

Background: Loss of PAR3 triggers accelerated growth and invasion of mammary tumors via STAT3 activity.
Results: PAR3 silencing induces aPKC activity, which triggers NF-κB-IL-6 signaling.
Conclusion: Regulation of aPKC activity is a key tumor suppressor function of PAR3.
Significance: This work contributes to our understanding of mechanisms by which polarity proteins restrain tumor progression.
Keywords: Breast Cancer, Cell Polarity, Cell Signaling, Signal Transduction, STAT3
Abstract
PAR3 suppresses tumor growth and metastasis in vivo and cell invasion through matrix in vitro. We propose that PAR3 organizes and limits multiple signaling pathways and that inappropriate activation of these pathways occurs without PAR3. Silencing Pard3 in conjunction with oncogenic activation promotes invasion and metastasis via constitutive STAT3 activity in mouse models, but the mechanism for this is unknown. We now show that loss of PAR3 triggers increased production of interleukin-6, which induces STAT3 signaling in an autocrine manner. Activation of atypical protein kinase C ι/λ (aPKCι/λ) mediates this effect by stimulating NF-κB signaling and IL-6 expression. Our results suggest that PAR3 restrains aPKCι/λ activity and thus prevents aPKCι/λ from activating an oncogenic signaling network.
Introduction
Breast cancers arise from epithelial cells or their progenitors and often retain epithelial characteristics as they progress (1). Although many of the signaling pathways involved in breast tumors have been elucidated, others are still being discovered. After many years of speculation that polarity disruption is fundamental to cancer, recent data have confirmed that defects in the epithelial polarity machinery accelerate solid tumor progression in mammals (2–6). The mechanisms by which polarity restrains tumor progression remain largely unknown. Understanding how polarity signaling impacts tumor biology will aid in the development of targeted therapies.
Three polarity networks are highly conserved in mammalian cells: the Par complex, consisting of PAR3, PAR6, and atypical protein kinase C (aPKC),2 is situated at tight junctions and the apical surface; the Crumbs complex, containing Crumbs (CRB), PALS1, and PATJ, is essential for specifying the apical membrane; and a group of proteins that includes Scribble (SCRB), Discs-large (DLG), and Lethal Giant Larvae (LGL), localize to the basolateral membrane (7). Members of all three groups have been implicated in tumorigenesis, although mainly through correlative evidence (6, 8–13). Recent papers have directly implicated PAR3 as a growth and metastasis suppressor in mammary and skin tumors (14–16).
PAR3 is an important regulator of mammary tissue structure. Loss of PAR3 in the developing mammary gland leads to disorganized ducts that lack apical-basal polarity and resemble atypical ductal hyperplasia, with increased proliferation offset by heightened apoptosis (17). When loss of PAR3 synergizes with an oncogene in mammary ducts, apoptosis is suppressed, growth of primary tumors is accelerated, and aggressive metastatic lesions arise in a manner dependent on signal transducer and activator of transcription 3 (STAT3) (15). Expression of the Notch-1 intracellular domain (NICD1) oncogene alone does not induce STAT3 in mouse mammary cells (mMECs) (15), showing that the effect depends on loss of PAR3. Moreover, NICD1-transformed mMECs (NICD1-mMECs) properly localize polarity markers, such as aPKC, but these markers become disrupted when PAR3 is silenced (15), recapitulating what is observed in other epithelial cells (16, 18).
STAT3 is a transcription factor that has been implicated in the initiation, progression, and metastasis of many cancers (19, 20). Our laboratory has found that STAT3 becomes active following loss of PAR3 in NICD1-transformed mammary cells and that this activation mediates local invasion and lung metastasis (15). However, the mechanism of STAT3 activation following loss of PAR3 has not been identified. In the present work, we sought to determine how loss of PAR3 activates STAT3 in a breast cancer cell model.
There is reason to suspect that aPKC mediates tumor-promoting effects, such as STAT3 activation, when PAR3 is lost. Loss of PAR3 leads to the mislocalization of aPKC in epithelial cells (15–18), accompanied in some contexts by activation (15). This observation, combined with evidence that aPKCι/λ has oncogenic functions (21), indicates that inappropriate aPKCι/λ activation may favor tumor aggression when PAR3 is silenced. Although the literature suggests several mechanisms by which aPKC activation could promote STAT3 signaling, such as potentiating TNF-α signaling (22), interacting with NF-κB (23), promoting ERK activity (24, 25), and transducing signals downstream of EGFR or RAS (16, 26, 27), it is not obvious which is involved when PAR3 expression is disrupted. In the present study, we use gene silencing to rigorously test the role of aPKCι/λ following Pard3 knockdown in transformed mouse mammary cells and to identify the mechanism through which it induces STAT3. Our results suggest that an important tumor suppressor function of PAR3 is to restrain signaling by key partners, such as aPKCι/λ.
EXPERIMENTAL PROCEDURES
Cell Culture, Constructs, and Transfections
Primary mammary epithelial cells were harvest from C3H mice, collagenase-digested, and purified by serial centrifugation as described previously (17). Following purification, these cells were infected with lentivirus expressing NICD1 at a multiplicity of infection of 5. These cells were then grown as mammospheres in ultralow adhesion dishes (Corning, Inc.) for 5 days, after which they were transferred to two-dimensional culture. These cells are referred to as NICD1-mMEC cells. They were cultured in DMEM/F-12 medium supplemented with 1% penicillin/streptomycin, 5% fetal bovine serum, 1% insulin-transferrin-selenium, 5 ng/ml EGF, and 2 μg/ml hydrocortisone. NMuMG cells were cultured in DMEM containing 10% FBS, 1% penicillin/streptomycin, and 10 μg/ml recombinant human insulin. Eph4 cells were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin.
All lentiviral transductions for protein expression were performed at a multiplicity of infection of 5, and all shRNA infections used a multiplicity of infection of 10. The shRNA vector against PAR3 has been described previously (17). The shRNA vector against Il6st was generated by cloning a hairpin with the targeting sequence GCACAGAGCTGACCGTGAA into the ClaI and MluI sites of the pLVTHM vector. shRNA vectors were purchased from Sigma-Aldrich for Il6 (catalog nos. TRCN0000067550 and TRCN0000067548), Prkci (catalog no. TRCN0000278129), and Nfkbia (catalog no. TRCN0000319455). The expression vector for GP130 was generated by cloning human Il6st cDNA into a multiple cloning site our laboratory created in the PmeI locus of the pWPI vector. TurboRFP (tRFP)-tagged constitutively active aPKCi/1 (aPKCi-CA) was cloned into the pLVTHM expression vector. Following knockdown or overexpression, cells were allowed to recover in culture for at least 48 h prior to further treatment or analysis.
