

Background: Exosomes show great promise as targeted therapeutic delivery vehicles.
Results: A strategy was identified for stabilizing targeting peptides on the surface of exosomes.
Conclusion: Glycosylation protects targeting ligands displayed on the surface of exosomes from proteolytic degradation.
Significance: Strategies for robust display of targeting peptides will enable targeted delivery of therapeutic exosomes.
Keywords: Endosome, Exosome, Glycosylation, Peptides, Protease
Abstract
Exosomes are secreted extracellular vesicles that mediate intercellular transfer of cellular contents and are attractive vehicles for therapeutic delivery of bimolecular cargo such as nucleic acids, proteins, and even drugs. Efficient exosome-mediated delivery in vivo requires targeting vesicles for uptake by specific recipient cells. Although exosomes have been successfully targeted to several cellular receptors by displaying peptides on the surface of the exosomes, identifying effective exosome-targeting peptides for other receptors has proven challenging. Furthermore, the biophysical rules governing targeting peptide success remain poorly understood. To evaluate one factor potentially limiting exosome delivery, we investigated whether peptides displayed on the exosome surface are degraded during exosome biogenesis, for example by endosomal proteases. Indeed, peptides fused to the N terminus of exosome-associated transmembrane protein Lamp2b were cleaved in samples derived from both cells and exosomes. To suppress peptide loss, we engineered targeting peptide-Lamp2b fusion proteins to include a glycosylation motif at various positions. Introduction of this glycosylation motif both protected the peptide from degradation and led to an increase in overall Lamp2b fusion protein expression in both cells and exosomes. Moreover, glycosylation-stabilized peptides enhanced targeted delivery of exosomes to neuroblastoma cells, demonstrating that such glycosylation does not ablate peptide-target interactions. Thus, we have identified a strategy for achieving robust display of targeting peptides on the surface of exosomes, which should facilitate the evaluation and development of new exosome-based therapeutics.
Introduction
Lipid nanoparticles display many properties that make them excellent drug delivery vehicles, including the ability to enhance drug stability and solubility and alter drug pharmacokinetics to achieve higher drug concentrations in target tissues (1). Biologically derived nanoparticles are an emerging subset of lipid nanoparticles that have been shown to effectively deliver a wide range of functional biomolecules, evade or dampen immune responses, and accumulate in tumors (2–4). In particular, exosomes, which are endosomally derived secreted vesicles, have shown great promise as therapeutic delivery vehicles (5, 6). Exosomes have been used to deliver therapeutic RNA to neurons (7), ovarian cancers (8), glioblastomas (9), and colon cancers (10); to deliver proteins to glioblastomas (9); and to deliver small molecule drugs to breast cancers (11) and glioblastomas (12). Based on these successes, exosomes are now being investigated in clinical trials as delivery vehicles for cancer vaccines and small molecule drugs (13).
An attractive property of lipid nanoparticle-based drug delivery is the potential to target lipid nanoparticles for uptake by specific recipient cells by functionalizing these particles with ligands that bind receptors on recipient cells. The addition of targeting ligands to lipid nanoparticles enhances their uptake and retention in the desired recipient cell type or tissue. The addition of peptide-based targeting ligands to synthetic lipid nanoparticles is nontrivial as peptide ligands affect the stability and material properties of the lipid nanoparticle and increase the complexity of synthesis (14). In contrast, displaying targeting ligands on exosomes is relatively simple because peptide ligands can be genetically fused to the extra-exosomal termini of exosomal membrane proteins. This strategy has been applied to target exosome uptake by neurons by fusing a rabies viral glycoprotein (RVG)3 peptide to the N terminus of lysosomal associated membrane protein 2b (Lamp2b) (7). Such a fusion resulted in RVG peptide being displayed on the surface of exosomes, leading to exosome uptake via the nicotinic acetylcholine receptor. Similarly, an internalizing RGD peptide fused to the N terminus of Lamp2b was used to target exosomes to breast cancer cells via αvβ3 integrins (11). One alternative to Lamp2b, the transmembrane domain of platelet-derived growth factor receptor, has also been used as a fusion partner to display peptides on the surface of exosomes (8). However, it is not known whether such platelet-derived growth factor receptor fusion proteins localize to endosomally derived exosomes or rather to extracellular vesicles that bud from the plasma membrane. Finally, fusion of peptides to the C1C2 domain of lactadherin has been used to display peptides on the surface of exosomes for vaccines (15). However, lactadherin is a membrane-associated protein (16), not an integral membrane protein. Thus, peptides fused to lactadherin may be closely associated with the membrane, rather than freely accessible to interact with cell receptors.
