

Abstract
Although the magnitude of a protein’s net charge (Z) can control its rate of self-assembly into amyloid, and its interactions with cellular membranes, the net charge of a protein is not viewed as a druggable parameter. This article demonstrates that aspirin (the quintessential acylating pharmacon) can inhibit the amyloidogenesis of superoxide dismutase (SOD1) by increasing the intrinsic net negative charge of the polypeptide, i.e., by acetylation (neutralization) of multiple lysines. The protective effects of acetylation were diminished (but not abolished) in 100 mM NaCl and were statistically significant: a total of 432 thioflavin-T amyloid assays were performed for all studied proteins. The acetylation of as few as three lysines by aspirin in A4V apo-SOD1—a variant that causes familial amyotrophic lateral sclerosis (ALS)—delayed amyloid nucleation by 38% and slowed amyloid propagation by twofold. Lysines in wild-type- and ALS-variant apo-SOD1 could also be peracetylated with aspirin after fibrillization, resulting in supercharged fibrils, with increases in formal net charge of ∼2 million units. Peracetylated SOD1 amyloid defibrillized at temperatures below unacetylated fibrils, and below the melting temperature of native Cu2,Zn2-SOD1 (e.g., fibril Tm = 84.49°C for acetylated D90A apo-SOD1 fibrils). Targeting the net charge of native or misfolded proteins with small molecules—analogous to how an enzyme’s Km or Vmax are medicinally targeted—holds promise as a strategy in the design of therapies for diseases linked to protein self-assembly.
Introduction
Coulombic forces drive the intermolecular repulsion and attraction of protein molecules at distances up to 10 Å in physiological buffer and possibly longer if the interaction occurs across the hydrophobic interior of a protein or lipid bilayer (1,2). Although these far-reaching forces scale with each protein’s magnitude of net positive or net negative charge (Z), the net charge of a protein has never been scrutinized as a medicinal target that can be permuted with a small molecule. Instead, small molecule drugs that target proteins are designed to affect other classical parameters such as Km, Vmax, Kd, and ΔG‡fold, by the introduction of hydrophobic, dipolar, or H-bonding interactions (3–5). Developing drugs that can increase the magnitude of net charge of a protein—for example, making an anionic protein more anionic—and thereby control intermolecular interactions electrostatically, might be effective in combating diseases that are linked to protein self-assembly, such as amyotrophic lateral sclerosis (ALS) (6). ALS, a currently untreatable and a fatal neurodegenerative disease, is characterized by the selective, progressive death of motor neurons. More than 150 different mutations in the gene encoding superoxide dismutase-1 (SOD1) cause familial ALS by a gain of function mechanism (6). What this function might be is still unclear but one leading hypothesis suggests that SOD1 mutations cause ALS by accelerating the rate of self-assembly of SOD1 into oligomers that are, for some unknown reason, toxic to motor neurons (6). The self-assembly of wild-type (WT) SOD1 is also suspected to cause certain cases of sporadic (nonfamilial) ALS (6). The exact stoichiometry, morphology, and structure of neurotoxic SOD1 oligomers (assuming that such oligomers cause ALS) have not been determined, although several studies point to amyloid-like SOD1 oligomers as toxic species (7).
In this article, we describe a first-order proof of this charge-boosting principle by demonstrating that lysine residues in WT- and ALS-linked SOD1 can be acetylated with an aryl ester drug (acetylsalicylic acid; aspirin), both before and after the fibrillization of SOD1 into amyloid (Fig. 1). We find that the nonspecific acetylation of as few as three lysines (out of 11) in a subunit of A4V apo-SOD1—a reaction that increases the magnitude of net negative charge of SOD1 (pI = 5.9)—delayed the nucleation of amyloid fibrillization by up to 5 h and decreased the rate of fibril propagation by twofold (Table 1). This diminished rate of nucleation is similar to, and the rate of propagation is slower than that of unacetylated WT apo-SOD1.
Figure 1.
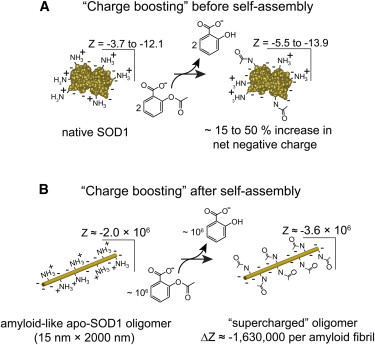
Chemically boosting the net charge (Z) of SOD1. The accumulation of acetyl modifications on native or oligomeric SOD1 by repeated doses of an acylating agent is plausible when considering the long lifetime of native SOD1 in motor neurons (27), and the generally long lifetime of amyloid oligomers in vivo (28,29). (A) The net charge of dimeric WT SOD1 has been measured to vary from ∼−3.7 to ∼−12.1, depending on the metalation state and subcellular pH (25). Acetylation of lysine residues in native SOD1 will increase the magnitude of net negative charge by between 15% and 50%, depending on subcellular pH and metalation state. (B) Peracetylating lysine residues in an amyloid fibril of SOD1 (d = 15 nm, l = 2 μm) with aspirin is expected to increase the magnitude of net negative charge by ∼106 formal units. To see this figure in color, go online.
Table 1.
Rate of nucleation and propagation of amyloid fibrils of WT- and ALS-variant apo-SOD1 as a function of lysine acetylation, Ac(N)
| SOD1a | Ac (∼N)b | 0 mM NaCl (n = 18) |
100 mM NaCl (n = 18) |
||||
|---|---|---|---|---|---|---|---|
| Lag time (h) (ΔLag time)c | 1/k (h)c (Δ1/k)d | P-valuee | Lag time (h) (ΔLag time)d | 1/k (h)b (Δ1/k)d | P-valuee | ||
| WT | 0 | 13.9 ± 1.1 | 1.1 ± 0.1 | 15.4 ± 1.1 | 1.8 ± 0.1 | ||
| 1 | 15.0 ± 1.2 (1.1 ± 1.6) | 2.2 ± 0.2 (1.1 ± 0.2) | 0.48 | 17.6 ± 1.6 (2.2 ± 1.9) | 2.7 ± 0.2 (0.9 ± 0.2) | 0.27 | |
| 3 | 16.5 ± 0.7 (2.6 ± 1.3) | 2.6 ± 0.1 (1.5 ± 0.1) | 0.06 | 16.9 ± 0.8 (1.5 ± 1.4) | 2.4 ± 0.1 (0.6 ± 0.1) | 0.28 | |
| 6 | 24.9 ± 1.3 (11.0 ± 1.7) | 3.3 ± 0.3 (2.2 ± 0.3) | <0.0001 | 25.5 ± 0.9 (10.1 ± 1.4) | 3.2 ± 0.3 (1.4 ± 0.3) | <0.0001 | |
| D90A | 0 | 11.9 ± 1.0 | 2.1 ± 0.2 | 9.3 ± 0.9 | 1.5 ± 0.2 | ||
| 2 | 11.1 ± 1.1 (−0.8 ± 2.1) | 3.3 ± 0.3 (1.2 ± 0.5) | 0.57 | 14.0 ± 1.1 (4.7 ± 1.4) | 2.1 ± 0.2 (0.6 ± 0.2) | 0.002 | |
| 3 | 9.8 ± 1.1 (−2.1 ± 2.1) | 3.4 ± 0.2 (1.3 ± 0.4) | 0.15 | 14.1 ± 1.1 (4.8 ± 1.4) | 2.2 ± 0.2 (0.7 ± 0.2) | 0.002 | |
| 5 | 12.4 ± 1.2 (0.5 ± 1.6) | 3.8 ± 0.2 (1.7 ± 0.2) | 0.76 | 15.2 ± 1.4 (5.9 ± 1.7) | 2.6 ± 0.2 (1.1 ± 0.2) | 0.001 | |
| A4V | 0 | 13.8 ± 1.7 | 2.2 ± 0.2 | 12.7 ± 1.3 | 2.2 ± 0.3 | ||
| 3 | 20.1 ± 2.1 (6.3 ± 2.7) | 4.1 ± 0.4 (1.9 ± 0.4) | 0.02 | 17.5 ± 1.3 (4.8 ± 1.3) | 4.8 ± 0.3 (2.6 ± 0.3) | 0.01 | |
| 4 | 11.5 ± 0.7 (−2.3 ± 1.8) | 3.2 ± 0.3 (1.0 ± 0.4) | 0.21 | 15.6 ± 2.2 (2.9 ± 2.5) | 4.6 ± 0.5 (2.4 ± 0.6) | 0.26 | |
| 9 | 28.3 ± 1.7 (14.5 ± 1.7) | 4.9 ± 0.3 (2.7 ± 0.4) | <0.0001 | 17.1 ± 1.2 (4.4 ± 1.3) | 4.7 ± 0.4 (2.5 ± 0.5) | 0.02 | |
All acetylated and unacetylated SOD1 proteins were in the metal free state with <0.06 ± 0.02 Zn2+ and < 0.03 ± 0.01 Cu2+ bound per SOD1 dimer.
Mean number of acetylated residues, per apo-SOD1 monomer, as inferred from CE and MS.
Rate of propagation expressed as inverse propagation constant (1/k).
ΔLag time and Δpropagation values are expressed relative to Ac(0) for each protein. All values and errors are listed as mean ± SE from 18 replicate ThT assays for each sample; the R2 values of all fits were >0.98.
