

Background: Excessive stimulation of endothelial cells by VEGF is a major cause of aberrant angiogenesis and leaky blood vessels in various medical conditions.
Results: The Small GTPase R-Ras inhibits VEGF-induced internalization and tyrosine phosphorylation of VEGF receptor 2.
Conclusion: R-Ras suppresses chronic and acute endothelial cell stimulation by VEGF.
Significance: Our study identifies a mechanism of attenuating VEGF signaling by a Ras homolog and offers mechanistic insights into the regulation of angiogenesis by this small GTPase.
Keywords: Angiogenesis, Cancer Biology, Endothelial Cell, Endothelium, Ras Protein, Tumor, Tumor Microenvironment, Vascular Biology, VEGF
Abstract
Abnormal angiogenesis is associated with a broad range of medical conditions, including cancer. The formation of neovasculature with functionally defective blood vessels significantly impacts tumor progression, metastasis, and the efficacy of anticancer therapies. Vascular endothelial growth factor (VEGF) potently induces vascular permeability and vessel growth in the tumor microenvironment, and its inhibition normalizes tumor vasculature. In contrast, the signaling of the small GTPase R-Ras inhibits excessive angiogenic growth and promotes the maturation of regenerating blood vessels. R-Ras signaling counteracts VEGF-induced vessel sprouting, permeability, and invasive activities of endothelial cells. In this study, we investigated the effect of R-Ras on VEGF receptor 2 (VEGFR2) activation by VEGF, the key mechanism for angiogenic stimulation. We show that tyrosine phosphorylation of VEGFR2 is significantly elevated in the tumor vasculature and dermal microvessels of VEGF-injected skin in R-Ras knockout mice. In cultured endothelial cells, R-Ras suppressed the internalization of VEGFR2, which is required for full activation of the receptor by VEGF. Consequently, R-Ras strongly suppressed autophosphorylation of the receptor at all five major tyrosine phosphorylation sites. Conversely, silencing of R-Ras resulted in increased VEGFR2 phosphorylation. This effect of R-Ras on VEGFR2 was, at least in part, dependent on vascular endothelial cadherin. These findings identify a novel function of R-Ras to control the response of endothelial cells to VEGF and suggest an underlying mechanism by which R-Ras regulates angiogenesis.
Introduction
The tumor microenvironment is abundant with VEGF produced by tumor and stromal cells in response to elevated tissue hypoxia (1, 2). The high levels of VEGF in tumors contribute significantly to the excessive vessel leakiness and constant stimulation of endothelial sprouting that leads to the aberrant regeneration of blood vessels. These overstimulated vessels fail to undergo the maturation processes necessary for establishing a functional vascular network. The leaky vasculature allows the penetration of tumor cells into the circulation, facilitating metastasis (3, 4).
The VEGF blockade by anti-VEGF therapies transiently normalizes the tumor vasculature by temporarily restoring the balance between pro- and antiangiogenic signals (5, 6). Normalization of the tumor vasculature improves the efficacy of radiation therapy (7) and the delivery of nanomedicines (8) in mice by improving tumor perfusion and oxygenation (sensitization to radiation), and it also substantially alleviates life-threatening cerebral edema in glioblastoma patients by minimizing the leakiness of tumor vessels (9). However, anti-VEGF treatment also appears to enhance tumor invasion and metastasis, potentially explaining the lack of a lasting clinical efficacy (10, 11). Furthermore, a recent study revealed an unexpected activity of VEGF to inhibit glioblastoma invasion through the inhibition of hepatocyte growth factor-dependent MET activation (12). These findings suggest that the suppression of VEGF signaling in tumor vessels without substantially reducing the bioavailability of the VEGF ligand in the tumor microenvironment may inhibit angiogenesis and provide the benefit of vascular normalization without potentiating the invasion pathways in tumor cells.
Previously, we found an activity of small GTPase R-Ras inhibiting tumor angiogenesis (13–15). R-Ras is a Ras family protein with 55% amino acid sequence homology to the Ras proteins K-Ras and H-Ras (16). These proteins share an identical sequence of the minimal effector binding region (17). However, the constitutively activated mutant of R-Ras does not transform rat fibroblasts, activate Raf, or mitogen-activated protein kinase, which is in contrast to the activities of the Ras mutants (18, 19). R-Ras activates PI3K (19) and integrin adhesion to the extracellular matrix (20), whereas H-Ras inhibits integrin adhesion (21). R-Ras differs from the other members of the Ras family in that it contains a putative Src homology 3 domain-binding proline motif, and this motif is required for the integrin-activating activity of R-Ras (22–24). Also, R-Ras can be tyrosine-phosphorylated by the Eph receptors and Src (22–24). This phosphorylation inhibits R-Ras function (22–24). The forced expression of constitutively active R-Ras increased the growth and invasiveness of a cervical carcinoma cell line (25, 26). In contrast, both stimulatory and inhibitory effects of R-Ras have been reported for breast cancer cell proliferation and migration (27–31). To date, few studies have examined R-Ras in spontaneously rising tumors, and available information is largely on the basis of cancer cell lines ectopically expressing activated R-Ras. Inhibition of R-Ras activity has been implicated in breast tumor malignancy in a study analyzing clinical samples (31). Therefore, the precise role and activity of R-Ras in human cancer cells remains unclear. In normal tissues, R-Ras is most strongly expressed in endothelial cells (ECs)2 and smooth muscle cells with accumulation at the cytoplasmic membrane (13).
A genetic study in mice revealed that ablation of R-Ras enhances angiogenesis in tumor implants (13). This enhancement of angiogenesis was accompanied by significant impairment of tumor vessel maturation, characterized by severe deformation and functional deficiency of the vessels (14). Conversely, the gain of function of R-Ras in ECs improved the structure and function of VEGF-induced blood vessels in Matrigel implants (14). These results demonstrated that R-Ras is essential for establishing functional vessels while limiting excessive vessel formation in tumors and that R-Ras normalizes pathologically regenerating vasculature. R-Ras is highly expressed in the blood vessels of normal adult tissues (13). The analyses of a panel of human breast adenocarcinomas as well as mouse and human tumor implants in mice showed that R-Ras is significantly down-regulated in the majority of tumor vessels (13, 14). The chronically reduced expression of R-Ras in tumor blood vessels is consistent with the immaturity and functional abnormalities of the tumor vasculature, which are the hallmark characteristics of malignant tumors.
At present, the molecular mechanism of R-Ras-mediated vessel regulation remains elusive. Accumulating evidence indicates that R-Ras counteracts VEGF-stimulated angiogenic activities and the permeability of ECs (13, 14). In this work, we explore a possible mechanism by which R-Ras inhibits tumor angiogenesis and promotes vessel maturation through attenuation of VEGF signaling in ECs.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies for the following proteins were purchased from Cell Signaling Technology, Inc. (Danvers, MA): VEGFR2, phospho-VEGFR2 Tyr-951, phospho-VEGFR2 Tyr-1175, p38MAPK, phospho-p38MAPK Thr-180/Tyr-182, and clathrin. Antibodies for R-Ras were obtained from Cell Signaling Technology or Abcam (Cambridge, MA). Anti-phospho-VEGFR2 Tyr-1054/1059, phospho-VEGFR2 Tyr-1214, neurophirin 1, and VEGF antibodies were purchased from EMD Millipore (Billerica, MA). Anti-ephrin-B2 antibody was purchased from Thermo Fisher Scientific (Waltham, MA). Recombinant human VEGF-A165 was purchased from R&D Systems (Minneapolis, MN).
