

Background: Hepatic overproduction of apoB-containing lipoproteins is a risk factor for atherosclerosis.
Results: Secretion of apoB:1000-containing lipoproteins is impaired in phospholipid transfer protein (PLTP)-deficient hepatocytes.
Conclusion: The initial step in the assembly of primordial apoB-containing lipoproteins is facilitated by PLTP.
Significance: Inhibition of PLTP may impede apoB-containing lipoprotein secretion without excessive hepatic lipid accumulation.
Keywords: Apolipoprotein, Biosynthesis, Hepatocyte, Lipid, Lipoprotein, Lipoprotein Secretion, Liver, Mouse, Phospholipid
Abstract
In this study, we tested the hypothesis that phospholipid transfer protein (PLTP) is a plausible mediator of phospholipid (PL) transfer to the N-terminal 1000 residues of apoB (apoB:1000) leading to the initiation of apoB-containing lipoprotein assembly. To this end, primary hepatocytes from wild type (WT) and PLTP knock-out (KO) mice were transduced with adenovirus-apoB:1000 with or without co-transduction with adenovirus-PLTP, and the assembly and secretion of apoB:1000-containing lipoproteins were assessed. PLTP deficiency resulted in a 65 and 72% reduction in the protein and lipid content, respectively, of secreted apoB:1000-containing lipoproteins. Particles secreted by WT hepatocytes contained 69% PL, 9% diacylglycerol (DAG), and 23% triacylglycerol (TAG) with a stoichiometry of 46 PL, 6 DAG, and 15 TAG molecules per apoB:1000. PLTP absence drastically altered the lipid composition of apoB:1000 lipoproteins; these particles contained 46% PL, 13% DAG, and 41% TAG with a stoichiometry of 27 PL, 10 DAG, and 23 TAG molecules per apoB:1000. Reintroduction of Pltp gene into PLTP-KO hepatocytes stimulated the lipidation and secretion of apoB:1000-containing lipoproteins by ∼3-fold; the lipid composition and stoichiometry of these particles were identical to those secreted by WT hepatocytes. In contrast to the WT, apoB:1000 in PLTP-KO hepatocytes was susceptible to intracellular degradation predominantly in the post-endoplasmic reticulum, presecretory compartment. Reintroduction of Pltp gene into PLTP-KO hepatocytes restored the stability of apoB:1000. These results provide compelling evidence that in hepatocytes initial recruitment of PL by apoB:1000 leading to the formation of the PL-rich apoB-containing initiation complex is mediated to a large extent by PLTP.
Introduction
Apolipoprotein (apo)2 B is synthesized primarily in hepatocytes and enterocytes and has a fundamental role in the transport and metabolism of plasma triacylglycerols (TAGs) and cholesterol (1). ApoB exists in two forms, apoB100 and apoB48 (1). ApoB100, the full-length protein, is one of the largest monomeric proteins known consisting of 4536 amino acid residues with a molecular mass of 550 kDa (1). ApoB100 is an essential structural component for the formation and secretion of very low density lipoproteins (VLDLs), the precursors of plasma low density lipoproteins (LDLs), and serves as a ligand for receptor-mediated uptake of LDL by a variety of cells (1). ApoB48, the N-terminal 48% of apoB100, is produced by a post-transcriptional modification of the apoB mRNA (1). ApoB48 is essential for the formation and secretion of chylomicrons and is expressed in mammalian intestine and in the liver of some non-human mammals, including mice (1). The processes involved in the assembly of apoB-containing lipoproteins in the liver are complex and are regulated at multiple levels by various factors throughout the secretory pathway. The addition of lipids to apoB is widely believed to occur in two steps (1, 2). The first step involves the addition of small amounts of lipids to apoB as it is translated and translocated into the lumen of endoplasmic reticulum (ER) and formation of a partially lipidated small pre-VLDL particle in the high density lipoprotein (HDL) density range (1, 2). In the second step, this pre-VLDL particle is believed to acquire the bulk of its core lipids and is converted to bona fide VLDL (1–3) presumably by fusing with a large, VLDL-sized, apoB-free TAG particle (3). Biochemical studies of VLDL assembly support the concept that the bulk of neutral lipids are added in the second step after apoB translation is completed (1, 2).
In our previous studies, based on experimentally derived results (4–6) and all atom molecular modeling of the βα1 domain (amino acid residues 1–1000) of apoB100 (7), we proposed that initiation of apoB particle assembly occurs when the βα1 domain, designated apoB:1000 (or apoB22.05 based on the percentage of full-length apoB), folds into a three-sided lipovitellin-like lipid binding cavity (8–10) to form the apoB “lipid pocket.” We demonstrated that the first 1000 amino acid residues of human apoB100 are required for the initiation of apoB-containing lipoprotein assembly (4, 5) and that this primordial apoB particle is phospholipid (PL)-rich (4, 5). We concluded that this apoB initiation complex is formed via a hairpin bridge mechanism without the structural requirement for MTP (6). Our studies, however, did not rule out the potential functional role, i.e. transfer of lipids to apoB:1000, of MTP (6). In subsequent comprehensive studies in rat hepatoma McA-RH7777 cells, we used MTP inhibitors (11) as well as microRNA-mediated MTP-deficient McA-RH7777 cells (12) and demonstrated that the initial addition of PL to apoB:1000 is independent of MTP lipid transfer activity. Based on these results, we hypothesized that phospholipid transfer protein (PLTP) is a plausible mediator of this very early step in apoB-containing particle assembly.
To test this hypothesis, we expressed apoB:1000 in primary cultures of hepatocytes isolated from wild type (WT) and PLTP knock-out (KO) mice with or without co-expression of PLTP. Metabolic labeling of hepatocytes with [35S]methionine/cysteine and [3H]glycerol demonstrated a marked reduction in apoB:1000 synthesis, lipidation, and secretion in hepatocytes from PLTP-KO mice when compared with the WT control. Reintroduction of Pltp gene into PLTP-KO hepatocytes reversed the suppression in the lipidation and secretion of apoB:1000-containing lipoproteins and restored their lipid composition to that observed for particles secreted by WT hepatocytes.
EXPERIMENTAL PROCEDURES
Materials
Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). Williams' medium, Hanks' balanced salt solution (HBSS), horse serum, and antibiotic-antimycotic were obtained from Gibco Life Technologies. Dulbecco's modified Eagle's medium (DMEM) and trypsin were purchased from Mediatech, Inc. (Herndon, VA). Sodium deoxycholate, Triton X-100, benzamidine, phenylmethylsulfonyl fluoride, leupeptin, aprotinin, pepstatin A, lactacystin, brefeldin A, and fatty acid-free bovine serum albumin (BSA) were from Sigma. Tris-glycine gels were obtained from Invitrogen-Novex. Protein G-Sepharose CL-4B, [3H]glycerol, [14C]oleic acid, and Amplify were from Amersham Biosciences. Collagenase Type I was purchased from Worthington. TRAN35S-LABEL [35S]methionine/cysteine ([35S]Met/Cys) was from MP Biomedicals, Inc. (Irvine, CA). Affinity-purified polyclonal antibody to human apoB100 was prepared in our laboratory and biotinylated as described previously (4). Monospecific polyclonal antibody to rat apoB was prepared in our laboratory as described previously (13). ApoB100 cDNA was a gift from Dr. Zemin Yao (University of Ottawa Heart Institute, Ottawa, Ontario, Canada). PLTP cDNA was a gift from Dr. Xian-Cheng Jiang (State University of New York Downstate Medical Center, Brooklyn, NY).
Animals
C57BL/6 WT and a pair of breeder mice heterozygous for Pltp gene (B6.129P2-Pltptm1Jia/J) were purchased from The Jackson Laboratory (Bar Harbor, ME). Animals were housed and bred in the Animal Resources Program facility of the University of Alabama at Birmingham. All protocols and experimental procedures were approved by the Institutional Animal Care and Use Committee prior to the start of studies. Mice homozygous for Pltp gene were identified by PCR and used for colony expansion. Animals were fed a chow diet (Research Diets, Inc.) and were used at 3–4 months of age. Both male and female mice were used in these studies.
Construction of Truncated ApoB:1000 Expression Plasmid for Transient Transfection
Truncated apoB cDNA spanning nucleotides 1–3081 of the full-length apoB100 cDNA was prepared from pB100L-L (14) as a PCR template and appropriate primers as described previously (4). Standard cloning procedures were used to identify clones with 100% correct sequence (4). The apoB fragment (3081 bp) (apoB:1000) was ligated into the mammalian expression vector, the Molony murine leukemia virus-based retrovirus pLNCX (15), and expression vectors were used for transformation. Clones harboring plasmids containing apoB gene with the correct orientation were identified by restriction enzyme digestion, confirmed by nucleotide sequencing, and used to transfect mouse primary hepatocytes as described in detail previously (4).