Despite these successes, achieving efficient exosome targeting via surface display of targeting peptides is nontrivial. Although Alvarez-Erviti et al. (7) achieved neuronal targeting of exosomes via display of the RVG peptide, they were unable to achieve muscle targeting via display of a muscle-specific peptide similarly fused to Lamp2b. This suggests that different target-binding peptides may have different utility as exosome-targeting peptides, possibly due to variations in target binding affinity or level of peptide display on the exosome surface, or a combination of these factors. In this study, we demonstrate that some peptides fused to the N terminus of Lamp2b are not displayed effectively on the surface of exosomes. However, this display can be enhanced by introducing frequently glycosylated motifs at particular locations within the engineered fusion protein. We hypothesize that engineered glycosylation protects the targeting peptides from degradation in the endosomal system during exosome biogenesis and secretion. We also demonstrate that some glycosylation-protected peptides retain the ability to bind their target proteins and that this peptide protection strategy can be applied to targeting peptides displayed on the surface of exosomes.
EXPERIMENTAL PROCEDURES
Plasmid Construction
Human Lamp2b cDNA was purchased from Open Biosystems and inserted into pcDNA3.1+ Hygro backbone. Peptide tags and glycine-serine amino acid spacers were added to the N and C termini of Lamp2b by PCR. The following tags were used: FLAG (DYKDDDDK), HA (YPYDVPDYA), and the glycosylation sequon (GNSTM) (17). Primer sequences are available upon request.
Cell Culture and Transfection
HEK293FT cells (Life Technologies) and Neuro2A cells (gift from Richard Morimoto) were maintained at 37 °C in 5% CO2 in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and 4 mm l-glutamine. HEK293FT cells were plated at ∼60% confluency in 10- or 15-cm dishes, and 1–1.5 μg of DNA/ml was transfected using the CaCl2-HEPES-buffered saline method.
Exosome Production and Characterization
Exosome-free medium was generated by pelleting FBS-derived exosomes from DMEM containing 20% FBS (see exosome pelleting protocol below) and combining the cleared supernatant with serum-free DMEM to achieve a final concentration of 10% FBS. HEK293FT cells were transfected with Lamp2b expression plasmids, and medium was changed to exosome-free medium 12–14 h after transfection. Conditioned medium was collected 2 days after medium change, and exosomes were concentrated by differential centrifugation. Conditioned medium was spun at 300 × g for 10 min, 2,000 × g for 10 min, and 10,000 × g for 30 min to remove cells, cell debris, and apoptotic bodies. From this supernatant, exosomes were pelleted at 120,416 × g for 135 min using an SW41 Ti rotor in an L-80 Optima XP ultracentrifuge (Beckman Coulter). Exosome pellets were washed in 10 ml of PBS and pelleted again via ultracentrifugation. Exosome morphology was evaluated by transmission electron microscopy using a 4% uranyl acetate negative stain. Exosome size distribution was profiled by NanoSight (Malvern) analysis.
Immunoblotting and Pulldown Assays
For Western blot analysis, cell extracts were prepared by lysis with radioimmunoprecipitation assay buffer. Exosomes were not lysed. Cell lysates, exosomes, and pulldowns were heated in Laemmli buffer at 70 °C. Equal quantities of protein, as measured by BCA assay (Pierce), were loaded in each lane of a 4–15% gradient polyacrylamide gel (Bio-Rad). After transfer to a PVDF membrane (Bio-Rad), membranes were blocked for 1 h in 1% milk at room temperature, and then blotted with anti-HA (Cell Signaling Technology, C29F4), anti-FLAG (Abcam, ab1162), or anti-β-actin (Cell Signaling Technology, 8H10D10) antibodies. Primary antibodies were detected with horseradish peroxidase-conjugated anti-mouse (Invitrogen) or anti-rabbit (Invitrogen and Abcam) immunoglobulin G secondary antibody. For FLAG pulldown experiments, cell lysates or exosomes were precleared with Sepharose beads (Sigma) and then pulled down with FLAG M2 Sepharose beads (Sigma) and eluted with 3× FLAG peptide. When indicated, cell lysates were diluted 1:5 in TBS.
