes in the percentage of mosaicism with increasing age.
(B) Events are plotted with zero origin and a mixed model for a subject with zero random-effect fit (solid black line), showing the estimated average change in mosaic proportion per year (1.44%, p value = 3.3 × 10−7).
A few limitations relating to the available data are worth considering. The studies used for analysis were primarily designed as GWASs of cancer (GENEVA, TGSI, and TGSII) and other traits (GENEVA), and study participants were drawn from cohort and case-control studies. Rate estimates of mosaicism, as provided by the combination of individuals included in these studies, could imperfectly represent underlying population prevalence. However, the consistency of effect estimates across GENEVA, TGSI, and TGSII suggests that our findings are robust. Additionally, although incomplete, adjustment for factors such as age group, gender, ancestry, cancer subtype, and contributing study minimized confounding effects. Because additional bioinformatics methods are needed to detect mosaicism on the sex chromosomes and because there is poor commercial array coverage of the Y chromosome, this report focused on detectable autosomal mosaicism. Another group has reported a similar association between Y mosaicism and increasing age.25
Our analysis is distinctive in that it involves a meta-analysis of 127,179 individuals and thus provides a more comprehensive portrait of the landscape of large structural mosaic genetic alterations. Our meta-analysis allowed for a more precise age effect, strengthened prior evidence of a gender effect, and found evidence of an ancestry effect. Additionally, we found evidence indicating that the number of mosaic events increases as event size decreases, suggesting that our detected events might represent the tip of the iceberg in relation to many smaller mosaic events that most likely exist and are currently undetected. Furthermore, we were able to analyze serial samples over multiple collection time points to show an overall increase in mosaic percentage as age increases.
The investigation of human clonal mosaicism can provide new insights into aging, as well as shed light on possible precursors of disease. Our results suggest that genome maintenance, particularly in relation to aging, could have pleotropic consequences, although it is not clear whether all mosaic events are necessarily deleterious. The long-held assumption that germline DNA remains static during the course of life is under reconsideration, which is particularly important for the comparative study of cancer genomes. As the detection of smaller mosaic events across tissue types improves, we can refine our understanding of the landscape of mosaic events across the spectrum of genetic events. In turn, this could represent an important step toward the investigation of how our once “stable” germline DNA might slowly erode into a complex mosaic over time and contribute to disease heterogeneity.
Acknowledgments
Acknowledgment of funding sources is available in the Supplemental Data. B.R.-S. and L.A.P.-J. are currently employee and scientific advisor, respectively, of qGenomics. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the NIH.
Supplemental Data
Web Resources
The URLs for data provided herein are as follows:
Genotype Library and Utilities (GLU), https://code.google.com/p/glu-genetics/
OMIM, http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Strachan T., Read A. Wiley-Liss; New York: 1999. Human Molecular Genetics. [Google Scholar]
- 2.Hall J.G. Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am. J. Hum. Genet. 1988;43:355–363. [PMC free article] [PubMed] [Google Scholar]
- 3.Machiela M.J., Chanock S.J. Detectable clonal mosaicism in the human genome. Semin. Hematol. 2013;50:348–359. doi: 10.1053/j.seminhematol.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youssoufian H., Pyeritz R.E. Mechanisms and consequences of somatic mosaicism in humans. Nat. Rev. Genet. 2002;3:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 5.Biesecker L.G., Spinner N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013;14:307–320. doi: 10.1038/nrg3424. [DOI] [PubMed] [Google Scholar]
- 6.Ainsworth P.J., Chakraborty P.K., Weksberg R. Example of somatic mosaicism in a series of de novo neurofibromatosis type 1 cases due to a maternally derived deletion. Hum. Mutat. 1997;9:452–457. doi: 10.1002/(SICI)1098-1004(1997)9:5<452::AID-HUMU12>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Papavassiliou P., York T.P., Gursoy N., Hill G., Nicely L.V., Sundaram U., McClain A., Aggen S.H., Eaves L., Riley B., Jackson-Cook C. The phenotype of persons having mosaicism for trisomy 21/Down syndrome reflects the percentage of trisomic cells present in different tissues. Am. J. Med. Genet. A. 2009;149A:573–583. doi: 10.1002/ajmg.a.32729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottlieb B., Beitel L.K., Trifiro M.A. Somatic mosaicism and variable expressivity. Trends Genet. 2001;17:79–82. doi: 10.1016/s0168-9525(00)02178-8. [DOI] [PubMed] [Google Scholar]
- 9.Amary M.F., Damato S., Halai D., Eskandarpour M., Berisha F., Bonar F., McCarthy S., Fantin V.R., Straley K.S., Lobo S. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet. 2011;43:1262–1265. doi: 10.1038/ng.994. [DOI] [PubMed] [Google Scholar]
