

Background: PDGF is a potent chemoattractant for cells.
Results: The PDGF-induced extracellular matrix component Cyr61 bridges the intracellular PDGF-ERK and JNK signaling pathways with integrin/FAK signaling, leading to cell migration.
Conclusion: Cyr61 is a key mediator of PDGF-induced cell migration via the Cyr61-integrin-FAK pathway.
Significance: Extracellular Cyr61 convergence with growth factor and integrin/FAK signaling is a new cell migration concept.
Keywords: Gene Expression, Signal Transduction, Signaling, Vascular Biology, Vascular Smooth Muscle Cells, Cyr61, PDGF
Abstract
Platelet-derived growth factor (PDGF), a potent chemoattractant, induces cell migration via the MAPK and PI3K/Akt pathways. However, the downstream mediators are still elusive. In particular, the role of extracellular mediators is largely unknown. In this study, we identified the matricellular protein Cyr61, which is de novo synthesized in response to PDGF stimulation, as the key downstream mediator of the ERK and JNK pathways, independent of the p38 MAPK and AKT pathways, and, thereby, it mediates PDGF-induced smooth muscle cell migration but not proliferation. Our results revealed that, when Cyr61 was newly synthesized by PDGF, it was promptly translocated to the extracellular matrix and physically interacted with the plasma membrane integrins α6β1 and αvβ3. We further demonstrate that Cyr61 and integrins are integral components of the PDGF signaling pathway via an “outside-in” signaling route to activate intracellular focal adhesion kinase (FAK), leading to cell migration. Therefore, this study provides the first evidence that the PDGF-induced endogenous extracellular matrix component Cyr61 is a key mediator in modulating cell migration by connecting intracellular PDGF-ERK and JNK signals with integrin/FAK signaling. Therefore, extracellular Cyr61 convergence with growth factor signaling and integrin/FAK signaling is a new concept of growth factor-induced cell migration. The discovered signaling pathway may represent an important therapeutic target in growth factor-mediated cell migration/invasion-related vascular diseases and tumorigenesis.
Introduction
Cell migration is a crucial process in vascular neointimal formation and cancer development, and PDGF plays an important role in the development of these diseases (1, 2). The PDGF family consists of five different disulfide-bonded homo- and heterodimers: PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD. In vivo evidence reveals that PDGF receptor β and PDGF-BB peptides are essential in neointimal formation and vascular remodeling (1, 3–8).
Previous studies have shown that various intracellular pathways mediate PDGF-induced cell migration. These pathways include MAPK, ERK, JNK, p38 MAPK, and PI3K/Akt kinase (9–11). However, how these kinases mediate PDGF-induced cell migration is still not well understood. In particular, whether activation of these kinases influences matrix protein expression is largely unknown, and whether PDGF-induced matricellular proteins interact with integrins and transduce the migratory signal back to intracellular focal adhesion kinase (FAK)2 activation and cell migration is currently unknown. We aimed to address these questions in this study. Cyr61 (CCN1), a cysteine-rich matricellular protein, has been reported to regulate a wide range of cellular processes, including proliferation, adhesion, survival, migration, and differentiation (12–17). Cyr61 was rapidly induced in vascular SMCs during vascular injury (18). PDGF-BB has been reported to be the most potent mediator of SMC migration in vascular injury (4–8), and, similar to vascular remodeling, certain glioblastoma cell lines express high levels of Cyr61 (19). Cyr61 has recently been considered as a tumor-promoting factor (20), and PDGF has been shown to promote tumorigenesis (2). However, the relationship between PDGF and matricellular Cyr61 in these diseases has not been revealed. We hypothesized that Cyr61 is a key regulator that is produced by PDGF and that, in turn, mediates PDGF signaling in the extracellular matrix (ECM) via integrin interaction, leading to cell migration.
In this study, we found that PDGF highly induces Cyr61 expression in SMCs. We identified the upstream signaling pathway controlling the production of Cyr61. Our data further point out that Cyr61 is a key molecule regulating PDGF-induced cell migration. Although three MAPKs (ERK, JNK, and p38) and AKT have been reported to regulate PDGF-mediated cell migration (9–11), we found that Cyr61 expression is specifically dependent on ERK and JNK activation independent of p38 and AKT activity. Furthermore, our results reveal that PDGF-induced Cyr61 interacts with specific integrins and that Cyr61 and integrins are integral components in PDGF signaling. Finally, we identified the Cyr61-integrin-FAK axis in the PDGF pathway. These data reveal, for the first time, that the PDGF/Cyr61/integrin pathway contributes to cell migration.
EXPERIMENTAL PROCEDURES
Reagents
Recombinant PDGF-BB and antibody against mouse Cyr61 were from R&D Systems (Minneapolis, MN). Antibody against β-actin was from Sigma. Antibodies against p-MEK, p-ERK, ERK, p-JNK, p-p38, AKT1, p-AKT-S473, and FAK; the integrins αV, α4, α5, β1, β3, β4, and β5; PDGF receptor β; and phospho-PDGF receptor β were from Cell Signaling Technology (Beverly, MA). Antibodies against integrin α6, Egr1, and rabbit IgG were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against α6β1 and ανβ3 were from Millipore (Danvers, MA). Antibodies against p-FAK-Tyr-397, p-FAK-Tyr-576, p-FAK-Tyr-577, p-FAK-Tyr-861, and p-FAK-Ser-910 were from Life Technologies, Inc. The MEK1/2-specific inhibitor U0126, the JNK-specific inhibitor SP600125, the p38-specific inhibitor SB203580, and the PI3K-specific inhibitors wortmannin and LY294002 were from Enzo Life Sciences (Farmingdale, NY). The FAK-specific inhibitor PF573228 was from TOCRIS Bioscience (Bristol, UK). Non-silencing siRNA, siRNAs for Cyr61, and the integrins α6, β1, β3, and FAK were from Qiagen (Gaithersburg, MD). The cross-linker 3,3′-dithiobis(sulfosuccinimidylpropionate) (DTSSP) was from Pierce.
Tissue Culture
Mouse aortic SMCs were prepared from explants of excised aortas of mice as described previously (21). Cells were maintained in DMEM containing 10% fetal bovine serum. Cells were made quiescent by incubation in serum-free DMEM for 24 h. PDGF-BB was dissolved in PBS.
Western Blot Analysis
Cultured mouse SMCs were rinsed with cold PBS and lysed in lysis buffer (0.5 m Tris-HCl (pH 6.8), 10 m urea, 10% SDS, 1 m DTT, a mixture of protease inhibitors (Roche), 0.05 m PMSF, 0.2 m Na3VO4, and 0.5 m NaF) with sonication for 30 s on ice. Cellular proteins were separated by 7%, 8%, and 10% SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Immobilon-P, Millipore). Membranes were then probed with specific antibodies, and the specific protein bands were viewed using ECL Plus (GE Healthcare).
siRNA Treatment
Cells were transfected with non-silencing or specific siRNA (Qiagen) for 48 h using Lipofectamine RNAiMAX reagent (Life Technologies) following the instructions provided by the manufacturer. On day 3, cells were starved for 24 h, followed by treatment either with or without PDGF-BB.