Analyses of Tumor Blood Vessels and Dermal Microvessels
R-Ras KO mice have been described previously (13). All animal experiments were performed in accordance with protocols approved by the institutional animal care and use committees of the Sanford-Burnham Medical Research Institute. B16F10 melanoma and Lewis lung carcinoma (LLC) cells (1 × 106 cells) were injected subcutaneously into the flank of R-Ras KO or wild-type control mice (14). Ten days later, tumor cells were collected, lysed in 0.5% SDS lysis buffer, and analyzed by Western blotting. The Western blot band intensity was determined for CD31, total VEGFR2, and phospho-VEGFR2 (Tyr(P)-1214) for each tumor lysate sample by densitometry. The level of total VEGFR2 or Tyr(P)-1214 VEGFR2 was standardized to CD31 or total VEGFR2, respectively, as follows: total VEGFR2 standardized to CD31 = (band intensity for total VEGFR2 of each tumor)/(CD31 band intensity of corresponding tumor); Tyr(P)-1214 VEGFR2 standardized to total VEGFR2 = (band intensity for Tyr(P)-1214 VEGFR2 of each tumor) / (total VEGFR2 band intensity of corresponding tumor). Data are presented as -fold increase from the wild-type animal group.
For immunofluorescence of tumor vessels, frozen histology sections were stained with anti-Tyr(P)-1214 VEGFR2, VEGR2, and CD31 antibodies and imaged using a Nikon Eclipse 90i microscope. For the analysis of dermal microvessels of the mouse ear, 30 μl of 50 ng/ml VEGF-A was injected intradermally into the ear using a 30-gauge needle and Hamilton syringe. The ears were collected 15 min later, and tissue lysate was prepared for Western blotting analyses. For immunofluorescence imaging, ears were fixed with 1% paraformaldehyde overnight. The ear specimens were permeabilized by 0.2% Triton X-100 in PBS for 1 h at 4 °C and stained with anti-Tyr(P)-1214 VEGFR2 and CD31 antibodies. The immunofluorescence of the whole-mounted ear was imaged by a Nikon A1R laser-scanning confocal microscope (Nikon Instrument Co.).
VEGF Expression in Tumors
The expression of VEGF-A, C, and D in tumors was determined by RT quantitative PCR using TaqMan probes (Mm00437304_m1, Mm00437310_m1, and Mm00438963_m1, respectively). To measure the VEGF-A protein level, tumor tissues were minced and lysed in CelLyticTM MT cell lysis reagent (Sigma-Aldrich) supplemented with protease inhibitor mixture (Sigma-Aldrich). After centrifugation at 15,000 × g at 4 °C for 20 min, the concentration of VEGF-A in the supernatant was measured using a mouse VEGF-A ELISA kit (Sigma-Aldrich).
Cell Culture, Lentivirus Transduction, and siRNA Transfection
Human umbilical cord vein endothelial cells and growth medium EGM-2 were purchased from Lonza (Basel, Switzerland). These cells were transduced with a constitutively active form of R-Ras (R-Ras38V), dominant negative R-Ras (R-Ras43N), or an insertless control using a pLenti6 lentivirus expression vector (Invitrogen) as described before (13). R-Ras knockdown was carried out by lentivirus transduction of shRNA that targets the R-Ras sequence 5′-GGA AAT ACC AGG AAC AAG A-3′, as described previously (14). The negative control shRNA, which does not target any known sequence of human, mouse, rat, or zebrafish origin, was obtained from COSMO BIO Co., Ltd. (Tokyo, Japan) (14).
Subconfluent cultures were used for in vitro cell signaling studies. Cells were starved of growth factors overnight with 2% horse serum in EBM-2 basal media, stimulated with VEGF-A165, and lysed at various time points for analyses. VE-cadherin siRNA (assay ID s2780), clathrin siRNA (assay ID s3190), and control siRNA were purchased from Ambion® (Life Technologies). Cells were transfected with Lipofectamine® RNAiMAX transfection reagent (Life Technologies).
VEGFR2 Internalization Assays
The VEGF-induced internalization of VEGFR2 was analyzed as described previously (19–21). Briefly, Human umbilical cord vein endothelial cells grown in Lab-TekTM chamber slides were starved of growth factors overnight in 2% horse serum in EBM-2 basal media. The next day, cells were incubated with monoclonal antibody that recognizes the extracellular domain of VEGFR2 (clone scFvA7, Fitzgerald, North Acton, MA) at 4 °C for 30 min to allow antibody binding to cell surface VEGFR2. Cells were then stimulated with 50 ng/ml VEGF-A at 37 °C to induce VEGFR2 internalization for 10 min. Subsequently, cells were washed with cold mild acid buffer (25 mm glycine 3% BSA in PBS (pH 2.5)) at 4 °C for 15 min to remove surface-bound antibodies and fixed with 4% paraformaldehyde for 10 min. For the VE-cadherin-silenced cells and control cells, chamber slides were spun using a swing bucket rotor at 100 × g for 5 min during fixation to avoid detachment of cells in subsequent staining steps. Cells were permeabilized by 0.1% Triton X-100 in PBS and stained with anti-mouse IgG-Alexa Fluor 488 secondary antibody or anti-E tag antibody (Abcam), followed by anti-rabbit IgG Alexa Fluor 594. At least 10 micrographs were taken with a Nikon Eclipse 90i fluorescence microscope (Nikon Instruments Inc.) equipped with a CoolSNAP HQ2 camera (Photometrics, Tucson, AZ), and the fluorescence images were analyzed by Volocity® software (PerkinElmer Life Sciences). The level of VEGFR2 internalization was quantified by measuring the integrated fluorescence signal intensity of the internalized antibody-VEGFR2 complex and normalized to the number of nuclei in each micrograph (integrated fluorescence signal intensity of internalized VEGFR2 per cell). To visualize the cell surface expression of VEGFR2, cells were incubated with anti-VEGFR2 antibody (clone scFvA7) at 4 °C for 30 min, rinsed with cold PBS, and fixed with 4% paraformaldehyde. A transferrin internalization assay was performed as described previously (22). Briefly, cells were incubated with 20 μg/ml Alexa Fluor 555-conjugated transferrin (Life Technologies) at 4 °C and then incubated at 37 °C for 5 min to allow cells to internalize transferrin/transferrin receptor. To inhibit the lysosomal degradation of internalized VEGFR2, cells were treated with 200 nm Bafilomycin A1 (Millipore) overnight before performing the internalization assay.
