

Background: The mechanism of filamin Ser2152 phosphorylation by PKA is unclear.
Results: Autoinhibitory filamin is resistant to phosphorylation despite exposed Ser2152, but ligand binding alters the filamin conformation, triggering PKA recognition.
Conclusion: Filamin Ser2152 phosphorylation is conformation-dependent on ligand binding.
Significance: The overall conformation of substrate, not just the exposed phosphorylation site, regulates the kinase substrate recognition in signaling.
Keywords: Enzyme Catalysis, Filamin, Nuclear Magnetic Resonance (NMR), Protein Kinase A (PKA), Protein Phosphorylation, Serine/Threonine Protein Kinase
Abstract
Protein phosphorylation mediates essentially all aspects of cellular life. In humans, this is achieved by ∼500 kinases, each recognizing a specific consensus motif (CM) in the substrates. The majority of CMs are surface-exposed and are thought to be accessible to kinases for phosphorylation. Here we investigated the archetypical protein kinase A (PKA)-mediated phosphorylation of filamin, a major cytoskeletal protein that can adopt an autoinhibited conformation. Surprisingly, autoinhibited filamin is refractory to phosphorylation by PKA on a known Ser2152 site despite its CM being exposed and the corresponding isolated peptide being readily phosphorylated. Structural analysis revealed that although the CM fits into the PKA active site its surrounding regions sterically clash with the kinase. However, upon ligand binding, filamin undergoes a conformational adjustment, allowing rapid phosphorylation on Ser2152. These data uncover a novel ligand-induced conformational switch to trigger filamin phosphorylation. They further suggest a substrate shape-dependent filtering mechanism that channels specific exposed CM/kinase recognition in diverse signaling responses.
Introduction
Phosphorylation is a biochemical phenomenon discovered more than a century ago (1). Since then, it has been intensively studied and established as a pivotal posttranslational modification step to control nearly all cellular processes such as metabolism, cytoskeleton remodeling, membrane trafficking, muscle contraction, and immune responses (2, 3). In a eukaryotic cell, >30% of total proteins are predicted to be phosphorylated on at least one residue (4, 5). Given that the human genome encodes ∼500 kinases (6), it was estimated that there would be ∼700,000 potential phosphorylation sites (P-sites)2 for any given kinase (7). However, at physiological conditions, a kinase may only phosphorylate several to hundreds of P-sites (8, 9). How kinases confer their exquisite specificity has been the subject of extensive studies for many decades (for selective reviews, see Refs. 5, 7, 10, and 11). It is now well understood that a kinase can be recruited to a subcellular compartment or directly to its substrate site to increase the probability of the kinase/substrate encounter (7). It is also understood that upon localization kinases may utilize their distinct active sites to recognize linear P-site-containing consensus motifs (CMs) in the substrates (5, 7, 10, 12, 13). Although distal site interactions may also enhance the kinase/substrate interaction in some cases, the CM-based recognition is considered to be the universal mechanism in all kinases to critically confer substrate specificity (7, 10). Consequently, significant effort has been made to build CM databases to help identify potential P-sites in many cellular proteins (14–17).
A large body of structural and functional data has shown that kinases can exhibit latent versus active conformational states to switch off versus switch on their binding to substrates, respectively, for regulating a variety of signaling pathways (18–21). By contrast, much less has been elucidated for the reverse, i.e. the gamut of global conformational changes within substrates that are required to accommodate the kinase binding to CM. This may be partly due to the fact that vast majority of CMs are exposed and located in disordered regions or flexible loops, leading to the assumption that they may readily fit into the kinase active sites without constraints of global substrate conformations (22–28). A small portion of substrates have buried or ordered CMs, which require local unfolding to allow the CM access to kinases as alluded by some recent studies (28–30). However, the role of global substrate conformation in dictating the disordered CM access to kinases remains completely unexplored.
In this study, we attempted to address this issue by using a combination of biochemical, functional, protein engineering, and structural approaches. We focused on analyzing the phosphorylation of filamin, a large actin-cross-linking protein (280 kDa) containing two N-terminal actin binding domains and 24 contiguous immunoglobulin (Ig)-like repeats that dimerize through its 24th Ig repeat (31–33). Selective filamin Ig repeats 4, 9, 12, 17, 19, 21, and 23 (Class A repeats) have been shown to engage with a wide range of ligands such as transmembrane receptors and cytoskeletal adaptors (34–36), thereby regulating a variety of cytoskeleton-related events such as cell attachment, cell shape change, and cell motility (31–33). It is well known that filamin can be phosphorylated at Ig20 Ser2152 by cAMP-dependent protein kinase A (PKA) (37), which significantly impacts filamin function (38–42). Interestingly, Ig20 can also structurally mask the ligand binding site on Ig21, resulting in an autoinhibited conformation (43). In the crystal structure of the autoinhibited filamin A (FLNa) Ig19–21 (43), Ig20 appears to be highly dynamic with several parts having weak or no electron densities (Protein Data Bank code 2J3S). Although a segment (Val2140–Arg2147) in Ig20 occupies the ligand binding site of Ig21 to form the autoinhibited conformation, the P-site Ser2152 is located in a nearby exposed loop containing the PKA CM sequence, i.e. 2148RRAPSVA2154 (44) (see Fig. 1A). Therefore, FLNa Ig19–21 serves as an ideal case to examine whether and how the variable three-dimensional substrate conformations of FLNa Ig19–21 affect its exposed CM phosphorylation and signaling.
FIGURE 1.