Immunofluorescence
Cells were plated on 8-well chamber slides (Lab-Tek) and grown to ∼75% confluence, at which point they were fixed with either methanol-acetone (for STAT3 staining) or 4% paraformaldehyde (for other stains). Following fixation, cells were permeabilized with 0.25% Triton X-100, blocked with 3% bovine serum albumin (BSA) in PBS for 1 h at room temperature, stained overnight at 4 °C with primary antibodies in 0.3% BSA in PBS, washed three times in 0.3% BSA in PBS for 5 min/wash, and stained with Alexa Fluor secondary antibodies in 0.3% BSA in PBS. Antibody dilutions used were as follows: phospho-STAT3, 1:400 (Cell Signaling); p65/RELA, 1:600 (Cell Signaling); Alexa Fluor secondary antibodies, 1:1000 (Life Technologies). After probing with secondary antibodies, cells were washed three times in PBS for 5 min/wash and then stained with DAPI and phalloidins as indicated. Images were obtained using a ×20 objective on an Eclipse TI microscope (Nikon) and analyzed in TIFF format using NIS Elements (Nikon) and ImageJ (National Institutes of Health) software.
Quantitative PCR (qPCR)
Total RNA was isolated from cells using RNAEasy kits (Qiagen), treated with RNase-free DNase (Qiagen), and reverse transcribed into cDNA with random hexamers (Invitrogen) and SuperScript II reverse transcriptase (Invitrogen) plus RNasin (Promega). qPCR of the reverse transcription products was performed using a CFX96 real-time system (Bio-Rad) and SYBR Green real-time PCR master mixes (Life Technologies). Primer sequences for Socs3, Myc, Junb, Stat3, Il6, and Stat1 were obtained from the Harvard Medical School PCR PrimerBank. The 18 S rRNA primer sequences were described previously (28).
Antibodies and Immunoblotting
Cells treated as indicated were collected by scraping in ice-cold PBS and centrifugation, followed by direct lysis in 4× Laemmli sample buffer supplemented with 1× protease inhibitors and phosphatase inhibitors (Roche Applied Science). Lysates were boiled for 5 min, briefly sonicated to break chromatin, and either frozen at −20 °C or immediately run out on 10% acrylamide gels and transferred to nitrocellulose membranes. Blocking was performed with 3% BSA in TBS-T. Primary antibodies used were as follows: anti-PAR3 developed by our laboratory and described previously (17), anti-GP130 (Cell Signaling 3732), anti-phospho-STAT3 (Cell Signaling 9145), anti-total STAT3 (Cell Signaling 9139), anti-phospho-aPKC (Cell Applications CG1453), anti-total aPKCι/λ (Transduction Laboratories 610175), anti-IκBα (Cell Signaling 4814), anti-phospho-IκB kinase (IKK) (Cell Signaling 2697), anti-total IKKβ (Cell Signaling 8943), anti-total IKKα (Cell Signaling 11930), anti-phospho-p65/RELA (Cell Signaling 3033), anti-total p65/RELA (Cell Signaling 8242), anti-GAPDH (Cell Signaling 2118), and anti-β-tubulin (Santa Cruz Biotechnology 9104). HRP-conjugated secondary antibodies (IgG; Jackson ImmunoResearch Laboratories) were used at a dilution of 1:5,000 in TBS-T with 3% milk. Blots were imaged with an ImageQuant device (GE Healthcare). Band intensities were quantified using ImageJ software (National Institutes of Health). To neutralize IL-6 activity, 200 ng/ml of an anti-IL-6 antibody (BD Pharmingen 554400) with validated neutralizing activity (29) was added to cell culture medium.
ELISA Test
Cells were plated at ∼75% confluence, and culture medium was replaced 16 h prior to collection. After collecting medium, cytokine levels were measured with a mouse interleukin-6 Quantikine ELISA kit (R&D Systems).
Luciferase Assay
Cells were grown to ∼75% confluence in 24-well culture plates and calcium phosphate-transfected with 100 ng of 8×NF-κB-GFP-luciferase reporter plasmid (gift of Fiona Yull, Vanderbilt University; described previously (30)) and 50 ng of Renilla luciferase plasmid. 24 h after transfection, cells were lysed and analyzed for luminosity with a Dual-Glu luciferase assay kit (Promega) and a GloMax luminometer (Promega).
Statistical Analysis
Two-tailed Student's t tests were used in statistical analyses. All statistical analysis and graphing were done using Excel for Mac version 14.3.9 (Microsoft).
RESULTS
STAT3 Activation following Loss of PAR3 in Transformed Mammary Epithelial Cells Requires GP130
Interestingly, Drosophila homologues of the IL-6 family, the Unpaired (Upd) genes, are induced when cell polarity is disrupted in ventral nerve cord tumors, with a consequent activation of STAT signaling (10). STAT3 activation in mammals commonly occurs by IL-6 family cytokines binding to the GP130 receptor (31). Therefore, we asked whether cytokine signaling via the GP130 receptor might activate STAT3 in mammary epithelial cells downstream of silencing the cell polarity protein PAR3. To block expression of GP130, we expressed shRNAs against the Il6st gene that encodes GP130 in NICD1-mMECs (Fig. 1A). Consistent with our prior findings (15), knockdown of PAR3 led to a robust activation of STAT3, as assessed by Tyr-705 phosphorylation (Fig. 1, A and 1B), but STAT3 activation returned to baseline levels when GP130 and PAR3 were silenced together (Fig. 1, A and B). We also performed immunofluorescence on cultured NICD1-mMECs. Following infection with virus encoding a hairpin against the genes for either luciferase, as a control, or GP130, less than 10% of nuclei displayed staining for phospho-Tyr-705 STAT3 (Fig. 1, C and D). However, knockdown of PAR3 led to more than a 10-fold increase in the proportion of nuclei positive for Tyr(P)-705 STAT3 (Fig. 1, C and D). Additional silencing of GP130 caused nuclear Tyr(P)-705 STAT3 accumulation to return to baseline levels (Fig. 1, C and D).
FIGURE 1.

Loss of PAR3 triggers STAT3 activation via GP130 in NICD1-mMEC cells. A, NICD1-mMECs, infected with lentivirus to express the indicated shRNAs, were harvested, and equal amounts of lysate were immunoblotted for PAR3, GP130, phospho-STAT3, total STAT3, and β-tubulin (loading control). B, quantitation of phospho-STAT3/total STAT3 from immunoblot experiments shown in A (n = 7). C, NICD1-mMECs expressing the shRNAs indicated were fixed with paraformaldehyde and immunostained for phospho-STAT3. DAPI was used to stain nuclei. Images were taken using a ×20 objective. D, quantitation of the proportion of cells displaying nuclear phospho-STAT3 staining (n = 6). E, mRNA was isolated from NICD1-mMECs infected with lentivurus expressing the shRNAs indicated and reverse transcribed into cDNA, and expression of the genes indicated was analyzed by qPCR. 18 S rRNA was used as a normalization control (n = 4). Error bars, S.E. p values were as follows: for shLuc versus shPAR3, p < 0.05 (*) and p < 0.01 (**); for shPAR3 versus shPAR3/shIl6st, p < 0.05 (#) and p < 0.01 (##). F, NICD1-mMECs infected with lentivirus to express the constructs indicated were harvested, and lysates were immunoblotted for GP130, phospho-STAT3, total STAT3, and β-tubulin (loading control). G, quantitation of the phospho-STAT3/total STAT3 from the immunoblot experiment shown in F (n = 3). Error bars, S.E.
