

Background: A Collybistin R290H mutation causes epilepsy and intellectual disability in humans.
Results: Collybistin R290H is defective in PI3P binding and Gephyrin clustering activity during neuronal synaptogenesis.
Conclusion: Deficient lipid binding by Collybistin R290H is a likely pathomechanism in epilepsy and intellectual disability.
Significance: The study describes a new mechanism of Collybistin dysfunction during synapse formation that likely causes epilepsy and mental retardation.
Keywords: Epilepsy, GABA Receptor, Genetic Disease, Guanine Nucleotide Exchange Factor (GEF), Synapse, Inhibitory Synapse, Mental Retardation
Abstract
Signaling at nerve cell synapses is a key determinant of proper brain function, and synaptic defects—or synaptopathies—are at the basis of many neurological and psychiatric disorders. In key areas of the mammalian brain, such as the hippocampus or the basolateral amygdala, the clustering of the scaffolding protein Gephyrin and of γ-aminobutyric acid type A receptors at inhibitory neuronal synapses is critically dependent upon the brain-specific guanine nucleotide exchange factor Collybistin (Cb). Accordingly, it was discovered recently that an R290H missense mutation in the diffuse B-cell lymphoma homology domain of Cb, which carries the guanine nucleotide exchange factor activity, leads to epilepsy and intellectual disability in human patients. In the present study, we determined the mechanism by which the CbR290H mutation perturbs inhibitory synapse formation and causes brain dysfunction. Based on a combination of biochemical, cell biological, and molecular dynamics simulation approaches, we demonstrate that the R290H mutation alters the strength of intramolecular interactions between the diffuse B-cell lymphoma homology domain and the pleckstrin homology domain of Cb. This defect reduces the phosphatidylinositol 3-phosphate binding affinity of Cb, which limits its normal synaptogenic activity. Our data indicate that impairment of the membrane lipid binding activity of Cb and a consequent defect in inhibitory synapse maturation represent a likely molecular pathomechanism of epilepsy and mental retardation in humans.
Introduction
Efficient signal transfer at chemical synapses between nerve cells is a key determinant of all brain functions. Correspondingly, defects in synapse formation or function, which are often referred to as synaptopathies, are at the basis of numerous neurological and psychiatric disorders. In view of this paramount importance of synapses for brain function and dysfunction, a detailed understanding of the molecular mechanisms of synapse formation and function is essential for basic neuroscience and translational neuroscience alike and must be considered a prerequisite for the development of diagnoses and treatment strategies for synaptopathies.
Synaptic inhibition in the brain is essential for the regulation of neuronal excitability, for the maintenance of the excitation/inhibition balance in neuronal circuits, and for the determination of the input-output relationship in nerve cells. Fast synaptic inhibition in the brain is mostly mediated by GABA acting upon GABAA receptors (GABAARs),2 which are ligand-gated chloride channels that play a key role in the control of neuronal and network function in the brain (1). Correspondingly, defects at inhibitory GABAergic synapses, e.g. caused by mutations of their core protein components, cause very diverse brain diseases, such as epilepsy, mental retardation, autism spectrum disorders, or schizophrenia (2–4).
Core components of many inhibitory GABAergic postsynapses are the cell adhesion protein Neuroligin-2 (NL2), the scaffolding protein Gephyrin, the guanine nucleotide exchange factor (GEF) Collybistin (Cb), and GABAARs (5). Cb is required for the postsynaptic clustering of Gephyrin and GABAARs in various regions of the mammalian forebrain, including the hippocampus and the basolateral amydgala (6). Accordingly, Cb KO mice exhibit a region-specific loss of Gephyrin and Gephyrin-dependent GABAARs at inhibitory synapses and a consequent increase in anxiety scores, impaired spatial learning, and generalized tonic-clonic convulsions (5, 6).
In humans, several mutations of the Cb gene (ARHGEF9; OMIM 300429) have been linked to epilepsy, X-linked mental retardation, aggressive behavior, anxiety, and hyperekplexia (7–13). The corresponding patients show microdeletions (10, 11), nonsense mutations (9, 11), chromosomal disruptions (8), or missense mutations (7, 12, 13) in the ARHGEF9 gene. Common clinical features of all these ARHGEF9 mutations are epileptic seizures and intellectual disability, whereas other symptoms appear to be dependent upon the different mutation types.
The clarification of the pathomechanisms that cause epilepsy and intellectual disability in human patients with Cb mutations requires a detailed understanding of the organization and interactions of protein domains in Cb. Most Cb splice variants that are detectable in vivo contain an N-terminal Src homology 3 (SH3) domain, followed by a diffuse B-cell lymphoma (Dbl) homology (DH) domain and a C-terminal pleckstrin homology (PH) domain (14). The DH domain of Cb acts as a GEF that activates the Rho-like GTPases Cdc42 and TC10 in vivo, albeit with very low efficacy (15, 16). In accord with its low GEF activity, GEF-deficient Cb mutants have an almost normal Gephyrin and GABAAR clustering activity, which indicates that the catalytic activity of the DH domain of Cb is not required for the Cb-dependent maturation of inhibitory synapses (17).
In contrast, loss of function mutation of the PH domain residues that are essential for the binding of Cb to phosphatidylinositol 3-phosphate (PI3P), the major phosphoinositide ligand of the Cb PH domain (9, 14), abolishes the Gephyrin and GABAAR clustering activity of Cb (17). In addition to phosphoinositide binding, the PH domain of Cb interacts with GTP-TC10, and likely also with GTP-Cdc42, in an effector-like manner, which enhances Cb-mediated Gephyrin clustering and GABAergic synaptic transmission in cultured neurons (16).
The N-terminal SH3 domain of Cb, which is present in most Cb splice variants detectable in vivo (7, 14, 18), mediates intramolecular interactions with the DH/PH tandem domain and thereby inhibits the ability of Cb to bind PI3P and to cluster Gephyin at nascent synapses (14). This intramolecular interaction and the consequent autoinhibition are absent in Cb splice variants lacking the SH3 domain (e.g. ΔSH3CbII), and they can be functionally relieved by NL2 binding to the Cb SH3 domain, which triggers the maturation of inhibitory postsynapses at NL2-containing nascent synaptic contacts (7, 14, 17, 19). In essence, depending on whether SH3 domain-containing Cb isoforms interact with NL2 or not, they can adopt an open/active conformation, like ΔSH3Cb, or a closed/inactive conformation and thus act as switchable adaptors that bind PI3P and cluster Gephyrin at inhibitory postsynaptic sites (14).
In the present study, we characterized an R290H missense mutation in Cb, which was discovered in three adult brothers suffering from epileptic seizures and intellectual disability (12). Based on the very high amino acid sequence identity between human and rat Cb and on the available high resolution crystal structure of the constitutively active ΔSH3CbII splice variant, which lacks the autoinhibitory SH3 domain, we used site-directed mutagenesis to substitute the corresponding arginine residue by a histidine in the sequences of two rat Cb splice variants, i.e. ΔSH3CbII and an autoinhibited full-length Cb variant carrying an N-terminal SH3 domain (SH3(+)CbII). Our biochemical and cell biological analyses show that the Arg → His (R/H) mutation leads to a significant impairment of Cb-dependent Gephyrin clustering in COS7 cells and cultured rat hippocampal neurons, as compared with neurons expressing WT CbII isoforms. This impairment of the mutant Cb proteins to efficiently cluster Gephyrin can be attributed to a PI3P binding defect caused by the R/H mutation. Based on complementary molecular dynamics (MD) simulations of the WT and R/H mutant Cb variants, we show further that the R/H mutation affects the strength of intermolecular interactions between the DH and PH domains of Cb, which leads to a conformation of the PH domain relative to the DH domain that significantly perturbs the interaction of Cb with PI3P.
EXPERIMENTAL PROCEDURES
Antibodies
The following primary antibodies were used for immunocytochemistry: monoclonal mouse anti-Gephyrin (mAb7a, 1:3,000; Connex, Martinsried, Germany), polyclonal rabbit anti-HA (1:2,000; Zymed Laboratories Inc., Invitrogen), polyclonal anti-c-Myc (C3956), and monoclonal mouse anti-c-Myc clone 9E10 (1:1,000; Sigma-Aldrich). The following secondary antibodies were used for immunocytochemistry: Alexa Fluor 488, 555, or 633 goat anti-mouse or goat anti-rabbit IgG (1:2,000; Invitrogen). The following primary antibodies were used for Western blotting: monoclonal mouse anti-Gephyrin (3B11, 1:3,000; Synaptic Systems, Göttingen, Germany), rabbit anti-TC10 T8950 (Sigma-Aldrich; 1:4,000), rat monoclonal anti-HA conjugated with peroxidase (Roche; 1:10,000), and polyclonal anti-c-Myc (C3956, 1:2,000; Sigma-Aldrich). The following secondary antibodies were used for Western blotting: HRP-conjugated AffiniPure goat anti-mouse or goat anti-rabbit IgGs (Jackson ImmunoResearch Laboratories, Inc., Hamburg, Germany, 1:10,000). For protein-lipid overlay assays, a goat anti-GST-HRP conjugate (1:10,000; GE Healthcare) was used.
cDNA Constructs
The pEGFP-C2-Gephyrin plasmid (20) and the N-terminally Myc-tagged CbII constructs (7) were described previously. The HA-tagged NL2 construct was described previously (14). The Myc-tagged SH3(+)- or ΔSH3CbII R/H mutants were generated by oligonucleotide-directed mutagenesis using the QuikChange mutagenesis kit (Stratagene). The N-terminally HA-tagged TC10 constructs (WT or constitutively active (CA)) were a generous gift from Dr. Jeffrey E. Pessin (New York, NY). His-TC10 in pRSET-A was kindly provided by Dr. Perihan Nalbant (Cardiff, UK). GST-tagged SH3(+)- and ΔSH3CbII were created by cloning the CbII cDNAs (18) into the EcoRI/XhoI sites of the pGEX-4T-1 vector (GE Healthcare). The GST-ΔSH3CbII R/H cDNA was generated in pGEX-4T-1 by oligonucleotide-directed mutagenesis using the QuikChange mutagenesis kit (Stratagene). All constructs used in the present study were confirmed by sequencing.
Purification of GST- and His6-tagged Proteins
GST-tagged and His6-tagged proteins were expressed and purified as described previously (16).