Preparation of Detached Cells and ECM
SMCs were grown in 60-mm dishes. After removal of the culture medium and rinsing with PBS, cells were detached from the dish by incubation with 1 mm EDTA. The dish was then rinsed twice with 1 mm EDTA to remove remaining cells. Cellular fractions were lysed as described under “Western Blot Analysis.” Extracellular material remaining on the dishes after removal of the cellular components was extracted by scraping at 90 °C in Laemmli sample buffer (60 mm Tris-HCI (pH 6.8), 2% SDS, 5% fl-β-mercaptoethanol, and 5% glycerol). These fractions were designated as ECM as described previously (22).
Cell Migration Assay
Cell migration was performed by trypsinizing SMCs and plating them onto transwell migration plates (Corning, New York, NY) for migration assays. 2 × 105 cells were added to the upper chamber. Cells were allowed to migrate through filters (8-μm pore size) precoated on both sides with gelatin in the presence of either medium (600 μl) alone or medium with PDGF-BB at designated concentrations in the lower chamber. Cell migration was carried out at 37 °C in 5% CO2 for 6 h. Cells remaining on the upper surface of the filter were carefully removed by mechanical scraping. Cells migrated to the lower side were fixed with methanol and then stained with Harris H&E. The cells that had migrated to the lower surface of the filter were counted in four random objective fields (×200 magnification) using a Nikon Eclipse E600 microscope.
Cross-linking
SMCs were grown in 60-mm dishes. After 24 h of starvation, the culture medium was removed, and SMCs were rinsed with PBS twice. Adherent cells were incubated in 2.0 mm DTSSP (Pierce) in PBS for 30 min at room temperature (23). Unreacted cross-linker was quenched for 15 min by addition of 1 m Tris-HCl (pH 7.5) to a final concentration of 20 mm. After quenching, samples were lysed and analyzed by Western blotting with antibodies against integrins α6, β1, and β3 or Cyr61.
Overexpression of Cyr61 Protein and Immunoprecipitation
Mouse aortic SMCs were cultured in DMEM supplemented with 10% fetal bovine serum. SMCs were infected for 24 h with either Ad-CMV-null adenovirus or Ad-CMV-mCyr61 adenovirus (Vector Biolabs, Philadelphia, PA). After infection, cells were lysed in immunoprecipitation lysis buffer (1% CHAPSO, 30 mm Tris-HCl (pH 8.0), 150 mm NaCl, 5 mm EDTA, and protease/phosphatase inhibitor mixture). After sonication for 20 s, total cell lysates were centrifuged at 14,000 × g for 5 min at 4 °C to remove cell debris, and the supernatants were incubated with Cyr61 antibody for 3 h with rotation at 4 °C. Protein A-Sepharose beads were then added and incubated with rotation overnight at 4 °C. After washing five times with cold PBS, the immunoprecipitates were separated by SDS-PAGE and probed with either Cyr61 antibody or specific integrin antibody.
[3H]Thymidine Incorporation
SMC proliferation was determined as described previously (24). Cells were transfected with non-silencing or Cyr61 siRNA for 48 h. After 24-h starvation, SMCs were stimulated with or without PDGF-BB for 24 h. During the last 6-h incubation period, cells were labeled with 1 μCi/ml methyl-[3H]thymidine. After labeling, cells were washed three times with ice-cold PBS and two times with 10% trichloroacetic acid. Trichloroacetic acid-insoluble material was hydrolyzed by 0.25 N NaOH, and radioactivity was assayed in a liquid scintillation counter. The proliferation of SMCs was assessed by determination of [3H]thymidine incorporation into DNA.
Statistical Analysis
Results are means ± S.E. Comparisons between multiple groups were performed using one-way analysis of variance with post-hoc Student's t tests. Single comparisons were made using two-tailed, unpaired Student's t tests. A p value of 0.05 was considered statistically significant.
RESULTS
PDGF-BB Highly Induces Expression of Matricellular Protein Cyr61, Which Mediates SMC Migration
To test our hypothesis that Cyr61 has a function in the PDGF signaling pathway, we first tested whether PDGF influences Cyr61 expression in SMCs. Cultured mouse aortic SMCs were serum-starved for 24 h and then treated with 10 ng/ml PDGF-BB for various time periods. Cell lysates were analyzed by SDS-PAGE, and Cyr61 protein expression was determined by Western blotting. As shown in Fig. 1A, we found that PDGF-BB markedly induced Cyr61 protein expression (over 17-fold), peaking at around 1 h. PDGF-BB induction of Cyr61 was in a dose-dependent manner (Fig. 1B). At concentrations above 2.5 ng/ml, PDGF-BB significantly induced Cyr61 protein expression. These data suggest a role of Cyr61 in PDGF-induced cellular function. Because it is well known that PDGF induces SMC migration and proliferation, we determined whether PDGF-induced Cyr61 mediates these functions. When Cyr61 expression was knocked down successfully (Fig. 1C) with specific siRNA, we observed a significant blockage of PDGF-induced cell migration (Fig. 1D) (60–70% inhibition in all PDGF concentration ranges used). In contrast, knockdown of Cyr61 expression had no detectable effect on PDGF-induced cell proliferation (Fig. 1E). Therefore, these data reveal a novel and specific role for Cyr61 in the PDGF signaling pathway. That is, Cyr61 regulates PDGF-induced cell migration rather than proliferation. We also observed that knockdown of Cyr61 expression has no significant effect on either expression or phosphorylation of PDGF receptor β (Fig. 1F), indicating that Cyr61 has influence downstream of the PDGF-BB receptor. The effect of the endogenously produced Cyr61 on SMC migration and proliferation was assessed further by overexpression of Cyr61 by adenovirus infection (Fig. 1, G–I). We observed that overexpression of Cyr61 enhances SMC migration (Fig. 1H) but has no significant effect on SMC proliferation (Fig. 1I). These data further support the role of Cyr61 in PDGF-induced SMC migration.
FIGURE 1.

Cyr61 mediated PDGF-induced SMC migration but not proliferation. A, PDGF-BB induced Cyr61 protein expression in mouse aortic SMCs in a time-dependent manner. Cultured cells were starved for 24 h prior to PDGF-BB stimulation for various times as indicated. Cell lysates were subjected to Western blot analysis. β-actin served as the loading control. Top panel, representative Western blot result from three independent experiments. Bottom panel, results of the Western blot analysis quantified by densitometry. *, p < 0.05; **, p < 0.01 versus control. B, PDGF-BB dose-dependently induced Cyr61 protein expression in SMCs. Top panel, representative Western blot result from three independent experiments. Bottom panel, results of Western blot analysis quantified by densitometry. **, p < 0.01 versus control. C, Western blot results of knockdown of Cyr61 expression. Non-silencing siRNA (Non) served as a control. D, PDGF-BB dose-dependently induced SMC migration, and knockdown of Cyr61 expression with the specific Cyr61 siRNA largely blocked PDGF-BB-induced SMC migration. *, p < 0.05; **, p < 0.01 versus control; ##, p < 0.01 versus non-silencing siRNA group. E, knockdown of Cyr61 expression with specific siRNA had no effect on PDGF-BB-induced SMC proliferation. *p < 0.05, **p < 0.01 versus controls. F, Western blot results of the effects of knockdown of Cyr61 expression on the expression or phosphorylation of PDGF receptor β. G, Western blot result of Cyr61 overexpression after cells were infected with Ad-CMV-mCyr61 adenovirus compared with control cells infected with Ad-CMV-null adenovirus. β-actin served as the loading control. IB, immunoblot. H, effect of overexpression of Cyr61 on SMC migration (transwell migration assay in serum-free medium). **, p < 0.01 versus control. I, effect of overexpression of Cyr61 on SMC proliferation.