To determine whether the repression of STAT3 activity observed following GP130 knockdown was functionally significant, we analyzed the expression of five genes, the transcription of which can be activated by STAT3: Socs3, Junb, c-Myc, Stat3, and Stat1 (32, 33). All five genes were induced when PAR3 was knocked down, and three of the five (Junb, Stat3, and Stat1) returned toward baseline levels of expression when PAR3 and GP130 were knocked down concurrently (Fig. 1E). We note that the increase in Stat3 mRNA was not accompanied by any significant increase in STAT3 protein levels, perhaps because of translational or post-translational regulation.
To ensure that the inactivation of STAT3 seen after shRNA-mediated silencing of GP130 was not due to nonspecific effects of the hairpin, we infected cells with lentivirus encoding the human GP130 protein, which can interact with the mouse isoforms of the interleukin-6 receptor to activate STAT3 (34). Expression of human GP130 successfully restored activation of STAT3 in NICD1-mMEC/shPAR3 in which murine Il6st had previously been silenced (Fig. 1, F and G). These experiments demonstrate that STAT3 activation following PAR3 depletion occurs via a GP130 receptor-mediated signaling pathway.
To determine whether GP130-mediated STAT3 activation is specific to transformed cells, we silenced Pard3 and Gp130 in freshly isolated primary murine mammary epithelial cells. These primary mMECs were grown for 5 days in three-dimensional mammosphere culture and then lysed and probed for STAT3 activation. Loss of PAR3 efficiently activated STAT3, whereas the simultaneous silencing of PAR3 and GP130 prevented STAT3 phosphorylation (Fig. 2A). Additionally, we observed a robust activation of STAT3 when Pard3 was silenced in the untransformed NMuMG and Eph4 mouse mammary cell lines, and knockdown of GP130 also prevented STAT3 activation in these cell lines (Fig. 2, B and C). These results demonstrate that STAT3 activation following PAR3 depletion occurs in primary murine mammary epithelial cells, transformed primary mammary epithelial cells, and untransformed mammary epithelial cell lines. We conclude that STAT3 activation via GP130 is probably a general response to loss of PAR3 in the mammary epithelium.
FIGURE 2.

Activation of GP130-JAK-STAT3 signaling occurs when Pard3 is silenced in multiple mammary cell lines. A, primary murine mammary cells were harvested and infected with lentivirus to express the constructs indicated and then cultured as mammospheres in non-adherent conditions for 5 days. After culture, the mammospheres were harvested and lysed, and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and β-tubulin (loading control). B, NMuMG cells were infected with lentivirus encoding shRNAs as indicated. Cells were lysed, and equal amounts of lysate were immunoblotted for PAR3, GP130, phospho-STAT3, total STAT3, and GAPDH (loading control). C, as in B, but with Eph4 cells. D–F, either NICD1-mMECs or the cells indicated were infected with lentivirus to express the indicated shRNAs and treated with the JAK-STAT pathway inhibitors indicated; cells were harvested; and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and GAPDH (loading control).
For additional evidence that loss of PAR3 triggers STAT3 by activating a GP130-JAK-STAT3 signaling pathway, we treated NICD1-mMEC, NMuMG, or Eph4 cells with inhibitors of the JAK-STAT pathway in addition to silencing Pard3 with shRNA. Treatment with either the JAK inhibitor pyridone 6 or the JAK-STAT pathway inhibitor Cucurbitacin I prevented STAT3 activation in all three cell types (Fig. 2, D–F).
Along with the other results shown in Figs. 1 and 2, these data strongly implicate the GP130-JAK-STAT pathway as being responsible for the STAT3 activation that we observe in mouse mammary cells that lack PAR3.
Atypical Protein Kinase C-ι/λ Is Necessary for STAT3 Activation
We next asked how loss of PAR3 triggers activation of STAT3 in mammary cells. In previous studies, our group has demonstrated that aPKC becomes activated in transformed mammary cells following the loss of PAR3. Moreover, we found that STAT3 activation could be blocked using pseudosubstrate inhibitors of aPKC (15). However, aPKC pseudosubstrate inhibitors have since been shown to have high nonspecific activities (35, 36), calling this result into question. Therefore, we used gene silencing to test the hypothesis that signaling via aPKC is necessary for STAT3 activation following PAR3 knockdown.
We first confirmed that aPKC is activated following loss of PAR3, as measured by phosphorylation of Thr-560. As previously reported, activation of aPKC is induced by PAR3 silencing in NICD1-mMECs (Fig. 3A). Silencing of the gene encoding aPKCι/λ, Prkci, reduced phospho-STAT3 to levels seen in shLuc control cells, demonstrating that aPKCι/λ is necessary to induce STAT3 (Fig. 3, B and C). To confirm that this effect was due to on-target silencing of Prkci by our shRNA, we re-expressed wild-type aPKCι/λ tagged with tRFP and observed rescue of STAT3 phosphorylation (Fig. 3D). A similar result was found in NMuMG cells as well, suggesting that it is a general mechanism in mouse mammary cells (Fig. 3E).
FIGURE 3.

Activation of aPKCι/λ triggers STAT3 activation when PAR3 is silenced. A, NICD1-mMECs infected with lentivirus to express the shRNAs indicated were harvested, and equal amounts of lysate were immunoblotted for PAR3, phosphothreonine 560 aPKC, total aPKCι/λ, and β-tubulin (loading control). B, NICD1-mMECs infected with lentivirus to express the shRNA constructs indicated were harvested, and equal amounts of lysate were immunoblotted for PAR3, aPKCι/λ, phospho-STAT3, total STAT3, and β-tubulin (loading control). C, quantitation of phospho-STAT3/total STAT3 in B (n = 4). n.s., not significant. D and E, NICD1-mMECs or NMuMGs infected with lentivirus to express the constructs indicated were harvested, and equal amounts of lysate were immunoblotted for PAR3, aPKCι/λ, phospho-STAT3, total STAT3, and GAPDH (loading control). Arrowhead, expressed tRFP-tagged aPKCι/λ. F, NICD1-mMECs expressing tRFP-tagged aPKCi-CA or tRFP (control) were lysed and immunoblotted for phospho-T560 aPKC, total aPKCι/λ, phospho-STAT3, total STAT3, and β-tubulin (loading control). Arrowheads, expressed tRFP-tagged aPKCi-CA. G, quantitation of phospho-STAT3/total STAT3 in F (n = 4). H, NICD1-mMECs infected with lentivirus to express the indicated constructs were harvested and immunoblotted for phospho-Thr-560 aPKC, total aPKCι/λ, GP130, phospho-STAT3, total STAT3, and β-tubulin (loading control). I, quantitation of phospho-STAT3/total STAT3 in H (n = 3). J, primary murine mammary cells were harvested and infected with lentivirus to express the constructs indicated and then cultured as mammospheres in non-adherent conditions for 5 days. After culture, the mammospheres were harvested and lysed, and equal amounts of lysate were immunoblotted for phospho-Thr-560 aPKC, total aPKCι/λ, and GAPDH (loading control). Error bars, S.E.