Construction of Recombinant Adenovirus
Truncated apoB cDNA spanning nucleotides 1–3081 of the full-length apoB100 cDNA was prepared from pB100L-L (14) as a PCR template and the appropriate primers described below. The NotI and MluI cloning sequences (shown as underlined, italic nucleotides), which are not present in the full-length apoB100 cDNA, were installed at the 5′-end of forward and reverse primers, respectively, to allow convenient cloning. For human apoB:1000, the sequence of the forward primer containing the NotI restriction site was 5′-TTGCGGCCGCATGGACCCGCCGAGGCCCGCG-3′; the sequence of the reverse primer containing the MluI restriction site and stop codon was 5′-CCACGCGTCTATCTGTCCTCTCTCTGGAGCTC-3′. For human PLTP, the sequence of the forward primer containing the NotI restriction site was 5′-TGCGGCCGCATCCCACGTGACCGCGCCGC-3′; the sequence of the reverse primer containing the MluI restriction site and FLAG was 5′-CACGCGTTCACTTGTCGTCATCGTCTTTGTAGTCGACAGCTGCTGTGGACGGTGT-3′. The amplified PCR products were digested with NotI and MluI restriction enzymes and inserted into the adenovirus shuttle vector pAdTrack-CMV, which also contains a GFP expression cassette. The resultant plasmid harboring apoB:1000 or Pltp gene were isolated and identified by restriction enzyme digestion and nucleotide sequencing of the entire open reading frames. Positive pAdTrack-CMV clones with the gene of interest were linearized by PmeI restriction enzyme and transformed into BJ5183 bacteria containing the adenoviral backbone plasmid pAdEasy-1. The recombinant clones were selected for kanamycin resistance and confirmed by restriction digestion analysis and sequencing. Only clones with 100% correct sequence were used in these studies. The desired adenoviral plasmids were digested by PacI to expose the inverted terminal repeats. The larger fragment was recovered using a gel extraction kit and transfected into 293 cells for packaging adenovirus (Adv)-apoB:1000 or Adv-PLTP. After 8 or 10 days, the recombinant adenovirus was collected by repeated freezing and thawing of the 293 cells and used to transduce mouse primary hepatocytes as described below.
Isolation of Primary Hepatocytes
Liver cells were isolated from 3–4-month-old WT or PLTP-KO mice essentially using the procedure of Berry and Friend (16) as modified for mice (17). Each liver was first perfused in situ in a non-recirculating system with 20 ml of calcium- and magnesium-free HBSS to remove the blood. Liver was then digested with 20 ml of collagenase Type I (100 units/ml) in calcium/magnesium-containing HBSS as described previously (13). At the termination of perfusion, the liver was placed in cold HBSS and minced. Liver cells were dispersed by swirling gently and filtered through a 70-μm cell strainer (BD Falcon) to remove cell debris and clumped cells. The filtrate was centrifuged, cells were washed twice with HBSS, and the final sedimented parenchymal cells were suspended in Williams' medium containing 10% FBS. The viability of isolated hepatocytes was assessed by trypan blue exclusion as described previously (13). Aliquots of the cell suspension were plated onto collagen-coated 35-mm dishes as described previously (13). After 4 h in culture, medium was removed, fresh medium containing 10% FBS was added, and monolayers were used for transfection or transduction studies under conditions described for each experiment.
Transient Transfection of ApoB:1000 in Primary Cultures of WT and PLTP-KO Mouse Hepatocytes
Cells were transfected with pLNCB:1000 or empty pLNCX using TransIT®-LT1 Transfection Reagent (Mirus, Madison, WI) according to the manufacturer's instructions as described previously (4–6). The expression of apoB:1000 was determined 48 h post-transfection using metabolic labeling of cells with [35S]Met/Cys as described below.
Transduction of Mouse Primary Hepatocytes with Adv-B:1000 and Adv-PLTP
After 4 h in culture, medium was removed, and fresh medium containing 10% FBS was added. Cells were transduced with either Adv-GFP as a control or Adv-apoB:1000 with or without co-transduction with Adv-PLTP; GFP allowed detection of transduction efficiency. Adv-apoB:1000 was first tested in McA-RH7777 cells to establish the optimum concentration of virus for transduction of primary hepatocytes. The expression of apoB:1000 in primary hepatocytes was determined 48 h post-transduction.
Isolation of Lipoproteins Secreted by Parental WT and PLTP Knock-out Hepatocytes in Primary Cultures
Maintenance medium was removed, cells were washed with phosphate-buffered saline (PBS), and serum-free DMEM containing [3H]glycerol (7 μCi/ml of medium) was added. Labeled conditioned medium was collected after overnight (17–20-h) incubation, a preservative mixture described previously (11) was added to prevent oxidative and proteolytic damage, and samples were centrifuged at 2,000 rpm for 30 min at 4 °C to remove broken cells and debris (11). The secreted 3H-labeled VLDLs (d < 1.006 g/ml), LDLs (d = 1.006–1.063 g/ml), and HDLs (d = 1.063–1.21 g/ml) were isolated by sequential preparative ultracentrifugation (18). The isolated lipoproteins were dialyzed against PBS (pH 7.4) and analyzed for lipid composition as described below.
De Novo Synthesis and Secretion of Mouse Endogenous ApoB and Human ApoB:1000
Parental WT and PLTP-KO hepatocytes were tested for the secretion of mouse endogenous apoB100 and apoB48 after 17 h in culture. The expression of human apoB:1000 in WT and PLTP-KO hepatocytes was determined 48 h after transduction of cells with Adv-apoB:1000 or Adv-apoB:1000 plus Adv-PLTP. At the start of experiments, culture medium was removed, and monolayers were washed twice with PBS. Serum-, methionine-, and cysteine-free DMEM was added, and cells were metabolically labeled with [35S]Met/Cys (70 μCi/ml of medium). The incorporation of [35S]Met/Cys into newly synthesized human apoB:1000 or mouse endogenous apoB100 and apoB48 was determined after 3.5 h of incubation. The 35S-labeled conditioned medium was collected, preservative mixture (11) was added, and the medium was processed as described above. Cell monolayers were resuspended in lysis buffer containing preservative mixture and processed as described previously (5, 6). The secreted 35S-labeled apoB in the medium and in the cell lysate were isolated by immunoprecipitation as described below.
Pulse-Chase Analysis
Forty-eight hours after transduction with Adv-apoB:1000 with or without co-transduction with Adv-PLTP, medium was removed, and monolayers were pulsed for 10 min in serum- and methionine/cysteine-free DMEM containing [35S]Met/Cys (100 μCi/ml) and chased for the indicated time as described in detail previously (6). Experiments designed to test the effects of lactacystin and brefeldin A were carried out as described previously (6). In all experiments, the secreted and cellular 35S-labeled apoB were immunoprecipitated using appropriate antibody as described below. Cell protein content was measured by the method of Lowry et al. (19).
Metabolic Labeling of the Lipid Content of ApoB:1000-containing Lipoprotein Particles
Culture medium was removed, and cells were washed twice with PBS and incubated for 17 h in serum-free DMEM containing 3H-labeled glycerol (7 μCi/ml of medium). The labeled lipids and lipoproteins secreted into the medium by parental untransduced cells were extracted directly and analyzed as described previously (5). The incorporation of 3H-labeled glycerol into various lipid moieties of the secreted apoB:1000-containing lipoproteins was determined by immunoprecipitation of intact particles under non-denaturing condition or by non-denaturing gradient gel electrophoresis of labeled conditioned medium as described previously (5). Cell monolayers were washed with PBS, scraped off the plate in 1.0 ml of PBS, and sonicated. The incorporation of 3H-labeled glycerol into cellular lipids was determined as described below.
Immunoprecipitation
Human 35S-labeled apoB:1000 secreted into the conditioned medium and accumulated in the cells was immunoprecipitated using monospecific polyclonal antibody to human apoB100 coupled to Protein G-Sepharose CL-4B as described previously (5, 6). Mouse 35S-labeled endogenous apoB100 and apoB48 were immunoprecipitated using polyclonal antibodies to rat apoB100 as described (11, 12). The 35S-labeled proteins were extracted from Protein G and resolved by 4–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by autoradiography (20). The [3H]glycerol-labeled apoB:1000-containing lipoproteins secreted into the conditioned medium were immunoprecipitated under non-denaturing conditions using monospecific polyclonal antibody to human apoB100, and lipids were extracted as described previously (5).
Lipid Analysis of Isolated ApoB:1000-containing Particles
Radioactive lipids associated with the immunoprecipitated apoB:1000-containing particles were extracted from Protein G with chloroform/methanol (2:1). In non-denaturing gradient gel electrophoresis studies, the band corresponding to labeled apoB:1000-containing particles was excised, and lipids were extracted with chloroform/methanol (2:1) as described in detail previously (5). Total labeled lipids extracted from immunoprecipitated and gel-isolated apoB:1000-containing lipoproteins were washed by the Folch method (21) and applied to a thin layer chromatography (TLC) plate. Bands corresponding to PL, DAG, and TAG identified by comparison with known standards were visualized with iodine, and each band was scraped off the plate and quantified by liquid scintillation counting. The number of PL, DAG, and TAG molecules per apoB:1000 particle was calculated as described previously (5, 6).
Statistical Analysis
Data between two groups were analyzed by unpaired, two-tailed Student's t test.