In Vivo GTPase Activation Assay
Glutathione-Sepharose-immobilized GST-PAK1 PBD, a GST fusion protein containing the p21-binding domain (PBD, residues 67–150) of human PAK-1, was purchased from Cytoskeleton (PAK02; Tebu-bio, Frankfurt, Germany). COS7 cells were grown and transfected as described previously (16). At 24 h after transfection, cells were washed twice with ice-cold PBS and lysed on the dish for 20 min by adding 0.8 ml of lysis buffer (25 mm HEPES, pH 7.5, 150 mm NaCl, 1% (w/v) Igepal CA-630, 2% (v/v) glycerol, 25 mm NaF, 10 mm MgCl2, 1 mm EDTA, 1 mm sodium orthovanadate, 10 μg/ml leupeptin, 10 μg/ml aprotinin). After centrifugation at 14,000 × g for 10 min at 4 °C, the supernatants of the lysates were incubated at 4 °C for 1 h with 15 μg of GST-PAK1 PBD under constant agitation. The beads were washed four times with lysis buffer and eluted directly with SDS-PAGE sample buffer. The input and bead-coupled fractions were separated on a 12% polyacrylamide gel. The amounts of total and bound recombinant HA-TC10 were detected by Western blotting using an antibody against the HA epitope tag. Western blotting was performed as described previously (6).
GST Pulldown Assays with Tissue Extracts
Adult (8 weeks old) mice were anesthetized by isoflurane and decapitated according to legal guidelines. Brains were removed and homogenized with a Potter homogenizer (Braun Melsungen, Melsungen, Germany) after addition of 5 volumes of cold 25 mm Tris/HCl, pH 7.5, 150 mm NaCl containing 1 μg/ml aprotinin, 0.5 μg/ml leupeptin, and 1.74 μg/ml PMSF. The homogenate was then centrifuged at 2,000 × g for 10 min at 4 °C. To the supernatant, Triton X-100 was added to a final concentration of 1% (v/v). After 30 min under mild mixing at 4 °C, samples were centrifuged at 12,000 × g for 30 min at 4 °C. The supernatant was used directly in the GST pulldown assays. The GST pulldown procedure was performed by incubating purified GST-tagged proteins coupled to glutathione-Sepharose beads with 0.5 ml of Triton X-100 extracts for 2 h at 4 °C. After washing four times with 0.5 ml of Triton X-100 extraction buffer, proteins were resuspended in 50 μl of SDS-PAGE sample buffer, heated at 95 °C for 10 min, and analyzed by SDS-PAGE and Western blotting. A MemCode staining (MemCodeTM reversible protein stain kit for nitrocellulose membrane; Thermo Scientific, Bonn, Germany) was performed to visualize proteins in the input (2% of total) and GST-tagged proteins. Native Gephyrin was visualized by immunostaining.
In Vitro GTPase Binding Assays
TC10 binding assays were performed by incubating purified GST-tagged proteins coupled to glutathione-Sepharose beads for 2 h at 4 °C in a buffer containing a final concentration of 50 mm Tris/HCl, pH 7.5, 1 mm DTT, 0.5% Triton X-100, 200 mm NaCl, and, for nucleotide-free conditions, 5 mm EDTA; for nucleotide-bound conditions 5 mm MgCl2 was used. After washing four times with at least 20 bead volumes, proteins were resuspended in 50 μl of SDS-PAGE sample buffer, heated at 60 °C for 30 min, and analyzed by SDS-PAGE and Western blotting. A MemCode staining (MemCodeTM reversible protein stain kit for nitrocellulose membrane; Thermo Scientific) was performed to visualize GST-tagged proteins, and TC10 was visualized by immunostaining.
Protein-Lipid Overlay Assays
To test relative PI3P binding affinity of the GST-ΔSH3CbII R/H mutant as compared with GST-ΔSH3CbII WT, custom-made PI3P strips were prepared by spotting 200, 100, 25, and 12.5 pmol of PI3P-diC16 (P-3016; Echelon, Mobitec, Göttingen, Germany) onto Hybond-C-extra membranes (GE Healthcare). Membranes were blocked with 3% (w/v) fatty acid-free BSA (A7030; Sigma-Aldrich) in TBS, pH 7.5, and incubated with 0.5 μg/ml (2.5 μg of total protein in 5 ml of TBST (0.1% Tween 20 in TBS), 3% BSA) of GST-tagged Cb proteins for 2 h at room temperature. After four washes with TBST and two washes with TBS, bound proteins were detected with standard dot-blot techniques using a goat anti-GST-HRP conjugate (GE Healthcare).
Transfection and Immunostaining of COS-7 Cells
COS7 cells were cultured in DMEM (Gibco, Life Technologies), 10% (v/v) FCS (Gibco, Life Technologies), 50 units/ml penicillin, and 50 units/ml streptomycin at 37 °C and 5% CO2. In the past, we have optimized the transfection conditions and post-transfection analysis time points for the optimal readout of Cb function in submembraneous Gephyrin clustering (14, 16, 19). For transfection, cells were plated in 24-well plates on 12-mm coverslips. Sterile coverslips were coated with poly-l-lysine (Sigma; 0.001%) diluted from 0.01% stock in Dulbecco's PBS (PAA Laboratories, Cölbe, Germany) for at least 2 h, washed three times with Dulbecco's PBS before plating COS-7 cells. Medium was exchanged to DMEM prior to transfection at ∼80% confluency. For transfection, 200 ng of GFP-Gephyrin, 100 ng of HA-TC10 (WT, CA), and 100 ng of Myc-Cb plasmids were used per well. pcDNA 3.1 was used to equalize amounts of DNA per transfection to 400 ng. Cells were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. DMEM containing 10% FCS and antibiotics were added 4 h after transfection. Cells were fixed in 4% PFA in PBS for 10 min at 10 h after transfection. After three washes in PBS, cells were permeabilized for 20 min using 0.2% Triton X-100 in PBS. Blocking was performed in 10% goat serum in PBS for at least 60 min. For the detection of epitopes, antibodies were diluted as indicated above in 10% goat serum in PBS and incubated for 1.5 h (primary antibodies) or 45 min (secondary antibodies). Cells were washed three times with PBS before mounting with Aqua Poly/Mount (Polysciences, Warrington PA). For microscopy, a Zeiss Imager Z1 with a Zeiss apochromat 63× objective was used. GFP-Gephyrin exposure was kept constant at 170 ms.
Transfection and Immunostaining of Cultured Rat Hippocampal Neurons
Rat hippocampal neurons were prepared from embryonic day 18 rats. Hippocampi were treated with trypsin in Hanks' balanced salt solution (Gibco, Life Technologies) for 15 min at 37 °C and mechanically triturated. Cells were plated on poly-l-lysine-coated glass coverslips at a density of 120,000 cells/ml in Neurobasal medium supplemented with B27 (Gibco, Life Technologies), GlutaMAX (Gibco, Life Technologies), and penicillin-streptomycin (Roche). Neurons were transfected at DIV 4 using the CalPhosTM mammalian transfection kit (Clontech) and 2 μg of plasmid DNA per well of a 12-well plate. Pictures of transfected hippocampal neurons were taken on an inverse Leica DMIRE2 microscope equipped with a 63× oil immersion objective and connected to a Leica TCS SP2 AOBS confocal laser scanning setup (Leica Microsystems, Bensheim, Germany). Acquired images were processed identically using the ImageJ software package. Single channels were recorded using exactly the same standardized threshold levels. Subsequently, a binary image was generated, and puncta were counted automatically by the ImageJ software. For colocalization analysis, perisomatic and dendritic areas were selected within the image, image channels (green, Gephyrin label; red, Myc label) were automatically thresholded and processed using the colocalization threshold algorithm of ImageJ, and a standard Pearson's correlation coefficient between the channels was calculated. The values are expressed as means ± S.E., and statistical significance was evaluated with Student's t test.
MD Simulations
MD simulations of the WT and the R/H mutant proteins were performed using the Gromacs software package (version 4.6) (21–23) and the amber99sb*ildn force field (24) using the tip3p water model (25). Electrostatic interactions were calculated using particle mesh Ewald (26). Simulations were carried out in an ensemble with constant pressure, temperature, and particle number, using the velocity rescaling method (27) for temperature coupling with a coupling time constant of 0.1 ps and a reference temperature of 300 K, and the Berendsen pressure coupling (28) with a 1-ps coupling time constant. All bonds were constrained using the LINCS algorithm (29). Initial coordinates were taken from the 2.3 Å crystal structure of the Cdc42-Cb II complex (PDB code 2DFK) (30). The R/H mutation in the DH domain was introduced by Sybyl (Sybyl 8.0.1; Tripos, St. Louis, MO). Protonation states were chosen according to pH 7, meaning that His-237 is not protonated. Because His-237 is largely solvated and has negatively as well as positively charged residues next to it, we assume that the pKa of His-237 remains nearly unchanged. Both the WT and the mutant protein complex were placed in a cubic simulation box (box size, ∼97 nm) with periodic boundary conditions. The box was filled with water and NaCl with a concentration of 0.15 m. Each system was then equilibrated for 5 ns applying position restraints (force constant, 1000 kJ/(mol*nm2)) on the non-hydrogen protein atoms. Subsequently, five ∼300-ns unconstrained simulations of WT and mutant protein were carried out. To characterize structural changes caused by the mutation, PH domain root mean square deviations of the PH domain backbone relative to the DH domain backbone were calculated. Additionally, we performed principal component analyses of the PH domain backbone relative to the DH domain backbone, of the PH domain backbone itself, as well as of the β3β4 loop region of the PH domain. For each simulation, the average structure was obtained by discarding the first 100 ns. To analyze the impact of the mutation on interdomain energetics, interaction enthalpies (Coulomb and Lennard-Jones interactions within 4 nm) between selected Cb groups were calculated for all trajectories, discarding the first 100 ns.
RESULTS
The R/H Mutation Results in Defective Cb-mediated Submembrane Clustering of GFP-Gephyrin in COS7 Cells
The R290H missense mutation was identified in three adult brothers suffering from epileptic seizures and mental retardation (12). We analyzed the effects of this R/H mutation in the rat CbII isoform because the three-dimensional structures of rat SH3(+)CbII (Fig. 1B; Ref. 14; PDB code 4MT6) and ΔSH3CbII (Fig. 1C; Ref. 30; PDB code 2DFK) are known, which supports data interpretation and MD simulations, and because the human Cb isoform (hPEM-2 or CB3; also present in rats and mice) and the SH3(+)CbII isoform (also named CB2SH3+; present in rats and mice but not in humans) are very similar, except for their very C termini and seven additional amino acid (aa) residues in the initial N-terminal sequence of SH3(+)CbII (Fig. 1A). Indeed, there is currently no evidence that the additional N-terminal aa and the C terminus of Cb are involved in Cb-mediated Gephyrin clustering at inhibitory postsynapses, and in hippocampal neurons, both isoforms target to GABAergic postsynapses and induce a similar increase in Gephyrin clustering (31).
FIGURE 1.