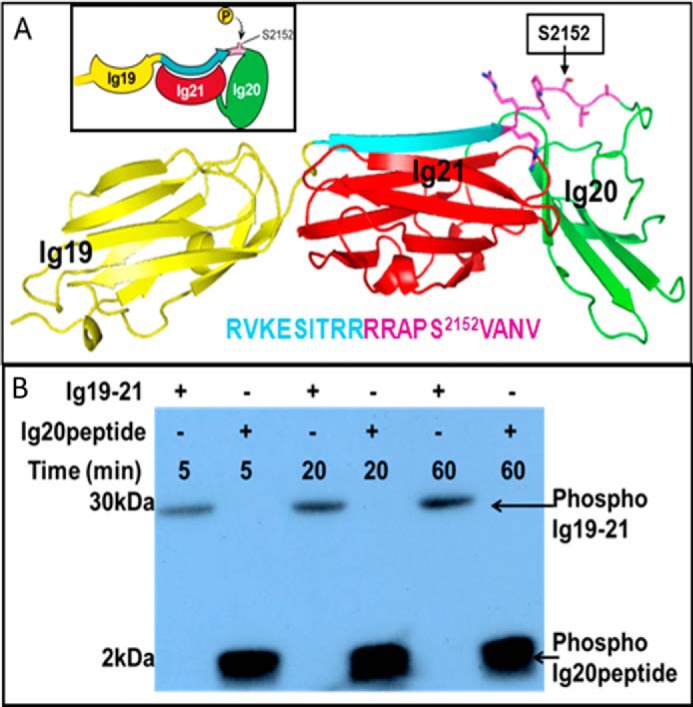
Autoinhibited conformation of FLNa Ig19–21 prevents Ser2152 phosphorylation by PKA. A, crystal structure of FLNa Ig19–21 (Protein Data Bank code 2J3S) showing the highly exposed CM in Ig20, 2148RRAPSVA2154 (pink), and its preceding autoinhibitory segment, Val2140–Arg2147 (cyan). Ig19 and Ig20 are colored in yellow and green, respectively. The corresponding peptide sequence (color-coded) is also shown. B, 32P autoradiogram of FLNa Ig19–21 and the Ig20 Arg2139–Val2156 peptide at equimolar ratios (10 μm) at various reaction times showing the weak phosphorylation of the former but the potent saturated phosphorylation of the latter by PKA at the 5-min point.
EXPERIMENTAL PROCEDURES
Plasmids and Reagents
Human filamin A (UniProt P21333) immunoglobulin domains Ig19–21 (amino acids 2045–2329) and Ig16–24 (amino acids 1772–2647) were cloned into pGST-parallel vectors and purified as described earlier (34). The “locked” and “active” mutants of filamin were made using the Agilent QuikChange II XL site-directed mutagenesis kit using primers (Integrated DNA Technologies) that changed the residues “VKESITRRRRAPS” in the autoinhibition loop of repeat 20 to “FRSSLFLWVRAPS” (locked) and “VKEAAARRRRAPS” (active). GST-fused proteins were induced overnight at room temperature in Escherichia coli BL21(DE3) using 0.3 mm isopropyl 1-thio-β-d-galactopyranoside. The fusion protein was purified using glutathione resin, and the filamin Ig fragments were released from GST using tobacco etch virus protease. Free GST was removed from the resultant mixture by binding it to glutathione resin to obtain >95% pure filamin Ig fragments. The buffer conditions, 15N labeling, and other procedures are described in detail in earlier work (34). The final purified proteins have a GAMDP sequence at the N terminus derived from the fusion tag. An optional anion exchange step using a ResourceQ (GE Healthcare) column was incorporated for Ig19–21 and Ig16–24 proteins when the fusion tag GST could not be eliminated by glutathione affinity and gel filtration chromatography. All proteins were greater than 95% pure on Coomassie-stained SDS-PAGE. The antibodies for phosphofilamin A (Ser2152) and filamin A were obtained from Cell Signaling Technology (Danvers, MA).
Protein Phosphorylation
Murine PKA was purchased from New England Biolabs and used with minor changes to the reaction conditions. The assay conditions for PKA reaction were 50 mm Tris, pH 7.5, 10 mm MgCl2, 10 μm filamin fragments as substrate, and 500 μm ATP and [γ-32P]ATP for autoradiography. [γ-32P]ATP was eliminated from the kinase reactions for the Western blots. 50/200 μm filamin binding peptides of β7 integrin/migfilin/GPIbα-M was used to release autoinhibition. Assays with only peptide as substrates had 10 or 50 μm peptide. For each 100-μl reaction, 1000 units of PKA was used. Protein and peptide phosphorylation was detected by Western blotting and autoradiography.
Peptide Synthesis
The following peptides were synthesized by the Biotechnology Core at the Lerner Research Institute of the Cleveland Clinic: 1) migfilin, MASKEPKRVASSVFITLAPPRRDV; 2) integrin β7, WKQDSNPLYKSAITTTINPRFQEADSPTL; 3) WT Ig20, RVKESITRRRRAPSVANV; 4) GPIbα, LRGSLPTFRSSLFLWVRPNGRV; 5) GPIbα-M, LRGSLPTFRSSLALAVRPNGRV; and 6) control GPIbα-filamin hybrid, LFLWVRAPSVANV. All the peptides were purified by HPLC and were >95% pure; their masses were confirmed by mass spectroscopy. Peptides were estimated using their extinction coefficient at A280 using ExPASy ProtParam. In cases where the peptide did not have a tryptophan residue, the thoroughly lyophilized peptide was weighed carefully, and 85% purity was assumed to estimate concentration.
Phosphorylation Analysis of Filamin by Mass Spectrometry
Filamin Ig16–24 was phosphorylated by PKA for 1 h in conditions described above at 50 μm. PKA-phosphorylated filamin Ig16–24 and locked Ig16–24 were excised from Coomassie-stained PAGE gels. For the protein digestion, the bands were cut to minimize excess polyacrylamide and divided into a number of smaller pieces. The gel pieces were washed with water and dehydrated in acetonitrile. The bands were then reduced with DTT and alkylated with iodoacetamide prior to in-gel digestion. All bands were digested in gel using trypsin (5 μl; 10 ng/μl or 20 μl; 25 ng/μl) or Glu-C (20 μl; 50 ng/μl) in 50 mm ammonium bicarbonate and incubated overnight at room temperature to achieve complete digestion. The peptides that were formed were extracted from the polyacrylamide in two aliquots of 30 μl of 50% acetonitrile with 5% formic acid. These extracts were combined and evaporated to <10 μl in a SpeedVac and then resuspended in 1% acetic acid to make up a final volume of ∼30 μl for LC-MS analysis.
