

Abstract
The myocardium undergoes extensive metabolic and energetic remodeling during the progression of cardiac disease. Central to remodeling are changes in the adenine nucleotide pool. Fluctuations in these pools can activate AMP-activated protein kinase (AMPK), the central regulator of cellular energetics. Binding of AMP to AMPK not only allosterically activates AMPK but also promotes phosphorylation of AMPK by an upstream kinase complex, LKB1/Mo25/STRAD (liver kinase B 1, mouse protein 25, STE-related adaptor protein). AMPK phosphorylation by the LKB1 complex results in a substantial increase in AMPK activity. Molecular targeting by the LKB1 complex depends on subcellular localization and transcriptional expression. Yet, little is known about the ability of the LKB1 complex to modulate targeting of AMPK after activation. Accordingly, we hypothesized that differing stoichiometric ratios of LKB1 activator complex to AMPK would uniquely impact myofilament function. Demembranated rat cardiac trabeculae were incubated with varying ratios of the LKB1 complex to AMPK or the LKB1 complex alone. After incubation, we measured the Ca2+ sensitivity of tension, rate constant for tension redevelopment, maximum tension generation, length-dependent activation, cooperativity, and sarcomeric protein phosphorylation status. We found that the Ca2+ sensitivity of tension and cross-bridge dynamics were dependent on the LKB1 complex/AMPK ratio. We also found that the LKB1 complex desensitizes and suppresses myofilament function independently of AMPK. A phospho-proteomic analysis of myofilament proteins revealed site-specific changes in cardiac Troponin I (cTnI) phosphorylation, as well as a unique distribution of cTnI phosphospecies that were dependent on the LKB1 complex/ AMPK ratio. Fibers treated with the LKB1 complex alone did not alter cTnI phosphorylation or phosphospecies distribution. However, LKB1 complex treatment independent of AMPK increased phosphorylation of myosin-binding protein C. Therefore, we conclude that the LKB1/AMPK signaling axis is able to alter muscle function through multiple mechanisms.
Introduction
During the progression of cardiac disease, the myocardium undergoes cellular and molecular remodeling, including a changing metabolic and energetic landscape (1,2). Central to energetic remodeling is an alteration in the production and use of ATP. The molecular underpinnings of the metabolic derangements that occur during cardiac disease involve changes in the mediators of ATP generation, utilization, and delivery. In general, creatine kinase (CK) reversibly and rapidly converts ADP and phosphocreatine (PCr) to ATP and creatine (Cr) (3). In a parallel reaction, adenylate kinase (AK) mediates a complementary intracellular phosphotransfer, promoting a high-energy Pi transfer from ADP to ATP (leaving an increasing [AMP] pool) via distinct isoforms with different cellular localizations (4,5).
As cardiac disease ensues, the loss of total Cr and PCr results in elevated [ADP] and [AMP] even if [ATP] is maintained (6). Further along the disease process, CK activity is reduced, leading to a gradual decrease in cellular [ATP] (7). Interestingly, the amount and activity of AK do not change in heart disease, which may be a compensatory mechanism in response to declining CK levels (4). Considering the relatively high rate of ATP synthesis in the heart (2), a gradual decrease in [ATP] can cause disproportionate energetic deficiencies (8,9). Such changes in energetics limit contractile reserve and the ability to power myocellular ATPases, which are necessary to support contractile function.
AMP-activated protein kinase (AMPK) has emerged as a key nongenomic, posttranslational regulator of cellular energy homeostasis. AMPK is a phylogenetically conserved heterotrimeric complex that consists of a catalytic α subunit and regulatory β and γ subunits (10). An increase in myocellular [AMP], as occurs with cardiac disease, allosterically activates AMPK and permits phosphorylation of the α catalytic subunit at thr172 by the upstream liver kinase B1 (LKB1) kinase complex (11–13). LKB1 acts in concert with Mo25 (mouse protein 25) and STRAD (ste-related adaptor protein) to phosphorylate AMPK, potentiating its activity and promoting ATP-producing pathways while inhibiting ATP-consuming pathways (12,13). In addition, AMPK targets ser150 of cardiac Troponin I (cTnI) and subsequently increases myofilament sensitivity to Ca2+ (14–16). Therefore, AMPK could not only respond to changes in CK and AK activity through changes in the ATP pool but could also directly tune myofilament function to the energetic demand through posttranslational modifications (PTMs).
Because of its energy-freeing reactions, AMPK is an attractive therapeutic target for heart failure. Indeed, AMPK activation promotes survival in ischemia-induced heart failure (17). We propose that a potential mechanism by which AMPK protects the heart during cardiac disease is by acting as a nodal point for sensing changes in cellular energetics and then appropriately targeting the contractile apparatus to alter contractility. However, exploring the mechanism of AMPK-based protection against heart failure is complicated by two critical findings: 1) CK and AK enzymes are localized to the myofilaments (4,18), and 2) stoichiometry of the upstream activator LKB1/Mo25/STRAD complex (LKB1 complex) and AMPK drives AMPK-target activity (19,20). This suggests that both localized adenine nucleotides pools and the stoichiometric activator complex are indispensable for driving AMPK-dependent targeting of myofilament proteins. Although we are pursuing the former concept, in this work we provide a novel (to our knowledge) insight into the latter one. In this study, we hypothesized that differing stoichiometric ratios of the LKB1 activator complex to AMPK would uniquely impact myofilament function. To examine this hypothesis, we assessed the Ca2+ sensitivity of tension, rate constant for tension redevelopment, maximum tension generation, length-dependent activation (LDA), and cooperativity in demembranated cardiac trabeculae after treatment with varying ratios of LKB1 complex to activated AMPK. In addition, we measured the phosphorylation pattern of myofilament proteins. We show that the Ca2+ sensitivity of tension and cross-bridge dynamics is dependent on the LKB1 complex/AMPK ratio. Unexpectedly, we show that the LKB1 complex can uniquely desensitize and suppress myofilament function independently of activating AMPK. The results of a phospho-proteomic analysis provide insight into the underlying mechanism of these observations.
Materials and Methods
Muscle mechanics
Cardiac muscle preparation
Two-month-old male Sprague-Dawley rats were anesthetized with isoflurane and their hearts were rapidly excised. The hearts were then retrogradely perfused with a modified Krebs-Henseleit solution (NaCl 118.5 mM, KCl 5 mM, MgSO4 1.2 mM, NaH2PO4 2 mM, D-(+)-glucose 10 mM, NaHCO3 25 mM, CaCl2 0.2 mM, and 2,3-butanedione monoxime 20 mM) (21). Thin, even, free-standing trabeculae were isolated from the right ventricular wall and left ventricular papillaries. After isolation, the trabeculae and cut papillaries were transferred to an ice-cold relaxing solution (Table S1 in the Supporting Material) containing 1% Triton X-100 for overnight demembranation at 4°C.