This table only lists p-values comparing the lag time of acetylated proteins with unacetylated protein; all p-values comparing 1/k values of acetylated vs. unacetylated protein were ∼p < 0.005.
We chose acetylsalicylic acid in this proof of concept study because it acetylates serine and lysine residues in multiple proteins in vivo (8,9) (in addition to acetylating serine in its primary target, cyclooxygenase-1; COX1) (9). Aspirin was recently found to acetylate lysine and serine in 120 different proteins in cultured cells (10), but its reaction with SOD1 has never been reported, either in vivo or in vitro. Aspirin also exhibits therapeutic effects in several diseases (9) and has mysteriously beneficial effects in ALS-SOD1 transgenic mouse models (11). The acetylation of lysine is a posttranslational modification that is involved in controlling multiple cellular processes such as cell signaling, nucleic acid binding, gene expression, protein-protein interactions, and enzymatic activity (12).
The charge-boosting hypothesis was inspired, and is supported by the following: 1) the inverse correlation between the magnitude of a protein’s net charge and its rate of self-assembly into amyloid and nonamyloid aggregates (13,14) (including the historical observation that proteins precipitate rapidly at their isoelectric point (15,16)); 2) the naturally small net charge of proteins (∼80% are predicted to have net charges between +10 and −10, with median Z = −3) (17); 3) the observation that lysine acetylation can prevent protein precipitation by increasing net negative charge (18,19); 4) observations that the permeability of a protein across glycated, anionic cellular membranes—a process that might be requisite for the cell-to-cell, prion-like propagation of aggregated SOD1 (20)—can be abolished by inverting the protein’s sign of net charge from positive to negative (21); and 5) the occurrence of cryptic ALS mutations in sod1 (e.g., D90A, D101N, E100K, and N139K) that do not appear to alter the structure, stability, enzymatic activity, or metal binding properties of the SOD1 native state, but do neutralize its net negative charge (22,23). Although these cryptic substitutions might promote aggregation by altering local patterns of net charge, their scattering across SOD1’s surface suggests that neutralizing the negative surface potential at multiple loci is sufficient to trigger ALS, or equally and alternately, that neutralizing the net negative charge of SOD1 is sufficient for pathogenesis (24).
The use of a small molecule to increase the net charge of a folded protein is complicated by the absence of experimentally determined values of net charge for nearly all folded proteins. We estimate that the net charge of <0.1% of proteins in the Protein Data Bank have been measured, at pI ≠ pH (24,25). Recently, we used protein charge ladders and capillary electrophoresis (CE) to show that the measured net charge (denoted ZCE) of WT- and ALS-variant SOD1 can deviate (in sign and magnitude) from predicted values (denoted Zseq) at certain metal stoichiometries and subcellular pH (24,25). At pH 7.4, the net charge of WT Cu2,Zn2-SOD1 was ZCE = −7.37 charge units per dimer (Zseq = −8.2); and the net charge of apo-SOD1 was ZCE = −12.13 units per dimer (Zseq = −13.0). At pH 5.0 (i.e., the pH of the lysosome, one of SOD1’s subcellular loci (26)), the net charge of WT Cu2,Zn2-SOD1 was only ZCE = −3.7 units per dimer, which is opposite in sign and eight units more negative than the predicted value of Zseq = +4.0 (25). The net charge of WT apo-SOD1 could not be determined at pH 5, presumably because SOD1 approached Z = 0 and precipitated during CE (25). Nevertheless, the acetylation of a single lysine in a subunit of WT SOD1 is expected to increase its magnitude of net negative charge by ∼15% to 50% (per subunit), depending on its state of metalation and subcellular locus (Fig. 1 A). The effects of lysine acetylation on certain ALS-variant SOD1 proteins (e.g., D90A, G93R, E100K) will be even larger, as these proteins teeter on the edge of electrostatic neutrality at physiological pH (24).
Materials and Methods
Purification of WT- and ALS-variant apo-SOD1
Detailed descriptions of all experimental procedures can be found in the Supporting Material. Briefly, human WT, D90A, and A4V SOD1 proteins were recombinantly expressed in Saccharomyces cerevisiae, purified, demetallated, and characterized as previously described (25). The absence of metals was confirmed after demetallation (and again after acetylation) with inductively coupled plasma mass spectrometry (ICP-MS), as previously described (25).
Acetylation of WT- and ALS-variant apo-SOD1 with aspirin
Crystalline acetylsalicylic acid was purchased from Sigma-Aldrich (purity > 99.0%) and used for the acetylation of lysine residues in WT- and ALS-variant apo-SOD1 (see Supporting Material for additional details). All solutions and buffers were made with ultrapure metal-free water with a measured resistance of 18.2 MΩ/cm (dispensed through a Millipore MilliQ system). All containers used for making buffers or other solutions were rinsed with 5 mM EDTA and pure water before use. Peracetylated apo-SOD1 fibrils were also prepared by adding crystalline acetylsalicylic acid to aqueous solutions of unacetylated amyloid fibrils of apo-SOD1. Reaction byproducts (acetic acid, salicylic acid) and unreacted aspirin were removed by centrifugation and the discarding of supernatant. An identical aliquot of unmodified fibrils was set aside and not reacted with aspirin and functioned as a negative control. The buffer in which fibrils were suspended was changed to 10 mM potassium phosphate, 5 mM EDTA, pH 7.4 (for thermal stability assays) via centrifugation, and removal of supernatant followed by addition of fresh buffer. After acetylation, all apo-SOD1 proteins were analyzed with ICP-MS to confirm the absence of metal ions.
Biophysical characterization and mass spectrometric analysis of native and fibrillar apo-SOD1 proteins
Analysis of acetylated and unacetylated apo-SOD1 proteins with CE, differential scanning calorimetry, transmission electron microscopy, mass spectrometry (MS), and tandem mass spectrometry (MS/MS) was performed as previously described (27,30). MS/MS analysis was performed to characterize the types of acetyl-regioisomers that likely formed on nonspecific acetylation with aspirin. The effect of lysine acetylation on the structure of SOD1 proteins was also analyzed with amide hydrogen/deuterium exchange (H/D) and MS, as previously described (see Supporting Material for additional details). Measuring H/D exchange with MS allowed each acetyl derivative to be analyzed simultaneously under identical pH, and temperature, thus eliminating the sample-to-sample experimental variation that often occurs during H/D exchange-MS. To determine the identity of acetylated residues on fibrillar apo-SOD1, LC-MS and MS/MS analysis was performed after fibril dissociation to monomeric SOD1. Fibrils were dissociated by incubation in 6.5 M guanidinium hydrochloride (Gdm-HCl), 20 mM dithiothreitol (DTT) at 50°C for 1 h. This thermochemical defibrillization protocol has been previously shown to be adequate for dissociating amyloid fibrils of apo-SOD1 (31).
Fluorescent kinetic assays for apo-SOD1 proteins
Thioflavin-T (ThT) fluorescence assays for apo-SOD1 fibrillization were performed in a sealed 96-well microplate as previously described (27). To acquire statistically meaningful lag times and propagation rates, we performed 18 replicates of ThT assays for each of the three apo-SOD1 proteins and their four sets of acetylated derivatives, in the absence and presence of 100 mM NaCl, amassing a total of 432 separate assays. For any set of acetylated proteins, each of the 18 replicates was performed simultaneously in the same 96-well plate, on solutions of SOD1 taken from the same stock of acetylated or unacetylated SOD1. Before aggregation assays, solutions were filtered with a syringe filter (0.2 μm hydrophilic polypropylene, Acrodisc Pall Co., Port Washington, NY) to remove any type of high molecular weight oligomers of apo-SOD1 (or other colloids) that might seed or catalyze fibrillization. The concentration and purity of apo-SOD1 was confirmed with reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) immediately before the initiation of each aggregation assay and at the completion of the assay. The volume of each well was also measured at the end of the assay (with a micropipette) to ensure that evaporation did not occur during the aggregation assay.
Thermal stability assays for supercharged fibrillar apo-SOD1 proteins
The thermal stability of the unacetylated and acetylated apo-SOD1 fibrils was determined with a thermal defibrillization assay that was adapted from a previous study by Shammas et al. (32). Fibril homogenate from both acetylated and unacetylated fibrils of apo-SOD1 (in 10 mM potassium phosphate, 5 mM EDTA, pH 7.4) were aliquoted into ten different tubes that were each heated to a specific temperature between 40°C to 130°C. The temperature was raised at an average rate of 0.5 to 1.0°C/min (similar to a typical heating rate during differential scanning calorimetry). To reach temperatures up to 100°C, solutions were heated using a PCR machine (Mastercycler, Eppendorf, Hamburg, Germany). For temperatures above 100°C, a heat block was used (VWR, Radnor, PA). To avoid solvent evaporation during heating above 100°C, we used a pressurized external lid that was designed to cover the sample vials (that reached pressures of ∼350 kPa). At each temperature, a tube of protein solution was cooled and centrifuged at 17,000 × g (4°C) for 30 min and the supernatants were analyzed with ultraviolet-visible spectrophotometry (240 to 300 nm). Thus, the thermal defibrillization of the fibrils was detected by the retention of soluble, nonsedimentable SOD1 in the supernatant. The same four-parameter sigmoidal function that was fitted to the ThT fibrillization data (Eq. 1 in the Supporting Material) was fitted to the data points from the thermal defibrillization assays using SigmaPlot, version 11.0 (Systat Software, Chicago, IL); see Fig. S1. The inflection point of the sigmoid, x0, was reported as the melting temperature (Tm) of the amyloid fibrils.