To determine whether aPKCι/λ activation is sufficient to trigger STAT3 activation, we expressed a constitutively active mutant of tRFP-tagged aPKCι/λ (aPKCi-CA) in mMECs. STAT3 was activated following expression of aPKCi-CA (Fig. 3, F and G), demonstrating that activation of aPKCι/λ is sufficient to induce STAT3 in our model. Moreover, silencing of Il6st was sufficient to abrogate the STAT3 activation seen when aPKCi-CA is expressed (Fig. 3, H and I), showing that aPKCι/λ acts upstream of the GP130 receptor. Finally, to confirm that aPKC activation upon loss of PAR3 is not an artifact of NICD1 expression, we immunoblotted for active aPKCι/λ in freshly isolated, primary mouse mammary epithelial cells grown in suspension culture for 5 days. Depletion of PAR3 was sufficient to activate aPKCι/λ, indicating that transformation is not necessary for this phenotype (Fig. 3J).
Increased Interleukin-6 Production Is Triggered by aPKCι/λ Activity and Induces STAT3 Activation
GP130 is the coreceptor for cytokines of the interleukin-6 (IL-6) family (20), and IL-6 has been implicated in numerous malignancies, including breast cancer (37, 38). We hypothesized, therefore, that loss of PAR3 might induce production of IL-6. When we measured IL-6 levels in culture medium conditioned with either NICD1-mMEC/shLuc or NICD1-mMEC/shPAR3 cells for 16 h, we observed a near doubling of IL-6 levels in the medium from shPAR3 cells (Fig. 4A). This increase in protein was matched by a similar, ∼2-fold increase in Il6 transcript level (Fig. 4B). To test whether this cytokine can activate STAT3 in our model, NICD1-mMECs were treated with increasing concentrations of recombinant murine IL-6. Treatment with IL-6 induced phosphorylation of STAT3, with activation beginning at some point between 10 and 100 pg/ml IL-6 (Fig. 4C). This result demonstrates that NICD1-mMECs can respond to IL-6 by activating STAT3, as predicted from the hypothesis that loss of PAR3 triggers STAT3 signaling via IL-6 induction.
FIGURE 4.

Increased interleukin-6 production triggers STAT3 signaling after Pard3 knockdown. A, medium was collected from cultures of NICD1-mMECs infected with lentivirus to express the shRNAs indicated and analyzed by ELISA for IL-6 levels (n = 3). B, mRNA was isolated from NICD1-mMECs infected with lentivurus expressing the shRNAs indicated and reverse transcribed into cDNA, and expression of Il6 mRNA was analyzed by qPCR. 18 S rRNA was used as a normalization control (n = 4). C, NICD1-mMECs were treated for 20 min with the indicated concentrations of recombinant IL-6 and then harvested, and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and β-tubulin (loading control). D, mRNA was isolated from NICD1-mMEC/shPAR3 cells infected with lentivirus to express shRNAs against Il6 and reverse transcribed into cDNA, and expression of the genes indicated was analyzed by qPCR. GAPDH was used as a normalization control (n = 3). E, medium was collected from cultures of NICD1-mMEC/shPAR3 cells infected with the shIl6 constructs indicated and analyzed by ELISA for IL-6 levels (n = 3). Error bars, S.E. F, cells infected with lentivirus to express the shRNA constructs indicated were harvested, and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and β-tubulin (loading control). G, quantification of immunoblot shown in E (n = 4). H, NICD1-mMECs expressing shRNAs indicated from lentiviral vectors were treated for 20 min with recombinant IL-6 as indicated, and lysates were immunoblotted for phospho-STAT3, total STAT3, and β-tubulin (loading control). I and J, NICD1-mMECs or NMuMGs expressing the shRNAs indicated from lentiviral vectors and treated with an IL-6-neutralizing antibody as indicated were harvested, and lysates were immunoblotted for phospho-STAT3, total STAT3, and GAPDH (loading control).
We next asked whether IL-6 is necessary for STAT3 activation. We tested several shRNA vectors against the Il6 gene. Two of these hairpins, designated shIl6-1 and shIl6-3, depleted Il6 mRNA and IL-6 cytokine levels in conditioned media by roughly 50% when expressed in NICD1-mMEC/shPAR3 cells (Fig. 4, D and E). Both hairpins significantly reduced STAT3 activation relative to the PAR3 knockdown condition, as assessed by Tyr-705 phosphorylation (Fig. 4, F and G). To prove that the reduction in STAT3 activation was due to reduced IL-6 production, cells were treated with 100 pg/ml recombinant IL-6 in addition to either Il6 hairpin. As expected, treatment with recombinant IL-6 was sufficient to restore STAT3 phosphorylation (Fig. 4H).
To test whether secreted IL-6 is required for STAT3 activation, we used an IL-6-neutralizing antibody. When either PAR3-depleted NICD1-mMEC or NMuMG cells were treated with this antibody, no activation of STAT3 was observed (Fig. 4, I and J). Therefore, loss of PAR3 induces Il6 gene expression, and in an autocrine loop, the secreted IL-6 binds to the GP130/IL-6 receptor and triggers STAT3 activation.
Based on our previous data, we hypothesized that the induction of IL-6 is mediated through aPKCι/λ and tested this idea by expression of a constitutively activated mutant of the kinase, aPKCi-CA. This mutant induced an ∼2-fold increase IL-6 cytokine expression in the cell culture medium (Fig. 5A).
FIGURE 5.

aPKCι/λ activity mediates increased IL-6 production. A, medium was collected from cultures of NICD1-mMECs expressing the constructs indicated and analyzed by ELISA for IL-6 levels (n = 3). B, NICD1-mMEC/shPAR3/shPrkci cells were treated with the indicated concentrations of recombinant murine IL-6 for 20 min and harvested, and then equal amounts of lysates from each were immunoblotted for phospho-STAT3, total STAT3, and β-tubulin (loading control). C, NICD1-mMECs infected with lentiviruses to express the constructs indicated were harvested, and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and β-tubulin (loading control). D, quantitation of phospho-STAT3/total STAT3 shown in C (n = 5). E, cells infected with lentivirus as in C were treated with recombinant IL-6 as indicated for 20 min prior to harvesting, and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and GAPDH (loading control). F–H, NICD1-mMECs, NMuMGs, or Eph4s were infected with lentivirus to express aPKCi-CA, treated with a neutralizing antibody against Il6, and harvested, and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and GAPDH (loading control). Error bars, S.E.