The R/H mutation impairs Cb-mediated submembraneous clustering of GFP-Gephyrin in COS7 cells. A, schematic representation of the human SH3(+)Cb R290H mutant and the corresponding mutants of rat ΔSH3CbII (R237H) and rat SH3(+)CbII (R297H) used in this study. Amino acids surrounding Arg-290 (red) in the primary sequence of human Cb and the corresponding residues in the rat isoforms are shown. The affected arginine residue is located within the catalytic core of the DH domain, in close proximity to the highly conserved Asn-285/Glu-286 residues (bold) that are essential for exchange activity. B and C, three-dimensional structures of rat SH3(+)CbII (B) according to Soykan et al. (Ref. 14; PDB code 4MT6) and ΔSH3CbII (C) according to Xiang et al. (Ref. 30; PDB code 2DFK). The arginine affected in the R/H mutation (red) interacts with Asp-377 in the PH domain of ΔSH3CbII (C). The corresponding aspartate within the PH domain of SH3(+)CbII is Asp-437 (B, red). Arginine residues within the β3-β4 loop of the PH domain (B and C, magenta) that were shown to be involved in PI3P binding (17) are indicated. The SH3 domain (B, orange) forms an intramolecular interface within the DH and PH domains that has been shown to interfere with PI3P binding (14). Cyan and gray residues within the SH3 domain contribute to SH3-DH/PH and SH3-NL2 interactions, respectively (14). Note that the locations of Asp-437 and Arg-297 are on opposite sides of the SH3-DH/PH interface (B). D–K, images of COS7 cells transfected as indicated. GFP-Gephyrin accumulates in large cytoplasmic aggregates when expressed together with Myc-SH3(+)CbII (F). In contrast, in the presence of Myc-ΔSH3CbII (D), GFP-Gephyrin forms microclusters at the plasma membrane. Similarly, Myc-SH3(+)CbII and constitutively active HA-TC10 (TC10 CA), or Myc-SH3(+)CbII and HA-NL2 jointly trigger GFP-Gephyrin microcluster formation (H and J), as described previously (16, 19). Notably, the R/H mutation prevents both the ability of ΔSH3CbII to induce GFP-Gephyrin microclusters (E) and the ability of TC10 CA and NL2 to trigger SH3(+)CbII-mediated Gephyrin redistribution (I and K). The following antibodies were used: polyclonal rabbit anti-HA (1:2,000; Zymed Laboratories Inc., Invitrogen) and monoclonal mouse anti-c-Myc clone 9E10 (1:1,000; Sigma-Aldrich). Dotted lines indicate the cell borders. Scale bar, 10 μm. L, percentages of GFP-Gephyrin cotransfected cells classified as displaying GFP-Gephyrin microclusters (> 50 puncta per cell; n = 3 independent transfections; n = 300 cells). The data represent means ± S.E. ***, p < 0.001; unpaired, two-tailed Student's t test.
The arginine residue affected in the R/H mutation is located within the catalytic core of the DH domain, in close proximity to the highly conserved Asn-285/Glu-286 residues that are essential for exchange activity (16, 17). However, according to the crystal structure of the rat CbII splice variant lacking the N-terminal SH3 domain (ΔSH3CbII) in complex with Cdc42 (Ref. 30; PDB code 2DFK), the Arg-290 residue (Arg-237 in the sequence of rat ΔSH3CbII and Arg-297 in the sequence of SH3(+)CbII; Fig. 1, A and B) does not interact directly with the GTPase but contacts instead Asp-377 in the PH domain of ΔSH3CbII (the corresponding residue in SH3(+)CbII is Asp-437; Fig. 1, A and B).
To assess the functional consequences of the R/H mutation, we first expressed Myc-tagged WT CbII splice variants with or without an N-terminal SH3 domain (SH3(+)CbII and ΔSH3CbII) or their corresponding R/H mutants (R/H mut) together with GFP-Gephyrin in COS7 cells. As described previously (18), in the majority of the double-transfected cells, ΔSH3CbII, but not SH3(+)CbII, induced the redistribution of GFP-Gephyrin from large intracellular aggregates to small, submembranous microclusters (>50 GFP-Gephyrin positive puncta per cell; Fig. 1, D and F). In the presence of the SH3 domain, SH3-DH/PH intramolecular interactions in Cb (Fig. 1B) favor a closed/inactive conformation of the protein, which prevents its interaction with membrane-bound PI3P (14), an essential step in Cb-mediated clustering of Gephyrin (17). Hence, the percentage of transfected cells displaying microclusters was strongly reduced in cells expressing SH3(+)CbII, as compared with those expressing the constitutively active, ΔSH3CbII isoform (19.66 ± 1.76% versus 93 ± 0.57%, respectively; Fig. 1L, top panel).
Coexpression of the ΔSH3CbII R/H or the SH3(+)CbII R/H mutant with GFP-Gephyrin in COS7 cells resulted in the formation of intracellular aggregates, in which both Gephyrin and the mutant proteins were perfectly colocalized (Fig. 1, E and G). The percentage of ΔSH3CbII R/H-expressing cells showing submembraneous microclusters was strongly reduced, as compared with cells expressing WT ΔSH3CbII (5.33 ± 1.76% versus 93 ± 0.57%, respectively; Fig. 1L, top panel). Furthermore, the quantifications indicated that the SH3(+)CbII R/H mutant was completely defective in redistributing GFP-Gephyrin into microclusters because only 0.66 ± 0.33% of transfected cells formed microclusters (Fig. 1L, top panel).
We showed previously that NL2 or active, GTP-loaded TC10 relieve SH3-DH/PH domain-mediated autoinhibition and enhance SH3(+)CbII-mediated Gephyrin clustering via an interaction of NL2 with the SH3 domain or of the active GTPase TC10 with the PH domain of Cb (16, 19). Accordingly, cotransfection of GFP-Gephyrin together with SH3(+)CbII and NL2 or together with SH3(+)CbII and the constitutively active GTP-bound TC10 Q75L mutant (TC10 CA) in COS7 cells induced the formation of Gephyrin microclusters, similar to those observed with the ΔSH3 isoform (Fig. 1, H and J). The fraction of cells displaying GFP-Gephyrin microclusters was significantly higher in the presence of TC10 CA or NL2 (42 ± 1.53 and 61 ± 3.46%, respectively; Fig. 1L, bottom panel) as compared with cells expressing only GFP-Gephyrin and SH3(+)CbII (19.66 ± 1.76%). In contrast, both TC10 CA and NL2 failed to trigger the redistribution of Gephyrin into microclusters when coexpressed with the SH3(+)CbII R/H mutant (Fig. 1, I, K, and L, bottom panel).
The R/H Mutation Does Not Affect the GEF Activity of Cb
The arginine residue affected in the R/H mutation is located within the catalytic core of the DH domain of Cb (Fig. 1A). The proximal asparagine residue in the sequence NERKRR forms a hydrogen bond with D65 of Cdc42 (30), which is located within the highly conserved YDRLRPL switch II-α2 motif of Cdc42 (32). The corresponding residue of TC10 within the YDRLRPL motif is Asp-79, and we recently showed that, similar to Cdc42, TC10 can be activated in cells by WT ΔSH3CbII WT but not by the GEF-deficient ΔSH3CbII NE/AA mutant (16).
To analyze whether the R/H mutation affects the GEF activity of Cb, we used an in vivo activation assay described previously (16). TC10 was expressed either alone or together with WT ΔSH3CbII or the R/H mutant in COS7 cells, and after cell lysis, the amount of activated TC10 was determined by cosedimentation with immobilized GST-PAK1-PBD, a known effector that specifically binds GTP-bound GTPases (33). Notably, GTP-TC10 levels did not differ significantly between extracts from cells coexpressing TC10 along with either WT ΔSH3CbII or the R/H mutant (Fig. 2, A and B). These data show that the affected arginine residue in the R/H mutant of Cb is not involved in catalysis.
FIGURE 2.
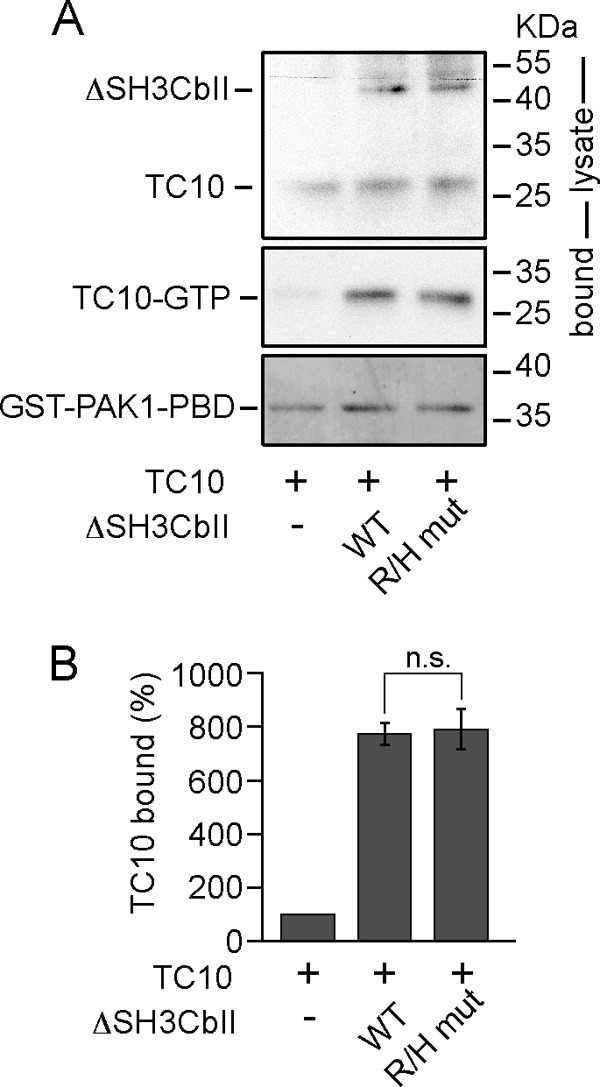
The R/H mutation does not affect the GEF activity of Cb. A, COS7 cells were transfected with HA-TC10 either alone (−) or together with Myc-ΔSH3CbII (WT or R/H mut). Cell lysates were used for cosedimentation with immobilized GST-PAK1-PBD. GTP-bound TC10 (middle panel) was detected by Western blotting with an anti-HA antibody coupled to HRP (1:10,000; Roche). MemCode staining (bottom panel) of the same membrane prior to TC10 immunoblotting was used to confirm that equal amounts of GST-PAK1-PBD had been added to each lysate. To measure HA-TC10 and Myc-ΔSH3CbII expression, 2% of the cell lysates used for GST-PAK1-PBD binding assays were in addition subjected to anti-HA (as described above) and anti-Myc (1:2,000, polyclonal anti-c-Myc C3956; Sigma-Aldrich) blotting, respectively. B, relative band intensities of TC10 bound to GST-PAK1-PBD (n = 3 experiments). Statistical significance was compared between ΔSH3CbII WT and ΔSH3CbII R/H mutant. The data represent means ± S.E. n.s., not significant (unpaired, two-tailed Student's t test).