RESULTS
Baseline Studies on PLTP Activity and Lipoprotein Levels in Plasma of WT and PLTP-KO Mice
Baseline studies (data not shown) demonstrated a 23 and 56% reduction in plasma levels of cholesterol and PL, respectively, in PLTP-KO mice when compared with WT control. Analysis of plasma lipoproteins by size exclusion chromatography demonstrated a major peak of cholesterol and PL in the HDL fractions of both WT and PLTP-KO mice. There was a near absence of cholesterol and PL in the VLDL and to a lesser extent LDL. Markedly lower amounts of cholesterol and PL were also detected in the HDL fraction of PLTP-KO mice. These results are consistent with studies reported previously by other investigators (22, 23).
PLTP Deficiency Decreases the Secretion of Lipids and Lipoproteins in Primary Hepatocytes
We first determined the de novo synthesis and secretion of lipids in primary hepatocytes of WT and PLTP-KO mice by metabolic labeling of cells with [3H]glycerol. Results showed a 60% decrease in the secretion of total lipids in PLTP-KO hepatocytes when compared with WT control (Table 1). TAG was the major lipid moiety secreted by WT hepatocytes, accounting for 62% of the total lipids; PL and DAG variably contributed 17–21% to the total lipids (Table 1). There was a marked reduction in the percent contribution of TAG to the total lipids and a corresponding increase in that of PL plus DAG in PLTP-KO hepatocytes, resulting in an equal contribution ranging from 36 to 39% of TAG and PL to the secreted total lipids in PLTP-KO hepatocytes (Table 1). Analysis of data demonstrated that the most profound inhibitory effect of PLTP deficiency was on the secretion of TAG, which was reduced by 77%, followed by DAG and PL, which were decreased by 44 and 30%, respectively (Table 1). The reduction in the secretion of 3H-labeled lipids in PLTP-KO hepatocytes was concurrent with their increased (25–40%) accumulation in the cells without any major change in their composition as compared with WT hepatocytes (Table 1).
TABLE 1.
Cellular and secreted [3H]glycerol-labeled lipids in primary cultures of hepatocytes isolated from wild type and PLTP knock-out mice
Primary hepatocytes were isolated from wild type and PLTP knock-out mice and cultured under conditions described in the text. The incorporation of 3H-labeled glycerol (7 μCi/ml of medium) into the secreted and cellular lipids was determined after 17-h incubation as described under “Experimental Procedures.” Values are means ± S.E. of triplicate dishes.
| Hepatocyte genotype | Secreted [3H]glycerol-labeled lipids |
Cellular [3H]glycerol-labeled lipids |
||||||
|---|---|---|---|---|---|---|---|---|
| Total lipids | PL | DAG | TAG | Total lipids | PL | DAG | TAG | |
| cpm/mg cell protein | % of total lipids | % of total lipids | % of total lipids | cpm/mg cell protein | % of total lipids | % of total lipids | % of total lipids | |
| Wild type | 15,503 ± 618 | 21.15 ± 4.28 | 17.25 ± 2.01 | 61.60 ± 5.60 | 111,761 ± 14,710 | 57.64 ± 2.95 | 10.51 ± 0.61 | 31.84 ± 2.65 |
| PLTP-KO | 5,998 ± 506a | 38.96 ± 5.38 | 24.78 ± 1.00b | 36.26 ± 5.39b | 138,692 ± 20,676 | 50.46 ± 3.00 | 9.72 ± 0.28 | 39.82 ± 2.94 |
a p < 0.001 wild type versus PLTP knock-out.
b p = 0.03 wild type versus PLTP knock-out.
Next, we examined the effect of PLTP deficiency on the level and composition of lipoproteins secreted by primary hepatocytes. Analysis of 3H-labeled lipoprotein fractions isolated from the conditioned medium demonstrated marked reduction in the secretion of VLDL (58%), LDL (82%), and HDL (46%) in PLTP-KO hepatocytes as compared with the WT control (Table 2). In WT hepatocytes, TAG was the major lipid moiety of VLDL, LDL, and HDL, accounting for 73–86% of the total lipids (Table 2). The VLDL, LDL, and HDL secreted by PLTP-KO hepatocytes contained a considerably lower content of TAG and a higher content of PL (Table 2).
TABLE 2.
Concentration and composition of [3H]glycerol-labeled lipids in VLDL, LDL, and HDL secreted by hepatocytes isolated from wild type and PLTP knock-out mice
Hepatocytes were isolated from wild type and PLTP knock-out mice and processed as described under “Experimental Procedures.” The incorporation of 3H-labeled glycerol (7 μCi/ml of medium) into the lipid moieties of secreted VLDL, LDL, and HDL was determined after 17-h incubation in the presence of 0.4 mm oleic acid bound to 0.75% BSA. Values are means ± S.E. of triplicate dishes.
| Lipoprotein fraction |
3H-Labeled lipids in lipoproteins secreted by wild type hepatocytes |
3H-Labeled lipids in lipoproteins secreted by PLTP knock-out hepatocytes |
||||||
|---|---|---|---|---|---|---|---|---|
| Total lipids | PL | DAG | TAG | Total lipids | PL | DAG | TAG | |
| cpm/mg cell protein | % of total lipids | % of total lipids | % of total lipids | cpm/mg cell protein | % of total lipids | % of total lipids | % of total lipids | |
| VLDL | 5,179 ± 370 | 8.84 ± 1.55 | 5.34 ± 0.44 | 85.82 ± 1.94 | 2,176 ± 38a | 18.43 ± 4.85 | 6.55 ± 1.85 | 75.02 ± 6.00 |
| LDL | 3,765 ± 436 | 10.53 ± 2.48 | 5.92 ± 0.53 | 83.55 ± 2.80 | 682 ± 82a | 36.46 ± 0.59b | 12.34 ± 0.22c | 51.21 ± 0.37c |
| HDL | 1,043 ± 11 | 19.41 ± 1.17 | 8.07 ± 1.28 | 72.52 ± 2.25 | 563 ± 50d | 36.90 ± 6.51e | 16.70 ± 0.83c | 46.40 ± 5.67f |
a p = 0.02 wild type versus PLTP knock-out.
b p = 0.002 wild type versus PLTP knock-out.
c p < 0.001 wild type versus PLTP knock-out.
d p = 0.01 wild type versus PLTP knock-out.
e p = 0.007 wild type versus PLTP knock-out.
f p = 0.003 wild type versus PLTP knock-out.
PLTP Deficiency Drastically Diminishes de Novo Synthesis and Secretion of ApoB:1000 in Primary Hepatocytes
We first validated the inhibitory effect of PLTP deficiency on the synthesis and secretion of rat endogenous apoB100 and apoB48. Results demonstrated a marked reduction in the secretion (Fig. 1A) and cellular accumulation (autoradiogram not shown due to a weak signal for apoB100) of both apoB100 and apoB48. Analysis of the autoradiogram demonstrated a 69 and 93% decrease in the secretion of endogenous 35S-labeled apoB100 (Fig. 1B) and apoB48 (Fig. 1C), respectively, in PLTP-KO hepatocytes when compared with WT control. PLTP deficiency also caused a 36 and 53% decrease in the cellular accumulation of apoB100 and apoB48, respectively. Thus, the total synthesis and secretion of endogenous apoB100 (Fig. 1B) and apoB48 (Fig. 1C) were reduced by 65 and 90%, respectively, in the absence of PLTP. The higher level of apoB48 than apoB100 observed in this study is consistent with that reported by other investigators in mouse plasma (24), primary cultures of mouse hepatocytes (22, 25), and rat hepatocytes and rat hepatoma McA-RH7777 cells (26–28).
FIGURE 1.