The LC-MS system was a Finnigan LTQ linear ion trap mass spectrometer system. The HPLC column was a self-packed 9-cm × 75-μm-inner diameter Phenomenex Jupiter C18 reversed-phase capillary chromatography column. Ten-microliter volumes of the extract were injected, and the peptides eluted from the column by an acetonitrile and 0.1% formic acid gradient at a flow rate of 0.25 μl/min were introduced into the source of the mass spectrometer on line. The data were analyzed by using all collision-induced dissociation spectra collected in the experiment to search the NCBI non-redundant database with the search program Mascot using a human taxonomy filter. All matching spectra were verified by manual interpretation. The interpretation process was aided by additional searches using the programs Sequest and BLAST as needed.
NMR Spectroscopy
HSQC spectra of all 15N-labeled protein samples were recorded in a Bruker Avance 600 MHz spectrometer at 30 °C. 15N-labeled proteins were extensively buffer-exchanged into 25 mm sodium phosphate, pH 6.4, 5 mm NaCl, and 1 mm DTT. All spectra were recorded using 0.1 mm Ig filamin repeats. Ligand-bound Ig spectra were recorded with a 2 m excess of peptides (0.2 mm) making sure that the pH did not change by more than 0.02 unit on addition of the ligand peptide. Spectral processing and analysis were done using NMRPipe and NMRView (45).
Isothermal Titration Calorimetry
A MicroCal iTC200 calorimeter from GE Healthcare was used for determining ligand affinities to filamin Ig repeats. Purified proteins were extensively buffer-exchanged into 25 mm sodium phosphate, pH 6.4, 5 mm NaCl, and 1 mm DTT. Ligand peptides were dissolved in the same buffer and estimated as described earlier (34). 50 μm protein in the sample cell was titrated against 1 mm peptide in the syringe at 30 °C in 1-μl increments. Affinities were determined by fitting the heat changes to a one-site binding model using the associated Origin package.
Solution Structural Analyses by Small Angle X-ray Scattering (SAXS)
Small angle x-ray scattering of various Ig19–21 filamin A protein fragments was done in 50 mm Tris, pH 7.5, 5 mm NaCl, and 1 mm DTT at a protein concentration of 2 mg/ml. Ligand peptides when present were at a 4-fold molar excess over Ig19–21. A typical experiment had 60 μm FLNa Ig19–21 and 250 μm ligand peptide. Each sample was also diluted 2× and 4× from this initial concentration, and SAXS data were collected to rule out aggregation or unusual oligomerization if any. X-ray scattering measurements were carried out at room temperature at the beamlines 12ID-B and -C of the Advanced Photon Source at the Argonne National Laboratory. The setups were adjusted to achieve scattering q values of 0.006 < q < 2.3 Å−1 where q = (4π/λ)sinθ and 2θ is the scattering angle. Twenty two-dimensional images were recorded for each buffer or sample solution using a flow cell with an exposure time of 1–2 s to minimize radiation damage and obtain good signal-to-noise ratio. No radiation damage was observed as confirmed by the absence of systematic signal changes in sequentially collected x-ray scattering images. The two-dimensional images were reduced to one-dimensional scattering profiles using the Matlab software package at the beamlines. The scattering profile of a sample solute was calculated by subtracting the buffer contribution from the sample-buffer profile using the program PRIMUS (46) and standard procedures. Concentration series measurements (4- and 2-fold dilution and stock solution) for the same sample were carried out to remove the scattering contribution due to interparticle interactions and to extrapolate the data to infinite dilution. The forward scattering intensity I(0) and the radius of gyration (Rg) were calculated from the data of infinite dilution at low q values in the range of qRg < 1.3 using the Guinier approximation: lnI(q) ≈ ln(I(0)) − Rg2q2/3. These parameters were also estimated from the scattering profile with a broader q range of 0.006–0.30 Å−1 using the indirect Fourier transform method implemented in the program GNOM (47) along with the pair distance distribution function, p(r), and the maximum dimension of the protein, Dmax. The parameter Dmax (the upper end of distance r) was chosen so that the resulting pair distance distribution function has a short, near zero value tail to avoid underestimation of the molecular dimension and consequent distortion in low resolution structural reconstruction. The molecular weights of solutes were calculated on a relative scale using SAXS MoW (48) as well as from AUTOPOROD (49) where estimation of molecular weights is independent of protein concentration and can be obtained with minimal user bias. The theoretical scattering intensity of the atomic structure model was calculated and fitted to the experimental scattering intensity using CRYSOL (50).
Low resolution ab initio shape envelopes were determined using the program DAMMIN, which generates models represented by an ensemble of densely packed beads (51), using scattering profiles within the q range of 0.006–0.30 Å−1. 32 independent runs were performed, and the resulting models were subjected to averaging by DAMAVER (52) and superimposed by SUPCOMB (53) based on the normalized spatial discrepancy criteria and filtered using DAMFILT to generate the final model. The models were manually fit to the autoinhibited Ig19–21 crystal structure (Protein Data Bank code 2J3S).
RESULTS
The Exposed CM in Autoinhibitory FLNa Ig19–21 Is Refractory to Phosphorylation by PKA
Using purified FLNa Ig19–21 (amino acids 2045–2329), we first performed a standard kinase assay to examine its phosphorylation by PKA. Fig. 1B shows that FLNa Ig19–21 was very weakly phosphorylated at 5- and 20-min reaction times (Fig. 1B). By contrast, under the same condition, a peptide derived from FLNa Ig20 containing the CM as well as the autoinhibitory segment (Arg2139–Val2156; Ig20 peptide) was potently phosphorylated by PKA (Fig. 1B). These data were rather surprising to us as the Ser2152-containing PKA CM in FLNa Ig19–21 is completely solvent-exposed (Fig. 1A). The immediate implication from the data was that the overall conformation of autoinhibited FLNa Ig19–21 plays a role in negatively regulating Ser2152 phosphorylation. Our failure to drive Ser2152 phosphorylation to completion in many assays in conjunction with mass spectrometry corroborated this hypothesis.