Activation and treatment of AMPK
Separate protocols were used to activate the AMPK complex. In one protocol, 20 nM of LKB1/Mo25/STRAD (LKB1 complex; Millipore, Darmstadt, Germany) was added to 100 nM of AMPK (α1β1γ1; Calbiochem, Darmstadt, Germany) in kinase reaction buffer (60 mM HEPES pH 7.5, 3 mM MgCl2, 3 μM Na3VO4, 1.2 mM DTT, 500 μM ATP, 10 mM EGTA, 5 μm NaF, 100 μM ADP, protease inhibitor cocktail (Sigma, St. Louis, MO), and 1 mM protein kinase A (PKA) inhibitor (Sigma) to prevent endogenous PKA activity) for 10 min at 37°C. In the second protocol, 100 nM of LKB1 complex was added to 100 nM of AMPK in the aforementioned kinase reaction buffer for 10 min at 37°C. Both protocols resulted in saturating levels of AMPK activation as measured by a direct AMPK kinase assay (data not shown) (20). As a control, 100 nM of the LKB1 complex in the absence of exogenous AMPK was used. To test how different stoichiometric ratios of the LKB1 to AMPK complex impact myofilament function, we incubated skinned trabeculae or papillaries with activated AMPK and either low LKB1 (10 nM LKB1:50 nM AMPK), high LKB1 (50 nM LKB1:50 nM AMPK), or the LKB1 complex alone (50 nM LKB1) in the kinase reaction buffer for 30 min at 30°C. After incubation, fibers were washed (15 min; repeated three times) with standard relaxing buffer on ice.
Experimental apparatus and protocol
The experimental apparatus used for mechanical measurements of cardiac trabeculae was similar to one described previously by Konhilas et al. (21) (Supporting Material). The Ca2+ sensitivity of tension development was determined by activating the muscle during a series of preactivating-activating-relaxation cycles using a range of free [Ca2+] in the activating solutions (Table S1) selected in random order (21). Sarcomere length (SL) was set at either 2.2 μm or 2.0 μm, as determined from the first-order He-Ne laser light diffraction band monitored by a 2048 pixel high-speed linear CCD sensor (Dexela Ltd., London, UK). Fibers reached steady-state tension and then were rapidly slackened by 20% of total fiber length. The difference between steady-state tension and slacked tension is the total tension (Fig. S1 A). Active tension is the difference between the total tension and relaxed, passive tension. To determine the rate constant for tension redevelopment (ktr), fibers reached steady-state tension at maximal and submaximal [Ca2+] under controlled SL (2.2 μm) and then subjected to a rapid release-restretch protocol (Fig. S1 B). Fibers that did not retain 85% of initial maximal tension or a diffraction pattern were discarded. Given these stringent criteria, the yields for mechanical experiments were ∼10%. Upon completion of each protocol, tissue was flash-frozen in liquid nitrogen and stored at −80°C for proteomic analysis.
Proteomic analysis
Sample preparation for SDS-PAGE and phospho-affinity SDS-PAGE
Myofibrillar proteins from frozen trabeculae or papillaries were isolated as detailed previously (22). An RC-DC assay (Bio-Rad, Hercules, CA) was performed to determine protein concentrations.
ProQ Diamond phosphoprotein stain
SDS-PAGE was used to separate myofibrillar proteins and measure phosphorylation status as detailed previously (23). After SDS-PAGE, gels were fixed and stained according to manufacturer’s (Invitrogen, Grand Island, NY) instructions. After phosphoprotein staining, gels were stained for total protein with Coomassie Brilliant Blue. Phosphorylated protein optical densities were quantified using LabImage 1D software and normalized to their respective Coomassie-stained total protein bands.
Western blot analysis
SDS-PAGE was used to separate myofibrillar samples. After transfer, total protein was measured with Ponceau S stain (Sigma). Antibodies were used to probe for total cTnI (AbCam Cambridge, UK; 1:5000), phospho-ser23/24 cTnI (Cell Signaling, Beverly, MA; 1:1000), phospho-ser150 cTnI (AbCam, 1:500), phospho-ser43cTnI (AbCam, 1:125), phospho-ser282 myosin-binding protein C (MBPC, 1:1000; Enzo Life Sciences, Farmingdale, NY), and total MBPC (Abcam, 1:1000). Protein optical densities were quantified using LabImage 1D software and normalized to total protein to adjust for alterations in loading parameters. Normalized optical densities from phospho-ser23/24cTnI, phospho-ser150cTnI, and phospho-ser43 cTnI were divided by the total cTnI optical density. In a similar manner, optical densities from phospho-ser282 MBPC were divided by the total protein C optical density.
All immunoblot analysis was performed from the semiquantitation of individual blots and was not compared across blots according to accepted guidelines. Some immunoblot images of a given target were cropped from the same blot to conserve figure space and avoid redundancy. All samples used for statistical analysis were obtained from different animals (no comparisons included two samples from the same rat).
Phosphate-affinity SDS-PAGE
Phosphate-affinity SDS-PAGE (Wako Pure Chemical Industries, Richmond, VA) was performed according to the manufacturer’s instructions and as described previously (24–26). Membranes were probed for total cTnI (1:5000), phospho-ser23/24cTnI (1:1000), and phospho-ser150cTnI (AbCam; 1:500). Total protein was measured by either SYPRO Ruby Protein Blot stain (Bio-Rad) or Ponceau S solution (Sigma). Human recombinant cTnI (hcTnI, kindly provided by Dr. Jil Tardiff) and rat cardiomyocytes treated with PKA (catalytic subunit, 1 U/μL) were used as relative standards to identify different phosphospecies populations (Supporting Material). The optical densities of the phosphospecies were quantified using LabImage 1D software and normalized to their SYPRO or Ponceau total protein bands. Each phosphospecies was normalized to their respective total cTnI optical density.
Data and statistical analysis
Tension in submaximally activating solutions was expressed as fractions (Frel) of the maximum tension (F0) at the same SL. The F0 value used to normalize submaximal tension was obtained by linear interpolation between successive maximal activations. Each individual Ca2+-tension relationship was fit to a modified Hill equation where Frel = [Ca2+]n/(EC50n + [Ca2+]n), Frel = relative tension, EC50 = [Ca2+] at which tension is half-maximal, n = slope of the Ca2+-tension relationship (Hill coefficient). The ΔEC50 was calculated as the difference in EC50 at SL = 2.0 μm and 2.2 μm for each experiment. Likewise, the ΔpCa50 was calculated as the difference in pCa50 (–log [EC50]) at SL = 2.0 μm and 2.2 μm. Tension redevelopment after the release and restretch protocol (ktr) was fit to a monoexponential function given by F = (F0 − Fres)(1 − ektrt) + Fres, where F0 is the steady-state isometric tension and Fres is the residual tension from which the fiber starts to redevelop tension. Results are presented as mean ± SEM. One-way ANOVA with Tukey’s post hoc test was used to determine differences in means. Multiple linear regression was also used to determine the relationship between ktr and tension, and p-values less than 0.05 were considered significant.
Animal subjects
All experiments were performed using protocols approved by the Institutional Animal Care and Use Committee of the University of Arizona, and conformed with NIH guidelines for the care and use of laboratory animals.