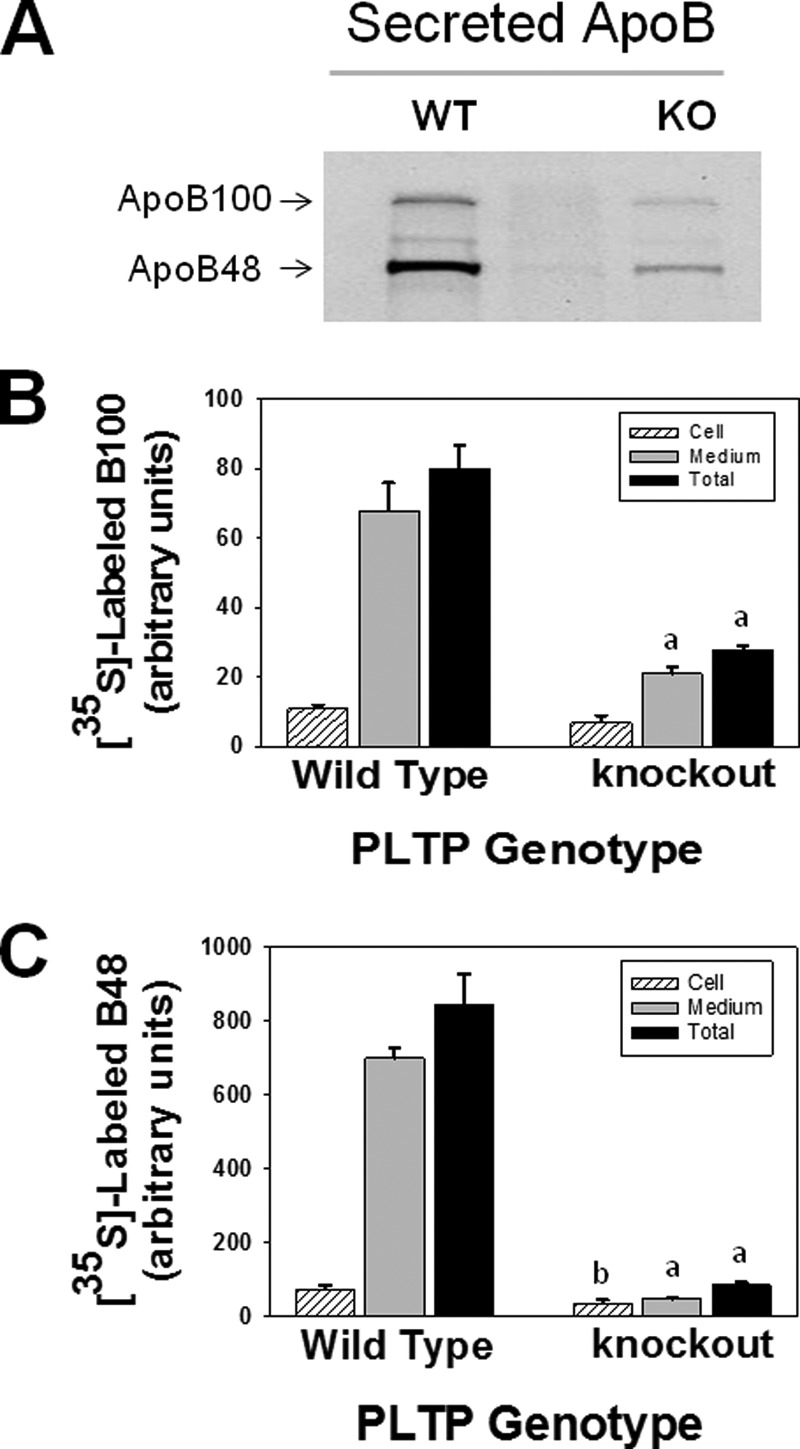
PLTP deficiency diminishes the synthesis and secretion of mouse endogenous apoB100 and apoB48. Primary cultures of hepatocytes isolated from wild type and PLTP knock-out mice were metabolically labeled with [35S]Met/Cys, and the secreted and cellular endogenous 35S-labeled apoB100 and apoB48 were immunoprecipitated using polyclonal antibody to rat apoB as described under “Experimental Procedures.” A, autoradiogram of secreted 35S-labeled apoB100 and apoB48. B and C, intensities of 35S-labeled apoB100 and apoB48 bands were determined by computer-assisted image processing, normalized for cell protein, and plotted as mean ± S.E. (error bars) of triplicate dishes. a, p < 0.001; b, p < 0.04 wild type versus PLTP knock-out.
To examine the effect of PLTP deficiency on the synthesis and secretion of apoB:1000, primary hepatocytes isolated from WT and PLTP-KO mice were transfected with pLNCB:1000 as described previously (5, 6, 11). As shown in Fig. 2, 35S-labeled apoB:1000 was found in cell lysate and culture medium of WT hepatocytes (Fig. 2A, lane 3) but not in the untransfected cells (Fig. 2A, lane 1) or cells transfected with control pLNCX (Fig. 2A, lane 2). In contrast to WT hepatocytes, there was no detectable 35S-labeled apoB:1000 in either cell lysate or culture medium of PLTP-KO hepatocytes (Fig. 2B, lanes 1 and 2). Although these results supported our hypothesis, the apparent low efficiency of transfection in primary hepatocytes hindered our planned studies to assess the effects of PLTP deficiency on lipidation, assembly, and secretion of intact apoB:1000-containing particles. To circumvent this inherent problem of low transfection efficiency of primary hepatocytes, cells were transduced with Adv-apoB:1000 with or without co-transduction with Adv-PLTP. The optimum concentration of each adenovirus was determined in a pilot study (data not shown). Results of metabolic labeling studies demonstrated a striking reduction in 35S-labeled apoB:1000 accumulation in the cells and secreted into the medium of PLTP-KO hepatocytes (Fig. 3, lanes 4–6) compared with WT hepatocytes (Fig. 3, lanes 1–3). In a series of 11 experiments, PLTP deficiency resulted in a 65 ± 3% decrease in the secretion and cellular accumulation of 35S-labeled apoB:1000.
FIGURE 2.
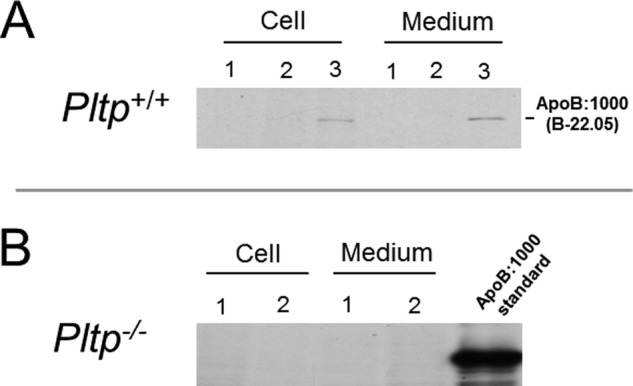
Synthesis and secretion of human apoB:1000 are dependent on PLTP activity. Primary cultures of hepatocytes isolated from wild type (A) and PLTP knock-out (B) mice were transfected with pLNCB:1000 or empty pLNCX and metabolically labeled with [35S]Met/Cys as described under “Experimental Procedures.” Secreted and cellular 35S-labeled human apoB:1000 was immunoprecipitated using antibody to human apoB100 and processed as described in the text. A, autoradiogram of 35S-labeled human apoB:1000 expressed in wild type hepatocytes (lane 3), untransfected cells (lane 1), and cells transfected with empty pLNCX (lane 2). B, autoradiogram of 35S-labeled apoB:1000 synthesized and secreted by PLTP-KO hepatocytes (lanes 1 and 2).
FIGURE 3.
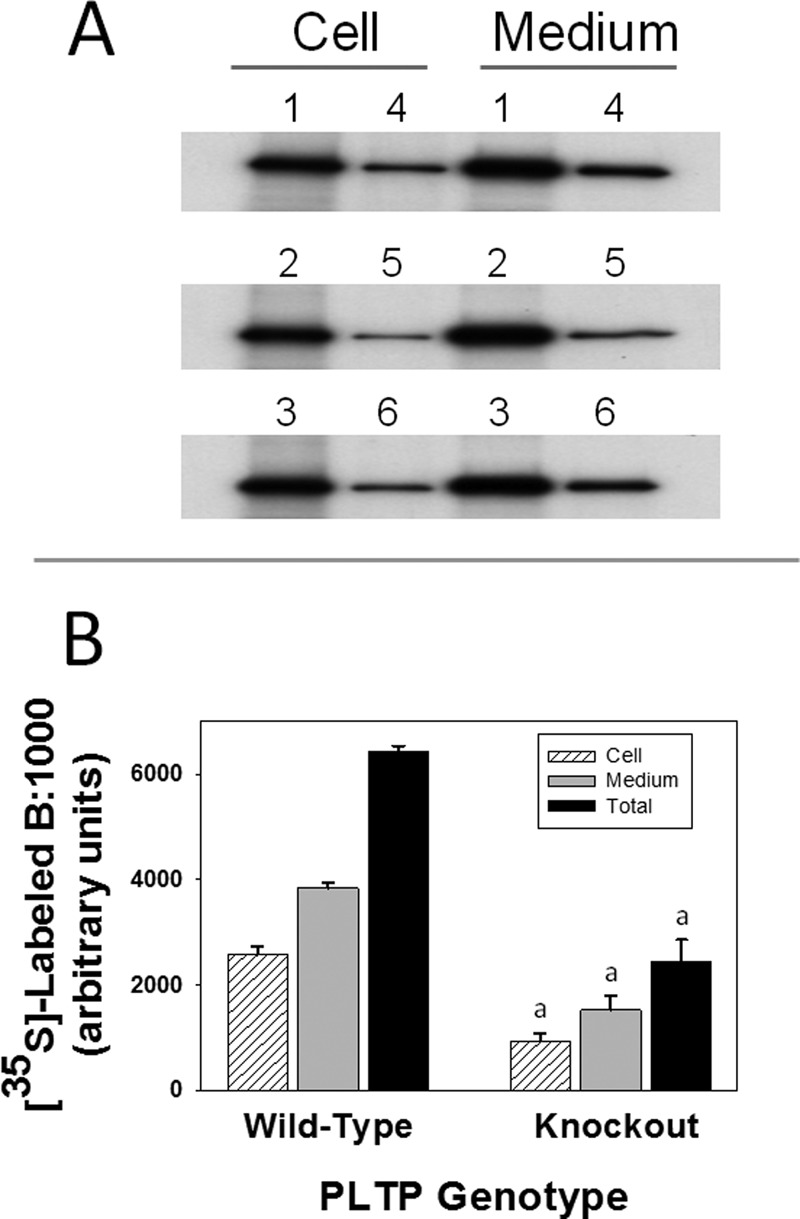
PLTP deficiency drastically decreases the synthesis and secretion of human apoB:1000. Primary hepatocytes from wild type and PLTP knock-out mice were transduced with Adv-apoB:1000 and metabolically labeled with [35S]Met/Cys. Secreted and cellular 35S-labeled apoB:1000 was immunoprecipitated using antibody to human apoB100 and processed as described in the text. Results are representative of 11 experiments. A, autoradiogram of 35S-labeled apoB:1000. B, intensities of 35S-labeled apoB:1000 bands were determined by computer-assisted image processing, normalized for cell protein, and plotted as mean ± S.E. (error bars) of triplicate dishes. a, p < 0.001 wild type versus PLTP knock-out.