Ligand Binding Triggers Filamin Phosphorylation
Because a range of filamin ligands such as integrin β7 and migfilin can displace the Ig20 segment to relieve autoinhibition (54), we decided to examine whether these ligands might alter the Ser2152 phosphorylation level by inducing a different filamin conformation. GPIbα peptide, the strongest known filamin ligand in complex with Ig19–21, resulted in loss of protein through precipitation. We therefore used a mutant peptide, GPIbα-M, where Phe and Trp of GPIbα were both substituted to Ala to increase the peptide-filamin complex solubility but retain relatively high affinity to filamin. Based on the crystal structure of the wild-type GPIbα peptide with Ig17, the peptide is a β strand wedged between β strands C and D of the Ig fold where Phe/Trp are not essential for the interface (55). Hence, this mutant peptide is likely to bind in the same mode as the WT. In support of this, the HSQC spectrum of 15N-labeled Ig21 with this mutant peptide clearly shows large chemical shift changes (Fig. 2A) reminiscent of tight binding partners of class A filamin repeats (34, 56). Furthermore, isothermal calorimetry experiments show that the mutant binds with a robust affinity of ∼3 μm (Fig. 2, B and C) that compares well with that of migfilin and integrin β7 peptides, albeit slightly weaker than the WT GPIbα (34).
FIGURE 2.

Mutant (FW to AA) GPIbα peptide retains binding to Ig21. A, HSQC of FLNa Ig21 in the absence (black) and presence (red) of GPIbα-M showing substantial chemical shift changes consistent with a strong binding affinity. B, isothermal calorimetry data showing the strong binding of GPIbα to FLNa Ig21 with a KD of ∼0.1 μm. C, isothermal calorimetry data showing that the strong binding of GPIbα-M to FLNa Ig21 (KD ∼ 2.88 μm) is largely retained as compared with the WT GPIbα in B.
These peptide ligands are listed in Fig. 3A, and the side chains of residues that contact the filamin Ig are marked by arrows. Using the same condition as in Fig. 1, we examined the PKA-mediated phosphorylation of FLNa Ig19–21 in the presence of migfilin and GPIbα-M, respectively. Remarkably, both ligands dramatically enhanced the rate of the PKA-mediated phosphorylation effect on FLNa Ig19–21 (Fig. 3B). Filamin dimerizes through its 24th Ig repeat, and integrins are a major ligand for filamin in multiple tissues (57). We therefore extended the analysis to a larger dimeric fragment, FLNa Ig16–24 (amino acids 1772–2647), along with the integrin β7 peptide that revealed the same trend (Fig. 3C). These data thus unravel for the first time that despite the exposed CM the autoinhibitory FLNa Ig19–21 is resistant to phosphorylation, and such resistance is removed upon FLNa Ig19–21 binding to various ligands that bind through β sheet augmentation of strands C and D of the filamin Ig fold.
FIGURE 3.
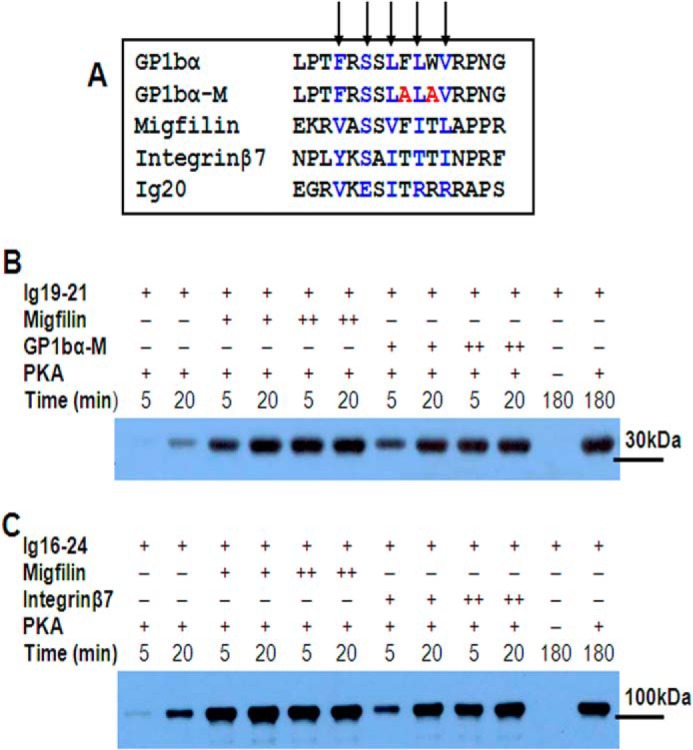
Ligand binding promotes filamin phosphorylation by PKA. A, sequence comparison of various filamin ligands (GPIbα, GPIbα-M, migfilin, integrin β7, and Ig20 autoinhibitory segment Val2140–Arg2148). The alternate residues that contribute maximally to binding are highlighted in blue. B, time-dependent phosphorylation of FLNa Ig19–21 as probed by a Ser2152 phosphospecific filamin antibody in the absence (−; lanes 1 and 2) and presence of migfilin (lanes 3–6) and GPIbα-M (lanes 7–10) at two different concentrations (+, 50 μm; ++, 200 μm). C, time-dependent phosphorylation of FLNa Ig16–24 (∼100 kDa) in the absence (lanes 1 and 2) and presence of migfilin (lanes 3–6) and integrin β7 (lanes 7–10) at two different concentrations (+, 50 μm; ++, 200 μm). In B and C, suitable “no PKA” controls were included.