Results
Muscle mechanics
Ca2+-sensitive tension development
AMPK targets cTnI by phosphorylation at ser150, increasing the sensitivity of the myofilaments to Ca2+ (14–16,27). In addition, Ca2+ sensitivity has been used to index the impact of exogenously administered kinases such as PKA (28–30). We asked whether the amount of LKB1 complex relative to the activated AMPK complex would differentially impact myofilament function. Therefore, we determined the Ca2+-sensitive tension development of demembranated cardiac fibers incubated with increasing amounts of LKB1 complex relative to activated AMPK complex. Fig. 1 displays the Ca2+-sensitive tension development (SL = 2.2 μM) of demembranated fibers from four treatment groups: 1) untreated, 2) recombinant AMPK complex (rAMPK) activated with a relatively low amount of LKB1 complex to AMPK (rAMPK/LKB1lo), 3) rAMPK activated with a relatively high amount of LKB1 complex to AMPK (rAMPK/LKB1hi), and 4) LKB1 complex alone (LKB1). Similar to what was observed in previous work (15), when demembranated fibers were treated with rAMPK/LKB1lo, the Ca2+-tension relationship was shifted left, indicative of an increase in Ca2+-sensitive tension development compared with untreated fibers (Fig. 1 A). In contrast, Ca2+ sensitivity was decreased in fibers treated with rAMPK/LKB1hi when compared with untreated fibers (Fig. 1 A). Unexpectedly, treatment with the LKB1 complex in the absence of activated rAMPK desensitized the fibers to Ca2+ compared with the untreated group and both the rAMPK/LKB1lo and rAMPK/LKB1hi treatment groups (Fig. 1 B).
Figure 1.
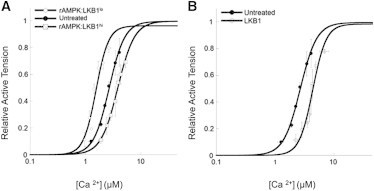
Ca2+ sensitivity of tension development in untreated and treated demembranated cardiac trabeculae. (A) Ca2+ sensitivity of tension in trabeculae treated with low levels of LKB1 complex activation of AMPK (open diamonds, rAMPK/LKB1lo) and high levels of LKB1 complex activation (open squares, rAMPK/LKB1hi) compared with untreated fibers (solid circles, untreated). (B) Ca2+ sensitivity of tension in trabeculae treated with only the LKB1 complex (open circles) compared with untreated fibers (solid circles). SL was set to 2.2 μm and all data were normalized to saturating Ca2+ (maximal) tension.
To test the response of the myofilaments to changes in length (LDA), the Ca2+ sensitivity of tension at a shorter SL (SL = 2.0 μM) was determined under each treatment condition. The impact of an SL change (ΔEC50 or ΔpCa50) on the Ca2+-tension relationship was not significantly different among all treatment groups (Table 1). However, treatment of fibers with the LKB1 complex alone significantly dampened the maximum tension relative to all other groups at both SLs. Cooperativity, as indexed by the Hill coefficient (Table 1), did not differ among the groups studied.
Table 1.
Ca2+ sensitivity of tension
| Untreated (n = 13) | rAMPK:LKB1lo (n = 8) | rAMPK:LKB1hi (n = 6) | LKB1 (n = 6) | |
|---|---|---|---|---|
| EC50 (μM) | 2.58 ± 0.11 | 1.56 ±. 06∗ | 3.71 ± .17∗ | 4.16 ± .12∗ |
| pCa50 | 5.59 ± 0.02 | 5.81 ± 0.02∗ | 5.43 ± 0.02∗ | 5.38 ± 0.01∗ |
| Hill coefficient | 5.04 ± 0.44 | 5.31 ± 0.61 | 3.98 ± 0.58 | 6.97 ± 2.29 |
| ΔEC50 | 0.58 ± 0.10 | 0.59 ± 0.06 | 0.60 ± 0.17 | 0.43 ± 0.08 |
| ΔpCa50 | 0.08 ± 0.02 | 0.13 ± 0.04 | 0.07 ± 0.03 | 0.03 ± 0.01 |
| Maximum tension (mN/mm2) | 33.98 ± 2.32 (n = 36) | 35.33 ± 4.07 (n = 14) | 30.02 ± 2.62 (n = 18) | 21.50 ± 1.71∗ (n = 27) |
| Tension Redevelopment | ||||
|---|---|---|---|---|
| Untreated (n = 10) | rAMPK:LKB1lo (n = 12) | rAMPK:LKB1hi (n = 10) | LKB1 (n = 11) | |
| ktr max (s−1) | 5.28 ± 0.27 | 6.14 ± 0.41 | 4.91 ± 0.25 | 5.35 ± 0.54 |
| Linear fit (s−1/P/P0) | 4.19 ± 0.48 | 6.16 ± 0.71∗ | 5.08 ± 0.46 | 3.59 ± 0.37 |
| Monoexponential fit (s−1/P/P0) | 2.26 ± 0.59 | 10.02 ± 2.37∗ | 4.71 ± 0.73 | 3.96 ± 1.11 |
Each column contains mean values ± SE. Cardiac trabeculae were untreated or treated with low amounts of activating LKB1 complex to AMPK (rAMPK/LKB1lo), high amounts of activating LKB1 complex to AMPK (rAMPK:LKB1hi), or exogenous LKB1 complex alone (LKB1). The Ca2+ sensitivity of tension for each group is indicated by EC50 (μM) and pCa50. There is an increase in Ca2+ sensitivity in the rAMPK/LKB1lo group (p < 0.001). There is a decrease in Ca2+ sensitivity in the rAMPK/LKB1hi group (p < 0.001), and an even further decrease in Ca2+ sensitivity is seen with the LKB1 complex alone (p < 0.001). A one-way ANOVA indicated that all EC50 values were different from one another. There was no difference in cooperativity (Hill coefficient) or LDA (ΔEC50, ΔpCa50) between SLs of 2.2 μm and 2.0 μm. Maximum tension generation was also determined; LKB1 complex treatment decreased tension generation (p < 0.05). In an additional set of skinned fibers, the relationship between ktr and tension was determined with both a linear and monoexponential fit to the data. Slopes for each fit are reported above. There is an increase in the slope between ktr and tension in the rAMPK/LKB1lo group regardless of linear or monoexponential fit (p < 0.05) compared with untreated fibers. ∗, statistically significant difference.
Rate of tension redevelopment: ktr
Considering the central role of AMPK as a regulator of cellular energy homeostasis, we wished to determine the impact of the different treatment protocols on measures of cross-bridge cycling. The rate constant for tension redevelopment (ktr) measures the sum of the apparent rates with which the actin-myosin cross-bridges enter and leave tension-generating states (31). Fig. S1 B shows representative tension recordings after a ktr protocol at activating [Ca2+] for each of the experimental conditions. There were no differences in maximal, or Ca2+ saturating, ktr (Table 1) between the treatment groups.