The relative molecular weights of nonsedimentable species that might be left over from the thermal defibrillization assays—i.e., melted fibrils, which still might exist as low molecular weight oligomers—were determined with size-exclusion liquid chromatography (SE-LC) and native PAGE. See Supporting Material for additional details.
Estimating the net charge of fibrillar apo-SOD1
The magnitude by which peracetylation affected the net charge of apo-SOD1 fibrils was estimated by approximating the number of SOD1 chains in an average-sized SOD1 fibril, and assuming that each lysine acetylation increased the net negative charge of the polypeptide by 0.9 units (instead of 1.0 unit), because of charge regulation (2). The number of SOD1 subunits in each fibril was approximated by estimating the molecular volume of misfolded SOD1 as described in the Supporting Material, based on a spherical approximation (33).
Results and Discussion
Acetylation of lysine in dimeric WT- and ALS-variant apo-SOD1 with acetylsalicylic acid
MS and CE showed that the reaction of 25 to 150 mM aspirin with native WT- or ALS-variant apo-SOD1 resulted in chemical arrays of apo-SOD1 proteins with varying degrees of acetylation per monomer (denoted “Ac(∼N),” Figs. 2 and S2). These aspirin concentrations are considerably higher than peak concentrations of intact (unhydrolyzed) acetylsalicylic acid in human plasma, which reach 30 to 150 μM (depending on dosage); concentrations of salicylic acid reach as high as 2.5 mM (9). The degree of acetylation for each mixture is expressed as the mean number of acetyl modifications in each mixture per monomer, as measured by MS and CE (Figs. 2 and S2). The mean number of acetylated lysines was determined from a Gaussian distribution of the MS charge ladders. Each acetylation is associated with a +42 Da increase in mass, Fig. 2, A–C. At 25 mM aspirin, approximately one lysine (mean) was acetylated in each WT apo-SOD1 monomer; two lysines per monomer in D90A; and three lysines per monomer in A4V apo-SOD1 (MS/MS sequencing is discussed below and in the Supporting Material). At 150 mM aspirin, the mean acetylation observed for WT apo-SOD1 was six lysines per monomer; five lysines per monomer for D90A; and nine lysines per monomer for A4V apo-SOD1 (Fig. 2 and Table S1).
Figure 2.
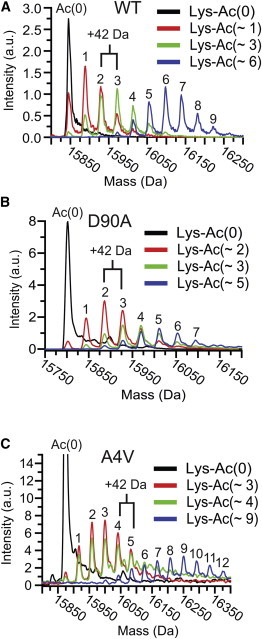
Acetylation of multiple lysines in WT- and ALS-variant apo-SOD1 by aspirin. Electrospray ionization mass spectra of soluble (A) WT, (B) D90A, and (C) A4V apo-SOD1 after reaction with different concentrations of aspirin (in aqueous buffer). The mean number of acetylated lysines are denoted as Lys-Ac(∼N), and are listed per apo-SOD1 monomer. To see this figure in color, go online.
Each of these acetylated arrays (charge ladders) of WT and D90A apo-SOD1 were partially resolvable with CE, wherein each acetylation increased the electrophoretic mobility, i.e., increased the net negative charge of SOD1 (Fig. S2, A–C). The capillary electropherogram of A4V apo-SOD1 exhibited poor resolution (Fig. S2 C), possibly because of its well-known dimer instability and dynamic structure (34). Proteolysis and MS/MS confirmed that each modification occurred on lysine; no other acetylated residues were detected in WT, D90A, or A4V apo-SOD1, with the following exception: in A4V apo-SOD1, we detected (via MS/MS analysis) a small amount of acetylation on Thr-88 and Ser-105, but only at [aspirin] = 150 mM (Fig. S4). We were not able to add sufficient aspirin to peracetylate any of the three SOD1 proteins because each protein began to precipitate after the addition of >150 mM aspirin.
The acetylation of nine lysines in WT and D90A apo-SOD1 was apparently nonspecific according to MS/MS (Table S1). For example, at least nine out of 11 lysines were acetylated in WT or D90A apo-SOD1 regardless of the mean acetylation. Lys-3 and Lys-91, however, were not acetylated in WT and D90A apo-SOD1 at any concentration of aspirin, except 150 mM aspirin, wherein Lys-91 was found to be acetylated in WT apo-SOD1 (Table S1). In contrast, the acetylation of all 11 lysines in A4V apo-SOD1 by aspirin was nonspecific. For example, a solution of A4V apo-SOD1 with a mean number of three acetylations per monomer was found with MS/MS to contain acetyl modifications on all 11 lysines (Table S1). See Supporting Material for additional discussion of the MS/MS data. Thus, each rung in the charge ladder consisted of a mixture of regioisomers. If we assume that all 11 lysines in a monomeric SOD1 protein are equally reactive (which is unlikely, considering that each lysine likely has its own specific pKa), then the maximum number of statistically possible regioisomers (r) can be expressed by r = 11!/[N!(11-N)!], where N is the number of acetylated lysines for that particular rung. For example, r = 11 for Ac (1); r = 55 for Ac (2), and r = 165 for Ac (3). The actual number of regioisomers that exist in solution is likely to be lower than the statistically possible maximum because of the disparate reactivity of each lysine (i.e., the unique solvent accessibility or pKa of each lysine), especially in the case of WT and D90A apo-SOD1 where Lys-3 and Lys-91 appear to be less reactive than the other nine lysines (Fig. S3).
The concentration of each rung could be approximated from its mass spectrum. For example, the A4V apo-SOD1-Ac(∼3) protein consisted of 3.6% Ac (0), 10.9% Ac (1), 19.5% Ac (2), 22.6% Ac (3), 16.8% Ac (4), 16.7% Ac (5), and 9.9% Ac (6) (Figs. S2 C, red spectrum, and S5 C).
Aspirin inhibits amyloidogenesis of WT- and ALS-variant apo-SOD1 by acetylation of lysine
The rates of amyloid formation for WT, A4V, and D90A apo-SOD1 (acetylated and unacetylated forms) were measured under reducing conditions (10 mM TCEP) with a thioflavin-T (ThT) fluorescence assay (35), in replicates of n = 18 for each derivative of each protein. The longitudinal plots of ThT fluorescence from each experiment exhibited a sigmoidal increase in fluorescence at 485 nm. A set of 18 of these raw, unnormalized sigmoids are shown, as an example, for WT-Ac(0) and WT-Ac(∼6) (Fig. 3). All other sets of sigmoidal plots for all three proteins (at different degrees of acetylation, with and without 100 mM NaCl) are presented in Figs. S6–S11. To increase visual clarity, each set of 18 replicate plots of ThT fluorescence were also averaged (combined) into a single sigmoid for each set of acetyl derivatives and are shown for all three SOD1 proteins in Figs. 4, A–C (0 mM NaCl) and 5, A–C (100 mM NaCl).
Figure 3.
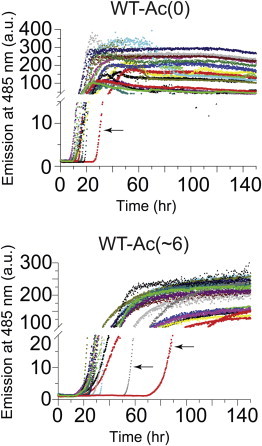
Fibrillization of unacetylated (top) and acetylated (bottom) apo-SOD1 (0 mM NaCl) as measured by thioflavin-T fluorescence in a 96-well microplate. Eighteen plots of thioflavin-T fluorescence from 18 replicate amyloid assays of WT apo-SOD1-Ac(0) (top) and WT apo-SOD1-Ac(∼6) (bottom) carried out simultaneously in the same 96-well microplate. Each of the 18 aliquots were taken from the same stock solution of acetylated or nonacetylated SOD1 and analyzed simultaneously in the same 96-well microplate to eliminate variations in solution conditions and minimize random error. Black arrows identify the outlier sigmoids in the data set. The raw plots of ThT fluorescence for other acetyl derivatives of WT apo-SOD1 (and D90A and A4V) can be found in the Supporting Material (Figs. S6–S11). The average number of acetylated lysines (Ac(∼N)) is listed per apo-SOD1 monomer. To see this figure in color, go online.
Figure 4.