Silencing of aPKCι/λ blocked STAT3 activation (Fig. 5B, leftmost lane), but the addition of recombinant IL-6 reversed this effect in a dose-dependent manner (Fig. 5B), proving that IL-6 stimulation of these cells can restore the STAT3 activation that is lost when Prkci is depleted. To confirm that aPKC activation triggers STAT3 activity through IL-6, we knocked down the Il6 gene in cells expressing aPKCi-CA. Silencing of Il6 eliminated STAT3 activation after aPKCi-CA expression (Fig. 5, C and D). Moreover, treatment of NICD1-mMEC/aPKCi-CA/shIl6 cells with 100 pg/ml recombinant IL-6 restored STAT3 phosphorylation (Fig. 5E).
Finally, we tested whether a neutralizing antibody against IL-6 could prevent STAT3 activation following aPKCi-CA expression. In NICD1-mMEC cells, NMuMG cells, and Eph4 cells, neutralization of IL-6 effectively inhibited STAT3 activation in the context of aPKCi-CA expression (Fig. 5, F–H). We conclude that loss of PAR3 from murine mammary cells activates aPKCι/λ, leading to production of IL-6 and activation of STAT3 via GP130.
aPKCι/λ-mediated NF-κB Activation Triggers STAT3 Signaling
One possible mechanism for induction of IL-6 by aPKCι/λ is through activation of NF-κB signaling. aPKCι/λ and aPKCζ interact with and activate multiple components of the NF-κB pathway (39). aPKCι/λ-mediated activation of NF-κB induces IL-6 production in prostate cancer (23), and silencing of PAR3 can activate NF-κB signaling in human intestinal epithelial cells (40). Moreover, the degree of IL-6 induction reported in prostate tumor cells following aPKC-mediated NF-κB activation is similar to what we have observed in mammary cells (23). To test whether activation of STAT3 requires NF-κB signaling in out system, we first treated NICD1-mMEC/shPAR3 cells with two chemical inhibitors of NF-κB, JSH-23 and caffeic acid phenethylester (CAPE). Both JSH-23 and CAPE eliminated the STAT3 phosphorylation triggered by loss of PAR3 (Fig. 6, A and B). In addition, treatment of cells with recombinant IL-6 along with either JSH-23 or CAPE restored STAT3 activation (Fig. 6C), strongly suggesting that NF-κB activity induces IL-6 following loss of PAR3. This result was also observed in NMuMG cells (Fig. 6D), suggesting that it is a general mechanism in mammary cells.
FIGURE 6.

STAT3 activation following aPKCι/λ activation is mediated by NF-κB signaling. A, NICD1-mMECs infected with lentivirus to express the shRNAs indicated and treated with the NF-κB inhibitors indicated at 30 mg/ml were harvested, and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and GAPDH (loading control). B, quantitation of phospho-STAT3/total STAT3 from A (n = 6). C, NICD1-mMEC/shPAR3 cells were treated with the NF-κB inhibitors indicated at 30 mg/ml and with recombinant IL-6, as indicated, and harvested, and equal amounts of lysate were immunoblotted for phospho-STAT3, total STAT3, and GAPDH (loading control). D, as in E, except that NMuMG cells were used. E, medium was collected from cultures of NICD1-mMECs infected with lentivirus to express shRNAs as indicated with or without treatment with CAPE and analyzed by ELISA for IL-6 levels (n = 3). F, as in C, except that NMuMG cells were used. G, NICD1-mMECs infected with lentivirus to express the shRNAs indicated were harvested, and equal amounts of lysate were immunoblotted for IκBα, phospho-IKK (which detects both α and β isoforms; β runs slightly higher), total IKKα, total IKKβ, phospho-p65/RELA, total p65/RELA, and GAPDH (loading control). H, NICD1-mMECs expressing the shRNA constructs indicated were fixed and immunostained for anti-p65/RELA and stained with phalloidin for actin and DAPI for DNA. Images were taken using a ×20 objective. I, NICD1-mMECs infected with lentiviruses to express the shRNA constructs indicated were calcium phosphate-transfected with an NF-κB reporter plasmid and with constitutively expressed Renilla luciferase (normalization control). Cells were lysed 24 h after transfection, and luciferase intensity was measured (n = 4). J, NICD1-mMECs infected with lentivirus expressing the constructs indicated were transfected with luciferase plasmids and analyzed as in I (n = 4). K, NICD1-mMECs infected with lentivirus expressing the shRNAs indicated were harvested, and equal amounts of lysate were immunoblotted for total IκBα, phospho-STAT3, total STAT3, and GAPDH (loading control). Error bars, S.E.
If our hypothesis is correct that activation of NF-κB mediations increases IL-6 production in mammary cells, inhibition of NF-κB signaling should prevent increased production of IL-6. Therefore, we treated both NICD1-mMEC cells with the NF-κB inhibitor CAPE in addition to silencing PAR3. In both cases, CAPE treatment prevented PAR3 silencing from triggering a rise in IL-6 cytokine production (Fig. 6, E and F).
We next asked directly whether the NF-κB pathway is activated after depletion of PAR3. Following loss of PAR3, the IκBα subunit is degraded (Fig. 6G), phosphorylation of both IKKβ and -α is increased, and p65 phosphorylation is slightly increased (Fig. 6G). These effects were all reversed by concurrent knockdown of aPKCι/λ (Fig. 6G). Additionally, we observed that the p65 subunit of NF-κB relocates to the nucleus of NICD1-mMEC cells after PAR3 silencing (Fig. 6H). To confirm transcriptional activation of NF-κB, cells were transfected with an NF-κB luciferase reporter plasmid (30). Consistent with our prior results, loss of PAR3 induced expression of the luciferase gene, and concurrent silencing of aPKCι/λ reverted luciferase expression toward baseline (Fig. 6I). In addition, expression of aPKCi-CA induced this luciferase construct (Fig. 6J), demonstrating that aPKC activation increases NF-κB signaling in these cells. Finally, we knocked down the Nfkbia gene, which encodes for the murine IκBα protein, and saw activation of STAT3 (Fig. 6K). We conclude that loss of PAR3 triggers NF-κB activity via aPKCι/λ and that NF-κB induces IL-6 expression and activation of STAT3 (Fig. 7), which, as we have shown previously, can promote invasive behavior and metastasis of NICD1-MECs (15).
FIGURE 7.