Biochemical Analyses of Receptor Internalization
The cell surface biotinylation-based analyses were performed as reported previously, with minor modifications (23, 24). Cells were starved of growth factors overnight in 2% horse serum in EBM-2 basal media. Cells were transferred to ice, rinsed twice in cold PBS, and surface-biotinylated using 0.25 mg/ml Sulfo-NHS-SS-Biotin (Pierce) for 20 min at 4 °C with gentle shaking. The biotinylated cells were rinsed twice with ice cold PBS and transferred to prewarmed EBM-2 media at 37 °C with or without 50 ng/ml VEGF-A to allow receptor internalization for 5–30 min as indicated. The cells were transferred to ice and rinsed twice with ice-cold PBS. The cell surface-bound biotin was removed using the membrane-impermeant reducing agent sodium 2-mercaptoethanesulphonate (Sigma-Aldrich,). Briefly, cells were incubated twice with ice-cold 250 mm sodium 2-mercaptoethanesulphonate in 50 mm Tris, 100 mm NaCl (pH 8.6) for 20 min at 4 °C. Unreacted sodium 2-mercaptoethanesulphonate was quenched by addition of 20 mm iodoacetamide for 10 min. Cells were then lysed in Triton X-100 lysis buffer and centrifuged at 10,000 × g for 10 min at 4 °C. Supernatant was collected, and protein concentration was measured by BCA protein assay (Pierce). The levels of biotinylated (internalized) VEGFR2 were determined in two ways: by avidin-agarose spin column followed by anti-VEGFR2 Western blot detection and by ELISA capture of VEGFR2 followed by avidin-HRP detection.
Isolation of Biotinylated Proteins with Avidin-Agarose
Biotinylated proteins were isolated from the cell lysate using NeutrAvidin agarose resin (Pierce) in a small spin column. The total protein concentration of the cell lysate was adjusted to 0.3 μg/μl, and 500 μl of this lysate was applied to the column and incubated for 1 h with rotation at room temperature. The column was then washed with lysis buffer, SDS-sample buffer containing 50 mm DTT was applied, and the column was incubated for 1 h at room temperature. The samples were then analyzed in SDS-PAGE followed by anti-VEGFR2 Western blotting. A small aliquot (100 μl) of cell lysate was taken before adding lysate to the avidin column and loaded directly onto SDS-PAGE to determine the total VEGFR2 level.
VEGFR2 Capture by ELISA
A Maxisorp 96-plate (Sigma-Aldrich) was coated overnight with 5 μg/ml anti-mouse IgG (clone A85-3) in 50 mm Na2CO3 (pH 9.6) at 4 °C, followed by another incubation with 5 μg/ml of anti-VEGFR2 (clone A-3, Santa Cruz Biotechnology) for 2 h at room temperature. The plate was then treated with blocking buffer, 5% BSA, 0.1% Tween 20 in PBS (PBS-T). A total of 50 μl of cell lysate (0.2 μg/μl) was added to a well and incubated overnight at 4 °C to capture VEGFR2. After rinsing the plate twice with PBS-T, the wells were incubated with avidin-HRP. The plate was rinsed with PBS-T twice, and 200 μl of the o-phenylenediamine dihydrochloride (OPD) substrate (SIGMAFASTTM OPD, Sigma-Aldrich) was added to each well. The plate was incubated at room temperature for 30 min. The reaction was stopped with 3 m HCl, and the absorbance was measured at 490 nm. The absorbance reading for each data point was normalized to the reading of the cells not treated with sodium 2-mercaptoethanesulphonate to determine the relative levels of VEGFR2 internalization. Four wells for each group were analyzed.
Statistics
Statistics were performed using two-tailed Student's t test to compare wild-type with R-Ras KO mice, and one-way analysis of variance with Tukey's multiple comparison test was used for the in vitro experiment. Error bars represent mean ± S.E.
RESULTS
R-Ras Suppresses VEGFR2 Activation in Tumor Blood Vessels
The level of VEGF is chronically elevated in tumors. We investigated the potential role of R-Ras in regulating the activation of VEGF signaling in the tumor vasculature. For this purpose, LLC or B16F10 melanoma was implanted subcutaneously into R-Ras KO or WT control mice. Histological sections of the tumor implants were analyzed by immunofluorescence for phospho-VEGFR2 Tyr-1214, total VEGFR2, and CD31 (Fig. 1A) or R-Ras and CD31 (Fig. 1B). The Tyr-1214 residue is one of the five major tyrosine sites for VEGFR2 autophosphorylation induced by VEGF. This analysis showed an increased level of phosphorylated VEGFR2 in the vasculature of LLC and B16F10 tumors implanted in R-Ras KO mice (Fig. 1A). Western blot analyses of tumor lysates confirmed the immunostaining results by showing an elevated level of phospho-VEGFR2 that was standardized to the level of total VEGFR2 (Fig. 1, C and D). To investigate the effect of R-Ras on VEGFR2 activation in a more direct manner, we analyzed VEGFR2 tyrosine phosphorylation in cultured ECs in response to a defined amount of VEGF-A (Fig. 1E). Cultured ECs (human umbilical cord vein endothelial cells) transduced with cDNAs for R-Ras mutants were serum-starved and then stimulated with 50 ng/ml VEGF-A. In this analysis, we found that R-Ras38V decreased VEGFR2 tyrosine phosphorylation induced by VEGF-A (Fig. 1E). Conversely, R-Ras43N increased it (Fig. 1E). These findings corroborate the in vivo observations of R-Ras KO and WT mice.
FIGURE 1.

R-Ras deficiency enhances VEGFR2 activation in the tumor vasculature. A, tumor sections were analyzed for VEGFR2 phosphorylation by immunofluorescence: CD31 (red), total VEGFR2 (purple), and phospho-VEGFR2 Tyr-1214 (green). Scale bar = 50 μm. B, R-Ras expression in the endothelium was assessed by coimmunostaining for R-Ras (green) and CD31 (red). Scale bar = 50 μm. C, Tyr(P)-1214 VEGFR2, total VEGFR2, and CD31 were analyzed by Western blotting of the tumor lysate. D, the level of VEGFR2 expression was quantified and normalized to CD31. The level of Tyr(P)-1214 VEGFR2 was normalized to the level of total VEGFR2 (pY1214-VEGFR2/VEGFR2) to assess VEGFR2 activation. Data are presented as -fold increase relative to the wild-type control animals. *, p < 0.05; **, p < 0.01; LLC tumors, n = 11 (WT) and 8 (KO); B16F10 tumors, n = 11 (WT) and 10 (KO). Error bars show mean ± S.E. E, endothelial cells transduced with R-Ras38V or R-Ras43N were stimulated with 50 ng/ml VEGF-A for 10 min in culture. VEGFR2 was immunoprecipitated (IP) from the cell lysate and analyzed for tyrosine autophosphorylation levels by anti-Tyr(P) Western blotting.