As ktr is dependent on both levels of activating Ca2+ and strongly bound cross-bridges, we determined ktr over a range of submaximal activating Ca2+, plotted against relative tension (Fig. 2) and fit by linear regression (Table 1). In addition to an increase in Ca2+ sensitivity, rAMPK/LKB1lo-treated fibers displayed a significantly steeper tension-ktr linear relationship compared with untreated fibers. However, the relationship between tension and ktr over the range of activating Ca2+ is curvilinear in striated muscle and is best fit with nonlinear regression (31). Even with a curvilinear fit to the tension-ktr relationship, rAMPK/LKB1lo-treated fibers displayed a significantly steeper slope compared with untreated fibers. Interestingly, the presence of activated rAMPK regardless of the amount of LKB1 complex resulted in a decreased ktr at submaximal tension (Fig. 2). However, LKB1-complex-treated fibers in the absence of activated rAMPK displayed a similar ktr at all activation levels.
Figure 2.
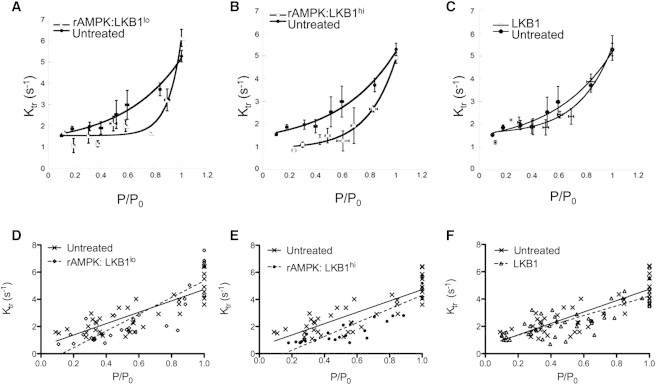
Rate constant for tension redevelopment. ktr is plotted as a function of normalized tension for fibers treated with low LKB1 activation of AMPK (rAMPK/LKB1lo), high LKB1 activation of AMPK (rAMPK/LKB1hi), and LKB1 complex alone (LKB1) plotted relative to untreated fibers. (A–C) Data were binned and fitted to a curvilinear fit. There is a significant increase in the slope of rAMPK/LKB1lo-treated fibers compared with untreated fibers (p < 0.05). rAMPK/LKB1hi-treated fibers tend to have an increased slope compared with untreated fibers. There is no difference between curvilinear slopes between LKB1-complex-treated fibers and untreated fibers. (D–F) Raw data were fitted with a linear regression, and multiple linear regression analysis was used to measure differences between slopes. There is a significant increase in the slope in fibers treated with rAMPK/LKB1lo (p < 0.05). There is a tendency for an increased slope with rAMPK/LKB1hi-treated fibers, with a significantly different intercept (p < 0.05) compared with untreated fibers. There is no difference in linear slopes between LKB1-complex-treated and untreated fibers.
Phosphorylation of sarcomeric proteins
Global phosphorylation status
Sarcomeric proteins are targets for multiple kinases and remain a central integration site for regulatory signaling cascades. As a result of these signaling cascades, PTM of myofilament proteins has known effects on Ca2+ sensitivity (28), the cross-bridge cycling rate (32) and Ca2+ binding characteristics (33). Considering the differential impact on Ca2+ sensitivity and tension redevelopment (ktr), we hypothesized that rAMPK/LKB1lo, rAMPK/LKB1hi, and LKB1 treatment would similarly have a differential effect on sarcomeric protein phosphorylation. Using SDS-PAGE followed by Pro-Q Diamond phosphoprotein staining, we were able to identify and quantify global phosphorylation levels of key myofilament proteins, including MBPC, desmin, cardiac troponin T (cTnT), cardiac tropomyosin (cTm), cTnI, and myosin light chain 1 (MLC-1). We found no measurable alteration in overall phosphorylation status in any of the activated rAMPK-treated groups. However, there was a detectable increase in MBPC phosphorylation in fibers treated with only the exogenous LKB1 complex (Fig. 3).
Figure 3.
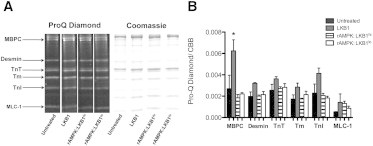
ProQ Diamond phosphoprotein stain after treatment with and without various activated AMPKs and the LKB1 complex. (A) Representative lanes (n = 4 per treatment group) from the same gel stained first for phosphorylation (ProQ Diamond) and then for total protein content (Coommassie Brilliant Blue). Protein identification as shown on the left was based on the molecular weights of known myofibrillar proteins. (B) The relative optical density of the phosphoprotein signal was normalized for loading by dividing the ProQ Diamond signal by the corresponding Coomassie Brilliant Blue signal per protein. There is a significant increase in phosphorylation of MBPC in skinned trabeculae treated with only exogenous LKB1 complex compared with all other groups (p < 0.05; n = 4 per group). In all other proteins, there is no significant change in global phosphorylation status.
Western blot site-specific phosphorylation status
cTnI
Although no differences in total phosphorylation were determined by Pro-Q Diamond phosphoprotein staining (Fig. 3), it remains possible that each treatment strategy resulted in a unique pattern of site-specific phosphorylation. Because AMPK is known to specifically target ser150 of cTnI (16), we first determined whether different amounts of the LKB1 complex could impact the ability of activating rAMPK to phosphorylate ser150 of cTnI by Western blot analysis using a phospho-ser150cTnI-specific antibody. As shown in Fig. 4, ser150-cTnI was robustly phosphorylated (phospho-ser150cTnI) in the presence of activated rAMPK independently of the LKB1 complex concentration. No difference in phospho-ser150cTnI was detected between untreated and LKB1-complex-treated groups.
Figure 4.
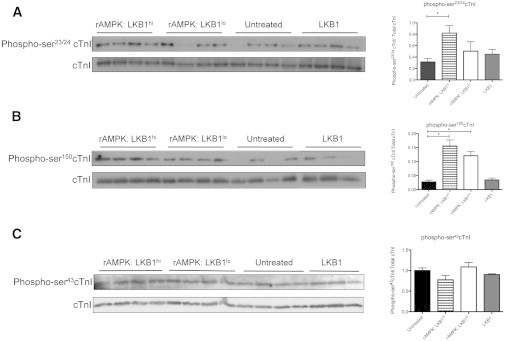
Western blot analysis of phosphorylated cTnI. The Western blot depicts phospho-cTnI in myofibrils treated with various stoichiometric LKB1 activations of AMPK and the LKB1 complex, with the corresponding normalized optical density plotted on the right. (A) Phospho-ser23/24 cTnI was measured in treated and untreated myofibrils. Fibers treated with a high level of LKB1 activation of AMPK (rAMPK/LKB1hi) had a significant increase in phosphorylated ser23/24 content relative to untreated myofibrils (p < 0.05; n = 4 for all groups). (B) Phospho-ser150 cTnI was measured in treated and untreated myofibrils. There was a significant increase in phosphorylated ser150 in rAMPK/LKB1hi- and rAMPK/LKB1lo-treated fibers (p < 0.05; n = 3–4 per group). (C) Phospho-ser43cTnI was measured in treated and untreated myofibrils. There was no change in phospho-ser43cTnI in any treatment group (p > 0.05).