- 10.Pansuriya T.C., van Eijk R., d’Adamo P., van Ruler M.A., Kuijjer M.L., Oosting J., Cleton-Jansen A.M., van Oosterwijk J.G., Verbeke S.L., Meijer D. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet. 2011;43:1256–1261. doi: 10.1038/ng.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwindinger W.F., Francomano C.A., Levine M.A. Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc. Natl. Acad. Sci. USA. 1992;89:5152–5156. doi: 10.1073/pnas.89.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groesser L., Herschberger E., Ruetten A., Ruivenkamp C., Lopriore E., Zutt M., Langmann T., Singer S., Klingseisen L., Schneider-Brachert W. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat. Genet. 2012;44:783–787. doi: 10.1038/ng.2316. [DOI] [PubMed] [Google Scholar]
- 13.Lindhurst M.J., Sapp J.C., Teer J.K., Johnston J.J., Finn E.M., Peters K., Turner J., Cannons J.L., Bick D., Blakemore L. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hafner C., Groesser L. Mosaic RASopathies. Cell Cycle. 2013;12:43–50. doi: 10.4161/cc.23108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heim S., Mitelman F. Nonrandom chromosome abnormailities in cancer-an overview. In: Heim S., Mitelman F., editors. Cancer Cytogenetics. John Wiley & Sons; Hoboken: 2009. pp. 25–44. [Google Scholar]
- 16.Bruder C.E., Piotrowski A., Gijsbers A.A., Andersson R., Erickson S., Diaz de Ståhl T., Menzel U., Sandgren J., von Tell D., Poplawski A. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am. J. Hum. Genet. 2008;82:763–771. doi: 10.1016/j.ajhg.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piotrowski A., Bruder C.E.G., Andersson R., Diaz de Ståhl T., Menzel U., Sandgren J., Poplawski A., von Tell D., Crasto C., Bogdan A. Somatic mosaicism for copy number variation in differentiated human tissues. Hum. Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- 18.Rodríguez-Santiago B., Malats N., Rothman N., Armengol L., Garcia-Closas M., Kogevinas M., Villa O., Hutchinson A., Earl J., Marenne G. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am. J. Hum. Genet. 2010;87:129–138. doi: 10.1016/j.ajhg.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.González J.R., Rodríguez-Santiago B., Cáceres A., Pique-Regi R., Rothman N., Chanock S.J., Armengol L., Pérez-Jurado L.A. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 2011;12:166. doi: 10.1186/1471-2105-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobs K.B., Yeager M., Zhou W., Wacholder S., Wang Z., Rodriguez-Santiago B., Hutchinson A., Deng X., Liu C., Horner M.-J. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 2012;44:651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laurie C.C., Laurie C.A., Rice K., Doheny K.F., Zelnick L.R., McHugh C.P., Ling H., Hetrick K.N., Pugh E.W., Amos C. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012;44:642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forsberg L.A., Rasi C., Razzaghian H.R., Pakalapati G., Waite L., Thilbeault K.S., Ronowicz A., Wineinger N.E., Tiwari H.K., Boomsma D. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Hum. Genet. 2012;90:217–228. doi: 10.1016/j.ajhg.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schick U.M., McDavid A., Crane P.K., Weston N., Ehrlich K., Newton K.M., Wallace R., Bookman E., Harrison T., Aragaki A. Confirmation of the reported association of clonal chromosomal mosaicism with an increased risk of incident hematologic cancer. PLoS ONE. 2013;8:e59823. doi: 10.1371/journal.pone.0059823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonnefond A., Skrobek B., Lobbens S., Eury E., Thuillier D., Cauchi S., Lantieri O., Balkau B., Riboli E., Marre M. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat. Genet. 2013;45:1040–1043. doi: 10.1038/ng.2700. [DOI] [PubMed] [Google Scholar]
- 25.Forsberg L.A., Rasi C., Malmqvist N., Davies H., Pasupulati S., Pakalapati G., Sandgren J., Diaz de Ståhl T., Zaghlool A., Giedraitis V. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat. Genet. 2014;46:624–628. doi: 10.1038/ng.2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campbell I.M., Yuan B., Robberecht C., Pfundt R., Szafranski P., McEntagart M.E., Nagamani S.C.S., Erez A., Bartnik M., Wiśniowiecka-Kowalnik B. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am. J. Hum. Genet. 2014;95:173–182. doi: 10.1016/j.ajhg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staaf J., Vallon-Christersson J., Lindgren D., Juliusson G., Rosenquist R., Höglund M., Borg A., Ringnér M. Normalization of Illumina Infinium whole-genome SNP data improves copy number estimates and allelic intensity ratios. BMC Bioinformatics. 2008;9:409. doi: 10.1186/1471-2105-9-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diskin S.J., Li M., Hou C., Yang S., Glessner J., Hakonarson H., Bucan M., Maris J.M., Wang K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008;36:e126. doi: 10.1093/nar/gkn556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pique-Regi R., Cáceres A., González J.R. R-Gada: a fast and flexible pipeline for copy number analysis in association studies. BMC Bioinformatics. 2010;11:380. doi: 10.1186/1471-2105-11-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurtin P.J., Dewald G.W., Shields D.J., Hanson C.A. Hematologic disorders associated with deletions of chromosome 20q: a clinicopathologic study of 107 patients. Am. J. Clin. Pathol. 1996;106:680–688. doi: 10.1093/ajcp/106.5.680. [DOI] [PubMed] [Google Scholar]
- 32.Döhner H., Stilgenbauer S., Benner A., Leupolt E., Kröber A., Bullinger L., Döhner K., Bentz M., Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000;343:1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 33.Shlush L.I., Zandi S., Mitchell A., Chen W.C., Brandwein J.M., Gupta V., Kennedy J.A., Schimmer A.D., Schuh A.C., Yee K.W., HALT Pan-Leukemia Gene Panel Consortium Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.R Development Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2013. R: A Language and Environment for Statistical Computing. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.