We also examined VEGF levels in the tumor implants. RT quantitative PCR analyses showed no difference in the expression levels of VEGF-A, VEGF-C, and VEGF-D between the tumors in wild-type and R-Ras KO mice (Fig. 2A). ELISA assays also showed comparable VEGF-A levels between LLC tumors in wild-type and KO mice. A moderate (∼30%) increase of VEGF-A was found in B16F10 tumors in R-Ras KO mice (Fig. 2B). However, because VEGF levels are already high in tumors in general (and particularly in B16F10 tumors compared with LLC tumors), it is unlikely that this moderate increase accounts for the significantly elevated activation of VEGFR2 in R-Ras KO mice.
FIGURE 2.

VEGF levels in B16F10 and LLC tumor implants. A, total RNA was isolated from B16F10 or LLC tumors, and RT quantitative PCR was performed to determine the mRNA expression levels of VEGF-A, VEGF-C, and VEGF-D in each tumor type. B, VEGF-A protein levels in the tumor lysate were determined by mouse VEGF-A ELISA. ***, p < 0.001.
Next, we examined the role of R-Ras in acute VEGF stimulation in normal tissue. We injected VEGF-A intradermally into the ear of R-Ras KO and WT control mice and analyzed the autophosphorylation of VEGFR2 Tyr-1214 by immunofluorescence. As with the analysis of tumors, we found a significant increase of VEGFR2 activation in the microvasculature of R-Ras KO mouse skin (Fig. 3A). The elevated VEGFR2 activation was confirmed by the quantification of Western blotting of skin tissue lysate (Fig. 3B). Therefore, endogenous R-Ras strongly attenuates acute stimulation by VEGF in normal blood vessels. The combined observations highlight the important in vivo role of R-Ras in counterbalancing both the chronic and acute VEGF stimulation of blood vessels.
FIGURE 3.

Acute activation of VEGFR2 is elevated in dermal microvessels of R-Ras KO mice. A, VEGF-A (30 μl of 50 ng/ml) was injected intradermally into mouse ears, and the ears were collected 15 min later. The whole-mount immunofluorescence for CD31 (red), total VEGFR2 (purple), and Tyr(P)-1214 VEGFR2 (green) was imaged by confocal microscopy. Scale bar = 100 μm. B, quantitative analysis was carried out by Western blotting of the ear tissue lysate as described for the tumor studies. Four (WT) or six (KO) animals were examined per group. Error bars shown mean ± S.E. *, p < 0.05.
R-Ras Inhibits Phosphorylation of VEGFR2 at All Major Tyrosine Phosphorylation Sites upon VEGF Stimulation
The major tyrosine phosphorylation sites of VEGFR2 important for the response of ECs to VEGF stimulation are Tyr-951 within the kinase insert domain, Tyr-1054 and Tyr-1059 within the kinase domain, and Tyr-1175 and Tyr-1214 in the C-terminal tail of the receptor (32–34). We determined which phosphorylation sites are regulated by R-Ras during VEGF stimulation in cultured ECs. R-Ras38V or mock-transduced ECs were starved of growth factors and stimulated with VEGF-A. The phosphorylation of individual tyrosine residues of VEGFR2 was determined by phospho-specific antibodies to each site. We found that the expression of R-Ras38V greatly diminishes VEGF-induced autophosphorylation at all five major tyrosine phosphorylation sites (Fig. 4, A and B). Autophosphorylation decreased by as much as 80% at the peak activation at 5 min post-VEGF (50 ng/ml) stimulation, demonstrating the profound effect of R-Ras signaling against VEGFR2 activation. In contrast, the suppression of R-Ras by dominant negative R-Ras43N (Fig. 4, C and D) or by R-Ras silencing using shRNA (Fig. 4, E and F) increased VEGFR2 autophosphorylation upon VEGF stimulation (only Tyr-1175 and Tyr-1214 were examined).
FIGURE 4.

R-Ras suppresses autophosphorylation of VEGFR2. A, time course analysis of VEGFR2 activation. ECs were stimulated with VEGF-A (50 ng/ml), and the cell lysate was collected at various time points. VEGFR2 phosphorylation was analyzed by Western blotting using phospho-specific antibodies for various tyrosine sites. B, the level of each tyrosine phosphorylation site was quantified and normalized to the total VEGFR2 expression level. The -fold differences are presented as relative values with the peak phosphorylation in mock control cells at 5 min as 1. The graphs present representative data of at least three experiments. C, ECs were stimulated with various doses of VEGF-A for 5 min, and phosphorylation levels at Tyr-1175 and Tyr-1214 were determined. D, quantification of C. *, p < 0.05; **, p < 0.01; ***, p < 0.001. E, endogenous R-Ras in ECs was knocked down by shRNA. ECs were then stimulated with various doses of VEGF-A for 5 min, and phosphorylation levels at Tyr-1175 and Tyr-1214 were determined by Western blotting. NC, negative control shRNA. F, quantification of E. *, p < 0.05; **, p < 0.01.
R-Ras Inhibits VEGF-dependent VEGFR2 Internalization
VEGF ligand binding induces the clathrin-dependent internalization of VEGFR2, and this internalization is required for the full activation of VEGFR2 (35–37). Because all major tyrosine autophosphorylation sites are down-regulated simultaneously by R-Ras, we investigated the effect of R-Ras on VEGFR2 internalization upon VEGF stimulation of ECs. To assess the internalization of VEGFR2 from the cell surface, we first conducted an antibody feeding assay (35, 36, 38). In this assay, VEGFR2 localization upon VEGF stimulation was analyzed using a monoclonal antibody that recognizes the extracellular domain of VEGFR2 (Fig. 5, A and B). When added to the culture, the antibody binds to cell surface VEGFR2 that is sensitive to a mild acid wash. The acid-resistant vesicular staining pattern represented VEGFR2 molecules that are internalized from the cell surface into intracellular vesicles upon VEGF stimulation (Fig. 5A). This analysis showed that R-Ras38V expression significantly reduced the internalization of VEGFR2 (Fig. 5, A and B). The R-Ras43N expression showed an opposite effect, increasing VEGFR2 internalization during VEGF-stimulation (Fig. 5, A and B).
FIGURE 5.

R-Ras inhibits VEGFR2 internalization induced by VEGF. A, ECs were first incubated with anti-VEGFR2 antibody (scFvA7) to allow antibody binding to the cell surface and subsequently stimulated with 50 ng/ml VEGF-A for ligand-induced VEGFR2 internalization. After washing away the cell surface-bound antibodies with mild acid buffer, the internalized antibody-VEGFR2 complex was visualized by staining with secondary antibody. Green, internalized anti-VEGFR2 antibody-VEGFR2 complex; red, phalloidin; blue, DAPI. Scale bar = 25 μm. B, VEGFR2 internalization was quantified by average integrated fluorescence signal intensity per cell and is presented as relative values. Ten fluorescent micrographs were analyzed for each cell type. The results were reproduced by three independent experiments. *, p < 0.05; ***, p < 0.001. C and D, study of VEGFR2 internalization with blockade of lysosomal degradation. ECs were treated with 200 nm Bafilomycin A1 overnight, and an antibody feeding assay was performed to assess VEGFR2 internalization. C, representative micrographs of cells cultured with or without bafilomycin showing internalized VEGFR2. Bafilomycin substantially increased the intracellular content of antibody-labeled VEGFR2 in mock-transduced control cells. R-Ras38V significantly reduced antibody-labeled VEGFR2 compared with control cells even in the presence of bafilomycin. Scale bar = 40 μm. D, internalized VEGFR2 was quantified by average integrated fluorescence signal intensity per cell and is presented as relative values. ***, p < 0.001.