Inhibition of Endosomal Degradation
HEK293FT cells were transfected with Lamp2b expression plasmids. Cells were treated with 50 nm bafilomycin A1 (Sigma) or an equivalent amount of dimethyl sulfoxide (DMSO) for 9 h, or treated with 50 μm leupeptin (Sigma) for 24 h. Cells were then lysed and evaluated by immunoblotting as described above.
Measuring Exosome Uptake
After the first ultracentrifuge spin of the exosome isolation procedure described above, exosome pellets (∼0.5 ml) were brought up to 1 ml in Diluent C (PKH67 kit, Sigma). This solution was mixed with 1 ml of Diluent C containing 6 μl of PKH67 dye and mixed via constant pipetting for 1 min. Next, 2 ml of 1% BSA was added to halt staining. 4.5 ml of serum-free medium was added to bring the mixture up to 8.5 ml. Excess PKH67 dye can form micelles similar in size to exosomes, and these micelles cannot be separated from exosomes by ultracentrifugation alone (18). We observed that these micelles are less dense than exosomes (in 0.971 m sucrose, dye micelles float and exosomes pellet).4 Thus, exosomes were purified via centrifugation through a 0.971 m sucrose cushion. Briefly, 1.5 ml of 0.971 m sucrose was slowly pipetted underneath the 8.5 ml of exosome solution containing BSA and excess dye. The entire mixture was centrifuged at 191,287 × g for 2 h, and then the upper layer and interface were carefully aspirated. To generate a negative control for estimating uptake of dye micelles, 0.5 ml of serum-free medium was treated as an exosome pellet, labeled with PKH67, and subsequently processed exactly as were the exosome pellet samples. After purification through the sucrose cushion, exosomes were diluted 1:10 in PBS and re-concentrated in 100-kDa cut-off centrifugal filter units (Millipore UFC910024) to reduce the concentration of sucrose. Exosome concentration was then counted via NanoSight, and an equal number of exosomes from each sample (or all of the medium negative control, to be conservative) were added to Neuro2A recipient cells. Recipient cells were plated at 50% confluency in a 48-well plate. Exosomes and cells were incubated for 2 h at 37 °C, as described (11). Cells were then washed with PBS and harvested for flow cytometry. Flow cytometry was performed on an LSRII flow cytometer (BD Bioscience) running FACSDiva software. Data were analyzed using FlowJo software (TreeStar). Live single cells were gated based upon scatter.
RESULTS
Peptides Fused to the N Terminus of Lamp2b Are Not Detected in Cells and Exosomes
We hypothesized that peptides fused to the N terminus of Lamp2b would be vulnerable to proteolysis due to their localization in the lumen of endosomes (Fig. 1A). Indeed, when glycosylation is artificially inhibited, Lamp2b is also vulnerable to degradation by endosomal proteases (19). To test this hypothesis, HEK293FT cells were transfected with Lamp2b containing a C-terminal HA tag (Lamp2b-HA), two C-terminal tags (Lamp2b-HA-FLAG), or an N-terminal FLAG tag and a C-terminal HA tag (FLAG-Lamp2b-HA). Cell lysates and exosomes were harvested from these cells, and N-and C-terminal tags were analyzed by Western blots. Exosomes harvested from HEK293FT cells exhibited expected morphology and size (Fig. 1, B and C). As compared with samples derived from whole cells, exosome samples were enriched in the exosomal protein CD63 relative to β-actin (Fig. 1D), as expected. The Lamp2b fusion proteins were expressed at similar levels in cells, as indicated by the HA Western blot. However, the FLAG peptide on FLAG-Lamp2b-HA could not be detected, although the C-terminal FLAG peptide of Lamp2b-HA-FLAG was detected (Fig. 1E). The same pattern was observed in exosomes (Fig. 1F). Together, these data indicate that the FLAG tag was lost from the N terminus of the Lamp2b protein.