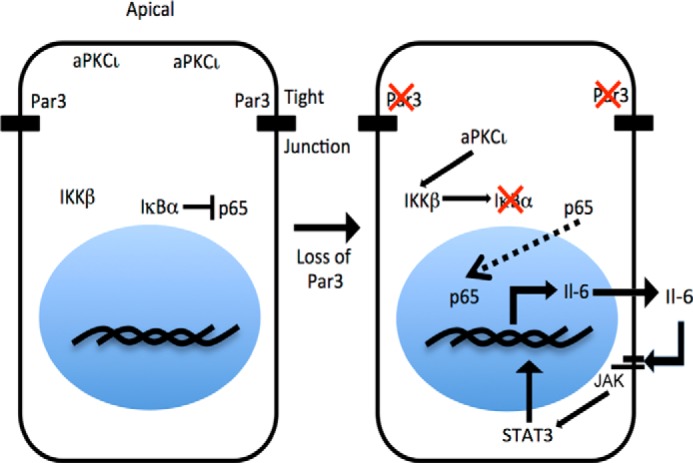
Model for activation of IL-6 production following loss of PAR3. When PAR3 is normally expressed, aPKCι/λ is restricted to the apical cortex, and IκBα binds to and restrains p65/RELA in the nucleus. When PAR3 expression is sufficiently diminished, aPKCι/λ diffuses in the cytoplasm and becomes activated. Activation of aPKCι/λ leads to activation of IKKβ, which phosphorylates IκBα and marks IκBα for degradation. Degradation of IκBα frees p65/RelA to enter the nucleus and induce transcription of target genes, including Il6. Increased Il6 transcription leads to production of the Il6 cytokine, which activates STAT3 through GP130.
DISCUSSION
In this study, we found that loss of PAR3 triggers STAT3 signaling through activation of aPKCι/λ. We propose that a major function of PAR3 is to restrict the activity of multiple signaling pathways, of which aPKC is one important example. Loss of PAR3 will result in inappropriate activity along these pathways, with sometimes deleterious consequences (15). The potential of polarity proteins as tumor suppressors has been recognized since early studies in Drosophila development found that they restrain tissue proliferation (41). Loss of polarity genes in Drosophila can also synergize with oncogenes to generate tumors that invade and metastasize aggressively (10, 42, 43). Although Pard3 is mutated or its expression is altered in several human cancers (44, 45), experimental confirmation for PAR3 as a mammalian tumor suppressor has only recently been reported (14–16). The molecular mechanisms underlying this tumor suppressor function remain partially obscure. The present work and previous studies (15) demonstrate that constitutive activation of aPKC can occur in the absence of PAR3. Functional significance of this aPKC activation was only established by our present work because prior studies relied on pharmacological aPKC inhibitors that have significant nonspecific activity (15, 35, 36). We propose that the tumor suppressor function of PAR3 stems, at least in part, from its role in restricting the activity of aPKC. This model is consistent with the reported oncogenic activity of aPKC (8, 46, 47).
Our data establish aPKC isoforms as key mediators of the increased malignancy that can be triggered by PAR3 silencing. In some aspects, this mechanism recapitulates what is seen in Drosophila models, where the loss of polarity genes cooperates with oncogenes to generate large, metastatic tumors (43, 48). However, the mechanism we propose differs from the Drosophila models in several important ways. First, although DaPKC may drive tumor growth following disruption of polarity (49), it is not known to do so through the fly NF-κB homologue. Second, the loss of Scrib plus RAS activation induces Upd genes, which are the fly homologues of IL-6 family cytokines (10), but this occurs though JNK signaling. We speculate that although some signaling components that become active following polarity disruption are conserved, the connectivities between these components are different in flies and mammals.
The NF-κB signaling pathway has been implicated in numerous human cancers (50). The canonical NF-κB pathway involves activation of IKKs, which phosphorylate IκBα and mark it for degradation. The degradation of IκBα frees p65/RELA and p50 protein dimers to translocate to the nucleus, where the dimers induce many target genes. The role of aPKC isoforms in NF-κB signaling has been appreciated for some time (51–53). In particular, both aPKCι/λ and aPKCζ have been shown to interact with and activate IKKβ (22, 54). Prior studies have also demonstrated that activated NF-κB induces IL-6 to promote malignancy (23, 55). These reports are consistent with our data, which indicate that loss of PAR3 induces preferential phosphorylation of IKKβ (Fig. 6D) and subsequent IL-6 production. Moreover, the magnitude of IL-6 induction that we observe in NICD1-mMEC cells (Figs. 4 (A and B), 5A, and 6E) is similar to the IL-6 induction reported following activation of aPKC and NF-κB in a prostate cancer model (23).
The loss of PAR3 has been reported to activate NF-κB in the Caco-2 cell line (40). However, in contrast with our results, NF-κB activation in Caco-2 cells was not mediated by active aPKCι/λ but rather was inhibited by it. The reasons for this discrepancy are not readily apparent, but different species and tissue origins of the cells used may be contributing factors. Another study recently demonstrated that polarity proteins, including PAR3, are required for NF-κB induction in MDCK cells exposed to Pseudomonas aeruginosa (56). When P. aeruginosa touches the apical membrane, it induces membrane protrusions that have basolateral characteristics, with PAR3 localizing to the boundary of the normal membrane and the basal-like protrusion. This process is associated with activation of NF-κB signaling in the host cell. If PAR3 is silenced, the NF-κB response is blunted, suggesting that in this context, PAR3 organizes signaling molecules that induce NF-κB. However, the role of aPKC was not investigated in this study.
Based on our results and prior reports, we propose a model for how loss of PAR3 initiates STAT3 signaling (Fig. 7). The present study confirms our prior report that loss of PAR3 activates aPKC in mouse mammary epithelial cells (15). The mislocalization of apical aPKC enrichment following PAR3 silencing has been described previously both by us (15) and others (16, 18). Therefore, we propose that the loss of PAR3 enables the inappropriate interaction of active aPKC with signaling networks, such as NF-κB, in mammary cells. This miswiring of signaling is common to wild type primary murine mammary cells, NICD-transformed mammary cells, and two independent, untransformed mouse mammary cell lines, a strong argument that it is a default response to the loss of PAR3 in mammary epithelia. It seems likely that a similar type of miswiring could occur in other epithelial tissues as a result of the mutation of PAR3 or suppression of PAR3 expression. Further experimentation will be required to test this theory.
In conclusion, the present study has uncovered a mechanism through which loss of the PAR3 polarity protein activates autocrine IL-6 signaling and triggers STAT3 activity. Important events downstream of PAR3 are mediated by aPKC, suggesting that restricting the activity of aPKC is a major tumor suppressor function for PAR3.
Acknowledgments
We thank Fiona Yull (Vanderbilt University) for generously providing reagents and Alex Andrews (graduate student in the Macara group) for assistance with molecular cloning for this project.
This work was supported by National Institutes of Health Grants CA132898 to Ian Macara, and T32-GM07347 to the Vanderbilt Medical-Scientist Training Program.
- aPKC
- atypical protein kinase C
- mMEC
- mouse mammary epithelial cell
- tRFP
- TurboRFP
- CA
- constitutively active
- qPCR
- quantitative PCR
- CAPE
- caffeic acid phenethylester
- IKK
- IκB kinase.