Bafilomycin, which inhibits lysosomal degradation of internalized proteins, significantly increased the intracellular content of internalized VEGFR2 in VEGF-stimulated mock-transduced control cells (Fig. 5, C and D). However, bafilomycin could not offset the effect of R-Ras, and the R-Ras38V-expressing cells still showed significantly reduced internalized VEGFR2 compared with the mock control cells treated with bafilomycin. These observations rule out the possibility that R-Ras induces a rapid degradation of internalized VEGFR2 in lysosomes, causing an apparent decrease of the receptor internalization.
We next employed biochemical approaches to further confirm the effect of R-Ras on VEGFR2 internalization (Fig. 6A). In these studies, the endothelial cell surface was biotinylated with Sulfo-NHS-SS-Biotin, and the cells were stimulated with VEGF to allow the internalization of VEGFR2. After removing surface-bound biotin with a cell-impermeant reducing agent, the cell lysate was analyzed by avidin affinity column followed by anti-VEGFR2 Western blot detection (Fig. 6B) or by ELISA capture of VEGFR2 followed by avidin-HRP detection (Fig. 6, C and D). The studies of gain (R-Ras38V) or loss (R-Ras shRNA) of function in these assays demonstrated the inhibitory effect of R-Ras on VEGF-induced VEGFR2 internalization, confirming the results of the antibody feeding assay.
FIGURE 6.

Biochemical analyses of VEGFR2 internalization. A, schematic of the assays. i, endothelial cell surface proteins were biotinylated by Sulfo-NHS-SS-Biotin. ii and iii, cells were stimulated with VEGF-A to allow VEGFR2 internalization. iv, cells were then treated with non-permeable reducing agent to remove surface-bound biotin. Cell lysates containing internalized biotinylated proteins were analyzed by avidin-agarose column, followed by anti-VEGFR2 Western blot detection (v) or by ELISA capture of VEGFR2 followed by avidin-HRP detection (vi). B, detection of internalized VEGFR2 using the avidin column method (A, v). ECs were stimulated with or without 50 ng/ml VEGF-A for 10 min. Bottom panel, an aliquot of cell lysate was loaded directly onto SDS-PAGE to assess total VEGFR2 (Input). C, time course analysis with the ELISA capture method (A, vi). The cell surface-biotinylated ECs were stimulated with 50 ng/ml VEGF-A for the indicated times. The relative levels of VEGFR2 internalization are presented for each data point compared with the mock control at 30 min. D, R-Ras knockdown by shRNA increases VEGFR2 internalization. Cells were stimulated with or without 50 ng/ml VEGF-A for 10 min, and the internalized VEGFR2 was measured by ELISA method (A, vi). The -fold increase of internalization is presented as a relative value. NC, negative control shRNA.
The VEGF-induced internalization of VEGFR2 is dependent on clathrin-mediated receptor endocytosis (35–37). Accordingly, clathrin knockdown has been shown to reduce VEGFR2 autophosphorylation (35). We showed that clathrin knockdown by siRNA reduced Tyr-1214 phosphorylation of VEGFR2 in control cells, as expected (Fig. 7, A and B). Clathrin knockdown also reduced the tyrosine phosphorylation level in R-Ras-silenced cells to a level equivalent to that of the R-Ras-expressing control cells with clathrin knockdown, therefore offsetting the elevated receptor phosphorylation caused by the R-Ras deficiency (Fig. 7B). These observations further support the hypothesis that R-Ras regulates VEGFR2 activity through the regulation of clathrin-dependent VEGFR2 internalization induced by VEGF.
FIGURE 7.
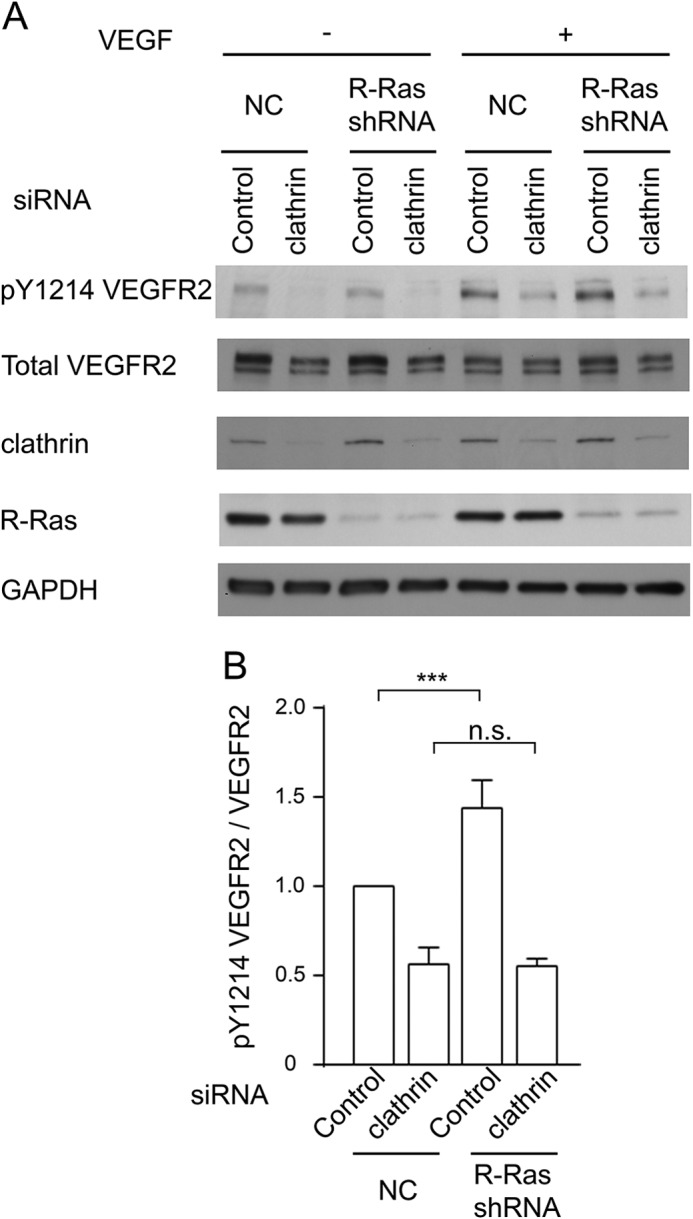
Clathrin knockdown inhibits autophosphorylation of VEGFR2. A, clathrin expression was knocked down in R-Ras38V or mock-transduced control ECs by siRNA. These cells were stimulated with 50 ng/ml VEGF-A for 5 min. The levels of clathrin expression and VEGFR2 Tyr-1214 phosphorylation in these cells were determined by Western blotting. B, quantification of the Tyr(P)-1214 VEGFR2:total VEGFR2 ratio in VEGF-stimulated ECs to determine receptor activation. The -fold differences are presented in comparison with mock cells treated with control siRNA. ***, p < 0.001; n.s., not significant.