AMPK, among other kinases, is capable of targeting ser23/24 of cTnI (16). In this study, we confirmed that activated rAMPK targeted and phosphorylated ser23/24-cTnI to a level that reached significance compared with controls only in the presence of high LKB1 (rAMPK/LKB1hi). Phospho-ser23/24cTnI levels in fibers treated with rAMPK/LKB1lo or LKB1 complex alone were unchanged from controls (Fig. 4). We also examined the PKC site, ser43cTnI, and found no changes in phospho-ser43cTnI content in any experimental group (Fig. 4).
MBPC
Pro-Q Diamond phosphoprotein staining suggests that the unique AMPK/LKB1 treatments impact the posttranslational pattern of MBPC phosphorylation. MBPC is targeted by many kinases at multiple phosphorylation sites (34), but to our knowledge, this study is the first to implicate MPBC as a potential target of the LKB1/AMPK signaling axis. Accordingly, we determined phosphorylation of MBPC at one of these sites (phospho-ser282MBPC) as an initial examination of this novel (to our knowledge) AMPK targeting. We found that fibers treated with rAMPK/LKB1lo showed a level of phospho-ser282MBPC that was significantly elevated compared with control fibers only (Fig. 5). Interestingly, this is different from the global elevation in MBPC phosphorylation in the LKB1-treated group as determined by Pro-Q Diamond phosphoprotein (Fig. 3). This is most likely due to potential differences in alternative MBPC phosphorylation sites as detailed in the Discussion.
Figure 5.
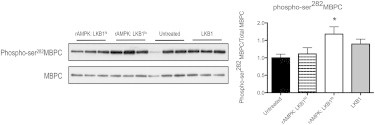
Western blot analysis of phosphorylated MBPC. The Western blot depicts phospho-ser282MPBC in myofibrils treated with activated AMPK and the LKB1 complex. The phosphorylated signal was normalized to total MBPC, and the corresponding normalized optical density is plotted on the right. Fibers treated with a low level of LKB1 activation of AMPK (rAMPK/LKB1lo) had a significant increase in phosphorylated ser282 content relative to untreated myofibrils (∗p < 0.05; n = 4 for all groups).
Phosphate-affinity SDS-PAGE
cTnI harbors multiple phosphorylation sites, including the PKA sites ser23/ser24, the protein kinase C sites ser43/45 and thr144, and the p-21 activated kinase/AMPK site ser150 (35). It has been suggested that the specific distribution of cTnI phosphorylation along its multiple phosphorylation sites can uniquely impact myofilament behavior (36,37). Therefore, we were interested in the overall distribution of cTnI phosphospecies in response to each treatment condition. We used the strategy of SDS-PAGE-Phos-tag coupled with phospho-specific antibodies to separate and measure the relative distribution of cTnI phosphospecies.
Using an antibody that recognizes total cTnI regardless of phosphorylation status, we found three distinct cTnI groups (Fig. 6) that have been designated by convention as the unphosphorylated (0P), monophosphorylated (1P), and bisphosphorylated (2P) species of phospho-cTnI. As expected, the summed densitometry of all three cTnI bands was not different among the groups studied. There was no change in cTnI phosphospecies distribution in fibers treated with LKB1 complex alone. Interestingly, when the same molar amount of LKB1 complex was added to the fibers in the presence of rAMPK, there was a shift in the distribution of cTnI (Fig. 6, rAMPK/LKB1hi). Fibers treated with rAMPK/LKB1hi had a shifted cTnI phosphospecies distribution away from the 0P unphosphorylated state toward the 2P bisphosphorylated state. The distribution away from the 0P state was also mirrored in the fibers treated with rAMPK/LKB1lo (Fig. 6, rAMPK/LKB1lo).
Figure 6.
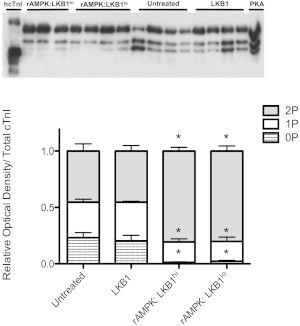
Phosphate-affinity SDS-PAGE (Phos-tag) of the total cTnI phosphospecies distribution. cTnI was separated into unphosphorylated [0P], monophosphorylated [1P], and bisphosphorylated [2P] phosphospecies by SDS-PAGE-Phos-tag followed by Western blot analysis with a total cTnI antibody. Top panel: representative SDS-PAGE-Phos-tag followed by Western blot, illustrating three bands corresponding to 0P, 1P, and 2P. Human recombinant cTnI (hcTnI) and PKA-treated (+PKA) cardiomyocytes were used to confirm each phosphospecies. Bottom panel: stacked bar graph indicating the relative amounts of each phosphospecies per experimental group. There is a significant shift in fibers treated with both high and low LKB1 activation of AMPK (rAMPK/LKB1hi and rAMPK:LKB1lo) away from the 0P state (p < 0.05). There is no change in total cTnI phosphospecies distribution in the LKB1-complex-treated group.
Phosphate-affinity SDS-PAGE was then performed again on cTnI using site-specific antibodies to phosphorylated ser23/24 and ser150 to determine the relative content of each cTnI phosphospecies. Cross-reactivity of the phospho-ser23/24 cTnI antibody with hcTnI at high concentrations (Fig. S2) identifies the 0P band and indicates the presence of phospho-ser23/24 cTnI in the 1P and 2P state. There was an increase in phosho-ser23/24 content in the 2P band for both rAMPK/LKB1hi and rAMPK/LKB1lo, with rAMPK/LKB1hi having the most overall phospho-ser23/24 in the 2P state (Fig. 7).
Figure 7.
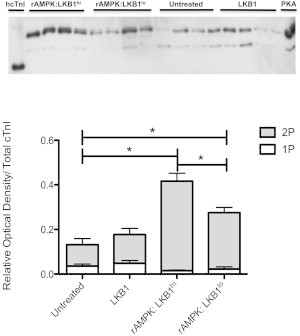
Phosphate-affinity SDS-PAGE (Phos-tag) of the phospho-ser23/24 cTnI phosphospecies distribution. cTnI was separated into unphosphorylated [0P], monophosphorylated [1P], and bisphosphorylated [2P] phosphospecies by SDS-PAGE-Phos-tag followed by Western blot analysis with a phospho-ser23/24 cTnI antibody. Top panel: representative SDS-PAGE-Phos-tag followed by Western blot, illustrating two distinct bands corresponding to 1P and 2P. Human recombinant cTnI (hcTnI) and PKA-treated (+PKA) cardiomyocytes were used to confirm each phosphospecies. Bottom panel: stacked bar graph indicating the relative amounts of each phosphospecies per experimental group. There is a significant increase in 2P phospho-ser23/24cTnI in rAMPK/LKB1hi- and rAMPK/LKB1lo-treated fibers (p < 0.05). In addition, rAMPK/LKB1hi-treated fibers contain more 2P phospho-ser23/24cTnI than rAMPK/LKB1lo-treated fibers. There was no change in the group treated only with LKB1 complex.