PLTP Deficiency Markedly Reduces the Lipid Content and Alters the Lipid Composition of ApoB:1000-containing Particles Secreted by Primary Hepatocytes
To test the effect of PLTP deficiency on the lipid content and composition of secreted apoB:1000-containing particles, WT and PLTP-KO primary hepatocytes were transduced with Adv-apoB:1000 followed by metabolic labeling with [3H]glycerol as described under “Experimental Procedures.” Results shown in Table 3 demonstrate a marked 72% reduction in the 3H-labeled lipids associated with apoB:1000-containing particles secreted by PLTP-KO primary hepatocytes when compared with WT controls. Consistent with our previous studies in rat McA-RH7777 cells (5), apoB:1000-containing particles secreted by WT primary hepatocytes were PL-rich and contained 69% PL, 9% DAG, and 23% TAG (Table 3). In WT hepatocytes, the calculated stoichiometries of PL, DAG, and TAG molecules per apoB:1000 were 46, 6, and 15, respectively; the surface to core lipid ratio of the secreted apoB:1000-containing particles was ∼3.5:1 (Table 3). PLTP deficiency not only decreased the total 3H-labeled lipids associated with secreted apoB:1000-containing particles, but it also altered their lipid composition. In the absence of PLTP, the secreted particles contained 46% PL, 13% DAG, and 41% TAG (Table 3). The calculated stoichiometries of PL, DAG, and TAG molecules per apoB:1000 were 27, 10, and 23, respectively; the surface to core lipid ratio of these particles was ∼1.6:1 (Table 3). Similar results were obtained when intact apoB:1000-containing particles were isolated by non-denaturing gradient gel electrophoresis (data not shown).
TABLE 3.
Effect of adenovirus-mediated expression of human phospholipid transfer protein on the concentration and composition of [3H]glycerol-labeled lipids associated with apoB:1000-containing particles secreted by primary hepatocytes isolated from wild type and PLTP knock-out mice
Hepatocytes were isolated from wild type and PLTP knock-out mice, cultured, and transduced with Adv-apoB:1000 with or without co-transduction with Adv-PLTP as described under “Experimental Procedures.” Cells were metabolically labeled with 3H-labeled glycerol (7 μCi/ml of medium) 48 h post-transduction, the secreted labeled apoB:1000-containing particles were isolated by immunoprecipitation under non-denaturing condition using polyclonal antibody to human apoB100, and processed as described in the text. Values are means ± S.E. of 9–15 samples from four experiments.
| PLTP genotype | Adv |
3H-Labeled lipids associated with secreted apoB:1000 particles |
Calculated number of lipid molecules per apoB:1000 particles |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Total lipids | PL | DAG | TAG | PL | DAG | TAG | Total molecules | ||
| cpm/mg cell protein | % of total lipids | % of total lipids | % of total lipids | ||||||
| Wild type | B:1000 | 3,239 ± 345 | 68.54 ± 1.75 | 8.71 ± 1.98 | 22.76 ± 2.26 | 46 ± 2 | 6 ± 1 | 15 ± 1 | 67 ± 1 |
| Knock-out | B:1000 | 900 ± 70a | 45.62 ± 1.26a | 13.41 ± 1.26 | 40.84 ± 2.02a | 27 ± 1a | 10 ± 1b | 23 ± 1a | 60 ± 1a |
| Knock-out | B:1000 and PLTP | 2,558 ± 311 | 70.06 ± 4.63 | 9.62 ± 1.42 | 20.26 ± 5.22 | 48 ± 2 | 8 ± 1 | 12 ± 3 | 68 ± 2 |
a p < 0.001 wild type versus PLTP knock-out.
b p = 0.004 wild type versus PLTP knock-out.
Reintroducing the Pltp Gene into PLTP-KO Primary Hepatocytes Restores the Synthesis and Secretion of ApoB:1000
To further validate the concept that PLTP is involved in PL transfer to apoB:1000, we reintroduced the Pltp gene into PLTP-KO hepatocytes by transducing the cells with Adv-B:1000 with or without co-transduction with Adv-PLTP followed by metabolic labeling with [35S]Met/Cys. Results demonstrated that PLTP expression in PLTP-KO hepatocytes increased cellular and secreted 35S-labeled apoB:1000 by 3-fold (Fig. 4, lane 2) as compared with PLTP-KO control (Fig. 4, lane 1).
FIGURE 4.
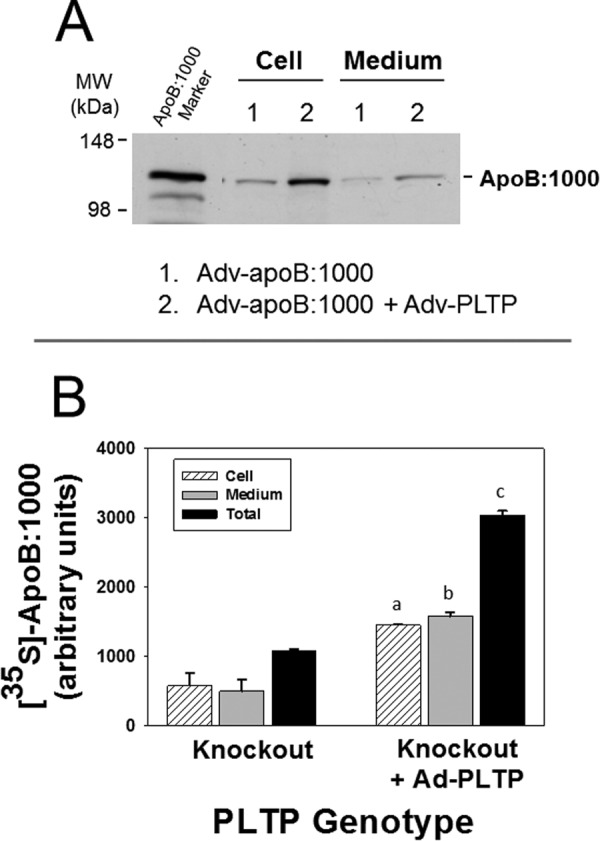
Re-introducing Pltp gene into PLTP-KO primary hepatocytes restores the synthesis and secretion of apoB:1000. Primary hepatocytes from PLTP knock-out mice were transduced with Adv-apoB:1000 with or without co-transduction with Adv-PLTP and metabolically labeled with [35S]Met/Cys. Secreted and cellular 35S-labeled apoB:1000 was immunoprecipitated using antibody to human apoB100 and processed as described in the text. A, autoradiogram of secreted and cellular 35S-labeled apoB:1000 of hepatocytes transduced with Adv-apoB:1000 only (lane 1) or Adv-apoB:1000 plus Adv-PLTP (lane 2). B, intensities of 35S-labeled apoB:1000 bands were normalized for cell protein and plotted as mean ± S.E. (error bars) of triplicate dishes. a, p = 0.04; b, p = 0.02; c, p = 0.002 PLTP knock-out versus PLTP knock-out plus PLTP.
PLTP Expression in PLTP-KO Primary Hepatocytes Demonstrates the Essential Role of PLTP in the Transfer of PL to ApoB:1000-containing Primordial Nascent Lipoproteins
Reintroduction of PLTP into PLTP-KO hepatocytes resulted in an ∼3-fold increase in the total 3H-labeled lipid content of the secreted apoB:1000-containing particles, restoring it to 80% of that observed in particles secreted by WT hepatocytes (Table 3). Importantly, PLTP expression reversed the changes in the composition of apoB:1000-containing particles caused by PLTP deficiency. Intact apoB:1000-containing particles secreted by PLTP-KO hepatocytes expressing exogenous PLTP contained 70% PL, 10% DAG, and 20% TAG, a composition similar to that observed for particles secreted by WT hepatocytes (Table 3). The calculated stoichiometries of PL, DAG, and TAG molecules per apoB:1000 were 48, 8, and 12, respectively, with a surface to core lipid ratio of ∼4.7:1 (Table 3). Similar results (not shown) were obtained when 3H-labeled apoB:1000-containing particles were isolated by non-denaturing gradient gel electrophoresis and analyzed for their lipid content and composition as described previously (5).