FIGURE 1.

Stability of exosome-targeting peptides. A, this graphic illustrates the orientation of Lamp2b in the endosomal membrane and exposure of N-terminal peptides (N.-term. peptide) to proteases. MVB, multivesicular body. C.-term., C terminus. B, transmission electron microscopy image of exosomes isolated by differential centrifugation from HEK293FT cell supernatant. C, size distribution of exosomes secreted by HEK293FT cells. D, enrichment of exosome-associated protein CD63 in exosome preparations relative to β-actin. E and F, expression of Lamp2b fusion proteins in cell lysates (E) and exosomes (F), as evaluated by HA (C-terminal) and FLAG (N-terminal) Western blots.
A Glycosylation Motif Protects Peptides on the N Terminus of Lamp2b and Enhances Expression of Lamp2b Fusion Proteins
Glycosylation of the Lamp2b protein protects it from proteolytic degradation (19). Therefore, we hypothesized that engineered glycosylation could also protect peptides fused to the N terminus of Lamp2b from proteolysis. To investigate, the amino acid sequence GNSTM was fused to the N terminus of FLAG-Lamp2b-HA. The NST sequence is a standard N-linked glycosylation sequon, and the amino acids G and M flanking the sequon may increase glycosylation frequency in mammals (17). To investigate how proximity to the GNSTM tag impacts targeting peptide stability and availability to bind cellular receptors, fusion proteins were engineered to include flexible amino acid spacers of various lengths between the GNSTM motif, the FLAG tag, and Lamp2b. Fusion protein expression was assessed in cells and exosomes. The GNSTM-tagged Lamp2b proteins accumulated to a greater level in cells than did Lamp2b-HA or FLAG-Lamp2b-HA (Fig. 2A), and this difference was not an artifact of differential transfection efficiency (Fig. 2B). The GNSTM-tagged Lamp2b proteins could also be detected by anti-FLAG Western blot in both cell lysates (Fig. 2C) and in exosomes (Fig. 2D). Notably, increased detection of the FLAG tag was not simply a consequence of increased protein expression (compare FLAG-Lamp2b-HA and GNSTM-3gs-FLAG-3gs-Lamp2b-HA in Fig. 2C, where 3gs indicates a flexible linker 3 amino acids in length comprising glycine and serine residues), although increased detection of the FLAG tag did correlate with increased protein expression. These results suggest that the GNSTM motif effectively confers protection of the N-terminal FLAG peptide. To test whether endosomal acidification was required for loss of the N-terminal FLAG peptide, cells were treated with bafilomycin A1. Bafilomycin A1 treatment preserved the FLAG peptide on FLAG-Lamp2b-HA but had no effect on either the FLAG peptide on the C terminus of Lamp2b-HA-FLAG or the glycosylated FLAG peptide. Inhibiting endosomal proteases with leupeptin yielded the same pattern (Fig. 2E). Together, these data confirm that peptides displayed on the N terminus of Lamp2b are degraded by acid-dependent proteolysis. Moreover, adding the GNSTM glycosylation motif protected such N-terminal peptides from this degradation.
FIGURE 2.

Glycosylation motif-mediated stabilization of Lamp2b fusion proteins. A, expression of Lamp2b fusion proteins including an engineered GNSTM glycosylation motif in cells. In this and subsequent figures, the abbreviation “Xgs” is used to indicate a flexible linker X amino acids in length, comprising glycine and serine residues. B, transfection efficiency of cells expressing Lamp2b fusion proteins. Error bars indicate mean ± S.D. C and D, expression of Lamp2b fusion proteins in cell lysates (C) and exosomes (D) measured by HA (C-terminal) and FLAG (N-terminal) Western blots. E, cells were treated with either bafilomycin A1 (Baf.), which blocks endosomal acidification, or leupeptin, which inhibits endosomal proteases.