Similarly to our previous study, we were able to detect phosphorylation at phospho-ser150 cTnI in the 0P, 1P, and 2P bands, indicating the existence of phospho-ser150 cTnI in at least three states of cTnI phosphorylation (26). However, there was no overall change in the distribution of phospho-ser150 cTnI phosphospecies among groups (Fig. 8). Therefore, the different ratios of LKB1 activation of AMPK will shift cTnI phosphospecies distribution toward the 2P state, but the fibers treated with rAMPK/LKB1hi will have a greater phospho-ser23/24 content in the bisphosphorylated state than fibers treated with rAMPK/LKB1lo. It should also be noted that fibers treated with LKB1 complex alone showed no change in phospho-ser23/24 and phospho-ser150 cTnI phosphospecies distribution (Figs. 7 and 8).
Figure 8.
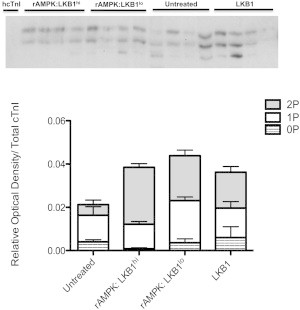
Phosphate-affinity SDS-PAGE (Phos-tag) of the phospho-ser150 cTnI phosphospecies distribution. cTnI was separated into unphosphorylated [0P], monophosphorylated [1P], and bisphosphorylated [2P] phosphospecies by SDS-PAGE-Phos-tag followed by Western blot analysis with a phospho-ser150 cTnI antibody. Top: representative SDS-PAGE-Phos-tag followed by Western blot illustrating two or three distinct bands corresponding to 0P, 1P, and 2P. hcTnI was used as the unphosphorylated cTnI standard. Bottom: stacked bar graph indicating the relative amounts of each phosphospecies per experimental group. There was no change in the distribution of phospho-ser150 cTnI phosphospecies among groups.
Discussion
Energetic remodeling is a hallmark of cardiac disease progression regardless of disease etiology. A disturbance in the CK/AK phosphotransfer system is observed early on and, importantly, is a stronger predictor of heart failure mortality than functional status (9). It is now appreciated that the driver for metabolic remodeling is the ATP requirement from myocellular ATP consumers such as myosin (2,38,39). Therefore, elucidating the underlying mechanisms of energetic remodeling requires a clear understanding of the costs of contraction. Consequently, we chose to examine the LKB1/AMPK signaling axis because it is uniquely positioned to respond to the changing energetic environment while modifying contractile function by directly targeting myofilament proteins. However, AMPK activity is subject to localized regulation by AMP pools. In addition, LKB1 target activation depends on its subcellular localization and transcriptional expression (4,18–20,40). Therefore, we hypothesized that the amount of the LKB1 complex relative to activated AMPK will uniquely impact myofilament function. We chose to use maximally activated AMPK and then vary the amount of LKB1, assuming that this would most appropriately represent in vivo conditions. The major findings from this study are that 1) the Ca2+ sensitivity of tension and cross-bridge cycling are dependent on the LKB1/AMPK ratio, 2) the LKB1 complex impacts contractile function independently of exogenous AMPK, 3) the pattern of PTMs indicates that the LKB1/AMPK ratio differentially alters cTnI phosphorylation, and 4) the LKB1 complex alone targets the myofilament, potentially through MBPC.
To best preserve the native state of myofilament proteins, we used intact demembranated cardiac fibers (trabeculae or cut papillaries) for incubation with each of the treatment protocols. Similar to what was observed in previous studies (15), we found that a lower LKB1/AMPK (rAMPK/LKB1lo) sensitizes the myofilaments to Ca2+, shifting the Ca2+-tension curve to the left. However, a higher molar ratio of LKB1 complex to AMPK (rAMPK/LKB1hi) desensitizes the myofilaments to Ca2+, shifting the Ca2+-tension curve to the right. Unexpectedly, when only exogenous LKB1 complex is incubated with cardiac trabeculae, the myofilaments are even further desensitized to Ca2+. In addition, fibers treated with rAMPK/LKB1lo demonstrate a reduced ktr compared with untreated fibers at submaximal Ca2+ concentrations (31). Because Ca2+-saturated (maximal) ktr is not different, the net result of a reduced submaximal ktr is a steeper tension-ktr relationship compared with all other groups.
The significance of this finding can be illustrated using a simplified two-state, cross-bridge model in which the rate of tension redevelopment, ktr, is proportional to both the attachment and detachment rates. Using this model, we predict that the dominant mechanism responsible for the lower, submaximal ktr is a decrease in the cross-bridge detachment rate. The implication from these data is a potential economizing of tension with AMPK treatment. In support of this contention, Oliveira et al. (14) demonstrated that treatment of ventricular myocytes with the AMPK activator 5-aminoimidazole-4-carboxamide ribonucleotide increases myocyte contractility without changing the amplitude of the Ca2+ transient. Although an increased sensitivity to Ca2+ may contribute to this observation, it is likely that a delay in cross-bridge detachment, evidenced by a decreased ktr as measured in this study, prolongs cross-bridge attachment and increases cell shortening. Similarly, a modest prolonged time to relaxation may indicate slower rates of cross-bridge detachment under AMPK activation. The ability of AMPK to provide an energetic benefit at the level of the myofilament is consistent with an energy-sparing function of AMPK activation, and may represent a fundamental mechanism by which AMPK impacts myofilament function and protects against a reduced ATP pool.
Still, the underlying mechanism that drives changes in myofilament function remains to be determined. PTMs of specific serine/threonine residues play a critical role in regulating parameters of myofilament function, including Ca2+ sensitivity, cross-bridge cycling, and passive tension (36). Consistent with an increase in Ca2+ sensitivity of tension and with previous studies (14,16), we found that incubation of rat cardiac trabeculae with activated AMPK regardless of the amount of LKB1 complex resulted in an increase in phospho-ser150cTnI relative to untreated controls.
Nixon et al. (15) previously demonstrated that the increase in Ca2+ sensitivity in fibers containing cTnI pseudophosphorylated at ser150 is blunted in the presence of cTnI pseudophosphorylated at ser23/24. Therefore, we also examined the phosphorylation status of ser23/24-cTnI. Western blot analysis indicated that there is also an overall increase in ser23/24 phosphorylation in rAMPK/LKB1hi-treated fibers. AMPK is capable of targeting ser23/24-cTnI, but the kinetic parameters of target-specific phosphorylation suggest that ser150 is the preferential site for activated AMPK (16). Our data indicate that increasing the amount of the LKB1 complex shifts the AMPK preferred site from ser150 to ser23/24. The implication is that the increase in phospho-ser23/24 blunts AMPK-dependent Ca2+ sensitization. However, our data also implicate an increase in LKB1 as a potential desensitization agent.