Activation of ERK and JNK, but Not p38 or AKT, Is Required for PDGF-BB-induced Cyr61 Protein Expression
To determine the molecular basis that mediates Cyr61 expression, we assessed whether activation of MAPKs or AKT has a role in Cyr61 expression because these kinases have been implicated in PDGF-induced cell migration (9–11). We first measured the activation of MEK, ERK, JNK, p38, and Akt stimulated by PDGF-BB in mouse primary SMCs. As shown in Fig. 2A, the activation of MEK, ERK, JNK, and p38 in response to PDGF-BB was dramatic and transient, peaking around 2–15 min. However, AKT had a long-lasting activation, from 2 min to 1 h (Fig. 2A), suggesting a possible role of these kinases in regulating Cyr61 expression.
FIGURE 2.

The ERK and JNK pathways, but not the p38 MAPK or AKT pathway, mediated PDGF-BB-induced Cyr61 expression. A, time course of PDGF-BB induction of phosphorylation of MEK, ERK, JNK, p38, and AKT in SMCs. Cultured SMCs were starved for 24 h prior to PDGF-BB (10 ng/ml) stimulation for various times. Cell lysates were analyzed by Western blotting using specific antibodies against p-MEK, p-ERK, p-JNK, p-p38, and p-AKT. β-actin served as the loading control. B, pretreatment with the specific MEK inhibitor U0126 (10 μm) or the specific JNK inhibitor SP600125 (SP) (10 μm), but not the specific p38 inhibitor SB203580 (SB, 10 μm) completely blocked PDGF-BB-induced Cyr61 expression. Quiescent SMCs were pretreated with inhibitors for 45 min, and then 10 ng/ml PDGF-BB was added for 1 h. Cyr61 protein level was determined by Western blotting. Top panel, representative Western blot result from three independent experiments. Bottom panel, results of Western blot analysis quantified by densitometry. **, p < 0.01 versus control (dimethyl sulfoxide (DMSO) alone); ##, p < 0.01 versus PDGF/dimethyl sulfoxide group. C, the specific PI3K inhibitor wortmannin (0.75 μm) or LY294002 (LY) (50 μm) had no significant effect on Cyr61 expression. Top panel, representative Western blot result from three independent experiments. Bottom panel, results of the Western blot analysis quantified by densitometry. **, p < 0.01 versus control. D and E, pretreatment with U0126 or SP600125 dose-dependently blocked PDGF-BB-induced SMC migration. **, p < 0.01 versus control; ##, p < 0.01 versus the PDGF alone group. F, Western blot analysis of the effects of knockdown of Cyr61 on the activation of AKT and p38. Non, non-silencing siRNA.
We next examined the regulatory relationship between these kinases and Cyr61 expression. As shown in Fig. 2B, 10 μm U0126, a specific MEK1/2 inhibitor, nearly completely blocked PDGF-induced Cyr61 expression. The same effect was also observed when the specific inhibitor of JNK (SP600125) was admitted. However, neither the specific inhibitor of p38, SB203580, nor the specific inhibitors of PI3K (wortmannin and LY294002) had significant inhibitory effects on PDGF-induced Cyr61 expression (Fig. 2, B and C). These data indicate that activation of ERK and JNK are required for de novo Cyr61 synthesis, but neither p38 nor PI3K/AKT play a significant role in Cyr61 expression. We observed a dose-dependent inhibition of U0126 or SP600125 on PDGF-induced cell migration (Fig. 2, D and E). The level of inhibition of cell migration by either U0126 or SP600125 is consistent with the level of knockdown of Cyr61 expression (Fig. 1D), supporting the notion that the ERK and JNK pathways mediate Cyr61 expression, leading to cell migration.
PDGF-BB-induced Cyr61 Has No Feedback Effect on the Activation of AKT and p38
Although p38 and PI3K/AKT do not contribute to Cyr61 expression (Fig. 2, B and C), previous studies showed that both p38 and AKT contribute to SMC migration (10, 11). We were curious about whether newly synthesized Cyr61 has a feedback effect on the intracellular activation of p38 and AKT. The effect of knockdown of Cyr61 on the activation of p38 and AKT was determined. As shown in Fig. 2F, knockdown of Cyr61 expression has no detectable effect on the activation of p38 and AKT, indicating that PDGF-induced Cyr61 contributes to SMC migration via a p38- and AKT-independent pathway.
PDGF-BB-induced Cyr61 Protein Is Located in the ECM
To determine whether PDGF-BB-induced Cyr61 is located in the ECM, we assessed Cyr61 accumulation in ECM compared with that accumulated intracellularly. At various time points, cells stimulated with or without PDGF-BB were detached from the cell culture dishes with 1 mm EDTA. After removal of the cellular fraction, the ECM was collected by incubation in Laemmli sample buffer at 90 °C, followed by scraping of the dish as described previously (22). Western blot analysis from the paired groups of detached cell lysates (Fig. 3A) and ECM (Fig. 3B) indicated that the de novo synthesized Cyr61 was highly accumulated intracellularly at 1 h and disappeared after 4 h. Accompanying this dynamic intracellular process, Cyr61 started to accumulate in the ECM at 1 h, peaking around 1–2 h. Therefore, PDGF-BB-induced Cyr61 protein went through the intracellular pathway and was promptly secreted into the ECM compartment and bound to the matrix.
FIGURE 3.
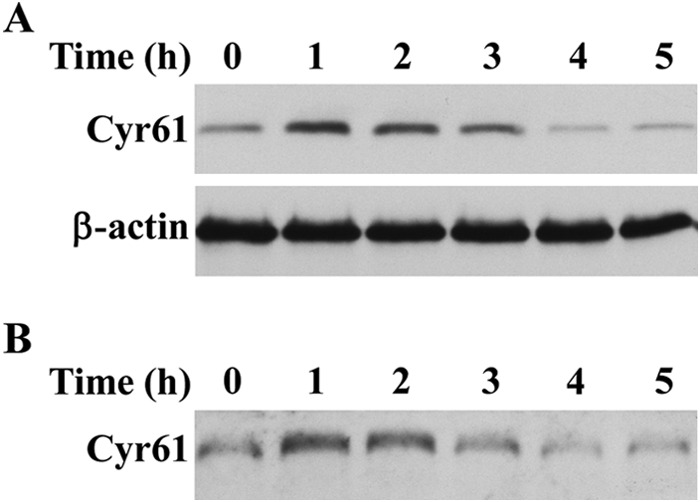
PDGF-BB-induced Cyr61 protein was secreted into the ECM. A, Western blot analysis of SMCs detached from culture dishes. Cells were detached from the dishes with 1 mm EDTA at various time points as indicated and lysed in the lysis buffer, followed by Western blot analysis. β-actin was used to assess protein loading. B, Western blot analysis of ECM proteins. ECM remaining on the culture dishes was extracted in Laemmli sample buffer (described under “Experimental Procedures”) and then subjected to Western blot analysis.