The R/H Mutation Does Not Affect Binding of Cb to Gephyrin
A previous study indicated that the DH domain of Cb is crucial for Gephyrin binding (34). To investigate whether the R/H mutation affects the interaction of Cb with Gephyrin, we performed a GST pulldown assay using Triton X-100 extracts from adult mouse brain and bacterially expressed and purified GST, GST-tagged WT ΔSH3CbII, or its R/H mutant coupled to glutathione-Sepharose beads. Western blotting followed by immunostaining with a Gephyrin-specific antibody indicated that native Gephyrin coprecipitated with the Cb R/H mutant at levels similar to those observed with Cb WT (Fig. 3, A and B). Thus, the R/H Cb mutant retains its affinity for Gephyrin.
FIGURE 3.

The ΔSH3CbII R/H mutant retains its binding to Gephyrin and to active TC10. A, Triton X-100 extracts from adult mouse brain were incubated with the indicated recombinant proteins bound to glutathione-Sepharose beads. Bound native Gephyrin was detected by Western blotting using the 3B11 antibody (1:3,000; Synaptic Systems). Top right panel, note that Gephyrin bound similarly well to GST-ΔSH3CbII WT and the GST-ΔSH3CbII R/H mutant, but not to GST alone. Bottom right panel, MemCode stainings of the same membrane prior to Gephyrin immunoblotting was used to assess the amounts of purified GST and GST-tagged bait proteins used. Left, MemCode staining (bottom panel) and immunoblotting (top panel) of the same membrane was used to ensure that similar amounts of total protein and total Gephyrin were present in all reaction mixtures. B, relative band intensities (Gephyrin pulled down/total Gephyrin, normalized to Cb in MemCode) of Gephyrin bound to GST, GST-ΔSH3CbII WT, or the GST-ΔSH3CbII R/H mutant (n = 3 experiments). Statistical significance was compared between ΔSH3CbII WT and ΔSH3CbII R/H mutant. No binding of Gephyrin to GST was observed. The data represent means ± S.E. n.d., not detectable; n.s., not significant (unpaired, two-tailed Student's t test). C, purified His-TC10, either nucleotide-free or preloaded with GTPγS or GDP, was incubated with the indicated recombinant proteins bound to glutathione-Sepharose beads. Bound His-TC10 was detected by Western blotting using a TC10-specific polyclonal antibody (T8950, 1:4,000; Sigma-Aldrich). Note that GST-ΔSH3CbII WT and the GST-ΔSH3CbII R/H mutant bound preferentially to GTPγS-TC10, as did GST-PAK1-PBD but not GST alone (top panel). Bottom panel, MemCode stainings of the same membranes prior to TC10 immunoblotting indicating the amounts of the GST-tagged bait proteins used in the actual reaction mixture. Middle panel, to ensure that similar amounts of His-TC10 were included in all reaction mixtures, 2% of the premixed reactions were stored and subsequently subjected to anti-TC10 Western blotting using the T8950 antibody. D, relative band intensities (pulled down His-TC10/total His-TC10, normalized to GST-tagged bait proteins in MemCode) of TC10 bound to GST or the GST-ΔSH3CbII R/H mutant, as compared with those of the corresponding TC10 (nucleotide-free, GDPγS, or GDP-loaded) bound to GST-ΔSH3CbII WT (n = 3 experiments). Statistical significance was compared between ΔSH3CbII WT and ΔSH3CbII R/H mutant. No binding of His-TC10 to GST was observed. The data represent means ± S.E. n.d., not detectable; n.s., not significant (unpaired, two-tailed Student's t test).
The R/H Mutation Does Not Affect the Preferential Binding of Cb to GTP-loaded TC10
As shown in Fig. 1 (I and K), Cb-mediated Gephyrin recruitment to the plasma membrane is defective upon coexpression of GFP-Gephyrin with the SH3(+)CbII R/H mutant and TC10 CA or with the SH3(+)CbII R/H mutant and NL2. We showed recently (14) that the SH3 domain of Cb binds autonomously to NL2 and that the SH3 domain residues that are involved in NL2 binding are on the opposite side of the DH domain helix that contains the mutated arginine within the R/H site (Fig. 1B), so that the R/H mutation is not expected to affect the interaction of the SH3 domain with NL2. In contrast, the interaction sites involved in the interaction between TC10 and the PH domain of Cb (16) have not yet been characterized in detail, and the Cb PH domain residues involved in TC10 binding are currently unknown. These data and considerations prompted us to test whether the R/H mutation affects the properties of TC10 binding by Cb.
To this end, we performed in vitro binding assays, using purified His-TC10 (preloaded with GTPγS, preloaded with GDP, or trapped in its nucleotide-free state) and GST-tagged WT ΔSH3CbII or its R/H mutant coupled to glutathione-Sepharose beads, as described previously (16). In agreement with our previous study on the Cb-TC10 interaction (16), we found a strong interaction of WT ΔSH3CbII with GTPγS-loaded TC10, but only weak interactions with GDP-loaded or nucleotide-free TC10 (Fig. 3C, right panel). Notably, the ΔSH3CbII R/H mutant also bound preferentially to GTPγS-loaded TC10 and exhibited WT-like binding efficacy (Fig. 3, C, right panel, and D). Control incubations with immobilized GST-PAK1-PBD, the binding domain of a known downstream effector of TC10, and GST alone confirmed the specificity of the binding (Fig. 3C, left panel). These results indicate that the R/H mutation does not affect the noncatalytic site in the PH domain of Cb that is responsible for the effector-like interaction with TC10 (16).
The R/H Mutation Impairs Cb Binding to PI3P
Given that the R/H mutation does not affect the GEF activity of Cb toward TC10 or the preferential binding of Cb to GTP-loaded TC10, we extended the functional characterization of this missense mutation by investigating the importance of the interaction between the DH domain helix bearing the mutated arginine residue and the PH domain of Cb for proper membrane targeting.
According to the crystal structure of ΔSH3CbII (30), the DH domain residue Arg-237 is in contact with the PH domain residue Asp-377 (Fig. 1C). The crystal structure of ΔSH3CbII was the first example where structural differences in the PH domain of a Dbl protein caused by variable interactions between the α6 region of the DH domain and the PH domain were observed (30). Depending on whether these variable interactions provide a less or more extensive interface, ΔSH3CbII adopts two different conformations (30). However, the functional implications of this structural flexibility have remained unclear. A recently published study of ours (14) indicated that in the presence of the SH3 domain, Cb displays an even more compact conformation, in which the SH3 domain is packed against the DH and PH domains (Fig. 1B). In this closed conformation, SH3(+)CbII binding to PI3P is strongly reduced, indicating that the inability of SH3(+)CbII to induce microcluster formation in cells is due to an orientation of the PH domain that impedes anchoring to membrane-bound PI3P (Fig. 1B). Binding of NL2 to the SH3 domain of Cb stabilizes the open/active conformation of Cb by competing with intramolecular SH3-DH/PH interactions within Cb. In this open/active conformation, the orientation of the β3-β4 loop of the PH domain, bearing the arginine residues that are involved in PI3P binding, is different from the one seen in the absence of NL2 (Fig. 1, B and C), which facilitates the interaction of the Cb PH domain with PI3P (14). In addition, we showed recently that NL2-induced activation of Cb and PI3P binding to the PH domain are both required for Gephyrin clustering, as indicated by the fact that SH3(+)CbII mutants deficient in PI3P binding (R363A/R364A and K358A/K359A; both aa pairs located in the β3-β4 loop) fail to redistribute Gephyrin to the plasma membrane in the presence of NL2 and remain tightly coclustered with intracellular Gephyrin aggregates (14). As also shown previously for ΔSH3CbII, which permanently adopts an open/active conformation (17), PI3P binding of Cb is essential for Gephyrin clustering, which is indicated by the finding that a PI3P binding-deficient ΔSH3CbII R303N/R304N mutant cannot redistribute GFP-Gephyrin into microclusters in heterologous cells and is unable to recruit Gephyrin to inhibitory postsynaptic sites in cultured neurons.
Because PI3P binding is essential for Cb function (17), because the activation of SH3 domain containing Cb variants by NL2, TC10, and functionally related proteins appears to operate upstream of PI3P binding and mainly results in an increase of PI3P binding by exposing the PH domain (14, 16), and because our results so far indicated a strong impairment of the ΔSH3CbII R/H mutant in mediating submembraneous GFP-Gephyrin clustering in COS7 cells (Fig. 1E), we next tested whether the R/H mutations affects the binding of ΔSH3CbII to PI3P. To this end, we used increasing amounts of immobilized PI3P (12.5–200 pmol) and bacterially expressed and purified WT GST-ΔSH3CbII or the GST-ΔSH3CbII R/H mutant in a protein-lipid overlay assay, as described previously (9, 17). This assay showed that WT ΔSH3CbII bound PI3P much more strongly than the ΔSH3CbII R/H mutant at all PI3P amounts tested (Fig. 4, B and C). Hence, the R/H mutation perturbs the interaction of the PH domain of Cb with PI3P, likely leading to a reduced tethering of Cb to lipid membranes. Furthermore, the impaired PI3P binding indicates that the R/H mutation may alter the conformation of Cb such that the PH domain shows a different degree of rotation relative to the DH domain as compared with that shown for WT ΔSH3CbII (Fig. 1C).
FIGURE 4.