Apart from the changes in Ca2+ sensitivity, the reduction of submaximal ktr cannot be wholly attributable to PTM of cTnI. An increase in cTnI-ser23/24 phosphorylation accelerates ktr in the murine myocardium (41). Our data suggest that the presence of AMPK and LKB1 blunts the impact of cTnI-ser23/24 phosphorylation on ktr. This may be due to PTM of alternative myofilament proteins or to a unique localization/association of the LKB1/AMPK with the myofilaments, altering the cross-bridge kinetics. Future studies are required to determine the molecular mechanisms of these observations. These studies support the notion that controlling the relative activation of AMPK through the LKB1 complex is a potential mechanism for fine-tuning contractile dynamics through phosphorylation.
To better illustrate the distribution of phosphorylation sites on cTnI, we implemented SDS-PAGE-Phos-tag followed by site-specific Western blotting with cTnI-specific antibodies. We detected three distinct bands of cTnI designated, by convention, as 0P, 1P, and 2P, with 2P containing the largest phospho-cTnI population (26,42). In general, the pattern of increased phosphorylation with activated AMPK treatment regardless of the amount of LKB1 parallels the Western blot data obtained using SDS-PAGE without Phos-tag: there is a shift in phosphospecies distribution away from the 0P toward the 2P state. Although there is a discrepancy when the total ser150-cTnI phosphospecies is examined by SDS-PAGE-Phos-tag, the ser150-cTnI phosphospecies tends to exist predominantly in the 2P band. The latter observation is most likely due to the increase in ser23/24-cTnI phosphorylation, which is clearly elevated whether it is determined by traditional Western blotting or SDS-PAGE-Phos-tag.
Because multiple kinase pathways converge on cTnI, resulting in at least 10 possible phosphorylation sites (36), we can conclude that the categorization of each band as un-, mono-, or bisphosphorylated may underrepresent the existence and number of alternative cTnI phosphospecies. The SDS-PAGE-Phos-tag technique identifies the most abundant phosphospecies within the densitometric range only. In this study, therefore, we cannot rule out the existence of other phosphospecies as indicated in previous work (25). This contention is supported by the presence of phospho-ser150cTnI in the presumably unphosphorylated 0P band. We previously demonstrated that a custom-made phospho-ser150cTnI antibody detects cTnI only when phosphorylated at ser150 of cTnI (26). In this study, using a different, commercially available phospho-ser150cTnI antibody (see Materials and Methods), we again detected phospho-ser150cTnI in the 0P band (Fig. 7). Nevertheless, SDS-PAGE-Phos-tag remains an extremely useful tool for highlighting unique and distinct patterns of cTnI phosphorylation (and other phospho-species such as MBPC (43)) in response to signaling pathway activation.
Interestingly, the LKB1 complex’s interaction with the myofilament seems to occur by two mechanisms: through activation of the AMPK complex (as described above) or through an AMPK-independent mechanism. In our study, the LKB1 complex alone could decrease the Ca2+ sensitivity of tension development and maximum tension generation independently of exogenous AMPK. In contrast to AMPK-treated groups, LKB1 complex treated fibers had no changes to the phosphorylation status of cTnI, both globally or a site-specific distribution of cTnI phosphospecies. We identify PTM of MBPC as a potential novel (to our knowledge) mechanism for LKB1 to act independently of AMPK. However, when we probed for changes in PTM at a known phosphorylation site on MBPC, we found a discrepancy between global phosphorylation as measured by Pro-Q Diamond phosphoprotein staining and a single phosphorylation site, specifically phospho-ser282MBPC. This is not surprising considering that MBPC harbors multiple phosphorylation sites, all of which contribute to the summed, global phosphorylation level. Again, we posit that the pattern of site-specific phosphorylation may better represent the summed effect of these many kinase pathways. Future studies to identify the impact of the LKB1/AMPK pathway on site-specific changes in MBPC phosphorylation are warranted.
It is also likely that LKB1-dependent changes in myofilament function may operate through direct association of the LKB1 complex with myofilament proteins. Indeed, certain isoforms of AMPK have been shown to target sarcomeres at the Z disc (44), which could affect localized molar ratios of LKB1 and activating AMPK complex. However, further investigation of LKB1’s association with the myofilament is required.
Conclusion
The LKB1/AMPK signaling axis is a promising target for heart failure, and thus a clear understanding of this complex regulatory system is required. We show that this signaling pathway has the capability to impact myofilament function through two independent means. One pathway is through an LKB1-dependent activation of AMPK. In this system, changing stoichiometric activation of AMPK will change the Ca2+ sensitivity of tension, cross-bridge cycling, and the specific content and distribution of cTnI phosphospecies. Another pathway can operate seemingly independently of AMPK: the LKB1 complex alone can target the myofilament (potentially through MBPC), altering tension generation and the Ca2+ sensitivity of tension. Future studies will be directed toward elucidating the mechanism of action for the LKB1 complex and understanding the complex relationship among adenine nucleotides, LKB1, AMPK, and PTM of myofilament proteins.
Author Contributions
S.M.B. conceived and designed experiments acquired, analyzed, and interpreted all data; and drafted the manuscript and revised it for intellectual content. M.A.L.-P. and C.L.B. acquired data. L.A.K.M. revised the manuscript for intellectual content. C.D. interpreted Western blot data and revised the manuscript for intellectual content. Z.K. revised the manuscript for intellectual content. J.P.K. conceived and designed experiments, interpreted data, and drafted the manuscript and revised it for intellectual content.
Acknowledgments
We thank Carolyn Smith for proofreading the manuscript.
This work was supported by National Institutes of Health grant HL098256, a National Mentored Research Science Development Award (K01 AR052840) and Independent Scientist Award (K02 HL105799) from the NIH to J.P.K., and an Interdisciplinary Training Grant in Cardiovascular Sciences (HL007249). Support was also received from the Sarver Heart Center at the University of Arizona and the Steven M. Gootter Foundation.