Cyr61 Mediates PDGF-induced FAK Activation, Which, in Turn, Contributes to SMC Migration
Intracellular FAK activation is important in cell migration because it regulates the dynamics of cell adhesion, actin polymerization, and cytoskeleton reorganization (25–27). Several sites of tyrosine phosphorylation have been identified in FAK to mediate FAK activity or FAK interaction with SH2 domain-containing proteins. The major autophosphorylation site Tyr-397 is essential for the majority of FAK functions (28). Phosphorylated Tyr-397, via other protein mediators, phosphorylates Tyr-576 and Tyr-577 in the activation loop of the FAK enzyme (29) and Tyr-861 at the C-terminal domain (30). We found that stimulation with PDGF induced biphasic activation of FAK. The first activation peak was weak and transient at 2 min of PDGF stimulation. A major late-phase FAK activation peaked at around 1–2 h (Fig. 4A). Strikingly, the late-phase FAK activation peak is concomitant with the extracellular matrix protein Cyr61 production peak. We hypothesized that Cyr61 mediates FAK activation in response to PDGF stimulation. To test our hypothesis, we examined whether depletion of Cyr61 using specific Cyr61 siRNA had an effect on FAK activation. As shown in Fig. 4B, down-regulation of Cyr61 effectively blocked the activation of FAK, indicating that PDGF-induced Cyr61 mediates FAK activation.
FIGURE 4.

The PDGF-BB-Cyr61 pathway, but not the p38 or Akt pathways, mediated PDGF-BB-induced FAK activation, which leads to SMC migration. A, time course of PDGF-BB-induced FAK activation. Quiescent SMCs were stimulated with PDGF-BB (10 ng/ml) for various times. Cell lysates were subjected to Western blot analysis using antibodies against phosphorylated FAK at specific sites. β-actin was used as the loading control. B, Western blotting data show that knockdown of Cyr61 expression with specific Cyr61 siRNA blocked PDGF-BB-induced FAK activation. Non, non-silencing siRNA. C, pretreatment with the specific FAK inhibitor PF573228 (PF) dose-dependently blocked PDGF-BB-induced SMC migration. **, p < 0.01 versus control; ##, p < 0.01 versus the PDGF alone group. D, knockdown of FAK expression with the specific FAK siRNA blocked PDGF-BB-induced SMC migration. FAK siRNA knockdown efficiency is shown (inset). **, p < 0.01 versus control; ##, p < 0.01 versus PDGF/non-silencing siRNA group. E, effects of knockdown of Cyr61, FAK, or both Cyr61 and FAK on PDGF-induced SMC migration. **, p < 0.01 versus control; ##, p < 0.01 versus PDGF/non-silencing siRNA group. F, pretreatment with U0126 (10 μm) or SP600125 (SP) (10 μm) blocked PDGF-BB-induced FAK activation (Western blot analysis). DMSO, dimethyl sulfoxide. G, neither the specific PI3K inhibitors wortmannin (0.75 μm) or LY294002 (LY) (50 μm) nor the specific p38 inhibitor SB203580 (SB) (10 μm) had any significant effect on PDGF-BB-induced FAK activation.
We next examined the functional role of FAK in PDGF-induced SMC migration. Pretreatment of SMCs with PF573228, the specific inhibitor of FAK, dose-dependently blocked PDGF-induced migration (Fig. 4C), suggesting that FAK is involved in PDGF-induced cell migration. To confirm the FAK role, we silenced FAK expression with the specific siRNA and found that knockdown of FAK expression blocked PDGF-induced SMC migration by 57% (Fig. 4D), indicating that FAK is a key regulatory molecule in the PDGF signaling pathway mediating SMC migration. To further confirm the upstream/downstream relationship of Cyr61 and FAK in PDGF-induced SMC migration, we examined whether knockdown of FAK has an additional influence on Cyr61 depletion-caused reduction of SMC migration. As shown in Fig. 4 E, knockdown of both Cyr61 and FAK blocked SMC migration to the same level as knockdown of either Cyr61 alone or FAK alone, indicating that the Cyr61-FAK axis contributes to PDGF-induced SMC migration. We next tested the role of specific upstream kinase(s) in FAK activation because our data revealed that MEK/ERK and JNK mediate PDGF-induced Cyr61 expression (Fig. 2) and that Cyr61 mediates FAK activation (Fig. 4B). As shown in Fig. 4F, the MEK/ERK-specific inhibitor U0126 and JNK specific inhibitor SP600125 completely blocked PDGF-induced FAK activation, whereas neither PI3K inhibitors (wortmannin and LY294002) nor the p38 MAPK inhibitor SB203580 had any effect on FAK activation (Fig. 4G). Taken together, these data indicate that the ERK- and JNK-regulated Cyr61 pathway mediates FAK activation, leading to cell migration.
The Integrins α6β1 and αvβ3 Interact Physically with Cyr61 and Are Functionally Involved in PDGF-induced Cell Migration
To pursue how the PDGF-induced matricellular protein Cyr61 transduces extracellular signals to the intracellular level to promote cell migration, we tested whether specific cell membrane receptor integrins are downstream regulators of Cyr61 and mediate extracellular Cyr61 signaling, leading to cell migration. It has been shown that Cyr61 is a ligand for certain integrins (12, 13, 31, 32). We screened the possible involvement of integrins by testing whether specific integrin antibodies can interfere with PDGF-induced cell migration and whether specific integrins interact with endogenous Cyr61. Integrins are a large family of cell transmembrane receptors. They are composed of integrin α (Itg α) and integrin β (Itg β) subunits, and each Itg αβ combination has its own signaling properties and binding specificity. A number of integrin subunits, such as αv, α4, α5, α6, β1, β3, β4, β5, α6β1, and ανβ3, are expressed in SMCs (33–35). We found that the specific antibodies against αv, α6, β1, β3, α6β1, and ανβ3 significantly blocked PDGF-BB-induced SMC migration, but the antibodies against α4, α5, β4, and β5 had no effect on PDGF-BB-induced SMC migration (Fig. 5A), suggesting that integrins α6β1 and ανβ3 are required for PDGF-BB-induced SMC migration. We next examined whether endogenously produced Cyr61 interacts with cell membrane integrins. Although it has been documented that the purified recombinant Cyr61 and its peptides interact with specific integrins (12–14), little is known about whether the endogenously produced Cyr61 in an intrinsic cellular microenvironment interacts with transmembrane integrins in physiological/pathological conditions. To address this question, we undertook two approaches: a protein cross-linking assay using DTSSP, a chemical reagent that has been used successfully to detect the interaction of transmembrane glycoproteins with the matrix protein collagen (36), and a coimmunoprecipitation assay in SMCs. As shown in Fig. 5B, when cultured SMCs were stimulated with PDGF-BB for 1 h without the cross-linking reagent DTSSP, we observed that Cyr61 (about 41 kD) was detected heavily in lane 2 compared with lane 1. Strikingly, we found that, with DTSSP cross-linking, Cyr61 was detected in two forms. One is the original 41-kD form, and the other is a complex (over 250 kD, Fig. 5B, lanes 3 and 4). Of note, in the complex, a huge amount of Cyr61 appeared in the PDGF-treated sample (compare Fig. 5B, lanes 4 and 3), indicating a strong binding capacity to other molecules because of the newly synthesized Cyr61. Similarly sized complexes were also detected when antibodies against Itg α6, β1, and β3 were used (Fig. 5B, lanes 7, 8, 11, 12, 15, and 16) but not detected when antibodies against Egr1 and ERK (negative controls) were used, suggesting a likelihood of binding of endogenous Cyr61 with the specific integrins α6β1 and ανβ3. Noticeably, when detected with an individual integrin antibody, the amount of complexes in both PDGF-induced and uninduced cross-linked samples was similar (Fig. 5B), indicating that dominant and consistently high levels of integrins are present in cell membranes.
FIGURE 5.