Peptides Protected by a Glycosylation Tag Retain the Capacity to Bind Cognate Peptide-binding Proteins
We next investigated whether GNSTM tag-stabilized peptide-Lamp2b fusion proteins retained the capacity to interact with binding partners. GNSTM-tagged FLAG-Lamp2b proteins were successfully pulled down by anti-FLAG beads in both cell lysates and intact exosomes (Fig. 3, A and B). To verify that pulldown was FLAG-specific and not an artifact due to increased loading of the highly expressed GNSTM-tagged proteins, lysates from cells expressing GNSTM-tagged proteins were diluted 1:5 such that the amount of this Lamp2b fusion protein loaded into the pulldown was comparable with the amounts in FLAG-Lamp2b-HA and Lamp2b-HA samples. This pulldown indicated that the FLAG peptide was indeed necessary for pulldown (Fig. 3C). Moreover, FLAG-dependent pulldown efficiency was much greater for some GNSTM-tagged Lamp2b constructs as compared with the FLAG-Lamp2b-HA control. Also, pulldown of intact exosomes required that FLAG be on the exosome exterior because exosomes from cells expressing Lamp2b-HA-FLAG were not pulled down (Fig. 3D). Collectively, these results indicate that the GNSTM motif can protect targeting peptides fused to the N terminus of Lamp2b from degradation without precluding interactions between these peptides and their cognate protein targets.
FIGURE 3.
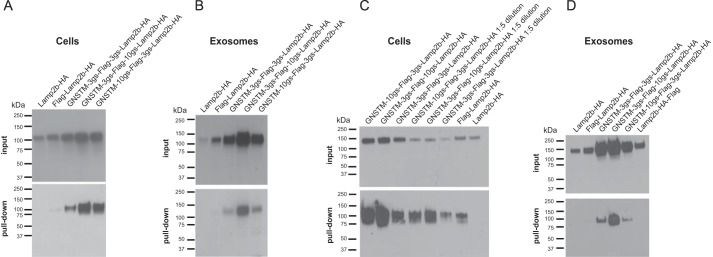
Impact of engineered glycosylation motif on targeting peptide binding interactions. A and B, expression of FLAG-Lamp2b fusion proteins in cell lysates (A) and exosomes (B) before and after pulldown with anti-FLAG beads. C, lysates from cells expressing NST-tagged FLAG-Lamp2b proteins were diluted 1:5 in TBS (where indicated) and pulled down to confirm that apparent FLAG-mediated pulldown was not an artifact of variable levels of protein in the pulldown assay load. D, pulldown of intact exosomes requires that the FLAG tag be expressed on the Lamp2b N terminus (exosome exterior).
Targeting Peptides Protected by a Glycosylation Tag Enhance Exosome Uptake by Recipient Cells
We next evaluated whether our engineered glycosylation approach is compatible with a demonstrated exosome targeting strategy. To this end, we utilized the system described by Alvarez-Erviti et al. (7), in which display of the RVG peptide on exosomes mediated delivery to Neuro2A neuroblastoma cells. The RVG peptide was fused to the N terminus of Lamp2b along with either a GNSTM glycosylation motif or a mutated motif, GASTM, which is not glycosylated. Exosomes displaying GNSTM-FLAG-Lamp2b-HA were also included as negative controls. All exosomes were labeled with the lipophilic dye PKH67, and equal numbers of exosomes from each sample were incubated with Neuro2A cells for 2 h. Notably, uptake of GNSTM-RVG exosomes exceeded uptake of either GASTM-RVG or GNSTM-FLAG exosomes (Fig. 4, A and B). Moreover, only the glycosylated RVG peptides mediated exosome uptake greater than was observed for negative control exosomes. Thus, in this system, engineered glycosylation of the targeting peptide did not prevent targeted exosome uptake, and indeed glycosylation was required to confer targeted exosome uptake.
FIGURE 4.