Autoinhibitory (Ligand-free) and Ligand-bound FLNa Ig19–21 Are Conformationally Different
The dramatically different responses to PKA by the ligand-free (autoinhibited) and ligand-bound (open) forms of FLNa Ig21 clearly suggest a filamin conformation-based on/off switch mechanism to regulate enzyme catalysis. To definitively evaluate this hypothesis, we performed an SAXS experiment of FLNa Ig19–21 in the absence and presence of the ligand migfilin peptide. Migfilin (Fig. 4A, red envelope) induced significant conformational change of FLNa Ig19–21 as compared with the ligand-free FLNa Ig19–21 (blue envelope). Interestingly, the ligand-bound form (Dmax ∼ 110 Å) is less elongated than the ligand-free form (Dmax ∼ 120 Å) (Fig. 4A and Table 1), suggesting that upon ligand binding FLNa Ig19–21 undergoes a distinct conformational rearrangement that is probably unique for PKA recognition. Surprisingly, when attempting to fit the back-calculated SAXS data of the crystal structure of FLNa Ig19–21 to the experimental SAXS data of FLNa Ig19–21, we found that there was significant deviation between the two data sets (Fig. 4, B and C). Given that FLNa Ig19–21 had limited phosphorylation by PKA (Figs. 1B and 3, B and C), we speculated that there is an “open/closed (autoinhibited)” conformational equilibrium of filamin where some small population of the “open” form may allow PKA catalysis. This would also explain why the crystal structure and SAXS data of FLNa Ig19–21 do not match well. Specifically, there are void areas in the SAXS envelope that we cannot fill with the crystal structure coordinates of Ig19–21 (Fig. 4A). Crystallization probably captured the closed form of the protein, whereas the SAXS data are the ensemble of the equilibrated open and closed forms. To rigorously investigate this possibility and to precisely evaluate how the two different conformations of filamin affect PKA activity, we designed two types of filamin constructs. (i) We engineered tightly autoinhibited (locked) filamin in which a strong ligand, GPIbα (FRSSLFLWV) (34), replaced the weak internal ligand, the Ig20 segment (2140VKESITRRR2148) (Fig. 3A). Consistent with the design, the SAXS data of the locked mutant fit very well with the crystal structure of FLNa Ig19–21 (Fig. 4, B, C, and D). To further validate the design of the locked mutant, we collected the HSQC spectrum of the locked Ig19–21, which was found to be well dispersed and folded, consistent with the SAXS data (Fig. 5A). To highlight the ligand binding property of the mutant, we selected Ser2088 from Ig19 and Ser2279 from Ig21 that are diagnostic markers for individual Ig19 and Ig21 occupation by ligands (54, 56, 58, 59). These two residues reside in the tight loop between C and D strands of the Ig repeats (Fig. 5A, inset), which are a convenient proxy for ligand engagement by β sheet augmentation. Fig. 5A, inset, shows that Ser2088 but not Ser2279 of the locked mutant underwent large chemical shift changes upon addition of migfilin peptide, demonstrating that although migfilin can readily bind to Ig21 of WT FLNa Ig19–21 (54) it failed to do so for the locked mutant. Correspondingly, although migfilin changed the overall shape of FLNa Ig19–21 (Fig. 4, A, B, and C), it had minimal effect on the locked mutant (Fig. 5B). This is consistent with the fact that the shape change in WT Ig19–21 upon migfilin binding is due to rearrangement of Ig20. (ii) We also engineered an active mutant where we made S2143A/I2144A/T2145A mutations in the Ig20 segment to weaken the autoinhibitory interaction. This is again best explained using Ser2279 (Ig21) and Ser2088 (Ig19) as indicator chemical shifts. Ser2088 remains unchanged in the HSQC spectrum of the active mutant compared with the WT (Fig. 5, C and inset). However, Ser2279 in the active mutant shifts toward that of Ig21 in which there is no autoinhibition whatsoever. This clearly shows that autoinhibition is substantially released in the active mutant of Ig19–21. Thus, we have obtained both locked autoinhibitory and active FLNa Ig19–21. In particular, the locked mutant allowed us to show that WT Ig19–21 clearly undergoes a shape change upon migfilin binding. To further confirm the migfilin-induced shape change, we further examined GPIbα-M binding to Ig19–21 by SAXS because the two peptides exhibit similar binding affinities to the protein. Fig. 5D and Table 1 show that both ligand peptides induce similar overall shapes of Ig19–21.
FIGURE 4.

Different conformations of FLNa Ig19–21. A, comparison of SAXS-based FLNa Ig19–21 shape in the absence (blue) and presence (red) of migfilin peptide. The crystal structure of Ig19–21 is superposed on top of the SAXS envelope. B, comparison of the back-calculated scattering curve (based on Protein Data Bank code 2J3S) (solid line) with experimental scattering curves (dotted line) for the locked FLNa Ig19–21 mutant (top), WT FLNa Ig19–21 (middle), and migfilin-bound FLNa Ig19–21 (bottom). C, the pair distance distribution functions (PDDF) of the locked Ig19–21 (red), the wild-type Ig19–21 (black), the wild-type Ig19–21 with migfilin bound (blue), and the back-calculated (backCal) scattering curve (green) from the crystal structure (Protein Data Bank code 2J3S). D, fitting of the crystal structure of autoinhibited FLNa Ig19–21 with the SAXS shape of the locked FLNa Ig19–21 mutant.
TABLE 1.
Basic structural parameters for filamin Ig19–21 samples
|
Rg (Å) |
Dmax | NSDa | ||
|---|---|---|---|---|
| Guinier fitting | GNOM fitting | |||
| Å | ||||
| WTb | 32.8 ± 0.7 | 33.5 ± 0.6 | 120 ± 3 | 0.688 ± 0.017 |
| WT + migfilinb | 30.1 ± 0.5 | 31.8 ± 0.4 | 110 ± 2 | 0.676 ± 0.013 |
| Lockedb | 28.2 ± 0.2 | 28.9 ± 0.1 | 100 ± 2 | 0.696 ± 0.015 |
| WTc | 32.3 ± 1.7 | 33.0 ± 0.8 | 120 ± 3 | 0.597 ± 0.009 |
| WT + GPIbα-Mc | 29.6 ± 0.7 | 29.8 ± 0.6 | 101 ± 2 | 0.658 ± 0.020 |
a Normalized spatial discrepancy from DAMMIN calculation.
b Different batches of protein; however, ligand binding consistently results in reduction in the overall size of filamin Ig19–21.
c Different batches of protein; however, ligand binding consistently results in reduction in the overall size of filamin Ig19–21.
FIGURE 5.

Conformations of the locked and active filamin mutants. A, HSQC of the locked FLNa Ig19–21 mutant in the absence (black) and presence (red) of migfilin. The top inset shows the representative shifts Ser2088 and Ser2279 in the locked form with (red) and without migfilin (black). The inset at the bottom shows the location of these ligand binding “indicator” serines in the Ig19–21 crystal structure. B, comparison of the SAXS data of the locked mutant in the absence (blue) and presence (red) of migfilin. C, HSQC spectra of Ig21 (blue), Ig19–21 (black), and active Ig19–21. The inset shows the diagnostic shifts Ser2088 and Ser2279. D, overlay of SAXS envelopes of migfilin-bound Ig19–21 (red) and GPIbα-M-bound Ig19–21 (green).