REFERENCES
- 1. Korpal M., Ell B. J., Buffa F. M., Ibrahim T., Blanco M. A., Celià-Terrassa T., Mercatali L., Khan Z., Goodarzi H., Hua Y., Wei Y., Hu G., Garcia B. A., Ragoussis J., Amadori D., Harris A. L., Kang Y. (2011) Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat. Med. 17, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ling C., Su V.-M.-T., Zuo D., Muller W. J. (2012) Loss of the 14-3-3σ tumor suppressor is a critical event in ErbB2-mediated tumor progression. Cancer Discov. 2, 68–81 [DOI] [PubMed] [Google Scholar]
- 3. Zhuang Z., Wang K., Cheng X., Qu X., Jiang B., Li Z., Luo J., Shao Z., Duan T. (2013) LKB1 inhibits breast cancer partially through repressing the Hedgehog signaling pathway. PLoS One 8, e67431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galvez A. S., Duran A., Linares J. F., Pathrose P., Castilla E. A., Abu-Baker S., Leitges M., Diaz-Meco M. T., Moscat J. (2009) Protein kinase Cζ represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol. Cell. Biol. 29, 104–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benhamouche S., Curto M., Saotome I., Gladden A. B., Liu C.-H., Giovannini M., McClatchey A. I. (2010) Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 24, 1718–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhan L., Rosenberg A., Bergami K. C., Yu M., Xuan Z., Jaffe A. B., Allred C., Muthuswamy S. K. (2008) Deregulation of Scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 135, 865–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chatterjee S. J., McCaffrey L. (2014) Emerging role of cell polarity proteins in breast cancer progression and metastasis. Breast Cancer 6, 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Regala R. P., Weems C., Jamieson L., Copland J. A., Thompson E. A., Fields A. P. (2005) Atypical protein kinase Cι plays a critical role in human lung cancer cell growth and tumorigenicity. J. Biol. Chem. 280, 31109–31115 [DOI] [PubMed] [Google Scholar]
- 9. Nolan M. E., Aranda V., Lee S., Lakshmi B., Basu S., Allred D. C., Muthuswamy S. K. (2008) The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res. 68, 8201–8209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu M., Pastor-Pareja J. C., Xu T. (2010) Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 463, 545–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agrawal N., Kango M., Mishra A., Sinha P. (1995) Neoplastic transformation and aberrant cell-cell interactions in genetic mosaics of lethal(2) giant larvae (lgl), a tumor suppressor gene of Drosophila. Dev. Biol. 172, 218–229 [DOI] [PubMed] [Google Scholar]
- 12. Timmons L., Hersperger E., Woodhouse E., Xu J., Liu L.-Z., Shearn A. (1993) The expression of the Drosophila awd gene during normal development and in neoplastic brain tumors caused by lgl mutations. Dev. Biol. 158, 364–379 [DOI] [PubMed] [Google Scholar]
- 13. Russ A., Louderbough J. M. V., Zarnescu D., Schroeder J. A. (2012) Hugl1 and Hugl2 in mammary epithelial cells: polarity, proliferation, and differentiation. PLoS One 7, e47734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xue B., Krishnamurthy K., Allred D. C., Muthuswamy S. K. (2013) Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nat. Cell Biol. 15, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCaffrey L. M., Montalbano J., Mihai C., Macara I. G. (2012) Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 22, 601–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iden S., van Riel W. E., Schäfer R., Song J.-Y., Hirose T., Ohno S., Collard J. G. (2012) Tumor type-dependent function of the Par3 polarity protein in skin tumorigenesis. Cancer Cell 22, 389–403 [DOI] [PubMed] [Google Scholar]
- 17. McCaffrey L. M., Macara I. G. (2009) The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev. 23, 1450–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hao Y., Du Q., Chen X., Zheng Z., Balsbaugh J. L., Maitra S., Shabanowitz J., Hunt D. F., Macara I. G. (2010) Par3 controls epithelial spindle orientation by aPKC-mediated phosphorylation of apical pins. Curr. Biol. 20, 1809–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barbieri I., Pensa S., Pannellini T., Quaglino E., Maritano D., Demaria M., Voster A., Turkson J., Cavallo F., Watson C. J., Provero P., Musiani P., Poli V. (2010) Constitutively active STAT3 enhances Neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 70, 2558–2567 [DOI] [PubMed] [Google Scholar]
- 20. Yu H., Pardoll D., Jove R. (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 9, 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fields A. P., Regala R. P. (2007) Protein kinase Cι: human oncogene, prognostic marker and therapeutic target. Pharmacol. Res. 55, 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lallena M.-J., Diaz-Meco M. T., Bren G., Payá C. V., Moscat J. (1999) Activation of IκB kinase β by protein kinase C isoforms. Mol. Cell. Biol. 19, 2180–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishiguro H., Akimoto K., Nagashima Y., Kojima Y., Sasaki T., Ishiguro-Imagawa Y., Nakaigawa N., Ohno S., Kubota Y., Uemura H. (2009) aPKCλ/ι promotes growth of prostate cancer cells in an autocrine manner through transcriptional activation of interleukin-6. Proc. Natl. Acad. Sci. U.S.A. 106, 16369–16374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chung J., Uchida E., Grammer T. C., Blenis J. (1997) STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 17, 6508–6516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Litherland G. J., Elias M. S., Hui W., Macdonald C. D., Catterall J. B., Barter M. J., Farren M. J., Jefferson M., Rowan A. D. (2010) Protein kinase C isoforms ζ and ι mediate collagenase expression and cartilage destruction via STAT3- and ERK-dependent c-fos induction. J. Biol. Chem. 285, 22414–22425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kusne Y., Carrera-Silva E. A., Perry A. S., Rushing E. J., Mandell E. K., Dietrich J. D., Errasti A. E., Gibbs D., Berens M. E., Loftus J. C., Hulme C., Yang W., Lu Z., Aldape K., Sanai N., Rothlin C. V., Ghosh S. (2014) Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFα in glioblastoma. Sci. Signal. 7, ra75–ra75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uberall F., Hellbert K., Kampfer S., Maly K., Villunger A., Spitaler M., Mwanjewe J., Baier-Bitterlich G., Baier G., Grunicke H. H. (1999) Evidence that atypical protein kinase C-λ and atypical protein kinase C-ζ participate in Ras-mediated reorganization of the F-actin cytoskeleton. J. Cell Biol. 144, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bouras T., Pal B., Vaillant F., Harburg G., Asselin-Labat M.-L., Oakes S. R., Lindeman G. J., Visvader J. E. (2008) Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 3, 429–441 [DOI] [PubMed] [Google Scholar]