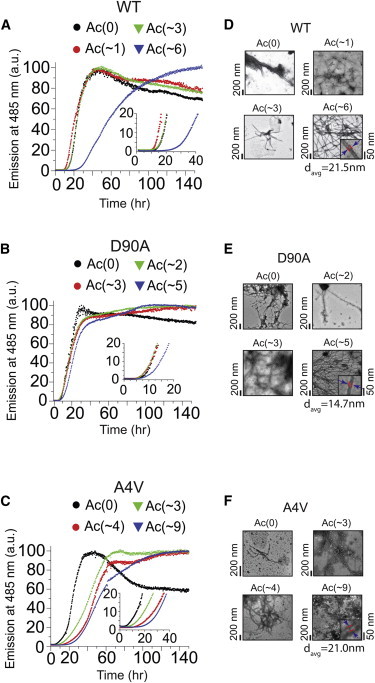
Effect of lysine acetylation on the rate of fibrillization of WT- and ALS-variant apo-SOD1 in 0 mM NaCl. Thioflavin-T fluorescence assays of unacetylated and acetylated (A) WT, (B) D90A, and (C) A4V apo-SOD1 proteins (pH 7.4, 37°C). Each curve is an average of normalized fluorescence measurements from 18 separate replicate experiments (of the sort shown in Fig. 3). Transmission electron micrographs of fibrillar forms of acetylated (D) WT, (E) D90A, and (F) A4V apo-SOD1 after the ThT assay. The mean number of acetylated lysines is denoted as Lys-Ac(∼N), per apo-SOD1 monomer. Table 1 and Fig. S12 list p-values for all kinetic analyses extracted from ThT fluorescence assays. To see this figure in color, go online.
Figure 5.
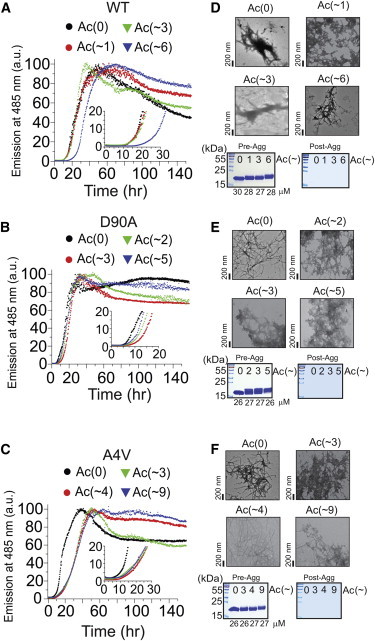
Effect of lysine acetylation on the rate of fibrillization of WT- and ALS-variant apo-SOD1 in 100 mM NaCl. Thioflavin-T fluorescence assays of unacetylated and acetylated (A) WT, (B) D90A, and (C) A4V apo-SOD1 proteins (pH 7.4, 37°C). Each curve is an average of data (normalized) from 18 separate replicate experiments. The mean number of acetylated lysines is denoted as Lys-Ac(∼N), per apo-SOD1 monomer. (D–F, upper panels) Transmission electron micrographs of fibrillar forms of acetylated WT, D90A, and A4V apo-SOD1 after the ThT assay in 100 mM NaCl. (D–F; lower panels) SDS-PAGE of samples before (left) and after (right) ThT aggregation assay in 100 mM NaCl. The gels on the right in each panel are from supernatants of samples after aggregation and centrifugation. Table 1 and Fig. S13 list p-values for all kinetic analyses extracted from ThT fluorescence assays. To see this figure in color, go online.
The high number of replicate assays (i.e., a total of 432 amyloid assays) was necessary to acquire statistically significant fibrillization rates because the fibrillization of apo-SOD1 in the microplate-based ThT assay is apparently stochastic (as previously observed for unacetylated SOD1 (27)). By stochastic, we do not mean entirely random, but rather nondeterministic, i.e., that the measured rates of amyloid formation in each well will fall within some type of mathematical distribution (e.g., Gaussian or Lorentzian) wherein the mean or maximum of the distribution is the best expression of the rate of the process. Whether or not the nucleation of amyloid can occur through a stochastic mechanism is apparently controversial. Stochastic nucleation is nevertheless observed in other self-assembled systems, at all scales of size and complexity, from the crystallization of water (36), to the cAMP-induced aggregation of social amoeba (37). The nucleation of amyloid fibrils has been observed (and described) to be stochastic for proteins including Aβ1-40, β2-microglobulin, and insulin (38–42). Even in reports that describe amyloid nucleation as a nonstochastic, deterministic process, these studies still report large, unexplained variations in lag times of replicate measurements (e.g., the lag time of nucleation of Aβ1-42 varied from 24 to 17 h in replicate measurements made by Hellstrand and coworkers (43)).
The rate of nucleation of amyloid fibrils is expressed as the lag time and the propagation of fibrils (elongation) is expressed as the inverse rate constant of propagation (1/k, i.e., the steepness of the sigmoid), as previously described (Table 1) (27). See Fig. S1 for a graphical illustration of this parameterization. The lag time and inverse propagation constants in Table 1 are average values calculated from 18 individual fits of the 18 replicate amyloid assays. An unpaired Student’s t-test was used to verify the statistical significance of any measured differences in the lag time and propagation rate for acetylated and unacetylated proteins (at a 95% confidence interval). Figs. S12 and S13 show p-values. Additional mathematical parameters from the sigmoidal fits of fluorescence data (i.e., a, x0, b, and x0-2b from Eq. 1 in the Supporting Material, and R2 values for fits) are also listed in Tables S2–S4.
We also experimented with calculating parameters such as lag time by first averaging all longitudinal fluorescence data into a single sigmoid and then performing a single nonlinear regression analysis of the average sigmoid (wherein each point in the sigmoid contains its own error). This method produced similar average values of lag time compared with the above method, but with error values that were, in our opinion, unrealistically small (e.g., tenfold smaller). For example, the average lag time for WT Ac(0) (0 mM NaCl) was calculated to be t = 13.9 ± 1.1 h when each sigmoid (data) set was analyzed individually with nonlinear regression analysis (Table 1), compared with t = 14.7 ± 0.1 h when individual sigmoidal data plots were averaged into a single sigmoid and analyzed with a single nonlinear regression analysis. Thus, we chose to report lag times (in Table 1) as averages of 18 separate nonlinear regression analyses for each derivative of each protein.
Many of the longitudinal plots of ThT fluorescence show a decrease in fluorescence after fibrillization is complete (Figs. S6–S11). This artifactual decrease has been previously observed (27) and was not included in sigmoidal fitting or the calculation of lag time and propagation rate. This decrease in fluorescence is likely caused by the adhesion of aggregated protein to the side of the polystyrene wells of the microplate, as inferred from visual inspection of each well at the end of the aggregation assay.
The elongation (propagation) of amyloid fibrils of acetylated WT, A4V, and D90A apo-SOD1 was consistently slower (i.e., 1/k was consistently larger) than the unacetylated protein, both in the presence and absence of 100 mM NaCl (Table 1 and Figs. 4 and 5). In general, the rate of elongation of amyloid fibrils of maximally acetylated apo-SOD1 proteins was slower than minimally acetylated forms (with the exception of A4V, wherein Ac(∼3) and Ac(∼9) elongated at similar rates). Acetylation of each apo-SOD1 protein also increased the lag time of amyloid, but with some acetyl derivatives, these effects were less uniform and not statistically significant (Table 1), e.g., with D90A in 0 mM NaCl (Table 1 and Fig. S12 C). Nevertheless, acetylation inhibited the formation of amyloid by either increasing the lag time or slowing fibril propagation, and in most cases by both increasing lag time and slowing propagation (Table 1 and Figs. S11 and S12).
When comparing the effects of acetylation on the average value of lag time and fibril propagation, it is important to remember that the set of 18 replicate sigmoidal plots of ThT fluorescence for each protein in Figs. 3 and S6–S11 includes, in some cases, outlier sigmoids. We did not use a Dixon’s Q-test to exclude outlier sigmoids with outlying lag times and/or propagation rates. Although such outliers (e.g., the far right sigmoids in Fig. 3) can skew averages, these outliers cannot be excluded because we have no reason to suspect that experimental errors occurred in the collection of these data. We do not suspect that the presence of outlier sigmoids is caused by differences in solution conditions because assays were carried out on identical solutions that were simply aliquoted into different wells of the sealed microplate. The presence of a heterogeneous distribution of acetyl derivatives or regioisomers in any particular solution cannot explain why different aliquots of the same solution exhibit outlier kinetics because even the unacetylated SOD1 protein—which is chemically monodisperse/homogenous—exhibited outlier kinetics. For example, wells that contained unacetylated WT SOD1-Ac(0) (in 0 mM NaCl) exhibited lag times from ∼6 to 28 h, within the same microplate and experiment (Fig. 3). Thus, it is necessary to not only compare average values of lag time and propagation, but to also compare probabilities (p-values) of similarity among different data sets (Table 1).
We do not know why we observe the nucleation and aggregation of SOD1 to be effectively stochastic, that is, why identical, aliquoted solutions of SOD1, analyzed at the same time in the same sealed microplate, with the same instrument, occur at different rates. The nucleation of amyloid SOD1 might be best described by nonclassical nucleation theory, wherein the ΔH and ΔS of the various nuclei that might form are different than the ΔH and ΔS of the final bulk assembly (27,44,45). A zeroth-order approximation (using the Arrhenius equation) would suggest that variations in the Ea of amyloid nuclei or the collision frequency of SOD1 proteins give rise to the nondeterministic rates. Variations in Ea might be caused by a random element within the apo-SOD1 protein, such as one or both of its two intrinsically disordered metal binding loops, which could result in heterogeneous nuclei, that form and elongate with different values of Ea (and thus whose relative concentrations vary from well to well). We also note that the self-assembly that we observe is likely to involve heterogeneous nucleation wherein ions, or surfaces of the apparatus (i.e., the Teflon bead or polystyrene microplate) contribute to the observed rate of aggregation.