With these above locked and active constructs, we performed the PKA assay again. Fig. 6A shows that the active FLNa Ig16–24 mutant dramatically increased the rate of phosphorylation by PKA as compared with WT FLNa Ig16–24 (lane 1 versus lane 5), but no phosphorylation was observed for the locked mutant (lanes 9–12), indicating that in the locked mutant the open/closed conformational equilibrium probably has completely shifted to the closed/autoinhibited form, which is strongly supported by the SAXS data (Fig. 4B). Mass spectrometric data further confirmed this finding (Tables 2 and 3). Fig. 6B shows that strong ligands GPIbα-M and integrin β7, which can enhance the phosphorylation in the WT, produced minimal phosphorylation in the locked form. By contrast, a control peptide derived from the locked form, LFLWVRAPSVANV containing Ser2152, is readily phosphorylated by PKA (Fig. 6C). These data provide strong supporting evidence for the dynamic open/closed conformational equilibrium of filamin in which PKA selectively phosphorylates the open but not the autoinhibited form of filamin A.
FIGURE 6.

Conformational basis of PKA-mediated phosphorylation on filamin. A, time-dependent phosphorylation of WT FLNa Ig16–24 (∼100 kDa) (lanes 1–4), active mutant (lanes 5–8), and locked mutant (lanes 9–12). Note that bands on the top are residual GST-FLNa Ig16–24, whereas bands on the bottom are possibly small amounts of degraded FLNa Ig16–24. B, 32P autoradiogram of WT FLNa Ig16–24 (lane 1), WT FLNa Ig16–24 with GPIbα-M (lane 2), WT FLNa Ig16–24 with integrin β7 (lane 3), locked mutant (lane 4), locked mutant with GPIbα-M (lane 5), and locked mutant with integrin β7 (lane 6). Peptide alone controls were also included (lanes 7 and 8). C, 32P autoradiogram of Ser2152-containing peptide LFLWVRAPSVANV from the locked mutant was phosphorylated by PKA for 1 h. D, superposition of “RRASI” of protein kinase inhibitor α (red) bound to PKA with FLNa Ig19–21 showing that the filamin RRAPSV (blue) loop fits well into the active site but that its surrounding regions are incompatible with PKA (yellow). Especially Ig20 has strong steric clash (mesh area) with PKA. E, modeling analysis suggests that the ligand (migfilin)-bound FLNa Ig19–21 may structurally match the PKA conformation without steric clash, thus explaining why it promotes catalysis.
TABLE 2.
Phosphopeptides identified in WT and locked mutant filamin A Ig16–24 samples
Numerical values correspond to the peak area ratio for the phosphorylated to unmodified peptides. No unmod, no unmodified form of the peptide was identified in the LC-MS analysis; NI, not identified; SRM, low abundance and determined in a specific reaction monitoring (SRM) experiment; Chymo, chymotrypsin; pT, phosphothreonine; pS, phosphoserine.
| [M + H]+ | m/z | z | Protease | Site | Sequence | WT | Locked |
|---|---|---|---|---|---|---|---|
| 1734.79 | 578.93, 867.85 | 3, 2 | Trypsin | Ser2152 | APpSVANVGSHCDLSLK | 0.18 | NI |
| 1890.89 | 631.30 | 3 | Trypsin | Ser2152 | RAPpSVANVGSHCDLSLK | 0.55 | NI |
| 3889.88 | 1297.97 | 3 | Trypsin | Ser2152 | RAPpSVANVGSHCDLSLKIPEISIQDMTAQVTSPSGK | SRM | NI |
| 2046.99 | 683.0 | 3 | Trypsin | Ser2152 | RRAPpSVANVGSHCDLSLK | No unmod | NI |
| 2219.06 | 555.77 | 4 | Glu-C | Ser2152 | SITRRRRAPpSVANVGSHCD | 0.71 | NI |
| 1497.76 | 749.39 | 2 | Trypsin | Thr2336 | RLpTVSSLQESGLK | 0.07 | 0.10 |
| 2981.54 | 994.86 | 3 | Trypsin | Thr2336 | RLpTVSSLQESGLKVNQPASFAVSLNGAK | SRM | SRM |
| 2505.27 | 1253.64 | 2 | Chymo | Thr2336 | VVPVASPSGDARRLpTVSSLQESGL | No unmod | No unmod |
| 3376.73 | 1126.92 | 3 | Chymo | Thr2336 | VVPVASPSGDARRLpTVSSLQESGLKVNQPASF | No unmod | No unmod |
| 1339.67 | 670.34 | 2 | Glu-C | Thr2336 | ARRLpTVSSLQE | No unmod | No unmod |
| 2579.29 | 860.77 | 3 | Glu-C | Thr2336 | SPFVVPVASPSGDARRLpTVSSLQE | SRM | SRM |
| 3973.97 | 1326.01 | 3 | Glu-C | Thr2336 | VSVKFNEEHIPDSPFVVPVASPSGDARRLpTVSSLQE | SRM | SRM |
TABLE 3.
Phosphopeptides targeted in specific reaction monitoring analysis of the tryptic digestions in WT and locked mutant filamin A Ig16–24 samples
Numerical values correspond to the peak area ratio for the phosphorylated to unmodified peptides. +, peptide identified; NI, not identified; pS, phosphoserine; pT, phosphothreonine.