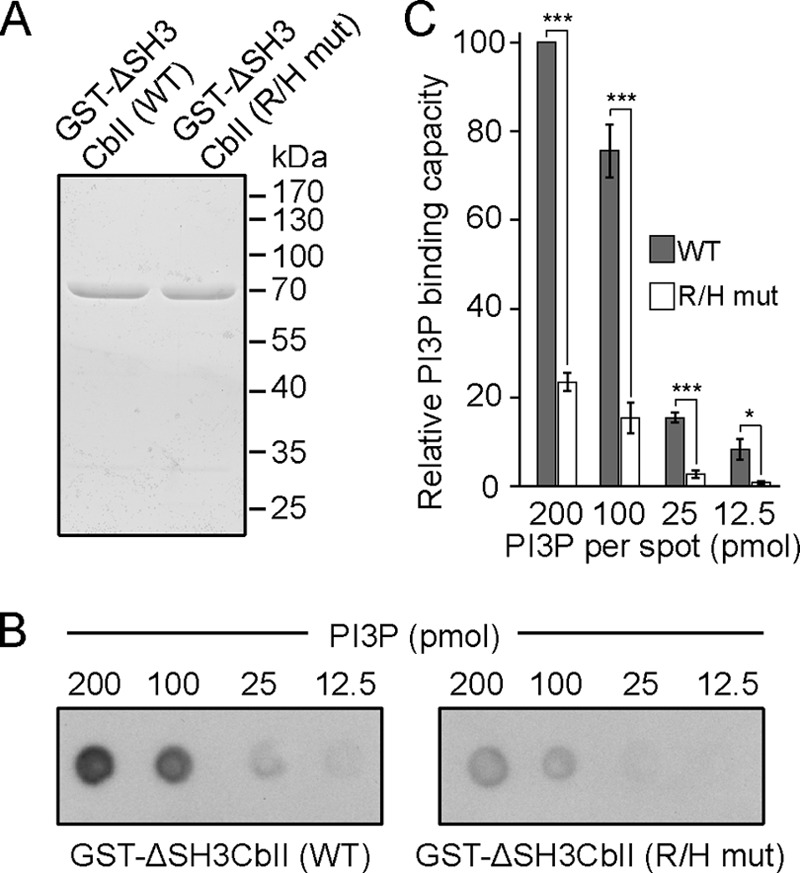
The R/H mutation changes the properties of PI3P binding by ΔSH3CbII. A, Coomassie Blue staining of SDS-PAGE of equal amounts of the purified GST-ΔSH3CbII WT and the GST-ΔSH3CbII R/H mutant preparations used in the protein-lipid overlay assay. B, protein-lipid overlay assay with GST-ΔSH3CbII WT (left panel) and the GST-ΔSH3CbII R/H mutant (right panel; 0.5 μg/μl protein, respectively). The amounts (in pmol) of PI3P spotted onto the membrane are indicated. Bound GST-tagged proteins were detected by incubating the membranes with a goat anti-GST-HRP conjugate (1:10,000; GE Healthcare). Note that PI3P binding of the R/H mutant is strongly reduced, as compared with WT Cb. C, relative binding of each protein (WT Cb and R/H mutant) for each PI3P amount, determined by measuring the intensity of the chemiluminescence. The data represent means ± S.E. of n = 3 experiments. *, p < 0.05; ***, p < 0.001 (unpaired, two-tailed Student's t test).
MD Simulations Show That the R/H Mutation Causes an Increased Conformational Heterogeneity in the Cb Structure
As mentioned above, previous studies (14, 30) support the view that altered interactions between the DH and PH domains of Cb can lead to changes in protein conformation, specifically in the orientation of the PH domain relative to the DH domain, which affect PI3P binding by the PH domain.
Our finding that the R/H mutation in the DH domain impairs the interaction of Cb with PI3P prompted us to examine WT ΔSH3CbII WT and the ΔSH3CbII R/H mutant using MD simulations. We decided to perform our MD simulations using the three-dimensional structure of the shorter, open/active, ΔSH3CbII isoform and not that of the SH3(+)CbII isoform, which is more closely related to human Cb, because we found previously that the SH3 domain moves freely in solution, resulting in an equilibrium of closed and open Cb states, with the closed/inactive state being favored in the presence of the SH3 domain (14). In contrast, ΔSH3CbII adopts permanently the open and active state. Furthermore, the intrinsic Gephyrin clustering activity of Cb increases upon release of the intramolecular SH3-DH/PH domain interactions (which is the case (i) in the presence of NL2, (ii) upon introduction of mutations that perturb SH3-DH/PH interactions (14), or (iii) upon removal of the SH3 domain). Based on these data and considerations, we decided that the structure of the ΔSH3CbII isoform would be the most appropriate for MD simulation-based analyses of the effects of the R/H mutation on the active Cb conformation.
Initial coordinates were taken from the 2.3 Å crystal structure of the Cdc42-ΔSH3CbII complex (Ref. 30; PDB code 2DFK), and MD simulations for the WT and the R/H mutant protein were performed as described under “Experimental Procedures.” Alignment of the average structures over five independent simulations for WT ΔSH3CbII and the ΔSH3CbII R/H mutant, respectively, show an increased conformational flexibility of the PH domain of the R/H mutant relative to the DH domain, as compared with the WT protein (Fig. 5A).
FIGURE 5.

The R/H mutation affects the orientation of the PH domain relative to the DH domain. A, ribbon backbone presentation of the superimposed average structures over five independent simulations of ΔSH3CbII WT (green) and the corresponding R/H mutant (red). For simplicity, the ribbon backbone of Cdc42 is omitted. The ΔSH3CbII DH and PH domains are indicated. The black dot and arrow indicate the position of the R/H mutation within the DH domain α-helix (left blue arrowed line). The right blue arrowed line indicates the neighboring PH domain α-helix. Note that the superimposed DH domains perfectly overlap, whereas the average PH domain of the R/H mutant shows a different orientation relative to the DH domain, as compared with WT. B, root mean square deviation (RMSD; WT, green; R/H mutant, red) of the whole PH domain backbone to the starting structure using the DH domain as the fitting group. Average structures of trajectories 1–5 (open triangles) and the average structures thereof (Av., closed triangles) are indicated. C, two-dimensional projections of the concatenated WT and R/H mutant trajectories, respectively, on the first and second eigenvector of the PH domain motion relative to the DH domain. D, two-dimensional projections on the first and second eigenvector of the internal PH domain motion. E, ribbon backbone presentation of the DH/PH α-helix (aa 213–251) and the PH domain α-helix (aa 357–381) of the average structures of the five independent ΔSH3CbII WT trajectories (colored) and the average structure thereof (black). F, Ribbon backbone presentation of the DH/PH α-helix (aa 213–251) and the PH domain α-helix (aa 357–381) of the average structures of the five independent ΔSH3CbII R/H mutant trajectories (colored) and the average structure thereof (black).
The slight shift in the relative DH/PH domain orientations between the average structures of the R/H mutant and the WT protein prompted us to measure the root mean square deviations of the whole PH domains relative to the whole DH domains for all five trajectories, as well as for the average structures of the WT protein and the R/H mutant. Although the average changes of the PH domain relative to the DH domain remained nearly unaltered for the WT protein, the trajectories of the R/H mutant showed larger changes and stronger fluctuations (Fig. 5B). To quantify the collective fluctuations of the R/H mutant relative to the WT protein and to decide whether these fluctuations are mainly interdomain (PH/DH domain) or intradomain (PH domain) motions, we concatenated all trajectories of the WT protein and the R/H mutant and performed a principal component analysis of both the motion of the PH domains relative to the DH domains (Fig. 5C) and of the internal PH domain motions (Fig. 5D) and the projected WT and R/H mutant protein motions on the first two eigenvectors of the principle component analysis. We found that the PH domain of the R/H mutant samples only few additional internal conformations, as compared with the WT protein (Fig. 5D). Moreover, a principle component analysis of the β3-β4 loop of the PH domain, bearing the Arg-303 and Arg-304 residues that are involved in PI3P binding (17), indicated that the β3-β4 loop of the R/H mutant adopts almost the same conformation as that of the WT protein (data not shown). In contrast, significantly enhanced PH motions relative to the DH domain are seen in the R/H mutant, which are not accessed by the WT protein (Fig. 5C) at the few 100-ns time scale of the MD simulations. Together, these results indicate that the PH domain of the R/H mutant mainly changes its orientation as a whole (rigid body movement), whereas the internal dynamics of the PH domain is not affected.
Next, we focused on the aa residues 213–251 of the long α-helix connecting the DH and PH domains and the aa 357–381 of the α-helix of the PH domain. The interface between these two helices was previously suggested to affect the mobility of the PH domain and its orientation relative to the DH domain (30). Ribbon backbone presentations of the average structures of the independent simulations indicate that the DH/PH domain helices (aa 213–251) nearly adopt the same conformations in both the WT and the R/H mutant (Fig. 5, E and F). Furthermore, the average PH domain helices (aa 357–381) of WT ΔSH3CbII show similar orientations relative to their corresponding DH domains (Fig. 5E). In contrast, the simulations of the R/H mutant revealed a much higher heterogeneity and stronger fluctuations of the average PH domain helices (Fig. 5F). Together, these results indicate that the R/H mutation might affect the strength of interactions between the DH/PH domain (aa 213–251) and the PH domain (aa 357–381) helices, thereby favoring alternative conformations of the PH domain relative to the DH domain.
To test this idea, the interaction energies between these two helices (helix 1 and helix 2), as well as between Arg/His-237 and selected interacting residues of the PH domain (Fig. 6, E and F) were calculated. Indeed, significantly weakened interactions between the mentioned helices were seen for the mutant (Fig. 6A) and also weaker interactions between His-237 and the aa Asp-377, Met-373, and Glu-265 compared with those of Arg-237 in the WT Cb (Fig. 6, B–D). Among these interactions, the Arg-237–Asp-377 interaction dominates (Fig. 6G) and thus mainly stabilizes the DH-PH conformation.
FIGURE 6.
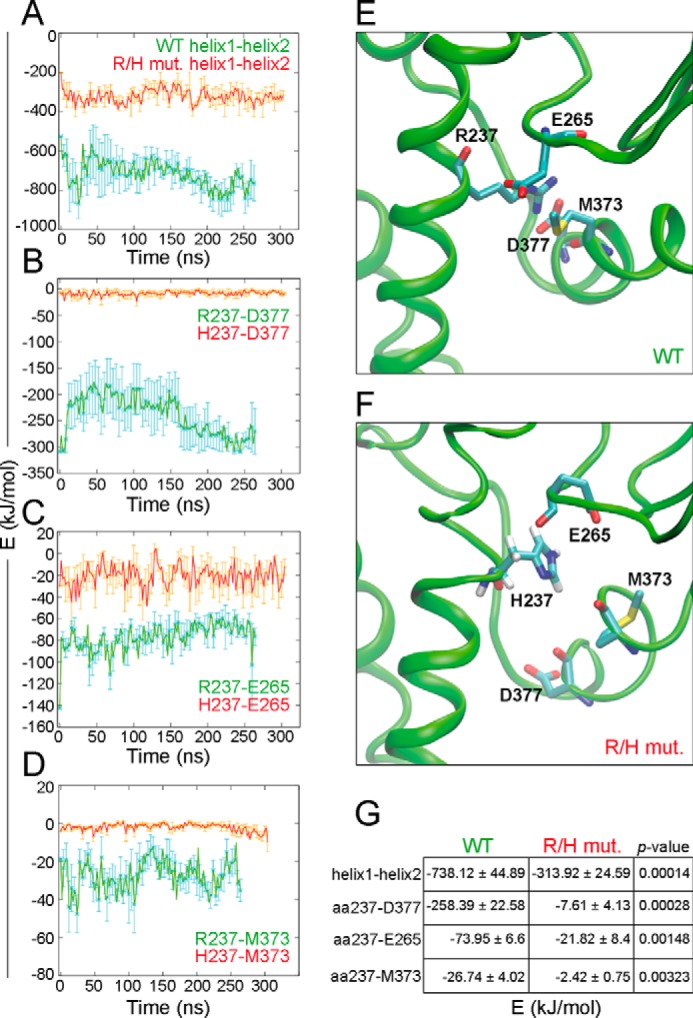
The R/H mutation affects the strength of interdomain interactions. A–D, interaction energies (Coulomb and Lennard-Jones energies within 4 nm) of the α-helices 1 (aa 213–251) and 2 (aa 357–381) of the WT and the R/H mutant Cb protein (A) averaged from five simulations each. Correspondingly, that of the selected aa pairs Arg/His-237–Asp-377 (B), Arg/His-237–Glu265 (C), and of Arg/His-237–Met-373 (D). E and F, close-up view of the interactions of Arg-237 in the averaged ΔSH3CbII WT structure with selected residues of the PH domain (E) and similarly, of His-237 in the R/H mutant (F). G, averaged interaction energies (sum of Coulomb and Lennard-Jones energies) for pairs of interacting helices and aa in ΔSH3CbII WT and the RH mutant. The data represent means ± S.E. of n = 5 simulations. p values are from unpaired, two-tailed Student's t test.