A variation in the collision frequency of reactants, in each well, might be caused by variations in the trajectory or velocity of the Teflon bead in each well. Variations in the mass of the machined Teflon bead (purchased from McMaster-Carr) is an unlikely source of variation, as these beads only vary in mass from 36.1 to 35.9 mg according to our measurements. Variations in the temperature of each well is an unlikely source of the kinetic variation as the plate is temperature controlled. The presence of a dust particle or preformed aggregate in some of the wells is also an unlikely explanation as we filtered each solution immediately before aggregation assay. Variations in volume (SOD1 concentration) cannot explain variations in rates of fibrillization because all replicate solutions are aliquoted from the same stock and the volume of each well was measured at the end of an assay to ensure no evaporation through the sealed lid. Varying degrees and types of nano-, micro-, or macro-scopic surface imperfections (e.g., scratches) on the surface of the bead or the well cannot yet be ruled out. Identifying the source of the stochastic nature of SOD1 amyloidogenesis must be the subject of a future study.
The protective effect of acetylation against amyloid formation was generally more pronounced in the absence of 100 mM NaCl (Table 1 and Fig. S12) than in the presence of salt (Table 1 and Fig. S13). This sensitivity to NaCl suggests that lysine acetylation inhibited the nucleation and propagation of amyloid by an electrostatic mechanism (at least in part). Acetylation had the largest inhibitory effect on the nucleation and propagation of A4V and WT apo-SOD1 fibrils, and the smallest effect on the self-assembly of D90A apo-SOD1 (regardless of NaCl concentration). For example, in the absence of 100 mM NaCl, the acetylation of ∼ three lysines per monomer in A4V and WT apo-SOD1 resulted in an approximate twofold increase in the inverse propagation constant for both proteins (i.e., a lower rate of propagation), ∗∗∗p < 0.0001 (Fig. S12). The lag time of A4V-Ac(∼3) fibrillization was also increased by 6.3 ± 2.7 h, ∗p = 0.0243 compared with A4V-Ac(0) (Table 1). Moreover, A4V-Ac(∼3) exhibited a lower lag time of fibrillization than the unacetylated WT apo-SOD1 protein. The rate of aggregation for WT apo-SOD1 is not necessarily a perfect benchmark for nontoxicity, because WT SOD1 is causally linked to sporadic ALS (46) (but might require chemical modification before aggregation (27)). Nevertheless, the slower aggregation of acetylated mutant SOD1 compared with WT SOD1 suggests that small increases in the magnitude of a mutant protein’s net negative charge, afforded by lysine acetylation, might impart a therapeutic effect in ALS (or other diseases linked to protein self-assembly).
In the absence of 100 mM NaCl, the maximally acetylated forms of WT and A4V (i.e., Ac > ∼5) exhibited increases in lag time of 11.0 ± 1.7 h and 14.5 ± 1.7 h, respectively, compared with their unacetylated forms (Table 1 and Figs. 4 and S12, ∗∗∗p < 0.0001). Maximal acetylation of WT and A4V apo-SOD1 also increased the inverse propagation constant by 2.2 ± 0.3 h (WT) and 2.7 ± 0.4 h (A4V), which corresponded to a 2.5-fold decrease in k (Table 1 and Figs. 4 and S12, ∗∗∗p < 0.0001). In the absence of 100 mM NaCl, the lag time of minimally acetylated D90A apo-SOD1, i.e., Ac(∼2), did not differ significantly from unacetylated D90A apo-SOD1 (Table 1, Figs. 4 and S12, p > 0.05), but the inverse propagation constant increased by approximately twofold (Table 1 and Figs. 4 and S12, ∗∗∗p = 0.0007).
In 0 mM NaCl, A4V-Ac(∼3) had a greater lag time of aggregation than A4V-Ac(∼4) by 8.6 ± 2.3 h (but had similar rates of propagation) (Table 1 and Fig. S12). This slower aggregation, despite slightly lower acetylation, might arise from difficulties in expressing mean acetylation. For example, the Gaussian fitting of the MS spectrum of A4V-Ac(∼4) resulted in a mean acetylation of 3.8 Ac. Inspection of the MS spectrum, however, demonstrated that the most abundant peak is that of Ac (2) (Fig. 2 C). This difference might explain why A4V-Ac(∼4) was found to aggregate faster than A4V-Ac(∼3) in 0 mM NaCl (albeit, the rates of A4V-Ac(∼4) and A4V-Ac(∼3) are statistically similar in 100 mM NaCl, Figs. 5 C and S13 F).
It is unlikely that the different rates of fibrillization that we observed among WT- and ALS-mutant SOD1, and their acetyl derivatives, are caused by Cu2+ or Zn2+ contamination. All SOD1 proteins were demetallated and contained < 0.06 ± 0.02 Zn2+ and < 0.03 ± 0.01 Cu2+ per SOD1 dimer after acetylation (according to ICP-MS). After acetylation and analysis with ICP-MS, proteins solutions were stored frozen until the initiation of the aggregation assay. Each aggregation assay was also carried out in the presence of 5 mM EDTA (to prevent metal contamination from reagents used during the assay). Moreover, all solutions and buffers were made with ultrapure metal-free water with a measured resistance of 18.2 MΩ/cm (dispensed through a Millipore MilliQ system). All containers used for making buffers or other solutions were rinsed with 5 mM EDTA and pure water before use.
The addition of 100 mM NaCl at pH 7.4 increased the overall rate of aggregation of each apo-SOD1 protein (regardless of its degree of acetylation), which has been reported previously for other proteins (47) (Figs. 5, A–C, and S13). Although the presence of 100 mM NaCl generally diminished—but did not abolish—the magnitude by which acetylation slowed the rate of fibril initiation and propagation, the D90A apo-SOD1 protein was one exception (Table 1). For example, the acetylation of five lysine residues had a negligible effect on the lag time of D90A fibrillization, in 0 mM NaCl, whereas the acetylation of five lysines increased the lag time by 5.9 ± 1.7 h in the presence of 100 mM NaCl (Table 1 and Figs. 5, A–C, and S13, ∗∗p = 0.0022). Thus, acetylation had a greater effect on inhibiting the fibrillization of D90A apo-SOD1 in 100 mM NaCl than in 0 mM NaCl. Notably, in the presence of 100 mM NaCl, the acetylation of A4V apo-SOD1 also slowed its fibrillization to a value below that of unacetylated WT apo-SOD1 (Fig. S13).
The inability of Na+ and Cl- to completely screen the electrostatic repulsions between negatively charged SOD1 proteins—i.e., to completely abolish the protective effects of acetylation against aggregation—is not surprising and strengthens the potential utility of the charge-boosting strategy. Free cations and anions do not screen all types of intermolecular electrostatic interactions involving proteins (2), especially, for example, when 1) the interaction occurs through the low dielectric interior of a protein; 2) the interaction depends thermodynamically on desolvation of charged groups; or 3) the electrostatic interaction occurs over a distance that is smaller than the Debye screening length (i.e., ∼1 nm at I = 0.1 M) (2). All three of these criteria might exist within an initiating or elongating amyloid fibril.
To ensure that the inhibitory effects of acetylation on the aggregation of apo-SOD1 were not attributable to differences (or errors) in protein concentration, and that solutions did not contain soluble SOD1 at the end of the fluorescence assay, we quantified the amount of nonsedimentable apo-SOD1 proteins in solution at the start of the aggregation assay and upon the completion of the assay. The concentration of all apo-SOD1 proteins, as measured by ultraviolet-visible spectrophotometry, only varied by ± 2 μM before the start of the assay (Fig. 5, D–F). SDS-PAGE also demonstrated that all proteins were initially present at similar concentration and purity at the start of the assay (Fig. 5, D–F, gels on left). The absence of SOD1 protein in the supernatant of solutions after completion of the aggregation assay (and centrifugation) demonstrated that all acetylated and unacetylated apo-SOD1 proteins underwent aggregation into a high molecular weight, sedimentable species (Fig. 5, D–F, gels on right). We note that the acetylation of lysine residues in apo-SOD1 decreased its electrophoretic mobility during reducing SDS-PAGE, most likely because of a decrease in bound SDS (as demonstrated by a recent study into the long-standing mystery of abnormal protein migration during SDS-PAGE (30)). Transmission electron microscopy demonstrated that the high molecular weight aggregates of acetylated and unacetylated apo-SOD1 were fibrillar in nature (Figs. 4, D–F, and 5, D–F). The typical diameter of unacetylated and acetylated fibrils was ∼15 to 20 nm, similar to previous studies (48).
Effect of lysine acetylation on the thermostability and structure of the native state of apo-SOD1
The acetylation of lysine in a folded protein does not necessarily lower its melting transition temperature (Tm) (18). To determine if the acetylation of lysine diminished the rate of aggregation of WT- and ALS-variant apo-SOD1 by increasing apo-SOD1 thermostability, in addition to by increasing net charge, we analyzed acetylated apo-SOD1 with differential scanning calorimetry, before fibrillization (Fig. S14 and Table S5). The acetylation of lysine in WT- and ALS-variant apo-SOD1 did not increase the thermostability of the native apo-protein, and therefore did not inhibit fibrillization by increasing the Tm of the native protein (Table S5). The acetylation of approximately six lysines (per monomer) in WT apo-SOD1, for example, lowered its Tm by ΔTm = −6.1°C (Fig. S14 A). These DSC data show that chemical modifications to a protein can inhibit self-assembly even if the modification diminishes Tm of the native state.