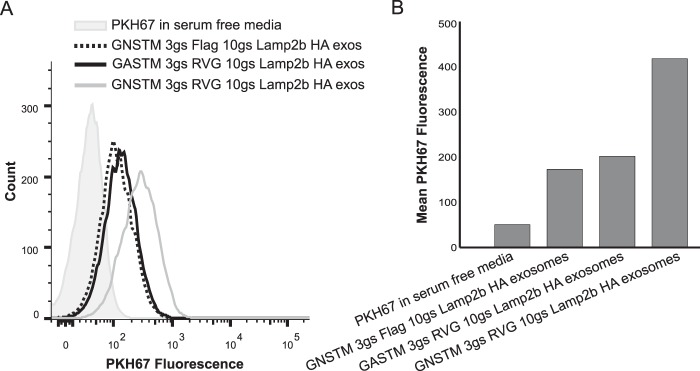
Glycosylation-enhanced targeted delivery of exosomes to neuroblastoma cells. A, equivalent numbers of PHK67-labeled exosomes were incubated with Neuro2A cells for 2 h (∼3 × 109 exosomes per 1 × 105 cells), and uptake was quantified by flow cytometry. The shaded histogram is the exosome-free negative control to evaluate excess dye and dye-derived micelles. exos, exosomes. B, quantification of peaks presented in panel A.
DISCUSSION
In this study, we found that peptides expressed on the N terminus of Lamp2b are susceptible to acid-dependent proteolytic degradation and that the glycosylation motif GNSTM protects such peptides from degradation. This strategy allows for the display of targeting peptides on the surface of exosomes. Furthermore, the GNSTM motif does not interfere with interactions between targeting peptides and their cognate protein targets. Indeed, in our hands, the GNSTM motif enhanced targeting peptide-mediated exosome uptake. In our investigation, the GNSTM motif conferred protection to peptides over a distance of at least 10 flexible amino acids, and larger spacers may also be feasible. Thus, there may exist a substantial design space for applying this strategy to many peptides of interest to ensure stable expression while avoiding interference with peptide-target interactions, although some optimization is likely to be required. Engineered glycosylation could also be useful for investigating and refining problematic exosome targeting strategies because without such modifications, candidate peptides may fail to enhance exosome uptake due to peptide loss rather than due to problems inherent to the targeting approach.
In addition to protecting N-terminal peptides from degradation, adding the GNSTM motif increased the total amount of Lamp2b fusion protein present in cells and exosomes. This increase could be due to decreased degradation because increasing the glycosylation of Lamp1 and Lamp2 during differentiation of the HL-60 promyelocytic cell line into granulocytes increases the half-life of these proteins (20). However, in this case, the complexity of the oligosaccharides on the proteins was increased, not the number of glycosylation sites. Because Lamp2b is already expected to include 16–20 N-linked glycosylation sites (19), whether adding one additional glycosylation site could increase the half-life of Lamp2b is unclear. Glycosylation also plays a role in the expression and sorting of other exosomal proteins. For example, sorting of the protein EWI-2 into exosomes is dependent on the presence of complex N-linked glycans on this protein (21). Mutation of even one of three N-linked glycosylation sites significantly decreased EWI-2 expression in extracellular vesicles. Furthermore, mutation of all three sites decreased both cellular and vesicle-associated EWI-2 levels. Thus, it is possible that altering the glycosylation of Lamp2b could have similar effects on expression in cells and exosomes.
Previous studies in which targeting peptides were fused to the N terminus of Lamp2b did not report targeting peptide degradation, although these characterizations focused on evaluating whether targeting peptide remained, rather than evaluating whether or not partial targeting peptide degradation occurred (7, 11). Notably, neither of the peptides used in these studies (RVG or internalizing RGD) contains any putative N-linked glycosylation sites that could protect the peptides from degradation. However, in these studies, exosomes were electroporated to load functional cargo molecules, and subsequent investigations have shown that electroporation can cause aggregation of exosomes as well as RNA (22, 23). Thus, it is possible that such aggregates may display different properties than did the exosomes used in our experiments. Furthermore, these prior studies utilized the mouse Lamp2b protein and isolated exosomes from murine dendritic cells, whereas we engineered human Lamp2b and isolated exosomes from human cells (HEK293FT). Thus, differences in protein trafficking and exosome secretion in different species and cell types may result in different susceptibilities of targeting peptides to degradation. Alternatively, any factors leading to increased stability of mouse Lamp2b-based constructs may lead to accumulation of higher levels of Lamp2b in cells and exosomes, such that some pool of intact peptide-Lamp2b escapes proteolytic cleavage prior to exosome secretion. Thus, the strategy proposed here may be especially important for engineering human Lamp2b and human cell-derived exosomes, and whether these benefits may extend to murine exosome engineering remains to be determined.