Supporting Material
References
- 1.Doenst T., Nguyen T.D., Abel E.D. Cardiac metabolism in heart failure: implications beyond ATP production. Circ. Res. 2013;113:709–724. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingwall J.S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009;81:412–419. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nascimben L., Ingwall J.S., Allen P.D. Creatine kinase system in failing and nonfailing human myocardium. Circulation. 1996;94:1894–1901. doi: 10.1161/01.cir.94.8.1894. [DOI] [PubMed] [Google Scholar]
- 4.Aksentijević D., Lygate C.A., Neubauer S. High-energy phosphotransfer in the failing mouse heart: role of adenylate kinase and glycolytic enzymes. Eur. J. Heart Fail. 2010;12:1282–1289. doi: 10.1093/eurjhf/hfq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dzeja P., Terzic A. Adenylate kinase and AMP signaling networks: metabolic monitoring, signal communication and body energy sensing. Int. J. Mol. Sci. 2009;10:1729–1772. doi: 10.3390/ijms10041729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingwall J.S. Kluwer Academic Publishers; Boston: 2002. ATP and the Heart. [Google Scholar]
- 7.Weiss R.G., Gerstenblith G., Bottomley P.A. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc. Natl. Acad. Sci. USA. 2005;102:808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen W., Asai K., Ingwall J.S. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation. 1999;100:2113–2118. doi: 10.1161/01.cir.100.20.2113. [DOI] [PubMed] [Google Scholar]
- 9.Neubauer S. The failing heart—an engine out of fuel. N. Engl. J. Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 10.Hardie D.G., Hawley S.A., Scott J.W. AMP-activated protein kinase—development of the energy sensor concept. J. Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hawley S.A., Davison M., Hardie D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 12.Suter M., Riek U., Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 13.Baron S.J., Li J., Young L.H. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ. Res. 2005;96:337–345. doi: 10.1161/01.RES.0000155723.53868.d2. [DOI] [PubMed] [Google Scholar]
- 14.Oliveira S.M., Zhang Y.H., Redwood C. AMP-activated protein kinase phosphorylates cardiac troponin I and alters contractility of murine ventricular myocytes. Circ. Res. 2012;110:1192–1201. doi: 10.1161/CIRCRESAHA.111.259952. [DOI] [PubMed] [Google Scholar]
- 15.Nixon B.R., Thawornkaiwong A., Biesiadecki B.J. AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J. Biol. Chem. 2012;287:19136–19147. doi: 10.1074/jbc.M111.323048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sancho Solis R., Ge Y., Walker J.W. A preferred AMPK phosphorylation site adjacent to the inhibitory loop of cardiac and skeletal troponin I. Protein Sci. 2011;20:894–907. doi: 10.1002/pro.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gundewar S., Calvert J.W., Lefer D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009;104:403–411. doi: 10.1161/CIRCRESAHA.108.190918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koons S., Cooke R. Function of creatine kinase localization in muscle contraction. Adv. Exp. Med. Biol. 1986;194:129–137. doi: 10.1007/978-1-4684-5107-8_10. [DOI] [PubMed] [Google Scholar]
- 19.Shaw R.J., Kosmatka M., Cantley L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H., Untiveros G.M., Konhilas J.P. Micro-RNA-195 and -451 regulate the LKB1/AMPK signaling axis by targeting MO25. PLoS ONE. 2012;7:e41574. doi: 10.1371/journal.pone.0041574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konhilas J.P., Irving T.C., de Tombe P.P. Length-dependent activation in three striated muscle types of the rat. J. Physiol. 2002;544:225–236. doi: 10.1113/jphysiol.2002.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Layland J., Solaro R.J., Shah A.M. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc. Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 23.van der Velden J., Merkus D., Duncker D.J. Transmural heterogeneity of myofilament function and sarcomeric protein phosphorylation in remodeled myocardium of pigs with a recent myocardial infarction. Front. Physiol. 2011;2:83. doi: 10.3389/fphys.2011.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamdani N., Borbély A., van der Velden J. More severe cellular phenotype in human idiopathic dilated cardiomyopathy compared with ischemic heart disease. J. Muscle Res. Cell Motil. 2010;31:289–301. doi: 10.1007/s10974-010-9231-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Messer A.E., Gallon C.E., Marston S.B. The use of phosphate-affinity SDS-PAGE to measure the cardiac troponin I phosphorylation site distribution in human heart muscle. Proteomics Clin. Appl. 2009;3:1371–1382. doi: 10.1002/prca.200900071. [DOI] [PubMed] [Google Scholar]
- 26.McKee L.A., Chen H., Konhilas J.P. Sexually dimorphic myofilament function and cardiac troponin I phosphospecies distribution in hypertrophic cardiomyopathy mice. Arch. Biochem. Biophys. 2013;535:39–48. doi: 10.1016/j.abb.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliveira, S. M., J. Davies, D. Carling, H. Watkins, and C. Redwood. 2009. Cardiac troponin I is a potential novel substrate for AMP-activated protein kinase. 2009 Biophysical Society Meeting Abstracts. 20a(Suppl):841.
- 28.Konhilas J.P., Wolska B., Martin A.F., Solaro R.J., de Tombe P.P. PKA modulates length-dependent activation in murine myocardium. Biophys. J. 2000;78:108A. [Google Scholar]
- 29.Solaro R.J., Moir A.J.G., Perry S.V. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 30.de Tombe P.P., Stienen G.J. Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ. Res. 1995;76:734–741. doi: 10.1161/01.res.76.5.734. [DOI] [PubMed] [Google Scholar]
- 31.Brenner B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. USA. 1988;85:3265–3269. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kentish J.C., McCloskey D.T., Solaro R.J. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ. Res. 2001;88:1059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 33.Biesiadecki B.J., Kobayashi T., de Tombe P.P. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 2007;100:1486–1493. doi: 10.1161/01.RES.0000267744.92677.7f. [DOI] [PubMed] [Google Scholar]
- 34.Sadayappan S., de Tombe P.P. Cardiac myosin binding protein-C as a central target of cardiac sarcomere signaling: a special mini review series. Pflugers Arch. 2014;466:195–200. doi: 10.1007/s00424-013-1396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solaro R.J., Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J. Biol. Chem. 2011;286:9935–9940. doi: 10.1074/jbc.R110.197731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solaro R.J., Henze M., Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ. Res. 2013;112:355–366. doi: 10.1161/CIRCRESAHA.112.268672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang P., Kirk J.A., Murphy A.M. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kammermeier H. Thermodynamic aspects of ATP homeostasis. Zoology. 1998;101:1–6. [Google Scholar]
- 39.Witjas-Paalberends E.R., Güçlü A., van der Velden J. Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc. Res. 2014;103:248–257. doi: 10.1093/cvr/cvu127. [DOI] [PubMed] [Google Scholar]
- 40.Fan D., Ma C., Zhang H. The molecular mechanisms that underlie the tumor suppressor function of LKB1. Acta Biochim. Biophys. Sin. (Shanghai) 2009;41:97–107. doi: 10.1093/abbs/gmn011. [DOI] [PubMed] [Google Scholar]
- 41.Patel J.R., Fitzsimons D.P., Moss R.L. PKA accelerates rate of force development in murine skinned myocardium expressing alpha- or beta-tropomyosin. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H2732–H2739. doi: 10.1152/ajpheart.2001.280.6.H2732. [DOI] [PubMed] [Google Scholar]
- 42.Kinoshita E., Kinoshita-Kikuta E., Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Kooij V., Holewinski R.J., Van Eyk J.E. Characterization of the cardiac myosin binding protein-C phosphoproteome in healthy and failing human hearts. J. Mol. Cell. Cardiol. 2013;60:116–120. doi: 10.1016/j.yjmcc.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pinter K., Grignani R.T., Redwood C. Localisation of AMPK γ subunits in cardiac and skeletal muscles. J. Muscle Res. Cell Motil. 2013;34:369–378. doi: 10.1007/s10974-013-9359-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.