The acetylation of lysine in SOD1 by aspirin did not disrupt the structure of the protein by a magnitude that resulted in an increased rate of amide H/D exchange. Instead, the acetylation of lysine in SOD1 proteins slightly diminished the rate of global amide H/D exchange by < 1 H per acetyl modification (Fig. S14, D–F). This result is consistent with previous reports showing that semi-random lysine acetylation slows the rate of amide H/D exchange in proteins (e.g., myoglobin, carbonic anhydrase, and SOD1) despite also diminishing thermostability (18,24,25,49,50). The acetylation of lysine has been hypothesized to slow amide H/D exchange by a purely electrostatic mechanism, i.e., by not altering H-bonding or solvent accessibility, but rather by lowering the local concentration of the hydroxide catalyst of H/D exchange and/or the activation energy of the divalent anionic intermediate (R—N:——R1) (47). In the case of A4V, the acetylated and unacetylated apo-protein exchanged most of its amide hydrogens after only 5 min, which is consistent with previous reports of its low thermostability.
Acetylation of lysine in soluble apo-SOD1 diminishes the thermostability of resulting amyloid fibrils
The cytotoxicity of an amyloid oligomer is thought to be caused, in part, by its high thermostability, which inhibits proteolysis by intracellular and extracellular proteases (51). One important caveat is, however, that the breakage of amyloid fibrils has been shown to increase cytotoxicity by seeding amyloidogenesis and/or increasing membrane permeability of oligomers (7). In the case of SOD1, this latter hypothesis has not yet been rigorously (quantitatively) tested. The thermostability and material properties of amyloid-like oligomers composed of WT and mutant SOD1 are unmeasured. In fact, we could find only one study that analyzed the thermostability of amyloid fibrils of any protein with DSC. This study showed that Tm = 83°C for amyloid fibrils of the N47A mutant of alpha-spectrin SH3 domain, compared with Tm = 49°C for the protein’s native state (52). Nevertheless, it is reasonable to hypothesize that an amyloid oligomer could be made less toxic by supercharging the fibril, assuming that supercharging destabilized the fibril to a level that rendered it a substrate for intra- or extracellular proteases, and diminished its permeability (electrostatically) across anionic cellular membranes. This type of successive supercharging after fibrillization, by repeated doses of an acylating agent, might represent a practical medicinal approach because the long half-life of amyloid-like oligomers would permit accumulation of chemical modifications (29).
The method that we used to determine the thermostability of acetylated and unacetylated apo-SOD1 fibrils was modified from Shammas and coworkers (32) and is illustrated in Fig. S15 A. We chose to measure the stability of fibrils using thermal defibrillization, instead of chemical defibrillization, because the interaction of chemical denaturants (such as guanidinium hydrochloride, sodium dodecyl sulfate, or urea) can be dependent on the net charge and hydrophobicity of proteins, both of which are altered by acetylation of Lys-NH3+ to Lys-NHCOCH3 (53,54). We assume that the low pressure generated in this thermal defibrillization assay (∼350 kPa) had a negligible effect on the thermostability of fibrils because it is 1000-fold lower than minimal values reported to induce protein aggregation (i.e., ∼400 MPa) (55). The Tm of SOD1 amyloid fibrils composed of unacetylated apo-SOD1 were 99.42 ± 0.89°C for WT, 104.69 ± 0.24°C for D90A, and 99.85 ± 0.48°C for A4V (Fig. S15, B–D, and Table S5). Maximal acetylation of lysine residues in apo-SOD1 with aspirin, before fibrillization, decreased the Tm of resulting fibrils by between ∼3°C and 7°C (Table S5): ΔTm = −3.15 ± 1.1°C for WT-Ac(0) and WT-Ac(∼6) amyloid; ΔTm = −7.07 ± 0.64°C for D90A-Ac(0) and D90A-Ac(∼5) amyloid; ΔTm = −4.78 ± 0.58°C for A4V-Ac(0) and A4V-Ac(∼9) amyloid SOD1. These differences in the Tm values of unacetylated and acetylated fibrils were statistically significant for all three proteins, with ∗∗∗p-values uniformly < 0.0001. In general, successive acetylation of native SOD1 diminished the Tm of resulting fibrils; however, the differences in the Tm of fibrils from proteins that differed in mean acetylation by less than approximately three lysines were often statistically indistinguishable (Table S5). This result might be caused by similarities (overlap) in the distribution of acetylated lysines among different samples. For example, the Tm of fibrillar D90A SOD1-Ac(∼3) = 96.02 ± 0.97°C, whereas the Tm of fibrillar D90A SOD1-Ac(∼5) = 97.62 ± 0.60°C (Table S5). Although these two sets of proteins have different mean numbers of acetylated lysine, these two sets have ∼70% overlap in the distribution of acetylated species (Fig. S5, A, B, and E), and this similar degree of acetylation might account for the statistically similar values of Tm.
To ensure that the total concentration of apo-SOD1 protein did not change during the course of heating (via solvent evaporation) we performed reducing SDS-PAGE on solutions before thermal defibrillization and after thermal defibrillization. We observed no significant difference in apo-SOD1 concentration before and after heating (Fig. S15 E), indicating that protein concentration did not change via solvent evaporation during thermal defibrillization.
Although the turbidity of each fibrillar solution was abolished at the end of each thermal scan, it is possible that fibrils of apo-SOD1 melted incompletely into small oligomeric forms that did not sediment or scatter light, but are nonetheless still in an amyloid state. To assay for such small oligomers, we performed size-exclusion liquid chromatography (SE-LC) and native PAGE on solutions at the end of thermal defibrillization experiment (Fig. S16, A–C). The supernatant solutions of melted fibrils from all three apo-SOD1 proteins contained SOD1 species that were <50 kDa, according to SE-LC calibration curve (Fig. S16 B). All three apo-SOD1 proteins did, however, migrate slower than the dimeric form of soluble WT apo-SOD1 during native PAGE (Fig. S16 C), which might be caused by improper refolding after cooling the thermally defibrillized and denatured polypeptides.
We also filtered the supernatants of melted fibrils with a 0.2 μm filter and performed SE-LC, native PAGE, and transmission electron microscopy (TEM) and found no significant difference in the chromatography, electrophoresis, or microscopy of nonfiltered and filtered samples, which suggested that oligomers that are >0.2 μm in diameter were not present after thermal defibrillization (Fig. S16, A, C, and D). Fibrillar species were not detected by TEM in these heated samples, even after examining ∼3.5 mm2 of the grid surface in three separate replicate analyses (Fig. S16 D). Thus, the assay we used to measure the Tm of fibrillar apo-SOD1 appears to be a valid method for determining the stability of the large SOD1 fibrils depicted in TEM images of Fig. 5 E. In conclusion, the predominant amyloid framework of fibrillar apo-SOD1 is not as thermostable as we expected and does not represent a thermodynamic black hole for SOD1. For example, the Tm of unacetylated WT apo-SOD1 fibrils is only a few degrees higher than the Tm of the properly folded and fully metalated WT protein (56). Moreover, the fibrils formed in this study did not appear to involve extensive intermolecular disulfide crosslinks (57), i.e., the thermal defibrillization assay did not include any reducing agent and fibrils nevertheless dissociated to species with molecular weights ≤50 kDa (Fig. S16).
Lysine, serine, and threonine residues in amyloid apo-SOD1 are reactive with aspirin
We also wanted to determine if lysine residues in amyloid apo-SOD1 could be acetylated with aspirin after fibrillization of unacetylated apo-SOD1, and the degree to which these modifications affect fibril thermostability.
To determine the reactivity of fibrils, we prepared amyloid fibrils of WT- and ALS-variant apo-SOD1 from each unacetylated protein and reacted the amyloid fibrils with aspirin in aqueous buffer. The number of acetylated lysines in fibrillized apo-SOD1 polypeptides was determined by thermochemically dissociating fibrils and analyzing the dissociated polypeptides with electrospray ionization MS (Fig. 6, A–C). Fibrils were gently dissociated by heating at 50°C, in 6.5 M guandinium hydrochloride (Gdm-HCl), 20 mM dithiothreitol (DTT), pH 8. This protocol has been proven to dissociate fibrils of SOD1 into monomeric polypeptides that can be analyzed with electrospray ionization mass spectrometry (31).
Figure 6.
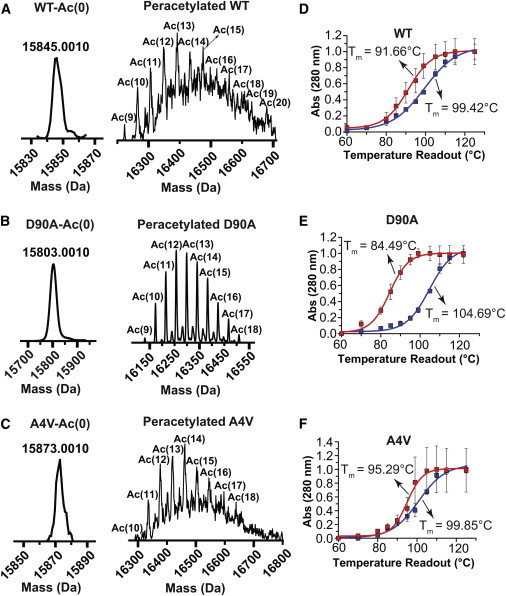
Effect of peracetylation of lysine in fibrillar WT- and ALS-variant apo-SOD1 on fibril thermostability (i.e., acetylation after fibrillization). (A–C) Mass spectra of unacetylated (left panel), and peracetylated (right panel) WT- and ALS-variant apo-SOD1 thermochemically dissociated from amyloid fibrils (acetylation was performed after fibrillization). (D–F) Thermal stability curves of unacetylated (blue solid squares) and peracetylated (red solid squares) WT, D90A, and A4V apo-SOD1 amyloid fibrils. To see this figure in color, go online.