Deficiency of PLTP in Hepatocytes Renders ApoB:1000 Susceptible to Intracellular Degradation
Regulation of apoB is thought to occur largely by intracellular degradation at both the co- and post-translational levels (1). To determine whether the drastic reduction in the secretion of apoB:1000 in PLTP-KO hepatocytes was due to its intracellular degradation, we performed pulse-chase experiments. Results showed that in contrast to WT hepatocytes apoB:1000 in PLTP-KO hepatocytes was susceptible to intracellular degradation (Fig. 5A). Importantly, reintroduction of Pltp gene into PLTP-KO hepatocytes rescued apoB:1000 from degradation (Fig. 5A).
FIGURE 5.
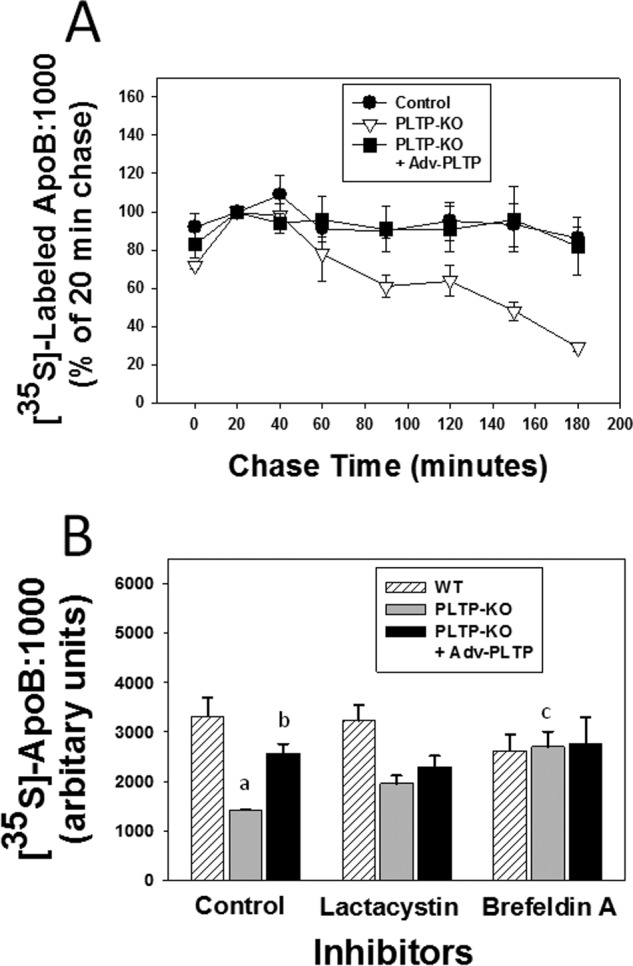
ApoB:1000 in PLTP knock-out hepatocytes is susceptible to intracellular degradation. Primary hepatocytes from wild type and PLTP knock-out mice were transduced with Adv-apoB:1000 with or without co-transduction with Adv-PLTP. A, cells were pulsed with [35S]Met/Cys for 10 min followed by 20–180-min chase. At the indicated chase period, secreted and cellular 35S-labeled apoB:1000 was immunoprecipitated using antibody to human apoB100 and processed as described in the text. Intensities of 35S-labeled apoB:1000 bands were normalized for cell protein and plotted as mean ± S.E. (error bars) of triplicate dishes. B, cells were metabolically labeled with [35S]Met/Cys in the presence or absence of either lactacystin (10 μm) or brefeldin A (2 μm). Secreted and cellular 35S-labeled apoB:1000 was immunoprecipitated using antibody to human apoB100 and processed as described in the text. The intensities of 35S-labeled apoB:1000 bands were normalized for cell protein and plotted as mean ± S.E. (error bars) of triplicate dishes. a, p = 0.03 control wild type versus control PLTP knock-out; b, p = 0.01 control PLTP knock-out versus control PLTP knock-out plus PLTP; c, p = 0.04 control PLTP knock-out versus PLTP knock-out with brefeldin A.
To assess the potential mechanism of intracellular degradation of apoB:1000, we tested the effects of lactacystin, a known inhibitor of proteasomal degradation, and brefeldin A, an inhibitor of ER-to-Golgi protein trafficking (1), on the synthesis and secretion of apoB:1000. Lactacystin and brefeldin A had no significant effect on the total recovered (cellular plus secreted) 35S-labeled apoB:1000 in WT hepatocytes (Fig. 5B). In the absence of inhibitors, the total recovered 35S-labeled apoB:1000 in PLTP-KO hepatocytes was significantly lower (57%; p = 0.03) than in WT hepatocytes (Fig. 5B). Lactacystin had no significant effect on the total recovered 35S-labeled apoB:1000 in PLTP-KO hepatocytes (Fig. 5B). Conversely, brefeldin A protected apoB:1000 from intracellular degradation and increased its total recovery by 90% (p = 0.04), restoring it to 82% of that observed in WT hepatocytes (Fig. 5B). Reintroduction of Pltp gene into PLTP-KO hepatocytes increased the total recovered 35S-labeled apoB:1000 by a significant 80% (p = 0.02), restoring it to 78% of that observed in WT hepatocytes (Fig. 5B). The effects of lactacystin and brefeldin A on the degradation of apoB:1000 in PLTP-KO hepatocytes expressing exogenous PLTP were similar to those observed in WT hepatocytes (Fig. 5B). These results suggest that the intracellular degradation of apoB:1000 in the absence of PLTP most likely occurred in the post-ER, presecretory compartment.
DISCUSSION
The processes involved in the assembly of apoB-containing lipoproteins in the liver are complex and are regulated at multiple levels by various factors throughout the secretory pathway. One of the most important factors in the assembly and secretion of apoB-containing lipoproteins is MTP (29). Despite numerous studies demonstrating the requirement for MTP in both the first step assembly of apoB and the second step bulk lipid addition to apoB and core expansion (29), information on its possible role in the initial step of apoB particle assembly, a process distinct from the conventional first step assembly of HDL-sized apoB-containing particles, is scarce. Although MTP has a distinct preference for TAG and cholesteryl esters, it also has a weak PL transfer activity (30), which has been suggested to be sufficient for the initiation of apoB particle assembly in non-hepatic COS7 cells (31). To test this potential role of MTP in cells of hepatic origin, we undertook comprehensive studies that included the use of MTP inhibitors (11) and rat hepatoma McA-RH7777 cells with 98% deficiency in MTP at both the mRNA and protein levels (12). We demonstrated that BMS-l97636 and BMS-200150, potent inhibitors of MTP activity (32), had no effect on the synthesis and secretion of apoB:1000-containing particles (11). In a subsequent study (12), we used MTP-deficient McA-RH7777 cells and demonstrated that the near complete absence of MTP had no effect on the synthesis and secretion of apoB:1000 (12). Likewise, identical levels of labeled lipids were found associated with apoB:1000-containing particles secreted by parental and MTP-deficient McA-RH7777 cells (12). Importantly, the lipid composition of the secreted apoB:1000-containing particles was the same regardless of the presence or absence of MTP expression (12). Based on these compelling results, we concluded that the initial addition of PL to apoB:1000 is independent of MTP lipid transfer activity and hypothesized that PLTP is a plausible mediator of this very early step in apoB particle assembly (11, 12).
PLTP plays an important role in the metabolism and remodeling of plasma lipoproteins (33), including modulating HDL size and composition and HDL-mediated cellular efflux of phospholipids and cholesterol resulting in the generation of large HDL2-like particles and small, lipid-poor pre-β-HDL (34–36). Although PLTP has been suggested to be antiatherogenic (33, 34), in vivo studies in mice heterozygous for LDL receptor (LDLR+/−), a model for atherosclerosis, demonstrated that elevation of PLTP resulted in a PLTP dose-dependent decrease in plasma HDL levels coinciding with an increased susceptibility to diet-induced atherosclerosis (37). The involvement of PLTP in VLDL synthesis and secretion was demonstrated in a landmark study by Jiang et al. (22). The absence of PLTP in various mouse strains with hyperlipidemia and high susceptibility for atherosclerosis reduced the production and plasma levels of apoB-containing lipoproteins and markedly decreased atherosclerosis in most mouse models (22). The decrease in atherosclerosis susceptibility in PLTP-deficient mice was explained by the reduction in hepatic VLDL secretion, which was corrected when PLTP was reintroduced by adenovirus (22). This was the first evidence for a novel, intracellular role of PLTP in hepatocytes (22).
In the present study, we tested the hypothesis that PLTP plays a key role in the early transfer of PL to apoB:1000 leading to the formation of apoB-containing initiation complex. The availability of the PLTP-KO mouse model facilitated the planned studies in primary cultures of hepatocytes. Consistent with studies reported previously (22, 23, 38), plasma levels of total cholesterol, PL, VLDL, LDL, and HDL were significantly lower in PLTP-KO mice compared with the WT control. PLTP deficiency also markedly decreased the secretion of total lipids by primary hepatocytes and altered their composition; the major change was the reduction in TAG. Furthermore, the absence of PLTP in hepatocytes resulted in a marked reduction in the secretion of all lipoproteins, predominantly the non-HDL fraction. Interestingly, in contrast to marked elevation in cellular TAG leading to hepatic steatosis observed in the absence of functional MTP (39), PLTP deficiency did not cause excessive lipid accumulation in hepatocytes.