When unstimulated with VEGF, R-Ras38V-transduced or R-Ras-silenced cells showed no differences in cell surface or total VEGFR2 expression levels from the control cells (Fig. 8, A–C and E). Also, we did not observe differences in cell surface or total expression of the VEGFR2-interacting proteins neuropilin 1 or ephrin B2 (Fig. 8, D and E). Neither R-Ras38V nor R-Ras43N expression affected the internalization of the transferrin receptor, demonstrating the specificity of the R-Ras effect without generally affecting the internalization of cell surface molecules (Fig. 8F).
FIGURE 8.

R-Ras does not alter receptor expression or transferrin internalization. A–E, R-Ras has no effect on cell surface and total expression of VEGFR2, neuropilin 1, and ephrin B2. A, cells without VEGF stimulation were incubated with anti-VEGFR2 antibody at 4 °C for immunofluorescence staining of cell surface, and fixed with paraformaldehyde. Scale bar = 25 μm. B, cell surface proteins were biotinylated, and cell lysate was prepared without VEGF stimulation or biotin stripping. The cell surface proteins were isolated from the lysate by avidin column, and anti-VEGFR2 Western blotting was performed to quantify the surface VEGFR2 levels. An aliquot of cell lysate was loaded directly onto the gel to determine total VEGFR2 and GAPDH (Input). C, quantification of surface and total VEGFR2 normalized to GAPDH, presented as the levels relative to mock control cells. D, the cell surface expression levels of neuropilin 1 and ephrin B2 were determined for R-Ras38V or mock control cells. The total expression levels of these proteins were also determined using the pre-avidin column lysate. E, similar analyses were conducted for R-Ras-silenced ECs. No significant differences were found in R-Ras38V or R-Ras shRNA-transduced cells compared with control cells. NC, negative control shRNA. F, Alexa Fluor 555-conjugated transferrin was allowed to internalize into the cells at 37 °C for 5 min. The level of transferrin internalization was quantified by average integrated fluorescence signal intensity per cell and is presented as relative values.
The Role of VE-cadherin in the R-Ras-mediated Inhibition of VEGFR2
VE-cadherin is essential for the regulation of endothelial function and homeostasis (39). VE-cadherin stabilized at endothelial adherens junctions blocks VEGFR2 internalization and inhibits VEGF signaling in ECs (35). This VE-cadherin-dependent mechanism of VEGFR2 inhibition is important for the contact inhibition of ECs that attenuates EC proliferation, and it counterbalances the effect of VEGF, a potent mitogen for ECs (40). The endothelial R-Ras stabilizes VE-cadherin at adherens junctions, thereby enhancing endothelial barrier function and reducing vascular permeability (14). We examined whether VE-cadherin plays a role in the regulation of VEGFR2 by R-Ras. VE-cadherin was knocked down in R-Ras38V or mock-transduced ECs by siRNA, and the internalization of VEGFR2 was analyzed upon VEGF stimulation (Fig. 9A). VE-cadherin knockdown significantly increased VEGFR2 internalization in mock-transduced ECs, in agreement with a previous report (35). VE-cadherin knockdown also substantially increased VEGFR2 internalization in R-Ras38V-transduced cells (Fig. 9, A and B). Hence, R-Ras38V reduced receptor internalization only by 20% in VE-cadherin knockdown cells compared with 58% in cells without VE-cadherin knockdown (Fig. 9B, Control), demonstrating the importance of VE-cadherin in this effect of R-Ras. These results were in agreement with the effect of VE-cadherin knockdown on VEGFR2 phosphorylation levels (Fig. 9D). Here, R-Ras38V reduced VEGFR2 phosphorylation by 49% in endothelial cells expressing the normal level of VE-cadherin (Fig. 9D, control). On the other hand, R-Ras38V reduced receptor phosphorylation only by 30% in VE-cadherin knockdown cells. The siRNA did not completely silence VE-cadherin (Fig. 9C). Therefore, some effect of R-Ras through the remaining VE-cadherin is expected to be present, potentially explaining the remaining inhibitory effect of R-Ras38V in these cells. Taken together, these data suggest a critical involvement of VE-cadherin in the inhibitory effect of R-Ras on VEGFR2 signaling.
FIGURE 9.

VE-cadherin is crucial to the R-Ras-dependent regulation of VEGFR2. A, VE-cadherin expression was knocked down in R-Ras38V or mock-transduced ECs by siRNA. Forty-eight hours later, cells were stimulated with 50 ng/ml VEGF-A for 10 min. VEGFR2 internalization was analyzed by antibody feeding assay. Scale bar = 50 μm. B, VEGFR2 internalization was quantified by average integrated fluorescence signal intensity per cell and is presented as relative values. ***, p < 0.001. VE-cad, VE-cadherin. C, the levels of VE-cadherin expression and VEGFR2 Tyr-1214 phosphorylation in these cells were determined by Western blotting. D, quantification of Tyr(P)-1214 VEGFR2. The -fold differences are presented in comparison with mock cells treated with control siRNA. **, p < 0.01.
DISCUSSION
R-Ras promotes the quiescence of ECs, pericytes, and vascular smooth muscle cells (13, 14). This R-Ras activity is important for limiting pathological vascular proliferation in conditions such as neointimal hyperplasia and tumor angiogenesis (13, 14). The effect of R-Ras is unique in that, unlike other antiangiogenic molecules, up-regulation of R-Ras does not lead to EC death, but, instead, it promotes the maturation of nascent blood vessels. Through this activity, R-Ras supports the structural and functional integrity of new vessels following angiogenic sprouting (12).
In this study, we investigated the molecular mechanism underlying the regulation of angiogenesis by R-Ras. In cultured ECs, we showed that R-Ras inhibits the internalization of VEGFR2, the process required for the full activation of VEGFR2 signaling by VEGF. Consistent with the inhibitory effect on receptor internalization, all five major tyrosine phosphorylation sites of VEGFR2, rather than a single specific site, were strongly down-regulated by R-Ras. Although these in vitro studies have a limitation in recapitulating the response of the endothelium, the results of the studies using R-Ras KO mice supported the in vivo relevance of our findings. We observed significantly elevated levels of tyrosine-phosphorylated VEGFR2 in the tumor vasculature in R-Ras KO mice. Our previous immunofluorescence analysis showed a moderate increase of average total VEGFR2 in R-Ras KO mice (14). This tendency was also noted in our Western blot analysis. However, the ratio of phosphorylated VEGFR2 to total VEGFR2 clearly indicated an elevated activation of the receptor in R-Ras KO mice. Furthermore, the endothelium of the normal dermal microvasculature stimulated with VEGF-A injection also showed significantly increased VEGFR2 activation in R-Ras KO mice. These observations demonstrated an important role of R-Ras in attenuating both chronic and acute VEGF stimulation of blood vessels.