The mass spectra of apo-SOD1 polypeptide chains from thermochemically dissociated fibrils demonstrated that each of the 11 lysine residues in apo-SOD1 amyloid fibrils could be acetylated by aspirin (Fig. 6, A–C, right panels). Thus, unlike the native soluble protein, we were able to acetylate all 11 lysine residues in fibrillar apo-SOD1; the formation of amyloid by SOD1 does not cause its lysines to be protected from chemical modification. MS also showed that a significant population of SOD1 proteins contained additional acetyl modifications (up to seven additional acetyl groups) on residues other than lysine (Fig. 6, A–C, right panels). We were able to identify acetyl modifications on each lysine with MS-MS analysis (Fig. S17 A), and also detected acetyl groups on Ser-134, Ser-142, Thr-135, and Thr-137 (Fig. S17 B). We hypothesize that these types of Ser and Thr modifications were not detected (and were presumably not present) in acetylated native WT and D90A apo-SOD1 because 1) native forms of these proteins precipitated before a sufficient excess of aspirin could be added to acetylate Ser and Thr (this impediment did not occur when acetylating amyloid fibrils); and/or 2) fibrillization protected ester groups on Ser and Thr from hydrolysis (that might occur during analysis).
Peracetylation of lysine in amyloid (after fibrillization) increases formal net charge by millions of units and lowers fibril thermostability
Thermostability assays for peracetylated amyloid fibrils of WT, D90A, and A4V apo-SOD1 (that were peracetylated after fibrillization) are shown in Fig. 6, D–F; fibril Tm values are listed in Table S5. Peracetylation lowered the Tm of D90A fibrils by three- to fourfold more than A4V or WT fibrils. Peracetylation lowered fibril Tm by ΔTm = −7.76 ± 1.18°C for WT fibrils; ΔTm = −20.2 ± 0.48°C for D90A fibrils; ΔTm = −4.56 ± 0.69°C for A4V fibrils (Fig. 6, D–F, and Table S5).
We point out that the Tm of peracetylated fibrils of D90A (Tm = 84.49 ± 0.42°C), A4V (Tm = 95.29 ± 0.50°C), and WT SOD1 (Tm = 91.66 ± 0.78°C) are near or below the reported Tm for the WT holo-SOD1 enzyme () in its native state (Tm ∼90°C to 95°C) (56). These low Tm values for supercharged fibrils raise the possibility that unstable, acetylated fibrils might have sufficient instability for proteolysis by intracellular or extracellular proteases (58). We also point out that any SOD1 oligomers or polypeptides that would dissociate from these unstable fibrils would be less prone to seed aggregation (than unacetylated polypeptides), because of their supercharged state.
The most straightforward explanation for why peracetylation diminished the thermostability of amyloid SOD1 is that peracetylation strengthened the repulsive Coulombic forces between similarly charged—now supercharged—SOD1 polypeptides. The peracetylation of an amyloid fibril with dimensions of 15 nm by 2 μm (composed of ∼1.9 × 105 SOD1 polypeptide chains) will increase the net negative charge of the fibril by ∼1.6 × 106 units of formal charge at pH 7.4. See Supporting Material for details of this calculation.
Conclusions
The results of this study do not suggest that an acylating agent such as aspirin—which nonspecifically acetylates SOD1—can function as a charge-boosting drug in the treatment of ALS. Instead, future work will be required to design and synthesize compounds that selectively boost the net charge of SOD1 (or its oligomers). We envision the general chemical anatomy of a selective charge booster to consist of a head for molecular recognition of the target protein or protein oligomer (Fig. S18), and a reactive acylating tail that acylates nearby lysine residues. One advantage to designing small reactive molecules that increase the net charge of a target protein, as opposed to designing conventional ligands that noncovalently stabilize the structure of the native or oligomeric protein, is that the former might be much easier to accomplish (medicinally). The acetylation of lysines in WT or mutant apo-SOD1 by aspirin inhibited the nucleation and propagation of amyloid fibrils, even though the acetylation was nonspecific. This result suggests that increasing the net surface potential of different surface loci—perhaps acetylating any surface lysine on a small anionic protein such as SOD1—will inhibit aggregation. This large number of chemical targets in a single protein is in stark contrast to the chemical constraints that typify conventional medicinal chemistry. It is also important to remember that this study demonstrated the basic tenet of the charge-boosting strategy by using the least potent, electrostatically speaking, type of chemistry, i.e., ΔZ = −1 per modification.
The interactions between highly charged proteins and cellular membranes were not examined in this study. Nevertheless, it is reasonable to hypothesize that a hyperanionic, acetylated SOD1 protein (oligomeric or native) will resist electrostatic interactions with inner and outer cellular membranes (which are generally anionic because of sialylated and sulfated glycans (59)) more than unacetylated SOD1 proteins. For example, it has been shown that supercharged green fluorescent protein with magnitudes of negative charge reaching Z = −30 are resistant to association with outer cellular membranes, whereas supercharged proteins with Z = +36 permeate the same cellular membranes (21). Increasing the electrostatic repulsion between cellular membranes and SOD1 proteins might diminish the cell-to-cell propagation of oligomeric SOD1 (20,60) and halt the progressive death of motor neurons in ALS.
Author Contributions
A.A. and B.F.S. organized the research. All the authors contributed to the design of the experiments, the analysis of the data, and editing of the manuscript.
Acknowledgments
Financial support for this research was provided to B.F.S. by the Department of Defense (W81XWH-11-1-0790), the National Science Foundation (CAREER CHE: 1352122), and the Welch Foundation (AA-1854).
Supporting Material
Supporting Citations
References (61–63) appear in the Supporting Material.
References
- 1.Roberts D., Keeling R., Curtis R. The role of electrostatics in protein-protein interactions of a monoclonal antibody. Mol. Pharm. 2014;11:2475–2489. doi: 10.1021/mp5002334. [DOI] [PubMed] [Google Scholar]
- 2.Gitlin I., Carbeck J.D., Whitesides G.M. Why are proteins charged? Networks of charge-charge interactions in proteins measured by charge ladders and capillary electrophoresis. Angew. Chem. Int. Ed. Engl. 2006;45:3022–3060. doi: 10.1002/anie.200502530. [DOI] [PubMed] [Google Scholar]
- 3.Lessene G., Czabotar P.E., Watson K.G. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013;9:390–397. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 4.Lee S., Zheng X., Lim M.H. Rational design of a structural framework with potential use to develop chemical reagents that target and modulate multiple facets of Alzheimer’s disease. J. Am. Chem. Soc. 2014;136:299–310. doi: 10.1021/ja409801p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shahian T., Lee G.M., Craik C.S. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat. Chem. Biol. 2009;5:640–646. doi: 10.1038/nchembio.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sreedharan J., Brown R.H., Jr. Amyotrophic lateral sclerosis: problems and prospects. Ann. Neurol. 2013;74:309–316. doi: 10.1002/ana.24012. [DOI] [PubMed] [Google Scholar]
- 7.Jiang L., Liu C., Eisenberg D.S. Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. eLife. 2013;2:e00857. doi: 10.7554/eLife.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grisolia S., Santos I., Mendelson J. Inactivation of enzymes by aspirin and salicylate. Nature. 1968;219:1252. doi: 10.1038/2191252a0. [DOI] [PubMed] [Google Scholar]
- 9.Alfonso L., Ai G., Bhat G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer. 2014;111:61–67. doi: 10.1038/bjc.2014.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bateman L.A., Zaro B.W., Pratt M.R. An alkyne-aspirin chemical reporter for the detection of aspirin-dependent protein modification in living cells. J. Am. Chem. Soc. 2013;135:14568–14573. doi: 10.1021/ja408322b. [DOI] [PubMed] [Google Scholar]
- 11.Barnéoud P., Curet O. Beneficial effects of lysine acetylsalicylate, a soluble salt of aspirin, on motor performance in a transgenic model of amyotrophic lateral sclerosis. Exp. Neurol. 1999;155:243–251. doi: 10.1006/exnr.1998.6984. [DOI] [PubMed] [Google Scholar]
- 12.Choudhary C., Weinert B.T., Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 13.Chiti F., Dobson C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence M.S., Phillips K.J., Liu D.R. Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osborne T.B. The basic character of the protein molecule and the reactions of edestin with definite quantities of acids and alkalies. J. Am. Chem. Soc. 1902;24:39–78. [Google Scholar]
- 16.Csonka F.A., Murphy J.C., Jones D.B. The iso-electric points of various proteins. J. Am. Chem. Soc. 1926;48:763–768. [Google Scholar]
- 17.Felder C.E., Prilusky J., Sussman J.L. A server and database for dipole moments of proteins. Nucleic Acids Res. 2007;35:W512–W521. doi: 10.1093/nar/gkm307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw B.F., Schneider G.F., Whitesides G.M. Lysine acetylation can generate highly charged enzymes with increased resistance toward irreversible inactivation. Protein Sci. 2008;17:1446–1455. doi: 10.1110/ps.035154.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw B.F., Moustakas D.T., Faull K.F. Taking charge of proteins: from neurodegeneration to industrial biotechnology. Adv. Protein Chem. Struct. Biol. 2010;79:127–164. doi: 10.1016/S1876-1623(10)79004-0. [DOI] [PubMed] [Google Scholar]