The R/H Mutation Abolishes the Effects of Overexpressed ΔSH3CbII or SH3(+)CbII on the Clustering of Endogenous Gephyrin in Cultured Hippocampal Neurons
To determine whether the R/H mutation affects the Cb-mediated clustering of endogenous Gephyrin in neurons, we employed an overexpression approach using WT rat hippocampal neurons. This assay has been used routinely in the past and provides a very robust, reliable, reproducible, and generally applicable readout of Cb function in synaptogenesis and synapse maturation (16, 17). We transfected cultured neurons at DIV 4 with either Myc-tagged ΔSH3CbII WT, SH3(+)CbII WT, or the corresponding R/H mutants. Neurons were fixed at DIV 14 and stained for Gephyrin and Myc immunoreactivities (Fig. 7, A–E). Overexpression of ΔSH3CbII R/H or SH3(+)CbII R/H resulted in an accumulation of Gephyrin within intracellular aggregates (indicated by arrows in Fig. 7, C and E) and a highly significant reduction of both perisomatic (Fig. 7F; ΔSH3CbII: WT, 26.47 ± 2.2 versus R/H mut, 16 ± 1.76; SH3(+)CbII: WT, 24.99 ± 1.72 versus R/H mut, 12.77 ± 1.62) and dendritic (Fig. 7H; ΔSH3CbII: WT, 40.2 ± 3.32 versus R/H mut, 27.2 ± 2.27; SH3(+)CbII: WT, 37.5 ± 1.76 versus R/H mut, 19.9 ± 1.65) Gephyrin clusters, as compared with overexpression of the respective WT variants. However, both ΔSH3CbII R/H and SH3(+)CbII R/H mutant values were similar to those of untransfected rat hippocampal neurons (Fig. 7F; untransfected: 14.24 ± 1.32; Fig. 7H; untransfected: 20.75 ± 1.91; significance levels compared with transfected neurons are shown within the bars), indicating that the recombinant R/H mutants have no substantial dominant negative effect on the ability of endogenous Cb to induce Gephyrin clustering in cultured neurons, at least not at the expression levels achieved here. In contrast, the ΔSH3CbII R/H isoform was still capable of inducing a slight but significant increase (p value: 0.042) in the density of perisomatic Gephyrin clusters, as compared with untransfected neurons (Fig. 7H), indicating that in the constitutively open conformation of Cb, the mutation retains some intrinsic Gephyrin clustering activity upon overexpression in cultured neurons.
FIGURE 7.

The R/H mutation abolishes the enhancing effects of overexpressed ΔSH3CbII or SH3(+)CbII on the clustering of endogenous Gephyrin in cultured hippocampal neurons. A–E, cultured rat hippocampal neurons were transfected at DIV 4 with Myc-ΔSH3CbII (B), Myc-SH3(+)CbII (D), or their corresponding R/H mutants (C and E). Untransfected neurons (A) served as control. At DIV 14, the cells were fixed and immunostained for Gephyrin (green) and Myc (red) using the mAb7a (1:3,000; Connex) and the polyclonal rabbit antibody C3956 (1:1,000; Sigma-Aldrich), respectively. Silhouettes of somata and dendrites of untransfected neurons (indicated by dotted lines in A) were identified by massively increasing the brightness of the Gephyrin staining using ImageJ so that the cellular background signal became visible. Note the accumulation of Gephyrin in somatic aggregates (indicated by arrows) in neurons expressing the R/H mutants, as compared with untransfected neurons and to neurons expressing the corresponding WT isoforms. Scale bar, 10 μm. F and G, bar diagrams of perisomatic Gephyrin cluster densities per 100-μm2 surface area (F) and average sizes of perisomatic Gephyrin clusters (G). H and I, bar diagrams of the number of Gephyrin immunoreactive clusters per 40-μm dendritic length (H) and average sizes of dendritic Gephyrin clusters (I). Bars correspond to values obtained from the perisomatic surface area and one randomly selected second order dendrite (60–100 μm distal to the soma) (F) per neuron, respectively (n = 10–20 individual neurons from three independent transfection experiments). The data represent means ± S.E. n.s., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001 (unpaired, two-tailed Student's t test). Significance levels compared with untransfected neurons are shown within the bars. J–M, exemplary overlapping pixel regions of selected somatic (top panels) and dendritic (bottom panels) areas from images of neurons expressing Myc-ΔSH3CbII (J), Myc-SH3(+)CbII (L), or their corresponding R/H mutants (K and M). Green (Gephyrin staining) and red (Myc staining) channels of selected image areas (as indicated by dotted lines) were automatically thresholded and processed using the colocalization threshold algorithm of the ImageJ software package The overlapping pixel regions were converted into a binary threshold mask (white) and superimposed on the original images as a colocalization map. Note the strong colocalization of the Myc-SH3(+)CbII R/H mutant with somatic Gephyrin aggregates (M, indicated by arrows) and the reduced colocalization of the Myc-ΔSH3CbII R/H mutant in both somatic and dendritic areas (K), as compared with WT ΔSH3CbII (J). N and O, Pearson's correlation coefficients were determined to quantify the levels of colocalization of each CbII variant. Red bars, Myc-ΔSH3CbII WT; orange bars, Myc-ΔSH3CbII R/H mutant; green bars, Myc-SH3(+)CbII WT; lime green bars, Myc-SH3(+)CbII R/H mutant) with Gephyrin in somatic/perisomatic (N) and dendritic (O) areas. The data represent means ± S.E. of n = 10 somata or dendrites of individual neurons. n.s., not significant; *, p < 0.05; **, p < 0.01 (unpaired, two-tailed Student's t test).
Previous studies demonstrated a significant increase in the size of Gephyrin clusters upon overexpression of ΔSH3CbII in cultured neurons (31, 35). In agreement with these studies, we found that the average sizes of perisomatic and dendritic clusters of endogenous Gephyrin were significantly increased in neurons expressing WT ΔSH3CbII as compared with untransfected neurons (Fig. 7G; untransfected: 0.12 ± 0.005 versus ΔSH3CbII WT: 0.3 ± 0.027; Fig. 7I; untransfected: 0.13 ± 0.007 versus ΔSH3CbII WT: 0.25 ± 0.022). Overexpression of the ΔSH3CbII R/H mutant, on the other hand, caused hardly any increase in the size of endogenous Gephyrin clusters (Fig. 7G; ΔSH3CbII R/H mut: 0.15 ± 0.016; Fig. 7I; ΔSH3CbII R/H mut: 0.16 ± 0.013). In contrast, the average sizes of Gephyrin puncta in neurons expressing the SH3(+)CbII isoform were not affected by the R/H mutation and were similar to those of untransfected neurons at both perisomatic and dendritic areas (Fig. 7, G and I). These results indicate that the increase in the size of Gephyrin clusters in neurons overexpressing the open/active ΔSH3CbII isoform might be a feature associated with the ability of this isoform to bind efficiently to PI3P-enriched membranes, because the impairment of Cb binding to PI3P was the only defect observed in the corresponding R/H mutant. In contrast, in SH3(+)CbII overexpressing neurons, the availability of endogenous NL2, GABAAR α2 subunit, or active TC10 and Cdc42, i.e. proteins that interact with either the SH3 domain or the PH domain of Cb and relieve autoinhibition (16, 19, 36), might be limiting factors that explain why the SH3(+)CbII isoform is not inducing a similar effect on the size of Gephyrin clusters as the effect observed with the ΔSH3CbII isoform.
We next tested whether the R/H mutation affects the colocalization of recombinant ΔSH3CbII and SH3(+)CbII with endogenous Gephyrin. To this end, we selected somatic/perisomatic and dendritic areas of neurons expressing the Myc-tagged CbII variants and their corresponding R/H mutants at similar levels (Fig. 7, J–M, and data not shown) and performed an analysis of colocalization between endogenous Gephyrin and the recombinant Myc-Cb labels, using the colocalization threshold algorithm provided by ImageJ. We found that the R/H mutants remained tightly coclustered with large intracellular Gephyrin aggregates, which were more frequent in neurons expressing the SH3(+)CbII R/H isoform (indicated by arrows in Fig. 7M). In contrast, Gephyrin aggregates were not observed in neurons expressing the WT CbII isoforms (Fig. 7, J and L). Furthermore, compared with WT ΔSH3CbII, the ΔSH3CbII R/H mutant showed a significantly reduced colocalization with Gephyrin in both somatic/perisomatic (Fig. 7N; WT: 0.34 ± 0.038 versus R/H mut: 0.17 ± 0.019;) and dendritic (Fig. 7O; WT: 0.39 ± 0.045 versus 0.2 ± 0.031) areas. In dendrites, also the SH3(+)CbII R/H mutant showed a significant reduction in its colocalization with Gephyrin, as compared with WT SH3(+)CbII (Fig. 7O; WT: 0.27 ± 0.019 versus R/H mut: 0.16 ± 0.043). In somatic/perisomatic areas, the colocalization of the SH3(+)CbII R/H mutant with Gephyrin was reduced (Fig. 7N; 0.26 ± 0.027), but this change did not reach statistical significance, which is probably due to the overlapping distributions of the Myc-tagged SH3(+)CbII R/H mutant and Gephyrin immunoreactivities in large somatic aggregates (Fig. 7, E and M). Taken together, these data indicate that the R/H mutation affects the ability of Cb to efficiently cocluster with postsynaptically enriched Gephyrin in the somata and dendrites of hippocampal neurons.