The blockade of VEGF (or VEGFR2) leads to normalization of the tumor vasculature (5, 7). The observed increase in VEGFR2 activation in the tumor vessels in R-Ras KO animals is in agreement with exacerbated abnormalities of these vessels (14). However, R-Ras may also affect vessel structure and function through additional mechanisms independent of VEGFR2 regulation. Further studies are needed to examine the direct causal relationship between elevated VEGFR2 activation and augmented deformation of tumor vessels in the R-Ras deficiency.
Ras GTPases have been shown to positively regulate endocytosis (41–45). However, accumulating evidence indicates that R-Ras is functionally distinct from other Ras-like proteins. In fact, R-Ras is known to function oppositely from H-Ras in the regulation of integrin adhesion (20, 21, 46). A few studies have reported that R-Ras promotes integrin endocytosis (47, 48). Another study has demonstrated that R-Ras promotes exocytosis (49). The current literature is unclear regarding how R-Ras affects VEGFR2 and other growth factor receptor internalization in ECs. In this study, we showed that R-Ras inhibits VEGFR2 internalization upon VEGF stimulation. Using RNAi, we showed the importance of VE-cadherin for this effect of R-Ras. These observations are in agreement with two key findings from previous studies: VE-cadherin stabilization at endothelial adherens junctions inhibits VEGFR2 internalization and activation by VEGF (35), and R-Ras stabilizes VE-cadherin at endothelial adherens junctions and protects endothelial integrity (14). The R-Ras deficiency in mice results in the disruption of VE-cadherin and endothelial adherens junctions of tumor vessels (14). In this study, we showed that R-Ras deficiency also elevates VEGFR2 activation in tumor vessels as well as in normal tissue vessels stimulated with VEGF, suggesting the in vivo relevance of our findings.
R-Ras may also affect VEGFR2 internalization through other mechanisms. For instance, R-Ras has a profound effect on integrin-mediated cell adhesion to the extracellular matrix. Altered focal adhesion and cytoskeletal architecture may contribute to the inhibition of VEGFR2 internalization. We showed, in this study, that R-Ras does not affect total or cell surface expression of neuropilin 1 or ephrin B2, which would otherwise affect VEGFR2 internalization (36, 38). However, these results do not rule out the functional modulation of these proteins as a possible regulatory mechanism for VEGFR2 internalization.
In summary, our results demonstrate the important function of R-Ras to attenuate EC stimulation by VEGF through suppressing VEGFR2 activation. The regulatory mechanism identified here may be important in cancer as well as in vascular complications of other medical conditions involving aberrant vascular proliferation and permeability caused by excessive VEGF.
Acknowledgments
Histological preparations were done at the Histology and Imaging core facilities and RT quantitative PCR at the Analytical Genomics Core of Sanford-Burnham Medical Research Institute at Lake Nona.
This work was supported, in whole or in part, by NCI/National Institutes of Health Grant CA125255. This work was also supported by Florida Department of Health Bankhead-Coley Cancer Research Program 4BB17 and by Florida Breast Cancer Foundation Grant 3021180.
- EC
- endothelial cell
- LLC
- Lewis lung carcinoma
- VE-cadherin
- vascular endothelial cadherin.
REFERENCES
- 1. De Bock K., Cauwenberghs S., Carmeliet P. (2011) Vessel abnormalization: another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr. Opin. Genet. Dev. 21, 73–79 [DOI] [PubMed] [Google Scholar]
- 2. Ferrara N., Kerbel R. S. (2005) Angiogenesis as a therapeutic target. Nature 438, 967–974 [DOI] [PubMed] [Google Scholar]
- 3. Weis S., Cui J., Barnes L., Cheresh D. (2004) Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J. Cell Biol. 167, 223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gerhardt H., Semb H. (2008) Pericytes: gatekeepers in tumour cell metastasis? J Mol Med 86, 135–144 [DOI] [PubMed] [Google Scholar]
- 5. Tong R. T., Boucher Y., Kozin S. V., Winkler F., Hicklin D. J., Jain R. K. (2004) Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 64, 3731–3736 [DOI] [PubMed] [Google Scholar]
- 6. Jain R. K. (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62 [DOI] [PubMed] [Google Scholar]
- 7. Winkler F., Kozin S. V., Tong R. T., Chae S. S., Booth M. F., Garkavtsev I., Xu L., Hicklin D. J., Fukumura D., di Tomaso E., Munn L. L., Jain R. K. (2004) Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 6, 553–563 [DOI] [PubMed] [Google Scholar]
- 8. Chauhan V. P., Stylianopoulos T., Martin J. D., Popović Z., Chen O., Kamoun W. S., Bawendi M. G., Fukumura D., Jain R. K. (2012) Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 7, 383–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Batchelor T. T., Sorensen A. G., di Tomaso E., Zhang W. T., Duda D. G., Cohen K. S., Kozak K. R., Cahill D. P., Chen P. J., Zhu M., Ancukiewicz M., Mrugala M. M., Plotkin S., Drappatz J., Louis D. N., Ivy P., Scadden D. T., Benner T., Loeffler J. S., Wen P. Y., Jain R. K. (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11, 83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ebos J. M., Lee C. R., Cruz-Munoz W., Bjarnason G. A., Christensen J. G., Kerbel R. S. (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pàez-Ribes M., Allen E., Hudock J., Takeda T., Okuyama H., Viñals F., Inoue M., Bergers G., Hanahan D., Casanovas O. (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu K. V., Chang J. P., Parachoniak C. A., Pandika M. M., Aghi M. K., Meyronet D., Isachenko N., Fouse S. D., Phillips J. J., Cheresh D. A., Park M., Bergers G. (2012) VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Komatsu M., Ruoslahti E. (2005) R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat. Med. 11, 1346–1350 [DOI] [PubMed] [Google Scholar]
- 14. Sawada J., Urakami T., Li F., Urakami A., Zhu W., Fukuda M., Li D. Y., Ruoslahti E., Komatsu M. (2012) Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell 22, 235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sawada J., Komatsu M. (2012) Normalization of tumor vasculature by R-Ras. Cell Cycle 11, 4285–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lowe D. G., Capon D. J., Delwart E., Sakaguchi A. Y., Naylor S. L., Goeddel D. V. (1987) Structure of the human and murine R-ras genes, novel genes closely related to ras proto-oncogenes. Cell 48, 137–146 [DOI] [PubMed] [Google Scholar]
- 17. Self A. J., Paterson H. F., Hall A. (1993) Different structural organization of Ras and Rho effector domains. Oncogene 8, 655–661 [PubMed] [Google Scholar]
- 18. Lowe D. G., Goeddel D. V. (1987) Heterologous expression and characterization of the human R-ras gene product. Mol. Cell. Biol. 7, 2845–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marte B. M., Rodriguez-Viciana P., Wennström S., Warne P. H., Downward J. (1997) R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr. Biol. 7, 63–70 [DOI] [PubMed] [Google Scholar]