The Integrins α6β1 and αvβ3 interacted with endogenous Cyr61 and were involved in PDGF-BB-induced cell migration. A, the specific antibodies against integrins α6β1, αvβ3, α6, αv, β1, or β3, but not the antibodies against integrins α4, α5, β4, or β5, blocked PDGF-BB-induced SMC migration. IgG was used as a control. **, p < 0.01 versus control; ##, p < 0.01 versus the PDGF-BB/IgG group. B, cross-link results show that integrins α6β1 and αvβ3 interacted with endogenous Cyr61 in cultured SMCs (Western blot analysis). A complex of over 250 kD was detected by antibodies against Cyr61 and integrins α6, β1, and β3 after cross-linker DTSSP treatment (described under “Experimental Procedures”). ERK and Egr-1 were used as internal controls under the same conditions. IB, immunoblot. C, immunoprecipitation results show that endogenous Cyr61 interacted with integrins α6β1 and αvβ3. Left panel, Western blot results of Cyr61 overexpression after cells were infected with Ad-CMV-mCyr61 adenovirus. Center panel, Western blot analysis. Cells infected with Ad-CMV-mCyr61 were immunoprecipitated (IP) with Cyr61 or Itg α6 antibody or IgG and immunoblotted with Cyr61 antibody. Right panel, Western blot analysis. Cells infected with Ad-CMV-mCyr61 were immunoprecipitated with Cyr61 antibody, Itg αv antibody, or IgG antibody and immunoblotted with Itg αv antibody.
To further validate our finding that Cyr61 interacts with integrins α6β1 and ανβ3, we overexpressed Cyr61 in SMCs (Fig. 5C, left panel) and performed immunoprecipitation experiments. As shown in Fig. 5C, when immunoprecipitated with the specific integrin α6 antibody, Cyr61 was detected in the immunoblot (center panel), indicating a specific interaction between Cyr61 and integrin α6. When immunoprecipitated with Cyr61 antibody, integrin αν was detected in the immunoblot (Fig. 5C, right panel), indicating a specific interaction between Cyr61 and integrin αν. Taken together, these results support the conclusion that endogenously produced Cyr61 interacts with integrins α6β1 and ανβ3 and mediates cell migration.
Specific Integrins Are Integral Signaling Molecules in PDGF-induced FAK Activation and Cell Migration
The results from the above experiments indicate that the endogenous Cyr61 interacts with α6β1 and ανβ3. Finally we examined whether integrins α6, β1, and β3 have an influence on PDGF-induced FAK activation by determining whether knockdown of the expression of integrins α6, β1, and β3 blocks FAK activation and diminishes PDGF-induced cell migration. As shown in Fig. 6, A and B, knockdown of expression of α6, β1, and β3 with the specific siRNA completely blocked FAK activation, indicating that these integrins mediate FAK activation in the PDGF pathway. Knockdown of expression of α6, β1, and β3 also largely blocked SMC migration (Fig. 6C). These data, together with the results presented in Fig. 5, support the conclusion that matricellular Cyr61 interacts with integrins α6β1 and ανβ3 to transduce PDGF signaling to activate FAK, leading to cell migration. Therefore, this study provides the first evidence of the MAPK-Cyr61-integrin-FAK axis in the PDGF pathway and of mediation of cell migration via this novel pathway.
FIGURE 6.
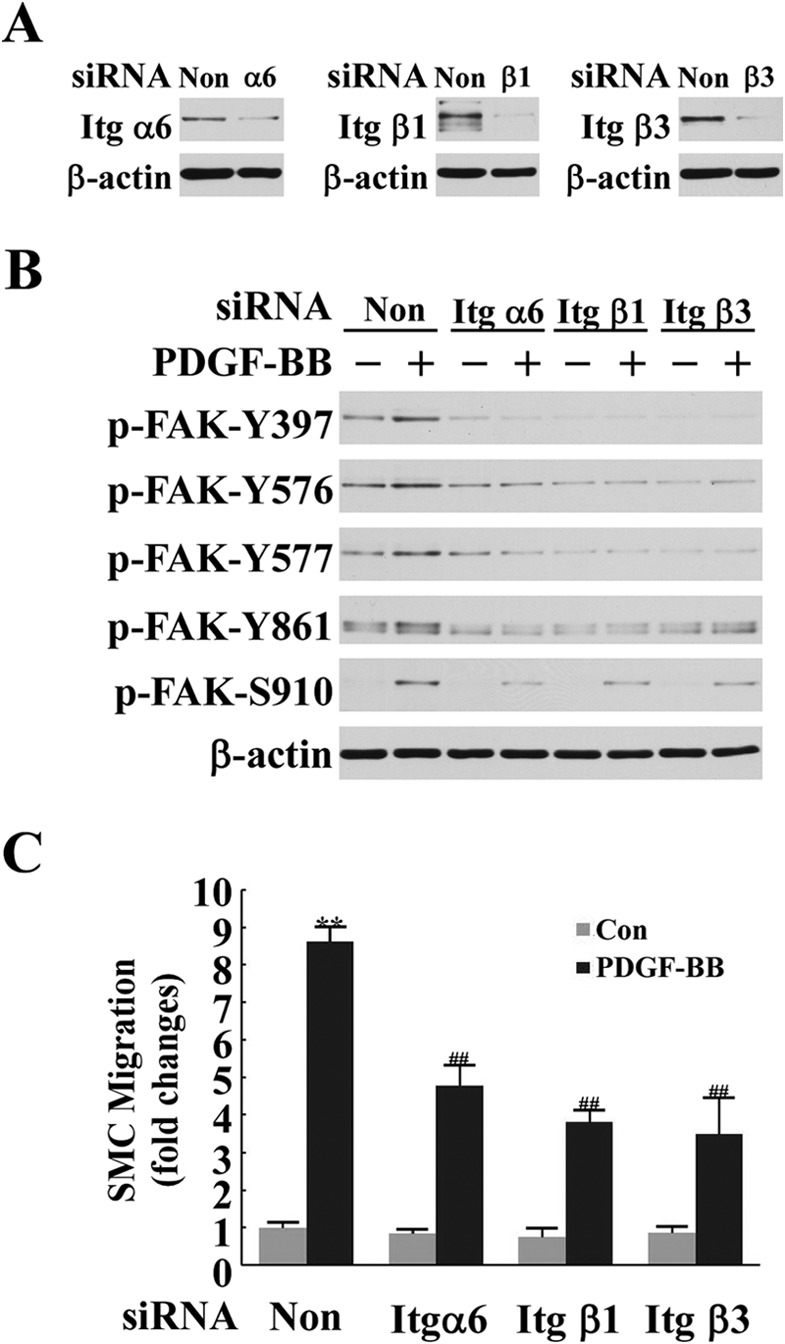
Integrins α6β1 and αvβ3 mediated PDGF-BB-induced FAK activation and SMC migration. A, knockdown efficiency of integrins α6, β1, and β3 with specific integrin siRNAs was assessed in a Western blot analysis. Non, non-silencing siRNA. B, knockdown of expression of integrins α6, β1, and β3 with specific integrin siRNAs blocked PDGF-BB-induced FAK activation (1 h). C, knockdown of integrin α6, β1, and β3 expression with specific integrin siRNAs significantly inhibited PDGF-BB-induced SMC migration. **, p < 0.01 versus control (Con); ##, p < 0.01 versus the PDGF/non-silencing RNA group.