DISCUSSION
In the present study, we employed biochemical, cell biological, and MD simulation approaches to elucidate the pathomechanism of an R290H missense mutation in human Cb, which was identified in three adult brothers suffering from epileptic seizures and intellectual disability (12). For this purpose, we used the CbII (rat- and mouse-specific) isoform and not the human (hPEM-2) Cb isoform. This choice was made for to the following reasons: (i) three-dimensional structures are currently available for the two CbII (ΔSH3 and SH3(+)) isoforms (14, 30), but not for hPEM-2, which allowed us to complement our cell biological and biochemical studies with MD simulations and detailed analyses of the DH/PH interface bearing the R/H mutations; (ii) a previous study indicated that hPEM-2 and SH3(+)CbII behave very similarly in targeting to GABAergic postsynapses and in inducing increases in Gephyrin clustering, when overexpressed in cultured neurons (31); and (iii) the two isoforms are identical except for their very C termini, which are extremely unlikely to influence the effects of the R/H mutation studied here and which do not appear to differentially affect Cb-mediated Gephyrin clustering.
Based on three lines of evidence, our present data define the R/H mutant of Cb as a loss of function mutant: (i) When expressed in COS7 cells together with GFP-Gephyrin, the ΔSH3CbII R/H mutant cannot mediate the redistribution of Gephyrin from large intracellular aggregates to membrane-associated microclusters. Indeed, the R/H mutation is the first known single mutation in the DH domain of the open/active ΔSH3CbII isoform that renders the protein completely inactive in redistributing Gephyrin in cells. Although the vast majority of Cb isoforms in mouse brain contains an SH3 domain (14) and the mRNAs encoding SH3 domain-containing isoforms are much more abundant than mRNAs encoding ΔSH3Cb isoforms in the rodent brain and spinal cord (7), some ΔSH3Cb splice variants appear to exist in vivo and may play an important role in synaptogenesis and synapse maturation (7). In this context, our approach to overexpress ΔSH3CbII variants in non-neuronal cells and hippocampal neurons proved to be very valuable for analyses of the functional importance of single aa residues for Cb-mediated Gephyrin clustering. The use of ΔSH3CbII variants circumvents the need for overexpressing additional Cb activators (i.e. NL2, the α2 subunit of GABAARs or active GTPases TC10 and Cdc42 (16, 19, 36)), which interact with the SH3 domain or the PH domain of Cb and thereby revert autoinhibition. In essence, ΔSH3CbII isoforms perfectly imitate the open/active states of SH3-containing Cb variants that are induced upon the interaction of the SH3 domain with NL2, the α2 subunit of GABAARs, or active TC10 or Cdc42. (ii) ΔSH3CbII R/H, as well as the longer, SH3 domain-containing, variant SH3(+)CbII R/H are unable to enhance perisomatic and dendritic clustering of endogenous Gephyrin in cultured neurons and instead lead to the somatic accumulation of Gephyrin. Interestingly, the clustering of endogenous Gephyrin in neurons overexpressing the R/H mutants was similar to the Gephyrin clustering seen in untransfected neurons, indicating that the overexpressed R/H mutants had no dominant negative effect on Gephyrin clustering in cultured neurons, at least not at the expression levels achieved here. (iii) Both NL2 and TC10 CA cannot induce the GFP-Gephyrin redistribution to submembraneous microclusters when coexpressed together with the SH3(+)CbII R/H mutant.
The arginine residue affected in the R/H mutation is located within the catalytic core of the DH domain of Cb, which mediates its GEF activity. However, according to the crystal structure of ΔSH3CbII in complex with Cdc42 (30), the affected arginine does not interact directly with the GTPase but contacts instead Asp-377 in the PH domain of Cb. The PH domain was previously shown to be involved in intramolecular interactions with the SH3 and DH domains (14) and to mediate the interaction with active, GTP-loaded TC10 (16) and with PI3P (9). The SH3 domain of Cb binds autonomously to NL2 (14), the SH3 domain residues involved in NL2 and PH/DH domain binding are on opposite sides of the SH3 structure (Fig. 1B), and the R/H site and the DH domain residues involved in SH3 domain interactions are facing in opposite directions, all of which indicates that the R/H mutation cannot affect the interaction of the SH3 domain with NL2. Whereas the residues of the PH domain that are involved in the GTP-TC10 interaction are currently not known, the interaction with PI3P is mediated by two arginines (Arg-303/Arg-304) located in the β3-β4 loop of the PH domain (17). Thus, the particular location and interacting residues of the affected arginine would be compatible with the notion that the R/H mutation might cause changes in the conformation of both the DH and PH domains of Cb, leading to altered GEF activity or altered binding to protein interactors. However, our biochemical and cell biological experiments showed that the R/H mutation affects neither the GEF activity of Cb nor its binding to Gephyrin or GTP-loaded TC10, as demonstrated by the in vivo activation assays, the GST pulldown assays, and the in vitro TC10 binding assays, respectively. Consistent with these findings, our MD simulations predicted no significant changes in the internal conformations of the average structures of the entire DH and PH domains of the ΔSH3CbII R/H mutant, as compared with WT ΔSH3CbII.
A key finding of the present study is that the R/H mutation impairs Cb binding to PI3P, as assessed in a protein-lipid overlay assay. Consequently, and in agreement with results obtained with the PI3P binding-deficient R303N/R304N mutant (17), the R/H mutation studied here abolishes the ability of the ΔSH3CbII splice variant, which adopts an intrinsically open/active conformation, to induce membrane clustering of Gephyrin in COS7 cells and synaptic clustering of Gephyrin in cultured neurons. Furthermore, GTP-TC10, which binds to the PH domain of Cb and thereby enables the SH3(+)CbII variant to adopt an open/active conformation (16), cannot induce Gephyrin targeting to the plasma membrane when coexpressed with the SH3(+)CbII R/H mutant. This finding is reminiscent of data obtained with the PI3P binding-deficient SH3(+)CbII RR/AA mutant (16) and indicates that the interaction of the PH domain of Cb with PI3P is an essential step in the Gephyrin clustering cascade that cannot be bypassed by GTPase overexpression.
But how can a single mutation in the DH domain of Cb affect PI3P binding to the distal β3-β4 loop of the PH domain? As mentioned above, the arginine residue mutated in the R/H mutant contacts an acidic side chain in the PH domain (30). Depending on whether the R/H mutation provides a less or more extensive interface between the DH and PH domains, the mutant might then adopt a more open or more closed conformation, respectively, thus increasing or decreasing the accessibility of the PH domain for membrane lipid binding. Indeed, a recently published study of ours (14) indicated that the autoinhibited SH3(+)CbII, which adopts a closed/inactive conformation, binds to PI3P only weakly, similar to the PI3P binding-deficient SH3(+)CbII RR/AA mutant. In contrast, an SH3(+)CbII W24A/E262A mutant, with two substitutions in the SH3-DH/PH interface that lead to a more open conformation of the protein, displayed an increased binding to PI3P and increased plasma-membrane targeting of Gephyrin as compared with SH3(+)CbII WT (14). Thus, conformational activation of Cb facilitates PI3P binding and subsequent Gephyrin clustering, and the R/H mutation might exert its effects in this context.
The PH domains of Dbl family proteins adopt multiple orientations with regard to the respective DH domains. Depending on variable interactions between the DH and PH domains, a flexibility in the orientation of the PH domain relative to the DH domain of ΔSH3CbII was suggested previously (30). However, the functional implications of this flexibility have remained unclear. Our MD simulations of WT ΔSH3CbII and its corresponding R/H mutant in complex with Cdc42 predict that the R/H mutation markedly increases orientational fluctuations of the entire PH domain relative to the DH domain, presumably because of changes in the strength of interactions between residues of the DH/PH α-helix (aa 213–251) and residues in the PH domain α-helix (aa 357–381). Together, these results indicate that the PH domain conformation that is favored in the WT Cb protein is the most efficient in mediating the binding to PI3P.
Although many mutations in the Cb gene have been identified in patients with epilepsy and intellectual disability, only very few of them have been characterized functionally. Our present analysis of the R/H mutation in Cb (12), together with a previously described balanced chromosomal translocation resulting in mRNAs that no longer encode the PH domain of Cb (9), provides evidence for the notion that an impairment of PI3P binding by Cb is a likely common pathomechanism for Cb dysfunction in the human brain.
Despite their low abundance, phosphoinositides are critical regulators of intracellular signaling and membrane compartmentalization. The functional roles of phosphoinositide metabolism have been studied in substantial detail at the presynaptic terminal, where phosphoinositide turnover is of critical importance for synaptic vesicle recycling and synapse function (37). Further, there is abundant evidence for the relevance of phosphoinositide pathways in synaptic plasticity (38). However, much less is known about specific roles of phosphoinositides at inhibitory postsynapses. PI3P is considered a lipid that is constitutively present on endosomes where it is mainly generated by class III phosphatidylinositol 3-kinase (39, 40). The specific recruitment of class III PI3K to endosomes and the local production of PI3P on endosomal membranes provide a mechanism for the recruitment of a specific group of proteins to endosomes, and most PI3P-binding proteins that have been functionally characterized so far are associated with different aspects of endosomal membrane sorting and trafficking (for review see Ref. 41). Thus, the accumulation of the Cb R/H mutants in large somatic aggregates together with endogenous Gephyrin might reflect a defect in the cotransport of both proteins via PI3P-enriched endosomal compartments to the plasma membrane, with consequent effects on inhibitory postsynapse formation. Alternatively, a signaling process involving plasma membrane PI3P might be involved in the control of Cb function. Over the past few years, it has become increasingly evident that pools of PI3P can be specifically generated at the plasma membrane upon cellular stimulation, and PI3P has emerged as a critical intracellular second messenger in various signaling pathways (42). Notably, PI3P is specifically produced downstream of TC10 activation to promote the plasma membrane translocation of the glucose transporter 4 (43). Moreover, TC10-mediated activation of a member of the class II PI3Ks, PI3K-C2α, has been shown to regulate the generation of PI3P at the plasma membrane (44). These findings, together with the previously described TC10-induced increase in GABAergic postsynaptic strength (16), strongly support the notion that the molecular mechanisms involved in local PI3P production on endomembranes or the plasma membrane play an important role in triggering inhibitory postsynapse formation and maturation. The present study indicates that the CbR290H mutant might cause epilepsy and intellectual disability because it is not responsive to the types of PI3P signaling described above, consequently lacks Gephyrin clustering activity, and therefore cannot support the proper formation and maturation of functional inhibitory synapses in the brain.
Acknowledgments
We thank Foteini Paraskevopoulou and Özge Demet Özcete for help with some of the experiments.