- 20. Zhang Z., Vuori K., Wang H., Reed J. C., Ruoslahti E. (1996) Integrin activation by R-ras. Cell 85, 61–69 [DOI] [PubMed] [Google Scholar]
- 21. Hughes P. E., Renshaw M. W., Pfaff M., Forsyth J., Keivens V. M., Schwartz M. A., Ginsberg M. H. (1997) Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell 88, 521–530 [DOI] [PubMed] [Google Scholar]
- 22. Wang B., Zou J. X., Ek-Rylander B., Ruoslahti E. (2000) R-Ras contains a proline-rich site that binds to SH3 domains and is required for integrin activation by R-Ras. J. Biol. Chem. 275, 5222–5227 [DOI] [PubMed] [Google Scholar]
- 23. Zou J. X., Wang B., Kalo M. S., Zisch A. H., Pasquale E. B., Ruoslahti E. (1999) An Eph receptor regulates integrin activity through R-Ras. Proc. Natl. Acad. Sci. U.S.A. 96, 13813–13818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zou J. X., Liu Y., Pasquale E. B., Ruoslahti E. (2002) Activated SRC oncogene phosphorylates R-ras and suppresses integrin activity. J. Biol. Chem. 277, 1824–1827 [DOI] [PubMed] [Google Scholar]
- 25. Rincón-Arano H., Rosales R., Mora N., Rodriguez-Castañeda A., Rosales C. (2003) R-Ras promotes tumor growth of cervical epithelial cells. Cancer 97, 575–585 [DOI] [PubMed] [Google Scholar]
- 26. Mora N., Rosales R., Rosales C. (2007) R-Ras promotes metastasis of cervical cancer epithelial cells. Cancer Immunol. Immunother. 56, 535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu Y., Feig L. A. (2002) Involvement of R-Ras and Ral GTPases in estrogen-independent proliferation of breast cancer cells. Oncogene 21, 7557–7568 [DOI] [PubMed] [Google Scholar]
- 28. Yu Y., Hao Y., Feig L. A. (2006) The R-Ras GTPase mediates cross talk between estrogen and insulin signaling in breast cancer cells. Mol. Cell. Biol. 26, 6372–6380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeong H. W., Nam J. O., Kim I. S. (2005) The COOH-terminal end of R-Ras alters the motility and morphology of breast epithelial cells through Rho/Rho-kinase. Cancer Res. 65, 507–515 [PubMed] [Google Scholar]
- 30. Wozniak M. A., Kwong L., Chodniewicz D., Klemke R. L., Keely P. J. (2005) R-Ras controls membrane protrusion and cell migration through the spatial regulation of Rac and Rho. Mol. Biol. Cell 16, 84–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song J., Zheng B., Bu X., Fei Y., Shi S. (2014) Negative association of R-Ras activation and breast cancer development. Oncol. Rep. 31, 2776–2784 [DOI] [PubMed] [Google Scholar]
- 32. Holmes K., Roberts O. L., Thomas A. M., Cross M. J. (2007) Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 19, 2003–2012 [DOI] [PubMed] [Google Scholar]
- 33. Matsumoto T., Bohman S., Dixelius J., Berge T., Dimberg A., Magnusson P., Wang L., Wikner C., Qi J. H., Wernstedt C., Wu J., Bruheim S., Mugishima H., Mukhopadhyay D., Spurkland A., Claesson-Welsh L. (2005) VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 24, 2342–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takahashi T., Yamaguchi S., Chida K., Shibuya M. (2001) A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-γ and DNA synthesis in vascular endothelial cells. EMBO J. 20, 2768–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lampugnani M. G., Orsenigo F., Gagliani M. C., Tacchetti C., Dejana E. (2006) Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 174, 593–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sawamiphak S., Seidel S., Essmann C. L., Wilkinson G. A., Pitulescu M. E., Acker T., Acker-Palmer A. (2010) Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 465, 487–491 [DOI] [PubMed] [Google Scholar]
- 37. Pitulescu M. E., Adams R. H. (2014) Regulation of signaling interactions and receptor endocytosis in growing blood vessels. Cell Adh. Migr. 8, 366–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lanahan A., Zhang X., Fantin A., Zhuang Z., Rivera-Molina F., Speichinger K., Prahst C., Zhang J., Wang Y., Davis G., Toomre D., Ruhrberg C., Simons M. (2013) The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell 25, 156–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carmeliet P., Lampugnani M. G., Moons L., Breviario F., Compernolle V., Bono F., Balconi G., Spagnuolo R., Oosthuyse B., Dewerchin M., Zanetti A., Angellilo A., Mattot V., Nuyens D., Lutgens E., Clotman F., de Ruiter M. C., Gittenberger-de Groot A., Poelmann R., Lupu F., Herbert J. M., Collen D., Dejana E. (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98, 147–157 [DOI] [PubMed] [Google Scholar]
- 40. Grazia Lampugnani M., Zanetti A., Corada M., Takahashi T., Balconi G., Breviario F., Orsenigo F., Cattelino A., Kemler R., Daniel T. O., Dejana E. (2003) Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, β-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161, 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barbieri M. A., Kohn A. D., Roth R. A., Stahl P. D. (1998) Protein kinase B/akt and rab5 mediate Ras activation of endocytosis. J. Biol. Chem. 273, 19367–19370 [DOI] [PubMed] [Google Scholar]
- 42. Jiang X., Sorkin A. (2002) Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol. Biol. Cell 13, 1522–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu A., Tebar F., Alvarez-Moya B., López-Alcalá C., Calvo M., Enrich C., Agell N., Nakamura T., Matsuda M., Bachs O. (2009) A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cell Biol. 184, 863–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fehrenbacher N., Philips M. (2009) Intracellular signaling: peripatetic Ras. Curr. Biol. 19, R454–R457 [DOI] [PubMed] [Google Scholar]
- 45. Misaki R., Morimatsu M., Uemura T., Waguri S., Miyoshi E., Taniguchi N., Matsuda M., Taguchi T. (2010) Palmitoylated Ras proteins traffic through recycling endosomes to the plasma membrane during exocytosis. J. Cell Biol. 191, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sethi T., Ginsberg M. H., Downward J., Hughes P. E. (1999) The small GTP-binding protein R-Ras can influence integrin activation by antagonizing a Ras/Raf-initiated integrin suppression pathway. Mol. Biol. Cell 10, 1799–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Conklin M. W., Ada-Nguema A., Parsons M., Riching K. M., Keely P. J. (2010) R-Ras regulates β1-integrin trafficking via effects on membrane ruffling and endocytosis. BMC Cell Biol. 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sandri C., Caccavari F., Valdembri D., Camillo C., Veltel S., Santambrogio M., Lanzetti L., Bussolino F., Ivaska J., Serini G. (2012) The R-Ras/RIN2/Rab5 complex controls endothelial cell adhesion and morphogenesis via active integrin endocytosis and Rac signaling. Cell Res. 22, 1479–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takaya A., Kamio T., Masuda M., Mochizuki N., Sawa H., Sato M., Nagashima K., Mizutani A., Matsuno A., Kiyokawa E., Matsuda M. (2007) R-Ras regulates exocytosis by Rgl2/Rlf-mediated activation of RalA on endosomes. Mol. Biol. Cell 18, 1850–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]