DISCUSSION
The major discoveries of the current study are that the matricellular protein Cyr61 is a key extracellular mediator of PDGF-induced cell migration, that endogenous Cyr61 interacts with specific integrins, and that Cyr61 bridges a PDGF-triggered intracellular signaling pathway with an integrin/FAK signaling pathway, leading to cell migration. These new observations point to a novel pathway of growth factor-mediated cell migration. The major findings are summarized and illustrated in Fig. 7.
FIGURE 7.
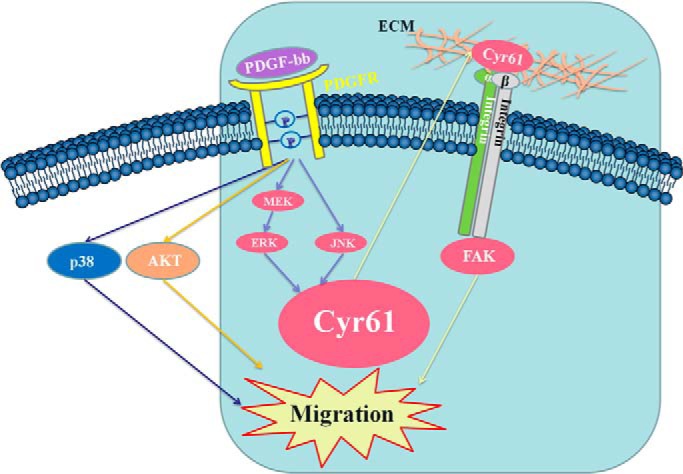
Summary illustration. De novo synthesized matricellular protein Cyr61 was up-regulated by the PDGF-BB-activated ERK and JNK pathways. Cyr61 bridges the PDGF-BB-triggered signaling pathway and the integrin signaling pathway, leading to SMC migration.
The identification of the matricellular protein Cyr61 as a key extracellular mediator of PDGF-induced cell migration also addressed the question of the downstream mediator of ERK and JNK pathways, which have been demonstrated previously to regulate PDGF-induced cell migration (9, 11). Our data indicate that the ERK and JNK pathways lead to the production of the immediate early gene product Cyr61 in the extracellular matrix. These results also indicate that the ERK- and JNK-mediated matricellular protein Cyr61-integrin-FAK pathway contributes to over 60% of PDGF-induced cell migration and that this pathway is independent of the activation of p38 and PI3K, which partially contributes to PDGF-induced SMC migration.
PDGF has been considered to be the top culprit for injury-induced vascular neointimal formation (1, 7, 37, 38) and to be a prominent factor for the development of atherosclerosis (6, 39, 40). PDGF also plays a major role in tumorigenesis and metastasis (2, 41). The identification of matrix protein Cyr61 as an extracellular molecule to bridge the immediate intracellular signaling pathway of PDGF and the cell membrane integrin pathway leading to cell function-migration may provide important therapeutic targets. This perspective is supported by recent findings that Cyr61 mediates vascular lesion formation and tumor development (42–44).
Serum and PDGF induction of Cyr61 expression in fibroblasts was originally reported by Lau and co-workers (45). However, the role of Cyr61 in mediation of PDGF-induced cell migration was not revealed. Our data demonstrate that knockdown of de novo Cyr61 expression blocked the majority of PDGF-induced SMC migration but had no effect on PDGF-induced cell proliferation, indicating a new and specific role of the newly synthesized Cyr61 in the PDGF pathway. We further revealed that the de novo synthesized Cyr61 is promptly translocated to the extracellular matrix, where Cyr61 interacts with the specific integrins α6β1 and ανβ3. Although the cysteine-rich Cyr61 does not have RGD sequences, purified recombinant Cyr61 has been demonstrated to bind to various integrins to mediate diverse cellular functions: adhesion, proliferation, migration, survival, and apoptosis (12–17). We sought to address whether endogenously produced Cyr61, under conditions similar to physiological and pathological conditions, could interact with cell membrane-specific integrins. The two complementary approaches used in this study demonstrated the physical interaction between the endogenously produced Cyr61 and specific integrins in a cellular/extracellular microenvironment, which is closer to physiological/pathological conditions. However, the details of exact components and the nature of the giant complex (over 250 kD) warrant further investigation.
The role of integrin in PDGF-induced cell migration, especially SMC migration, has been demonstrated in the last two decades. Studies have indicated that αVβ3, β1, and αVβ5 mediate PDGF-induced SMC and fibroblast migration (46–50) in various contexts. However, how these integrins interact with their ligands in the extracellular matrix to mediate PDGF signaling has been elusive. Our findings showing that the newly synthesized matricellular protein Cyr61 mediates PDGF signaling and interacts with integrins α6β1 and αVβ3 provide a novel concept for the PDGF signaling pathway. Therefore, Cyr61 seems to be the long-missing, important extracellular mediator in the PDGF pathway. Our recent study has demonstrated that Cyr61 is a key mediator for lysophosphatidic acid-induced cell migration (51). One previous article showed that Cyr61 mediates thrombin-induced cell proliferation (52). It is our perspective that the interaction of de novo Cyr61 with various cell membrane integrins and other effectors is a key mechanism for an array of growth factor-mediated cell migration- and invasion-related diseases.
FAK activation is important in cell migration because of its regulation of the dynamics of cell adhesion, actin polymerization, and cytoskeleton reorganization (27–29). One interesting observation is the biphasic peaks of FAK activation induced by PDGF: one weak, transient peak at around 2 min and another major peak at 60–120 min. The early weak activation of FAK (at 2 min) is apparently irrelevant to protein synthesis. However, the latter major activation peak was completely blocked by either Cyr61 siRNA silencing of the newly synthesized Cyr61 or by integrin α6, β1, or β3 siRNA silencing. These results present the first demonstration that a Cyr61-integrin-FAK axis in the PDGF pathway mediates cell function. Previous studies have reported that PDGF activates FAK in various cell types (53–55), and there are also studies demonstrating that integrin mediates FAK activation in the PDGF pathway (56, 57). The newly identified Cyr61 in the extracellular matrix may be the long sought after key mediator converging the immediate PDGF intracellular signal to integrin and FAK, leading to cell function migration.
In summary, our study presents the first evidence that the de novo synthesized Cyr61 is the key mediator required for PDGF-induced cell migration. Our data reveal that the ERK and JNK pathways control Cyr61 synthesis and that the interaction of Cyr61 with integrins α6β1 and ανβ3 mediates intracellular FAK activation, leading to PDGF-induced cell migration. The identified Cyr61-integrin-FAK axis may represent the new concept that the matricellular protein Cyr61 is a key extracellular mediator for growth factor-induced cell migration via a mechanism involving interaction with specific cell membrane receptor integrins and activation of intracellular FAK. Cyr61 and the effectors in this pathway may be markers/mediators in many migration-related vascular diseases and cancers.
Acknowledgment
We thank Misty Bailey for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL107466 (to M. Z. C.) and AG026640 (to X. X.).
- FAK
- focal adhesion kinase
- SMC
- smooth muscle cell
- ECM
- extracellular matrix
- DTSSP
- 3,3′-dithiobis(sulfosuccinimidylpropionate)
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid
- Itg
- integrin.