| [M + H]+ | m/z | z | Protease | Site | Sequence | WT | Locked |
|---|---|---|---|---|---|---|---|
| 1654.8 | 552.3 | 3 | Trypsin | APSVANVGSHCDLSLK | + | + | |
| 1734.79 | 578.93 | 3 | Trypsin | Ser2152 | APpSVANVGSHCDLSLK | 0.15 | NI |
| 1810.9 | 604.3 | 3 | Trypsin | RAPSVANVGSHCDLSLK | + | NI | |
| 1890.89 | 631.30 | 3 | Trypsin | Ser2152 | RAPpSVANVGSHCDLSLK | 0.55 | NI |
| 1967.0 | 656.3 | 3 | Trypsin | RRAPSVANVGSHCDLSLK | + | NI | |
| 2046.99 | 683.0 | 3 | Trypsin | Ser2152 | RRAPpSVANVGSHCDLSLK | 1.2 | NI |
| 1417.8 | 709.4 | 2 | Trypsin | RLTVSSLQESGLK | + | + | |
| 1497.76 | 749.39 | 2 | Trypsin | Thr2336 | RLpTVSSLQESGLK | 0.07 | 0.10 |
| 1901.6 | 967.9 | 3 | Trypsin | Thr2336 | RLTVSSLQESGLKVNQPASFAVSLNGAK | + | + |
| 2981.54 | 994.86 | 3 | Trypsin | Thr2336 | RLpTVSSLQESGLKVNQPASFAVSLNGAK | 0.16 | 0.20 |
Filamin/PKA Recognition Is Mediated by a Mutual Global Shape-matching Mechanism
How would the closed/autoinhibited filamin prevent PKA catalysis? This is a conceptually important issue because the Ser2152-containing CM is highly exposed (Fig. 1A) like many other exposed CMs that are thought to be accessible by kinase-active sites (22–28). To resolve this puzzle, we carefully examined the crystal structure of FLNa Ig19–21 by superimposing it with the phosphopeptide site “RRApSI” of a PKA inhibitor (protein kinase inhibitor α)-bound active PKA (60) (Protein Data Bank code 1JLU). Fig. 6D reveals that although the filamin RRAPSV loop fits well into the active site its overall conformation is incompatible with PKA. In particular, both filamin Ig20 and Ig21 would have strong steric clash with the PKA catalytic domain (Fig. 6D). By contrast, ligand binding removed such structural restraint with an altered conformation of FLNa Ig19–21 (Fig. 4A) that appears to fit well with the PKA conformation (Fig. 6E). Thus, global filamin conformation appears to act as a filter to only present ligand-induced filamin conformation for effective PKA catalysis.
In regard to P-sites of other proteins and their accessibility to various kinases, examination of known CMs in many crystal structures revealed that most of them have weak or no electron densities, consistent with the findings that the majority of P-sites are in disordered regions or flexible loops (22–28). Table 4 lists examples of PKA CM sites (61–71) that are all located in the loops, but their corresponding overall protein conformations are clearly incompatible with the PKA conformation. Thus, despite the exposed CMs, these proteins may use their distinct overall conformations to prevent their access to relevant kinases, and mechanisms may exist to temporally alter the protein conformations to allow their access to kinases, thereby leading to regulated phosphorylation and subsequent specific cellular responses.
TABLE 4.
Representative CM sites that are structurally incompatible with PKA
Data are derived from PhosphoSitePlus (77). RARA, retinoic acid receptor alpha; PKACA, PKA catalytic subunit; CSK, c-Src kinase; ER, estrogen receptor.
| Protein | Phosphoresidue | Structural location | Phosphosite | Protein Data Bank code | Ref. |
|---|---|---|---|---|---|
| ETV1 | Ser334 | Loop | PTYQRRGSLQLWQFL | 4AVP | 61 |
| Pin1 | Ser16 | Loop | PGWEKRMSRSSGRVY | 1F8A | 62 |
| RARA | Ser369 | Loop | YVRKRRPSRPHMFPK | 1DKF | 63 |
| PKACA | Thr198 | Loop | RVKGRTWTLCGTPEY | 4AE6 | 64 |
| NFκB-p105 | Ser335 | Loop | FVQLRRKSDLETSEP | 2O61 | 65 |
| NFκB-p65 | Ser276 | Loop | SMQLRRPSDRELSEP | 1VKX | 66 |
| SF2 | Ser19 | Loop | YGPPSRRSENRVVVS | 2M8D | 67 |
| DNAJC5 | Ser10 | Loop | DQRQRSLSTSGESLY | 2CTW | 68 |
| ER-α | Ser236 | Loop | IDKNRRKSCQACRLR | 1HCP | 69 |
| CSK | Ser364 | Loop | ALREKKFSTKSDVWS | 1BYG | 70 |
| CDKN2D | Thr141 | Loop | RRDARGLTPLELALQ | 1BD8 | 71 |
DISCUSSION
Substrate recognition is a key step in enzyme catalysis. The classic “induced fit” (72) and “conformational selection” (73, 74) models have long emphasized how a specific enzyme conformation is either induced or selected, respectively, to recognize the substrate but not vice versa (75). This is exemplified in the kinase field where there is a long list of examples of how kinases are conformationally activated to recognize substrates (for representative reviews, see Refs. 18–21). By contrast, only a few studies have reported how the buried/ordered CMs might undergo local substrate unfolding to access the kinase active site (29, 30), and there are no studies on how global substrate conformation affects the exposed CM access to the kinase. This gap in knowledge may be due to two reasons. (i) Enzyme active sites are small, which only allows short linear motifs of the substrates to enter for binding and catalysis. Because CMs are typically located in unstructured regions, this led to the notion that the CMs readily fit into the enzyme active sites in a global substrate shape-independent manner (for reviews, see Refs. 22, 23, and 25). (ii) It is technically difficult to determine the origin of phosphorylation from phosphorylatable or unphosphorylatable conformations because substrates with exposed CMs or even buried CMs may be in dynamic equilibrium between the two conformations. Because of such equilibrium, a phosphorylation effect is often detected to a certain extent as found in the case of filamin (Fig. 1). This scenario also applies to autophosphorylation in which a kinase may phosphorylate itself, e.g. on the activation loop. However, because a kinase with an active conformation (not autophosphorylated) is in dynamic equilibrium with its inactive form, it is difficult to determine whether the kinase with active conformation may phosphorylate itself or its inactive form. Thus, the exact conformational mechanism for kinase autophosphorylation remains elusive. In our study, using the protein engineering approach, we designed the locked filamin mutant, which showed unambiguously as compared with the open form that the autoinhibited conformation is unphosphorylatable by active PKA despite the exposed CM (Fig. 6). Structural analysis further confirmed that the active PKA conformation is incompatible with the autoinhibited filamin conformation (Fig. 6D) and other target substrates (Table 4) despite the exposed CMs. These data thus establish for the first time that despite having exposed CMs the global conformations of the substrates are crucial for fine-tuning the kinase specificity at least in certain biological systems.