Because targeting exosomes to specific receptors is required for effective delivery of therapeutic molecules in vivo (7, 11), our strategy for increasing the expression of targeting peptides on exosomes may be particularly useful for translating promising exosome-based therapeutic strategies from preclinical investigations to human trials. Even if mouse Lamp2b could confer enhanced targeting peptide display when expressed in human exosome-producing cells (which remains undetermined), utilizing human Lamp2b is desirable to increase the immune compatibility of exosome-based therapeutics (24). Our strategy for enhancing peptide display via human Lamp2b could also increase peptide display via other exosomal membrane proteins because all such peptide fusions may be susceptible to degradation by endosomal proteases. Given the need for robust technologies for effectively directing exosomes to traffic to specific destinations in vivo, this strategy for enhancing display of targeting peptides could generally enable and enhance exosome-based therapeutics.
Acknowledgments
We acknowledge Charlene Wilke for assistance with transmission electron microscopy, as well as Myrick Dennis and Dr. Arabela Grigorescu for assistance with NanoSight analysis. We thank the laboratory of Richard Morimoto for Neuro2A cells. NanoSight analysis was performed at the Northwestern University Keck Biophysics Facility. TEM was performed at the Northwestern University Biological Imaging Facility on a JEOL 3200 FETEM supported by the Northwestern University Office for Research and National Center for Research Resources Grant 1S10RR025092. Traditional sequencing services were performed at the Northwestern University Genomics Core Facility. BCA assays were performed at the Northwestern University High Throughput Analysis Lab, which is supported by the Chicago Biomedical Consortium and The Searle Funds at The Chicago Community Trust.
This work was supported by the Defense Advanced Research Projects Agency, Award W911NF-11-2-0066. This work was supported by the Northwestern University Flow Cytometry Facility and a Cancer Center Support Grant from the National Institutes of Health (NCI CA060553); a 3M Non-tenured Faculty Award and the Northwestern University Prostate Cancer Specialized Program of Research Excellence (SPORE) through National Institutes of Health Award P50 CA090386 (to J. N. L.); and the National Science Foundation Graduate Research Fellowship Program (NSF GRFP) award DGE-0824162 (to M. E. H.).
M. E. Hung and J. N. Leonard, unpublished observation.
- RVG
- rabies viral glycoprotein
- Lamp2b
- lysosomal associated membrane protein 2b.
REFERENCES
- 1. van der Meel R., Fens M. H., Vader P., van Solinge W. W., Eniola-Adefeso O., Schiffelers R. M. (2014) Extracellular vesicles as drug delivery systems: lessons from the liposome field. J. Control. Release 195, 72–85 [DOI] [PubMed] [Google Scholar]
- 2. Lai C. P., Mardini O., Ericsson M., Prabhakar S., Maguire C. A., Chen J. W., Tannous B. A., Breakefield X. O. (2014) Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 8, 483–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li X., Li J. J., Yang J. Y., Wang D. S., Zhao W., Song W. J., Li W. M., Wang J. F., Han W., Zhang Z. C., Yu Y., Cao D. Y., Dou K. F. (2012) Tolerance induction by exosomes from immature dendritic cells and rapamycin in a mouse cardiac allograft model. PLoS One 7, e44045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Montecalvo A., Shufesky W. J., Stolz D. B., Sullivan M. G., Wang Z., Divito S. J., Papworth G. D., Watkins S. C., Robbins P. D., Larregina A. T., Morelli A. E. (2008) Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J. Immunol. 180, 3081–3090 [DOI] [PubMed] [Google Scholar]
- 5. Marcus M. E., Leonard J. N. (2013) FedExosomes: engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals 6, 659–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. EL Andaloussi S., Mäger I., Breakefield X. O., Wood M. J. (2013) Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347–357 [DOI] [PubMed] [Google Scholar]