- 20.Münch C., O’Brien J., Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA. 2011;108:3548–3553. doi: 10.1073/pnas.1017275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNaughton B.R., Cronican J.J., Liu D.R. Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. Proc. Natl. Acad. Sci. USA. 2009;106:6111–6116. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandelin E., Nordlund A., Oliveberg M. Amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase mutations preferentially reduce the repulsive charge of the proteins. J. Biol. Chem. 2007;282:21230–21236. doi: 10.1074/jbc.M700765200. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez J.A., Shaw B.F., Valentine J.S. Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc. Natl. Acad. Sci. USA. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y., Abdolvahabi A., Shaw B.F. Protein charge ladders reveal that the net charge of ALS-linked superoxide dismutase can be different in sign and magnitude from predicted values. Protein Sci. 2014;23:1417–1433. doi: 10.1002/pro.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Y., Mowery R.A., Shaw B.F. Effect of metal loading and subcellular pH on net charge of superoxide dismutase-1. J. Mol. Biol. 2013;425:4388–4404. doi: 10.1016/j.jmb.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Della Valle M.C., Sleat D.E., Lobel P. Classification of subcellular location by comparative proteomic analysis of native and density-shifted lysosomes. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M110.006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y., Rhodes N.R., Shaw B.F. Deamidation of asparagine to aspartate destabilizes Cu, Zn superoxide dismutase, accelerates fibrillization, and mirrors ALS-linked mutations. J. Am. Chem. Soc. 2013;135:15897–15908. doi: 10.1021/ja407801x. [DOI] [PubMed] [Google Scholar]
- 28.Pepys M.B. Amyloidosis. Annu. Rev. Med. 2006;57:223–241. doi: 10.1146/annurev.med.57.121304.131243. [DOI] [PubMed] [Google Scholar]
- 29.Maji S.K., Schubert D., Riek R. Amyloid as a depot for the formulation of long-acting drugs. PLoS Biol. 2008;6:e17. doi: 10.1371/journal.pbio.0060017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Y., Mowery R.A., Shaw B.F. Abnormal SDS-PAGE migration of cytosolic proteins can identify domains and mechanisms that control surfactant binding. Protein Sci. 2012;21:1197–1209. doi: 10.1002/pro.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chattopadhyay M., Durazo A., Valentine J.S. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. USA. 2008;105:18663–18668. doi: 10.1073/pnas.0807058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shammas S.L., Knowles T.P., Devlin G.L. Perturbation of the stability of amyloid fibrils through alteration of electrostatic interactions. Biophys. J. 2011;100:2783–2791. doi: 10.1016/j.bpj.2011.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erickson H.P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online. 2009;11:32–51. doi: 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaw B.F., Durazo A., Valentine J.S. Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J. Biol. Chem. 2006;281:18167–18176. doi: 10.1074/jbc.M600623200. [DOI] [PubMed] [Google Scholar]
- 35.LeVine H., 3rd Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 36.Vali G. Repeatability and randomness in heterogeneous freezing nucleation. Atmos. Chem. Phys. 2008;8:5017–5031. [Google Scholar]
- 37.Prindle A., Hasty J. Biochemistry. Stochastic emergence of groupthink. Science. 2010;328:987–988. doi: 10.1126/science.1190372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xue W.F., Homans S.W., Radford S.E. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. USA. 2008;105:8926–8931. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foderà V., Librizzi F., Leone M. Secondary nucleation and accessible surface in insulin amyloid fibril formation. J. Phys. Chem. B. 2008;112:3853–3858. doi: 10.1021/jp710131u. [DOI] [PubMed] [Google Scholar]
- 40.Morris R.J., Eden K., MacPhee C.E. Mechanistic and environmental control of the prevalence and lifetime of amyloid oligomers. Nat. Commun. 2013;4:1891. doi: 10.1038/ncomms2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hortschansky P., Schroeckh V., Fändrich M. The aggregation kinetics of Alzheimer’s beta-amyloid peptide is controlled by stochastic nucleation. Protein Sci. 2005;14:1753–1759. doi: 10.1110/ps.041266605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghag G., Ghosh P., Vaidya A. Stability analysis of 4-species Aβ aggregation model: a novel approach to obtaining physically meaningful rate constants. Appl. Math. Comput. 2013;224:205–215. doi: 10.1016/j.amc.2013.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hellstrand E., Boland B., Linse S. Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 2010;1:13–18. doi: 10.1021/cn900015v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oxtoby D.W. Nonclassical nucleation theory: an exactly soluble model. Phys. Scr. 1993;1993:65. [Google Scholar]
- 45.Oxtoby D.W. Nucleation of first-order phase transitions. Acc. Chem. Res. 1998;31:91–97. [Google Scholar]
- 46.Rotunno M.S., Bosco D.A. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front. Cell. Neurosci. 2013;7:253. doi: 10.3389/fncel.2013.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buell A.K., Hung P., Knowles T.P. Electrostatic effects in filamentous protein aggregation. Biophys. J. 2013;104:1116–1126. doi: 10.1016/j.bpj.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan P.K., Chattopadhyay M., Valentine J.S. Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc. Natl. Acad. Sci. USA. 2013;110:10934–10939. doi: 10.1073/pnas.1309613110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaw B.F., Arthanari H., Whitesides G.M. Neutralizing positive charges at the surface of a protein lowers its rate of amide hydrogen exchange without altering its structure or increasing its thermostability. J. Am. Chem. Soc. 2010;132:17411–17425. doi: 10.1021/ja9067035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdolvahabi A., Gober J.L., Shaw B.F. Metal-ion-specific screening of charge effects in protein amide H/D exchange and the Hofmeister series. Anal. Chem. 2014;86:10303–10310. doi: 10.1021/ac502714v. [DOI] [PubMed] [Google Scholar]
- 51.Lorenzen N., Otzen D.E. Inhibition of amyloid formation by small molecules. In: Otzen D.E., editor. Amyloid Fibrils and Prefibrillar Aggregates: Molecular and Biological Properties. Wiley-VCH Verlag; Weinheim, Germany: 2013. pp. 350–356. [Google Scholar]
- 52.Morel B., Varela L., Conejero-Lara F. The thermodynamic stability of amyloid fibrils studied by differential scanning calorimetry. J. Phys. Chem. B. 2010;114:4010–4019. doi: 10.1021/jp9102993. [DOI] [PubMed] [Google Scholar]
- 53.Gitlin I., Gudiksen K.L., Whitesides G.M. Effects of surface charge on denaturation of bovine carbonic anhydrase. ChemBioChem. 2006;7:1241–1250. doi: 10.1002/cbic.200600191. [DOI] [PubMed] [Google Scholar]
- 54.Shaw B.F., Schneider G.F., Whitesides G.M. Complexes of native ubiquitin and dodecyl sulfate illustrate the nature of hydrophobic and electrostatic interactions in the binding of proteins and surfactants. J. Am. Chem. Soc. 2011;133:17681–17695. doi: 10.1021/ja205735q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seefeldt M.B., Kim Y.S., Randolph T.W. High-pressure studies of aggregation of recombinant human interleukin-1 receptor antagonist: thermodynamics, kinetics, and application to accelerated formulation studies. Protein Sci. 2005;14:2258–2266. doi: 10.1110/ps.051490205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodriguez J.A., Valentine J.S., Hayward L.J. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J. Biol. Chem. 2002;277:15932–15937. doi: 10.1074/jbc.M112088200. [DOI] [PubMed] [Google Scholar]
- 57.Banci L., Bertini I., Whitelegge J.P. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. USA. 2007;104:11263–11267. doi: 10.1073/pnas.0704307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nijholt D.A., De Kimpe L., Scheper W. Removing protein aggregates: the role of proteolysis in neurodegeneration. Curr. Med. Chem. 2011;18:2459–2476. doi: 10.2174/092986711795843236. [DOI] [PubMed] [Google Scholar]
- 59.Cooper G.M., Hausman R.E. Sinauer Associates; Sunderland, MA: 2013. The Cell: A Molecular Approach. [Google Scholar]
- 60.Grad L.I., Yerbury J.J., Cashman N.R. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA. 2014;111:3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kokot Z., Burda K. Simultaneous determination of salicylic acid and acetylsalicylic acid in aspirin delayed-release tablet formulations by second-derivative UV spectrophotometry. J. Pharm. Biomed. Anal. 1998;18:871–875. doi: 10.1016/s0731-7085(98)00225-8. [DOI] [PubMed] [Google Scholar]
- 62.Anderson J.R., Chemiavskaya O., Whitesides G.M. Analysis by capillary electrophoresis of the kinetics of charge ladder formation for bovine carbonic anhydrase. Anal. Chem. 2002;74:1870–1878. doi: 10.1021/ac0109927. [DOI] [PubMed] [Google Scholar]
- 63.Furukawa Y., O’Halloran T.V. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 2005;280:17266–17274. doi: 10.1074/jbc.M500482200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.