- 29. Starnes H. F., Jr., Pearce M. K., Tewari A., Yim J. H., Zou J. C., Abrams J. S. (1990) Anti-IL-6 monoclonal antibodies protect against lethal Escherichia coli infection and lethal tumor necrosis factor-α challenge in mice. J. Immunol. 145, 4185–4191 [PubMed] [Google Scholar]
- 30. Everhart M. B., Han W., Sherrill T. P., Arutiunov M., Polosukhin V. V., Burke J. R., Sadikot R. T., Christman J. W., Yull F. E., Blackwell T. S. (2006) Duration and intensity of NF-κB activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 176, 4995–5005 [DOI] [PubMed] [Google Scholar]
- 31. Jones S. A., Scheller J., Rose-John S. (2011) Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Invest. 121, 3375–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dauer D. J., Ferraro B., Song L., Yu B., Mora L., Buettner R., Enkemann S., Jove R., Haura E. B. (2005) Stat3 regulates genes common to both wound healing and cancer. Oncogene 24, 3397–3408 [DOI] [PubMed] [Google Scholar]
- 33. Kim E., Kim M., Woo D.-H., Shin Y., Shin J., Chang N., Oh Y. T., Kim H., Rheey J., Nakano I., Lee C., Joo K. M., Rich J. N., Nam D.-H., Lee J. (2013) Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23, 839–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hammacher A., Richardson R. T., Layton J. E., Smith D. K., Angus L. J. L., Hilton D. J., Nicola N. A., Wijdenes J., Simpson R. J. (1998) The immunoglobulin-like module of gp130 is required for signaling by interleukin-6, but not by leukemia inhibitory factor. J. Biol. Chem. 273, 22701–22707 [DOI] [PubMed] [Google Scholar]
- 35. Lee A. M., Kanter B. R., Wang D., Lim J. P., Zou M. E., Qiu C., McMahon T., Dadgar J., Fischbach-Weiss S. C., Messing R. O. (2013) Prkcz null mice show normal learning and memory. Nature 493, 416–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Volk L. J., Bachman J. L., Johnson R., Yu Y., Huganir R. L. (2013) PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 493, 420–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taniguchi K., Karin M. (2014) IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 26, 54–74 [DOI] [PubMed] [Google Scholar]
- 38. Guo Y., Xu F., Lu T., Duan Z., Zhang Z. (2012) Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 38, 904–910 [DOI] [PubMed] [Google Scholar]
- 39. Wooten M. W. (1999) Function for NF-κB in neuronal survival: regulation by atypical protein kinase C. J. Neurosci. Res. 58, 607–611 [DOI] [PubMed] [Google Scholar]
- 40. Forteza R., Wald F. A., Mashukova A., Kozhekbaeva Z., Salas P. J. (2013) Par-complex aPKC and Par3 cross-talk with innate immunity NF-κB pathway in epithelial cells. Biol. Open 2, 1264–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gateff E., Schneiderman H. A. (1974) Developmental capacities of benign and malignant neoplasms of Drosophila. Wilhelm Roux Arch. 10.1007/BF00577830 [DOI] [PubMed] [Google Scholar]
- 42. Cordero J. B., Macagno J. P., Stefanatos R. K., Strathdee K. E., Cagan R. L., Vidal M. (2010) Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev. Cell. 18, 999–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pagliarini R. A., Xu T. (2003) A genetic screen in Drosophila for metastatic behavior. Science 302, 1227–1231 [DOI] [PubMed] [Google Scholar]
- 44. Rothenberg S. M., Mohapatra G., Rivera M. N., Winokur D., Greninger P., Nitta M., Sadow P. M., Sooriyakumar G., Brannigan B. W., Ulman M. J., Perera R. M., Wang R., Tam A., Ma X.-J., Erlander M., Sgroi D. C., Rocco J. W., Lingen M. W., Cohen E. E. W., Louis D. N., Settleman J., Haber D. A. (2010) A genome-wide screen for microdeletions reveals disruption of polarity complex genes in diverse human cancers. Cancer Res. 70, 2158–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zen K., Yasui K., Gen Y., Dohi O., Wakabayashi N., Mitsufuji S., Itoh Y., Zen Y., Nakanuma Y., Taniwaki M., Okanoue T., Yoshikawa T. (2009) Defective expression of polarity protein PAR-3 gene (PARD3) in esophageal squamous cell carcinoma. Oncogene 28, 2910–2918 [DOI] [PubMed] [Google Scholar]
- 46. Takagawa R., Akimoto K., Ichikawa Y., Akiyama H., Kojima Y., Ishiguro H., Inayama Y., Aoki I., Kunisaki C., Endo I., Nagashima Y., Ohno S. (2010) High expression of atypical protein kinase C λ/ι in gastric cancer as a prognostic factor for recurrence. Ann. Surg. Oncol. 17, 81–88 [DOI] [PubMed] [Google Scholar]
- 47. Linch M., Sanz-Garcia M., Rosse C., Riou P., Peel N., Madsen C. D., Sahai E., Downward J., Khwaja A., Dillon C., Roffey J., Cameron A. J. M., Parker P. J. (2014) Regulation of polarized morphogenesis by protein kinase C iota in oncogenic epithelial spheroids. Carcinogenesis 35, 396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brumby A. M., Richardson H. E. (2003) scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22, 5769–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leong G. R., Goulding K. R., Amin N., Richardson H. E., Brumby A. M. (2009) scribble mutants promote aPKC and JNK-dependent epithelial neoplasia independently of Crumbs. BMC Biol. 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bassères D. S., Baldwin A. S. (2006) Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 25, 6817–6830 [DOI] [PubMed] [Google Scholar]
- 51. Ghosh S., Baltimore D. (1990) Activation in vitro of NF-κB. Nature 344, 678–682 [DOI] [PubMed] [Google Scholar]
- 52. Shirakawa F., Mizel S. B. (1989) In vitro activation and nuclear translocation of NF-κB catalyzed by cyclic AMP-dependent protein kinase and protein kinase C. Mol. Cell. Biol. 9, 2424–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dominguez I., Sanz L., Arenzana-Seisdedos F., Diaz-Meco M. T., Virelizier J. L., Moscat J. (1993) Inhibition of protein kinase C zeta subspecies blocks the activation of an NF-κB-like activity in Xenopus laevis oocytes. Mol. Cell. Biol. 13, 1290–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Win H. Y., Acevedo-Duncan M. (2008) Atypical protein kinase C phosphorylates IKKαβ in transformed non-malignant and malignant prostate cell survival. Cancer Lett. 270, 302–311 [DOI] [PubMed] [Google Scholar]
- 55. Maeda S., Hikiba Y., Sakamoto K., Nakagawa H., Hirata Y., Hayakawa Y., Yanai A., Ogura K., Karin M., Omata M. (2009) IκB kinase β/nuclear factor-κB activation controls the development of liver metastasis by way of interleukin-6 expression. Hepatology 50, 1851–1860 [DOI] [PubMed] [Google Scholar]
- 56. Tran C. S., Eran Y., Ruch T. R., Bryant D. M., Datta A., Brakeman P., Kierbel A., Wittmann T., Metzger R. J., Mostov K. E., Engel J. N. (2014) Host cell polarity proteins participate in innate immunity to Pseudomonas aeruginosa infection. Cell Host Microbe 15, 636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]