REFERENCES
- 1. Raines E. W. (2004) PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 15, 237–254 [DOI] [PubMed] [Google Scholar]
- 2. Heldin C. H. (2013) Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 11, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leppänen O., Janjic N., Carlsson M. A., Pietras K., Levin M., Vargeese C., Green L. S., Bergqvist D., Ostman A., Heldin C. H. (2000) Intimal hyperplasia recurs after removal of PDGF-AB and -BB inhibition in the rat carotid artery injury model. Arterioscler. Thromb. Vasc. Biol. 20, E89–E95 [DOI] [PubMed] [Google Scholar]
- 4. Noiseux N., Boucher C. H., Cartier R., Sirois M. G. (2000) Bolus endovascular PDGFR-β antisense treatment suppressed intimal hyperplasia in a rat carotid injury model. Circulation 102, 1330–1336 [DOI] [PubMed] [Google Scholar]
- 5. Sirois M. G., Simons M., Edelman E. R. (1997) Antisense oligonucleotide inhibition of PDGFR-β receptor subunit expression directs suppression of intimal thickening. Circulation 95, 669–676 [DOI] [PubMed] [Google Scholar]
- 6. Sano H., Sudo T., Yokode M., Murayama T., Kataoka H., Takakura N., Nishikawa S., Nishikawa S. I., Kita T. (2001) Functional blockade of platelet-derived growth factor receptor-β but not of receptor-α prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation 103, 2955–2960 [DOI] [PubMed] [Google Scholar]
- 7. Jawien A., Bowen-Pope D. F., Lindner V., Schwartz S. M., Clowes A. W. (1992) Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J. Clin. Invest. 89, 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buetow B. S., Tappan K. A., Crosby J. R., Seifert R. A., Bowen-Pope D. F. (2003) Chimera analysis supports a predominant role of PDGFRβ in promoting smooth-muscle cell chemotaxis after arterial injury. Am. J. Pathol. 163, 979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Graf K., Xi X. P., Yang D., Fleck E., Hsueh W. A., Law R. E. (1997) Mitogen-activated protein kinase activation is involved in platelet-derived growth factor-directed migration by vascular smooth muscle cells. Hypertension 29, 334–339 [DOI] [PubMed] [Google Scholar]
- 10. Choudhury G. G., Karamitsos C., Hernandez J., Gentilini A., Bardgette J., Abboud H. E. (1997) PI-3-kinase and MAPK regulate mesangial cell proliferation and migration in response to PDGF. Am. J. Physiol. 273, F931–F938 [DOI] [PubMed] [Google Scholar]
- 11. Zhan Y., Kim S., Izumi Y., Izumiya Y., Nakao T., Miyazaki H., Iwao H. (2003) Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler. Thromb. Vasc. Biol. 23, 795–801 [DOI] [PubMed] [Google Scholar]
- 12. Kireeva M. L., Lam S. C., Lau L. F. (1998) Adhesion of human umbilical vein endothelial cells to the immediate-early gene product Cyr61 is mediated through integrin αvβ3. J. Biol. Chem. 273, 3090–3096 [DOI] [PubMed] [Google Scholar]
- 13. Chen N., Chen C. C., Lau L. F. (2000) Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin α6β1 and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 275, 24953–24961 [DOI] [PubMed] [Google Scholar]
- 14. Grzeszkiewicz T. M., Kirschling D. J., Chen N., Lau L. F. (2001) CYR61 stimulates human skin fibroblast migration through Integrin α vβ 5 and enhances mitogenesis through integrin α vβ 3, independent of its carboxyl-terminal domain. J. Biol. Chem. 276, 21943–21950 [DOI] [PubMed] [Google Scholar]
- 15. Grzeszkiewicz T. M., Lindner V., Chen N., Lam S. C., Lau L. F. (2002) The angiogenic factor cysteine-rich 61 (CYR61, CCN1) supports vascular smooth muscle cell adhesion and stimulates chemotaxis through integrin α(6)β(1) and cell surface heparan sulfate proteoglycans. Endocrinology 143, 1441–1450 [DOI] [PubMed] [Google Scholar]
- 16. Leu S. J., Lam S. C., Lau L. F. (2002) Pro-angiogenic activities of CYR61 (CCN1) mediated through integrins αvβ3 and 6β1 in human umbilical vein endothelial cells. J. Biol. Chem. 277, 46248–46255 [DOI] [PubMed] [Google Scholar]
- 17. Chen C. C., Young J. L., Monzon R. I., Chen N., Todorović V., Lau L. F. (2007) Cytotoxicity of TNFα is regulated by integrin-mediated matrix signaling. EMBO J. 26, 1257–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee H. Y., Chung J. W., Youn S. W., Kim J. Y., Park K. W., Koo B. K., Oh B. H., Park Y. B., Chaqour B., Walsh K., Kim H. S. (2007) Forkhead transcription factor FOXO3a is a negative regulator of angiogenic immediate early gene CYR61, leading to inhibition of vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 100, 372–380 [DOI] [PubMed] [Google Scholar]
- 19. Xie D., Yin D., Tong X., O'Kelly J., Mori A., Miller C., Black K., Gui D., Said J. W., Koeffler H. P. (2004) Cyr61 is overexpressed in gliomas and involved in integrin-linked kinase-mediated Akt and β-catenin-TCF/Lef signaling pathways. Cancer Res. 64, 1987–1996 [DOI] [PubMed] [Google Scholar]
- 20. Bleau A. M., Planque N., Perbal B. (2005) CCN proteins and cancer: two to tango. Front. Biosci. 10, 998–1009 [DOI] [PubMed] [Google Scholar]
- 21. Brock T. A., Alexander R. W., Ekstein L. S., Atkinson W. J., Gimbrone M. A., Jr. (1985) Angiotensin increases cytosolic free calcium in cultured vascular smooth muscle cells. Hypertension 7, I105–109 [DOI] [PubMed] [Google Scholar]
- 22. Bradley R. S., Brown A. M. (1990) The proto-oncogene int-1 encodes a secreted protein associated with the extracellular matrix. EMBO J. 9, 1569–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. García A. J., Vega M. D., Boettiger D. (1999) Modulation of cell proliferation and differentiation through substrate-dependent changes in fibronectin conformation. Mol. Biol. Cell 10, 785–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chai Y. C., Howe P. H., DiCorleto P. E., Chisolm G. M. (1996) Oxidized low density lipoprotein and lysophosphatidylcholine stimulate cell cycle entry in vascular smooth muscle cells. Evidence for release of fibroblast growth factor-2. J. Biol. Chem. 271, 17791–17797 [DOI] [PubMed] [Google Scholar]
- 25. Tomar A., Schlaepfer D. D. (2009) Focal adhesion kinase: switching between GAPs and GEFs in the regulation of cell motility. Curr. Opin. Cell Biol. 21, 676–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schaller M. D. (2010) Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J. Cell Sci. 123, 1007–1013 [DOI] [PubMed] [Google Scholar]
- 27. Zhao X., Guan J. L. (2011) Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Delivery Rev. 63, 610–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schaller M. D., Hildebrand J. D., Shannon J. D., Fox J. W., Vines R. R., Parsons J. T. (1994) Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 14, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Calalb M. B., Polte T. R., Hanks S. K. (1995) Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol. Cell. Biol. 15, 954–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Calalb M. B., Zhang X., Polte T. R., Hanks S. K. (1996) Focal adhesion kinase tyrosine-861 is a major site of phosphorylation by Src. Biochem. Biophys. Res. Commun. 228, 662–668 [DOI] [PubMed] [Google Scholar]