- 7. Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M. J. (2011) Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345 [DOI] [PubMed] [Google Scholar]
- 8. Ohno S., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T., Gotoh N., Kuroda M. (2013) Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 21, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mizrak A., Bolukbasi M. F., Ozdener G. B., Brenner G. J., Madlener S., Erkan E. P., Ströbel T., Breakefield X. O., Saydam O. (2013) Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol. Ther. 21, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akao Y., Iio A., Itoh T., Noguchi S., Itoh Y., Ohtsuki Y., Naoe T. (2011) Microvesicle-mediated RNA molecule delivery system using monocytes/macrophages. Mol. Ther. 19, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tian Y., Li S., Song J., Ji T., Zhu M., Anderson G. J., Wei J., Nie G. (2014) A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 35, 2383–2390 [DOI] [PubMed] [Google Scholar]
- 12. Sun D., Zhuang X., Xiang X., Liu Y., Zhang S., Liu C., Barnes S., Grizzle W., Miller D., Zhang H. G. (2010) A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 18, 1606–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. György B., Hung M. E., Breakefield X. O., Leonard J. N. (2015) Therapeutic applications of extracellular vesicles: clinical promise and open questions. Annu. Rev. Pharmacol. Toxicol. 55, 439–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kraft J. C., Freeling J. P., Wang Z., Ho R. J. Y. (2014) Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J. Pharm. Sci. 103, 29–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeelenberg I. S., Ostrowski M., Krumeich S., Bobrie A., Jancic C., Boissonnas A., Delcayre A., Le Pecq J. B., Combadière B., Amigorena S., Théry C. (2008) Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 68, 1228–1235 [DOI] [PubMed] [Google Scholar]
- 16. Véron P., Segura E., Sugano G., Amigorena S., Théry C. (2005) Accumulation of MFG-E8/lactadherin on exosomes from immature dendritic cells. Blood Cells Mol. Dis. 35, 81–88 [DOI] [PubMed] [Google Scholar]
- 17. Bañó-Polo M., Baldin F., Tamborero S., Marti-Renom M. A., Mingarro I. (2011) N-Glycosylation efficiency is determined by the distance to the C-terminus and the amino acid preceding an Asn-Ser-Thr sequon. Protein Sci. 20, 179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van der Vlist E. J., Nolte-'t Hoen E. N., Stoorvogel W., Arkesteijn G. J., Wauben M. H. (2012) Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat. Protoc. 7, 1311–1326 [DOI] [PubMed] [Google Scholar]
- 19. Kundra R., Kornfeld S. (1999) Asparagine-linked oligosaccharides protect Lamp-1 and Lamp-2 from intracellular proteolysis. J. Biol. Chem. 274, 31039–31046 [DOI] [PubMed] [Google Scholar]
- 20. Lee N., Wang W. C., Fukuda M. (1990) Granulocytic differentiation of HL-60 cells is associated with increase of poly-N-acetyllactosamine in Asn-linked oligosaccharides attached to human lysosomal membrane glycoproteins. J. Biol. Chem. 265, 20476–20487 [PubMed] [Google Scholar]
- 21. Liang Y., Eng W. S., Colquhoun D. R., Dinglasan R. R., Graham D. R., Mahal L. K. (2014) Complex N-linked glycans serve as a determinant for exosome/microvesicle cargo recruitment. J. Biol. Chem. 289, 32526–32537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hood J. L., Scott M. J., Wickline S. A. (2014) Maximizing exosome colloidal stability following electroporation. Anal. Biochem. 448, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kooijmans S. A., Stremersch S., Braeckmans K., de Smedt S. C., Hendrix A., Wood M. J., Schiffelers R. M., Raemdonck K., Vader P. (2013) Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 172, 229–238 [DOI] [PubMed] [Google Scholar]
- 24. Neefjes J., Ovaa H. (2013) A peptide's perspective on antigen presentation to the immune system. Nat. Chem. Biol. 9, 769–775 [DOI] [PubMed] [Google Scholar]