Results of the present study consistently demonstrated a major role of PLTP in the addition of phospholipids to apoB:1000 and formation of the PL-rich initiation complex. PLTP deficiency in hepatocytes caused a striking 3-fold reduction in the de novo synthesis, lipidation, and secretion of apoB:1000-containing particles as compared with WT hepatocytes. Importantly, PLTP deficiency markedly altered the lipid composition of the secreted particles. Consistent with our previous studies in rat McA-RH7777 cells (5), apoB:1000-containing particles secreted by WT hepatocytes were PL-rich and had calculated stoichiometries of PL, DAG, and TAG molecules per apoB:1000 of 46, 6, and 15, respectively. ApoB:1000-containing particles secreted in the absence of PLTP had 40% fewer PL molecules per particle; distinct stoichiometries of 27 PL, 10 DAG, and 23 TAG molecules per particle; and a surface to core lipid ratio of ∼1.6:1. Thus, PLTP deficiency resulted in the secretion of fewer apoB:1000 particles that were also PL-poor when compared with their counterparts secreted by WT hepatocytes. These results demonstrate that, in striking contrast to MTP deficiency (11, 12), PLTP absence in primary hepatocytes causes a marked reduction in the lipidation and secretion of apoB:1000-containing lipoproteins and alters their lipid composition.
The reintroduction of Pltp gene into PLTP-deficient hepatocytes resulted in a ∼3-fold increase in both the protein and lipid content of the secreted apoB:1000-containing particles, restoring their levels to 80% of that observed in WT hepatocytes. The similar increase in protein and lipid contents of particles indicates that PLTP increased the number of apoB:1000-containing particles. Notably, PLTP expression in PLTP-KO hepatocytes reversed the changes in the lipid composition and stoichiometry of secreted apoB:1000-containing particles caused by PLTP deficiency. These particles were PL-rich and had a lipid composition and surface to core lipid ratio similar to their counterparts secreted by WT hepatocytes. To put these ratios in perspective, the surface to core lipid ratio of spheroidal HDL3 particles is 1.5:1 (40), supporting the concept that the lipids in the apoB:1000-containing particles secreted by WT hepatocytes and PLTP-KO hepatocytes expressing exogenous PLTP are in the form of a bilayer assembly (7). Based entirely on the calculated stoichiometry and the surface to core lipid ratio, we speculate that apoB:1000-containing particles secreted by PLTP-KO hepatocytes might be in the form of a mixed micelle assembly like HDL3.
The question arises as to what could be the reason(s) for the drastic reduction in the synthesis and secretion of apoB:1000 in the absence of PLTP? The general consensus is that apoB secretion is regulated predominantly through intracellular degradation by multiple mechanisms, including ER-associated degradation; post-ER, presecretory degradation; and autophagy (41). Our pulse-chase studies demonstrated that, in sharp contrast to WT hepatocytes, apoB:1000 in PLTP-KO hepatocytes was highly susceptible to intracellular degradation. The combined results of studies using lactacystin and brefeldin A indicated that the degradation of apoB:1000 occurred predominantly at the post-ER, presecretory level. We further demonstrated that reintroduction of Pltp gene into PLTP-KO hepatocytes restored the lipidation of apoB:1000 and rescued the polypeptide from intracellular degradation. We speculate that the lower PL content of apoB:1000-containing lipoprotein produced in the absence of PLTP may induce conformational changes in the particle that could impart a propensity to form large aggregates leading to subsequent degradation in the post-ER, presecretory compartment. Based on the current results, however, we cannot make any further speculation on the nature of the pathway involved in this process.
At present, the mechanism(s) involved in PLTP-mediated transfer of PL to apoB:1000 is not known. We do not as yet have a structural model for apoB:1000-containing particles secreted by PLTP-KO hepatocytes for comparison with our previously reported (7) all atom molecular model of the wild type apoB:1000-containing particle. Furthermore, the structural domain(s) and amino acid sequences in PLTP that might be involved in PL transfer to apoB:1000 are not known. Studies by Oram et al. (42) have indicated that the helix 144–163 located at the tip of the N-terminal barrel of PLTP is critical for interacting with phospholipids and promoting ABCA1-dependent cholesterol efflux. It has also been shown that specific charged residues in the N- and C-terminal pockets of PLTP may be critical to the PLTP transfer activity (43). Future studies are required to identify the structural motifs in both PLTP and apoB:1000 that are critical for the transfer of PL to the apoB lipid pocket.
In conclusion, our previous comprehensive studies (11, 12) provided strong evidence that in cells of hepatic origin the acquisition of PL by apoB:1000 occurs independently of MTP lipid transfer activity. Results of the current study provide a compelling argument in support of our concept that in hepatocytes the initial transfer of PL to apoB:1000 is mediated to a large extent by PLTP. Our results do not rule out the possibility that other mechanism(s), e.g. direct desorption of PL from the ER membrane by apoB:1000 or another as yet unidentified transfer protein might contribute to this process. Several studies have demonstrated the role of PLTP in the secretion of VLDL (23, 44, 45). It has been proposed that PLTP-mediated VLDL production occurs at the second step core expansion of particles by facilitating the fusion of apoB-free lipid droplets with primordial apoB-containing lipoproteins (45). We propose that PLTP participates in at least two stages of apoB-containing lipoprotein assembly and secretion. First, PLTP plays a major role in the initial acquisition of PL by apoB:1000 leading to the formation of a PL-rich apoB-containing initiation complex. Second, PLTP in concert with MTP assists in the second step core expansion of apoB-containing particles leading to the formation and secretion of bona fide VLDL. Together, our results suggest that PLTP might be a more effective target than MTP for therapeutic intervention to attenuate hepatic production of apoB-containing lipoproteins at the very early stage of their assembly.
Acknowledgments
We thank Dr. Zamin Yao for providing the human apoB100 cDNA and Dr. Xian-Cheng Jiang for providing the human PLTP cDNA.
Note Added in Proof
Fig. 4A was not correct in the version of this article that was published on January 31, 2015 as a Paper in Press. The correct image is now shown.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 HL-112647.
- apo
- apolipoprotein
- Adv
- adenovirus
- apoB:1000
- N-terminal 22.05% (residues 1–1000) of the mature protein
- DAG
- diacylglycerol
- ER
- endoplasmic reticulum
- MTP
- microsomal triglyceride transfer protein
- PL
- phospholipid
- PLTP
- phospholipid transfer protein
- TAG
- triacylglycerol
- HBSS
- Hanks' balanced salt solution.