- 31. Jedsadayanmata A., Chen C. C., Kireeva M. L., Lau L. F., Lam S. C. (1999) Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/mouse connective tissue growth factor is mediated through integrin α(IIb)β(3). J. Biol. Chem. 274, 24321–24327 [DOI] [PubMed] [Google Scholar]
- 32. Chen C. C., Chen N., Lau L. F. (2001) The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J. Biol. Chem. 276, 10443–10452 [DOI] [PubMed] [Google Scholar]
- 33. Witzenbichler B., Kureishi Y., Luo Z., Le Roux A., Branellec D., Walsh K. (1999) Regulation of smooth muscle cell migration and integrin expression by the Gax transcription factor. J. Clin. Invest. 104, 1469–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marek I., Volkert G., Jahn A., Fahlbusch F., Zürn C., Ozcan Z., Goppelt-Struebe M., Hilgers K. F., Rascher W., Hartner A. (2010) Lack of α8 integrin leads to morphological changes in renal mesangial cells, but not in vascular smooth muscle cells. BMC Cell Biol. 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moiseeva E. P. (2001) Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc. Res. 52, 372–386 [DOI] [PubMed] [Google Scholar]
- 36. Kotite N. J., Staros J. V., Cunningham L. W. (1984) Interaction of specific platelet membrane proteins with collagen: evidence from chemical cross-linking. Biochemistry 23, 3099–3104 [DOI] [PubMed] [Google Scholar]
- 37. Ferns G. A., Raines E. W., Sprugel K. H., Motani A. S., Reidy M. A., Ross R. (1991) Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science 253, 1129–1132 [DOI] [PubMed] [Google Scholar]
- 38. Jackson C. L., Raines E. W., Ross R., Reidy M. A. (1993) Role of endogenous platelet-derived growth factor in arterial smooth muscle cell migration after balloon catheter injury. Arterioscler. Thromb. 13, 1218–1226 [DOI] [PubMed] [Google Scholar]
- 39. Evanko S. P., Raines E. W., Ross R., Gold L. I., Wight T. N. (1998) Proteoglycan distribution in lesions of atherosclerosis depends on lesion severity, structural characteristics, and the proximity of platelet-derived growth factor and transforming growth factor-β. Am. J. Pathol. 152, 533–546 [PMC free article] [PubMed] [Google Scholar]
- 40. Changsirikulchai S., Hudkins K. L., Goodpaster T. A., Volpone J., Topouzis S., Gilbertson D. G., Alpers C. E. (2002) Platelet-derived growth factor-D expression in developing and mature human kidneys. Kidney Int. 62, 2043–2054 [DOI] [PubMed] [Google Scholar]
- 41. Cao Y. (2013) Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol. Med. 19, 460–473 [DOI] [PubMed] [Google Scholar]
- 42. Hilfiker A., Hilfiker-Kleiner D., Fuchs M., Kaminski K., Lichtenberg A., Rothkötter H. J., Schieffer B., Drexler H. (2002) Expression of CYR61, an angiogenic immediate early gene, in arteriosclerosis and its regulation by angiotensin II. Circulation 106, 254–260 [DOI] [PubMed] [Google Scholar]
- 43. O'Kelly J., Chung A., Lemp N., Chumakova K., Yin D., Wang H. J., Said J., Gui D., Miller C. W., Karlan B. Y., Koeffler H. P. (2008) Functional domains of CCN1 (Cyr61) regulate breast cancer progression. Int. J. Oncol. 33, 59–67 [PubMed] [Google Scholar]
- 44. Babic A. M., Kireeva M. L., Kolesnikova T. V., Lau L. F. (1998) CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. U.S.A. 95, 6355–6360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Brien T. P., Yang G. P., Sanders L., Lau L. F. (1990) Expression of cyr61, a growth factor-inducible immediate-early gene. Mol. Cell. Biol. 10, 3569–3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Choi E. T., Engel L., Callow A. D., Sun S., Trachtenberg J., Santoro S., Ryan U. S. (1994) Inhibition of neointimal hyperplasia by blocking α V β 3 integrin with a small peptide antagonist GpenGRGDSPCA. J. Vasc. Surg. 19, 125–134 [DOI] [PubMed] [Google Scholar]
- 47. Seki J., Koyama N., Kovach N. L., Yednock T., Clowes A. W., Harlan J. M. (1996) Regulation of β1-integrin function in cultured human vascular smooth muscle cells. Circ. Res. 78, 596–605 [DOI] [PubMed] [Google Scholar]
- 48. Bilato C., Curto K. A., Monticone R. E., Pauly R. R., White A. J., Crow M. T. (1997) The inhibition of vascular smooth muscle cell migration by peptide and antibody antagonists of the αvβ3 integrin complex is reversed by activated calcium/calmodulin- dependent protein kinase II. J. Clin. Invest. 100, 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kappert K., Blaschke F., Meehan W. P., Kawano H., Grill M., Fleck E., Hsueh W. A., Law R. E., Graf K. (2001) Integrins αvβ3 and αvβ5 mediate VSMC migration and are elevated during neointima formation in the rat aorta. Basic Res. Cardiol. 96, 42–49 [DOI] [PubMed] [Google Scholar]
- 50. Kirchberg K., Lange T. S., Klein E. C., Jungtäubl H., Heinen G., Meyer-Ingold W., Scharffetter-Kochanek K. (1995) Induction of β 1 integrin synthesis by recombinant platelet-derived growth factor (PDGF-AB) correlates with an enhanced migratory response of human dermal fibroblasts to various extracellular matrix proteins. Exp. Cell Res. 220, 29–35 [DOI] [PubMed] [Google Scholar]
- 51. Wu D. D., Zhang F., Hao F., Chun J., Xu X., Cui M. Z. (2014) Matricellular protein Cyr61 bridges lysophosphatidic acid and integrin pathways leading to cell migration. J. Biol. Chem. 289, 5774–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Walsh C. T., Radeff-Huang J., Matteo R., Hsiao A., Subramaniam S., Stupack D., Brown J. H. (2008) Thrombin receptor and RhoA mediate cell proliferation through integrins and cysteine-rich protein 61. FASEB J. 22, 4011–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rankin S., Rozengurt E. (1994) Platelet-derived growth factor modulation of focal adhesion kinase (p125FAK) and paxillin tyrosine phosphorylation in Swiss 3T3 cells. Bell-shaped dose response and cross-talk with bombesin. J. Biol. Chem. 269, 704–710 [PubMed] [Google Scholar]
- 54. Chen H. C., Guan J. L. (1994) Stimulation of phosphatidylinositol 3′-kinase association with focal adhesion kinase by platelet-derived growth factor. J. Biol. Chem. 269, 31229–31233 [PubMed] [Google Scholar]
- 55. Abedi H., Dawes K. E., Zachary I. (1995) Differential effects of platelet-derived growth factor BB on p125 focal adhesion kinase and paxillin tyrosine phosphorylation and on cell migration in rabbit aortic vascular smooth muscle cells and Swiss 3T3 fibroblasts. J. Biol. Chem. 270, 11367–11376 [DOI] [PubMed] [Google Scholar]
- 56. Grundström G., Mosher D. F., Sakai T., Rubin K. (2003) Integrin αvβ3 mediates platelet-derived growth factor-BB-stimulated collagen gel contraction in cells expressing signaling deficient integrin α2β1. Exp. Cell Res. 291, 463–473 [DOI] [PubMed] [Google Scholar]
- 57. Varadarajulu J., Laser M., Hupp M., Wu R., Hauck C. R. (2005) Targeting of α(v) integrins interferes with FAK activation and smooth muscle cell migration and invasion. Biochem. Biophys. Res. Commun. 331, 404–412 [DOI] [PubMed] [Google Scholar]