This work was supported by the Max Planck Society (to N. B.), the German Research Foundation (Center of Nanoscale Microscopy and Molecular Physiology of the Brain grant to N. B. and Grant PA2087/1-1 to T. P.) and European Commission Innovative Medicines Initiative FP7-115300 (to N. B.).
- GABAAR
- GABAA receptor
- aa
- amino acid(s)
- CA
- constitutively active
- Cb
- Collybistin
- Dbl
- diffuse B-cell lymphoma
- DH
- Dbl homology
- GEF
- guanine nucleotide exchange factor
- HMD
- molecular dynamics
- NL2
- Neuroligin-2
- PBD
- p21-binding domain
- PH
- pleckstrin homology
- PI3P
- phosphatidylinositol-3-phosphate
- SH3
- Src homology 3
- R/H
- Arg → His
- mut
- mutant(s)
- PDB
- Protein Data Bank
- DIV
- day in vitro.
REFERENCES
- 1. Luscher B., Fuchs T., Kilpatrick C. L. (2011) GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith K. R., Kittler J. T. (2010) The cell biology of synaptic inhibition in health and disease. Curr. Opin. Neurobiol. 20, 550–556 [DOI] [PubMed] [Google Scholar]
- 3. Ramamoorthi K., Lin Y. (2011) The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends Mol. Med. 17, 452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Verpelli C., Galimberti I., Gomez-Mancilla B., Sala C. (2014) Molecular basis for prospective pharmacological treatment strategies in intellectual disability syndromes. Dev. Neurobiol. 74, 197–206 [DOI] [PubMed] [Google Scholar]
- 5. Papadopoulos T., Soykan T. (2011) The role of collybistin in gephyrin clustering at inhibitory synapses: facts and open questions. Front. Cell. Neurosci. 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papadopoulos T., Korte M., Eulenburg V., Kubota H., Retiounskaia M., Harvey R. J., Harvey K., O'Sullivan G. A., Laube B., Hülsmann S., Geiger J. R., Betz H. (2007) Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J. 26, 3888–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harvey K., Duguid I. C., Alldred M. J., Beatty S. E., Ward H., Keep N. H., Lingenfelter S. E., Pearce B. R., Lundgren J., Owen M. J., Smart T. G., Lüscher B., Rees M. I., Harvey R. J. (2004) The GDP-GTP exchange factor collybistin: An essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marco E. J., Abidi F. E., Bristow J., Dean W. B., Cotter P., Jeremy R. J., Schwartz C. E., Sherr E. H. (2008) ARHGEF9 disruption in a female patient is associated with X linked mental retardation and sensory hyperarousal. J. Med. Genet. 45, 100–105 [DOI] [PubMed] [Google Scholar]
- 9. Kalscheuer V. M., Musante L., Fang C., Hoffmann K., Fuchs C., Carta E., Deas E., Venkateswarlu K., Menzel C., Ullmann R., Tommerup N., Dalprà L., Tzschach A., Selicorni A., Lüscher B., Ropers H. H., Harvey K., Harvey R. J. (2009) A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 30, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lesca G., Till M., Labalme A., Vallee D., Hugonenq C., Philip N., Edery P., Sanlaville D. (2011) De novo Xq11.11 microdeletion including ARHGEF9 in a boy with mental retardation, epilepsy, macrosomia, and dysmorphic features. Am. J. Med. Genet. 155A, 1706–1711 [DOI] [PubMed] [Google Scholar]
- 11. Shimojima K., Sugawara M., Shichiji M., Mukaida S., Takayama R., Imai K., Yamamoto T. (2011) Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J. Hum. Genet. 56, 561–565 [DOI] [PubMed] [Google Scholar]
- 12. Lemke J. R., Riesch E., Scheurenbrand T., Schubach M., Wilhelm C., Steiner I., Hansen J., Courage C., Gallati S., Bürki S., Strozzi S., Simonetti B. G., Grunt S., Steinlin M., Alber M., Wolff M., Klopstock T., Prott E. C., Lorenz R., Spaich C., Rona S., Lakshminarasimhan M., Kröll J., Dorn T., Krämer G., Synofzik M., Becker F., Weber Y. G., Lerche H., Böhm D., Biskup S. (2012) Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 53, 1387–1398 [DOI] [PubMed] [Google Scholar]
- 13. de Ligt J., Willemsen M. H., van Bon B. W., Kleefstra T., Yntema H. G., Kroes T., Vulto-van Silfhout A. T., Koolen D. A., de Vries P., Gilissen C., del Rosario M., Hoischen A., Scheffer H., de Vries B. B., Brunner H. G., Veltman J. A., Vissers L. E. (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929 [DOI] [PubMed] [Google Scholar]
- 14. Soykan T., Schneeberger D., Tria G., Buechner C., Bader N., Svergun D., Tessmer I., Poulopoulos A., Papadopoulos T., Varoqueaux F., Schindelin H., Brose N. (2014) A conformational switch in collybistin determines the differentiation of inhibitory postsynapses. EMBO J. 33, 2113–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reid T., Bathoorn A., Ahmadian M. R., Collard J. G. (1999) Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. J. Biol. Chem. 274, 33587–33593 [DOI] [PubMed] [Google Scholar]
- 16. Mayer S., Kumar R., Jaiswal M., Soykan T., Ahmadian M. R., Brose N., Betz H., Rhee J. S., Papadopoulos T. (2013) Collybistin activation by GTP-TC10 enhances postsynaptic gephyrin clustering and hippocampal GABAergic neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 110, 20795–20800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reddy-Alla S., Schmitt B., Birkenfeld J., Eulenburg V., Dutertre S., Böhringer C., Götz M., Betz H., Papadopoulos T. (2010) PH-domain-driven targeting of collybistin but not Cdc42 activation is required for synaptic gephyrin clustering. Eur. J. Neurosci. 31, 1173–1184 [DOI] [PubMed] [Google Scholar]
- 18. Kins S., Betz H., Kirsch J. (2000) Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat. Neurosci. 3, 22–29 [DOI] [PubMed] [Google Scholar]
- 19. Poulopoulos A., Aramuni G., Meyer G., Soykan T., Hoon M., Papadopoulos T., Zhang M., Paarmann I., Fuchs C., Harvey K., Jedlicka P., Schwarzacher S. W., Betz H., Harvey R. J., Brose N., Zhang W., Varoqueaux F. (2009) Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642 [DOI] [PubMed] [Google Scholar]
- 20. Fuhrmann J. C., Kins S., Rostaing P., El Far O., Kirsch J., Sheng M., Triller A., Betz H., Kneussel M. (2002) Gephyrin interacts with dynein light chains 1 and 2, components of motor protein complexes. J. Neurosci. 22, 5393–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 22. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 23. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 26. Darden T. A., Pedersen L. G. (1993) Molecular modeling: an experimental tool. Environ. Health Perspect. 101, 410–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bussi G., Donadio D., Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 28. Berendsen H. J. C., Postma J. P. M., Vangunsteren W. F., Dinola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 29. Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 30. Xiang S., Kim E. Y., Connelly J. J., Nassar N., Kirsch J., Winking J., Schwarz G., Schindelin H. (2006) The crystal structure of Cdc42 in complex with collybistin II, a gephyrin-interacting guanine nucleotide exchange factor. J. Mol. Biol. 359, 35–46 [DOI] [PubMed] [Google Scholar]
- 31. Chiou T. T., Bonhomme B., Jin H., Miralles C. P., Xiao H., Fu Z., Harvey R. J., Harvey K., Vicini S., De Blas A. L. (2011) Differential regulation of the postsynaptic clustering of γ-aminobutyric acid type A (GABAA) receptors by collybistin isoforms. J. Biol. Chem. 286, 22456–22468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hemsath L., Dvorsky R., Fiegen D., Carlier M. F., Ahmadian M. R. (2005) An electrostatic steering mechanism of Cdc42 recognition by Wiskott-Aldrich syndrome proteins. Mol. Cell 20, 313–324 [DOI] [PubMed] [Google Scholar]
- 33. Neudauer C. L., Joberty G., Tatsis N., Macara I. G. (1998) Distinct cellular effects and interactions of the Rho-family GTPase TC10. Curr. Biol. 8, 1151–1160 [DOI] [PubMed] [Google Scholar]
- 34. Grosskreutz Y., Hermann A., Kins S., Fuhrmann J. C., Betz H., Kneussel M. (2001) Identification of a gephyrin-binding motif in the GDP/GTP exchange factor collybistin. Biol. Chem. 382, 1455–1462 [DOI] [PubMed] [Google Scholar]
- 35. Tyagarajan S. K., Ghosh H., Harvey K., Fritschy J. M. (2011) Collybistin splice variants differentially interact with gephyrin and Cdc42 to regulate gephyrin clustering at GABAergic synapses. J. Cell Sci. 124, 2786–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saiepour L., Fuchs C., Patrizi A., Sassoè-Pognetto M., Harvey R. J., Harvey K. (2010) Complex role of collybistin and gephyrin in GABAA receptor clustering. J. Biol. Chem. 285, 29623–29631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wenk M. R., De Camilli P. (2004) Protein-lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc. Natl. Acad. Sci. U.S.A. 101, 8262–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Man H. Y., Wang Q., Lu W. Y., Ju W., Ahmadian G., Liu L., D'Souza S., Wong T. P., Taghibiglou C., Lu J., Becker L. E., Pei L., Liu F., Wymann M. P., MacDonald J. F., Wang Y. T. (2003) Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron 38, 611–624 [DOI] [PubMed] [Google Scholar]
- 39. Gillooly D. J., Morrow I. C., Lindsay M., Gould R., Bryant N. J., Gaullier J. M., Parton R. G., Stenmark H. (2000) Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19, 4577–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vicinanza M., D'Angelo G., Di Campli A., De Matteis M. A. (2008) Function and dysfunction of the PI system in membrane trafficking. EMBO J. 27, 2457–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Raiborg C., Schink K. O., Stenmark H. (2013) Class III phosphatidylinositol 3-kinase and its catalytic product PtdIns3P in regulation of endocytic membrane traffic. FEBS J. 280, 2730–2742 [DOI] [PubMed] [Google Scholar]
- 42. Falasca M., Maffucci T. (2006) Emerging roles of phosphatidylinositol 3-monophosphate as a dynamic lipid second messenger. Arch. Physiol. Biochem. 112, 274–284 [DOI] [PubMed] [Google Scholar]
- 43. Maffucci T., Brancaccio A., Piccolo E., Stein R. C., Falasca M. (2003) Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J. 22, 4178–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Falasca M., Hughes W. E., Dominguez V., Sala G., Fostira F., Fang M. Q., Cazzolli R., Shepherd P. R., James D. E., Maffucci T. (2007) The role of phosphoinositide 3-kinase C2alpha in insulin signaling. J. Biol. Chem. 282, 28226–28236 [DOI] [PubMed] [Google Scholar]