REFERENCES
- 1. Fisher E. A., Ginsberg H. N. (2002) Complexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteins. J. Biol. Chem. 277, 17377–17380 [DOI] [PubMed] [Google Scholar]
- 2. Olofsson S. O., Stillemark-Billton P., Asp L. (2000) Intracellular assembly of VLDL: two major steps in separate cell compartments. Trends Cardiovasc. Med. 10, 338–345 [DOI] [PubMed] [Google Scholar]
- 3. Hamilton R. L., Wong J. S., Cham C. M., Nielsen L. B., Young S. G. (1998) Chylomicron-sized lipid particles are formed in the setting of apolipoprotein B deficiency. J. Lipid Res. 39, 1543–1557 [PubMed] [Google Scholar]
- 4. Dashti N., Gandhi M., Liu X., Lin X., Segrest J. P. (2002) The N-terminal 1000 residues of apolipoprotein B associate with microsomal triglyceride transfer protein to create a lipid transfer pocket required for lipoprotein assembly. Biochemistry 41, 6978–6987 [DOI] [PubMed] [Google Scholar]
- 5. Manchekar M., Richardson P. E., Forte T. M., Datta G., Segrest J. P., Dashti N. (2004) Apolipoprotein B-containing lipoprotein particle assembly: lipid capacity of the nascent lipoprotein particle. J. Biol. Chem. 279, 39757–39766 [DOI] [PubMed] [Google Scholar]
- 6. Manchekar M., Richardson P. E., Sun Z., Liu Y., Segrest J. P., Dashti N. (2008) Charged amino acid residues 997–1000 of human apolipoprotein B100 are critical for the initiation of lipoprotein assembly and the formation of a stable lipidated primordial particle in McA-RH7777 cells. J. Biol. Chem. 283, 29251–29265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richardson P. E., Manchekar M., Dashti N., Jones M. K., Beigneux A., Young S. G., Harvey S. C., Segrest J. P. (2005) Assembly of lipoprotein particles containing apolipoprotein-B: structural model for the nascent lipoprotein particle. Biophys. J. 88, 2789–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raag R., Appelt K., Xuong N. H., Banaszak L. (1988) Structure of the lamprey yolk lipid-protein complex lipovitellin-phosvitin at 2.8 Å resolution. J. Mol. Biol. 200, 553–569 [DOI] [PubMed] [Google Scholar]
- 9. Baker M. E. (1988) Is vitellogenin an ancestor of apolipoprotein B-100 of human low-density lipoprotein and human lipoprotein lipase? Biochem. J. 255, 1057–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shoulders C. C., Narcisi T. M., Read J., Chester A., Brett D. J., Scott J., Anderson T. A., Levitt D. G., Banaszak L. J. (1994) The abetalipoproteinemia gene is a member of the vitellogenin family and encodes an α-helical domain. Nat. Struct. Biol. 1, 285–286 [DOI] [PubMed] [Google Scholar]
- 11. Dashti N., Manchekar M., Liu Y., Sun Z., Segrest J. P. (2007) Microsomal triglyceride transfer protein activity is not required for the initiation of apolipoprotein B-containing lipoprotein assembly in McA-RH7777 cells. J. Biol. Chem. 282, 28597–28608 [DOI] [PubMed] [Google Scholar]
- 12. Liu Y., Manchekar M., Sun Z., Richardson P. E., Dashti N. (2010) Apolipoprotein B-containing lipoprotein assembly in microsomal triglyceride transfer protein-deficient McA-RH7777 cells. J. Lipid Res. 51, 2253–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dashti N., McConathy W. J., Ontko J. A. (1980) Production of apolipoproteins E and A-I by rat hepatocytes in primary culture. Biochim. Biophys. Acta 618, 347–358 [DOI] [PubMed] [Google Scholar]
- 14. Yao Z. M., Blackhart B. D., Johnson D. F., Taylor S. M., Haubold K. W., McCarthy B. J. (1992) Elimination of apolipoprotein B48 formation in rat hepatoma cell lines transfected with mutant human apolipoprotein B cDNA constructs. J. Biol. Chem. 267, 1175–1182 [PubMed] [Google Scholar]
- 15. Miller A. D., Miller D. G., Garcia J. V., Lynch C. M. (1993) Use of retroviral vectors for gene transfer and expression. Methods Enzymol. 217, 581–599 [DOI] [PubMed] [Google Scholar]
- 16. Berry M. N., Friend D. S. (1969) High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J. Cell Biol. 43, 506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Honkakoski P., Negishi M. (1998) Protein serine/threonine phosphatase inhibitors suppress phenobarbital-induced Cyp2b10 gene transcription in mouse primary hepatocytes. Biochem. J. 330, 889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alaupovic P., Lee D. M., McConathy W. J. (1972) Studies on the composition and structure of plasma lipoproteins. Distribution of lipoprotein families in major density classes of normal human plasma lipoproteins. Biochim. Biophys. Acta 260, 689–707 [PubMed] [Google Scholar]
- 19. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 20. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 21. Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 22. Jiang X. C., Qin S., Qiao C., Kawano K., Lin M., Skold A., Xiao X., Tall A. R. (2001) Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat. Med. 7, 847–852 [DOI] [PubMed] [Google Scholar]
- 23. Siggins S., Bykov I., Hermansson M., Somerharju P., Lindros K., Miettinen T. A., Jauhiainen M., Olkkonen V. M., Ehnholm C. (2007) Altered hepatic lipid status and apolipoprotein A-I metabolism in mice lacking phospholipid transfer protein. Atherosclerosis 190, 114–123 [DOI] [PubMed] [Google Scholar]
- 24. Chang B. H., Liao W., Li L., Nakamuta M., Mack D., Chan L. (1999) Liver-specific inactivation of the abetalipoproteinemia gene completely abrogates very low density lipoprotein/low density lipoprotein production in a viable conditional knockout mouse. J. Biol. Chem. 274, 6051–6055 [DOI] [PubMed] [Google Scholar]
- 25. Luo Y., Shelly L., Sand T., Reidich B., Chang G., Macdougall M., Peakman M. C., Jiang X. C. (2010) Pharmacologic inhibition of phospholipid transfer protein activity reduces apolipoprotein-B secretion from hepatocytes. J. Pharmacol. Exp. Ther. 332, 1100–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davis R. A., Boogaerts J. R. (1982) Intrahepatic assembly of very low density lipoproteins. Effect of fatty acids on triacylglycerol and apolipoprotein synthesis. J. Biol. Chem. 257, 10908–10913 [PubMed] [Google Scholar]
- 27. Wang Y., Tran K., Yao Z. (1999) The activity of microsomal triglyceride transfer protein is essential for accumulation of triglyceride within microsomes in McA-RH7777 cells. A unified model for the assembly of very low density lipoproteins. J. Biol. Chem. 274, 27793–27800 [DOI] [PubMed] [Google Scholar]
- 28. Tran K., Wang Y., DeLong C. J., Cui Z., Yao Z. (2000) The assembly of very low density lipoproteins in rat hepatoma McA-RH7777 cells is inhibited by phospholipase A2 antagonists. J. Biol. Chem. 275, 25023–25030 [DOI] [PubMed] [Google Scholar]
- 29. Hussain M. M., Shi J., Dreizen P. (2003) Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res. 44, 22–32 [DOI] [PubMed] [Google Scholar]
- 30. Wetterau J. R., Aggerbeck L. P., Bouma M. E., Eisenberg C., Munck A., Hermier M., Schmitz J., Gay G., Rader D. J., Gregg R. E. (1992) Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science 258, 999–1001 [DOI] [PubMed] [Google Scholar]
- 31. Rava P., Ojakian G. K., Shelness G. S., Hussain M. M. (2006) Phospholipid transfer activity of microsomal triacylglycerol transfer protein is sufficient for the assembly and secretion of apolipoprotein B lipoproteins. J. Biol. Chem. 281, 11019–11027 [DOI] [PubMed] [Google Scholar]
- 32. Wetterau J. R., Gregg R. E., Harrity T. W., Arbeeny C., Cap M., Connolly F., Chu C. H., George R. J., Gordon D. A., Jamil H., Jolibois K. G., Kunselman L. K., Lan S. J., Maccagnan T. J., Ricci B., Yan M., Young D., Chen Y., Fryszman O. M., Logan J. V., Musial C. L., Poss M. A., Robl J. A., Simpkins L. M., Slusarchyk W. A., Sulsky R., Taunk P., Magnin D. R., Tino J. A., Lawrence R. M., Dickson J. K., Jr., Biller S. A. (1998) An MTP inhibitor that normalizes atherogenic lipoprotein levels in WHHL rabbits. Science 282, 751–754 [DOI] [PubMed] [Google Scholar]
- 33. van Tol A. (2002) Phospholipid transfer protein. Curr. Opin. Lipidol. 13, 135–139 [DOI] [PubMed] [Google Scholar]
- 34. Jiang X. C., Jin W., Hussain M. M. (2012) The impact of phospholipid transfer protein (PLTP) on lipoprotein metabolism. Nutr. Metab. 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang X. C., Bruce C., Mar J., Lin M., Ji Y., Francone O. L., Tall A. R. (1999) Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J. Clin. Investig. 103, 907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rye K. A., Clay M. A., Barter P. J. (1999) Remodelling of high density lipoproteins by plasma factors. Atherosclerosis 145, 227–238 [DOI] [PubMed] [Google Scholar]
- 37. van Haperen R., van Tol A., van Gent T., Scheek L., Visser P., van der Kamp A., Grosveld F., de Crom R. (2002) Increased risk of atherosclerosis by elevated plasma levels of phospholipid transfer protein. J. Biol. Chem. 277, 48938–48943 [DOI] [PubMed] [Google Scholar]
- 38. Yazdanyar A., Quan W., Jin W., Jiang X. C. (2013) Liver-specific phospholipid transfer protein deficiency reduces high-density lipoprotein and non-high-density lipoprotein production in mice. Arterioscler. Thromb. Vasc. Biol. 33, 2058–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hussain M. M., Bakillah A. (2008) New approaches to target microsomal triglyceride transfer protein. Curr. Opin. Lipidol. 19, 572–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edelstein C., Kézdy F. J., Scanu A. M., Shen B. W. (1979) Apolipoproteins and the structural organization of plasma lipoproteins: human plasma high density lipoprotein-3. J. Lipid Res. 20, 143–153 [PubMed] [Google Scholar]
- 41. Ginsberg H. N., Fisher E. A. (2009) The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J. Lipid Res. 50, (suppl.) S162–S166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oram J. F., Wolfbauer G., Tang C., Davidson W. S., Albers J. J. (2008) An amphipathic helical region of the N-terminal barrel of phospholipid transfer protein is critical for ABCA1-dependent cholesterol efflux. J. Biol. Chem. 283, 11541–11549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ponsin G., Qu S. J., Fan H. Z., Pownall H. J. (2003) Structural and functional determinants of human plasma phospholipid transfer protein activity as revealed by site-directed mutagenesis of charged amino acids. Biochemistry 42, 4444–4451 [DOI] [PubMed] [Google Scholar]
- 44. Okazaki H., Goldstein J. L., Brown M. S., Liang G. (2010) LXR-SREBP-1c-phospholipid transfer protein axis controls very low density lipoprotein (VLDL) particle size. J. Biol. Chem. 285, 6801–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yazdanyar A., Jiang X. C. (2012) Liver phospholipid transfer protein (PLTP) expression with a PLTP-null background promotes very low-density lipoprotein production in mice. Hepatology 56, 576–584 [DOI] [PMC free article] [PubMed] [Google Scholar]