What are the fundamental and mechanistic implications for the global substrate shape-dependent CM/kinase recognition? At the conceptual level, we believe that such a recognition mode calls upon revision of the conventional induced fit or conformational selection model that only stresses the importance of enzyme conformation in catalysis. An extended mutual conformational selection model suggesting that local unfolding of buried or ordered CMs is necessary to fit into the enzyme active sites has been described recently (30, 76). Here, we propose a new mutual global conformational selection model that emphasizes the overall three-dimensional shape-specific matching of the enzyme with its protein substrates. Such a model may appear to be apparent for elucidating non-enzymatic protein/protein interactions involving large interfaces. However, it has not been perceived previously in enzyme catalysis probably due to the above mentioned notion that the majority of CMs are in disordered regions and thus are adaptable to the catalytic pockets of enzymes without constraints of global substrate conformations (22, 23, 25). Our data now strongly endorse such a model at least for certain enzyme interactions with substrates containing exposed CMs. At the mechanistic level, such a model may help identify novel kinase-mediated phosphorylation and signaling mechanisms. Because tens of thousands of cellular proteins are phosphorylated in diverse cellular activities, any kinase, even if it is localized and activated, may encounter multiple exposed CMs in nearby proteins, in different subunits of a protein complex, or in the same protein (see Fig. 7). In these scenarios, overall substrate conformational restriction might be an effective filter to eliminate nonspecific phosphorylation (see Fig. 7) and thus help maintain cellular status. Specific stimuli such as ligand binding may induce a conformational change for one of the CM-containing regions or proteins to match the kinase conformation, thereby temporally triggering the phosphorylation and a particular downstream pathway (see Fig. 7). In the case of filamin, a broad range of filamin ligands, which correspond to diverse cellular events mediated by this phosphorylation event such as platelet aggregation (39), cytoskeletal rearrangement (41), mechanoprotection (40), cancer metastasis (38) and defective neuronal migration (42), were found to promote filamin Ser2152 phosphorylation. It is conceivable that different ligands, depending on their spatiotemporal localization with filamin, may promote its phosphorylation and modulate specific downstream signaling events. In this regard, ligand-dependent phosphorylation of filamin Ser2152 is a unique chemomechanical switch for regulating diverse filamin-mediated cytoskeletal remodeling events.
FIGURE 7.
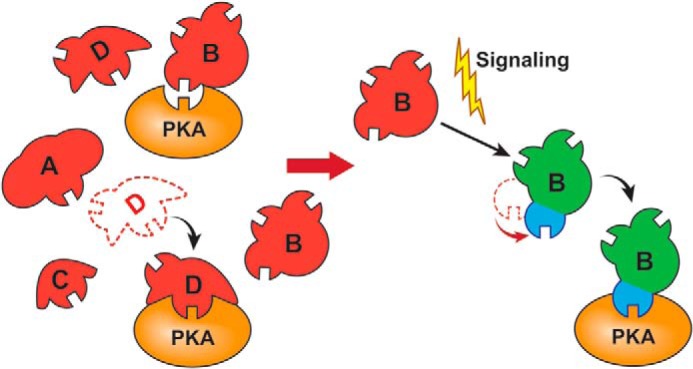
Graphic model illustrating shape-specific motif selection of substrate site by PKA.
In summary, we have discovered a novel regulatory pathway of phosphorylation at filamin Ser2152. Although this phosphorylation has long been known to be crucial for a range of cellular responses such as cytoskeleton remodeling, platelet aggregation, and cell migration, the pathway highlights for the first time how the phosphorylation is temporally controlled by ligand binding-induced conformational change of filamin. In broad terms, our findings suggest that in some complex signaling networks global substrate shapes may play a filtering role in channeling the specific exposed CM access to kinases, thereby triggering distinct signaling responses. Our findings also challenge the widely used induced fit or conformational selection model in enzymology, indicating that mutual global conformational matching between enzyme and macromolecular substrate may be crucial in controlling the catalytic specificity at least in certain biological events. Further investigations in this area are needed to help understand the spatiotemporal regulation of the enzyme specificity. At the experimental level, significant caution should now be taken to examine whether intact versus corresponding CM peptide substrates are phosphorylated at different rates, which may reveal novel substrate shape-dependent phosphorylation pathways that are otherwise overlooked but are critical for mediating a variety of physiological and pathological responses.
Acknowledgments
We thank Jun Yang, Xi-an Mao, and Jianmin Liu for technical assistance. We are grateful to Satya Yadav, Weizhen Shen, Belinda Willard, and Ling Li of the Lerner Research Institute Research Core for peptide synthesis and mass spectral analysis. We thank David Schumick of the Centre for Medical Art and Photography at the Cleveland Clinic for help with illustrations.
This work was supported, in whole or in part, by National Institutes of Health Grants GM062823, HL057470, and HL089473 (to J. Q., S. S. K., and S. V. N. P.).
- P-site
- phosphorylation site
- CM
- consensus motif
- FLNa
- filamin A
- GPIbα
- glycoprotein Ibα
- HSQC
- heteronuclear single quantum coherence
- SAXS
- small angle x-ray scattering
- GPIbα-M
- GPIbα mutant.
REFERENCES
- 1. Levene P. A., Alsberg C. L. (1906) The cleavage products of vitellin. J. Biol. Chem. 2, 127–133 [Google Scholar]
- 2. Cohen P. (2002) Protein kinases—the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315 [DOI] [PubMed] [Google Scholar]
- 3. Hunter T. (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80, 225–236 [DOI] [PubMed] [Google Scholar]
- 4. Cohen P. (2000) The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem. Sci. 25, 596–601 [DOI] [PubMed] [Google Scholar]
- 5. Pinna L. A., Ruzzene M. (1996) How do protein kinases recognize their substrates? Biochim. Biophys. Acta 1314, 191–225 [DOI] [PubMed] [Google Scholar]
- 6. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 7. Ubersax J. A., Ferrell J. E., Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol .Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 8. Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J., Guo H., Jona G., Breitkreutz A., Sopko R., McCartney R. R., Schmidt M. C., Rachidi N., Lee S. J., Mah A. S., Meng L., Stark M. J., Stern D. F., De Virgilio C., Tyers M., Andrews B., Gerstein M., Schweitzer B., Predki P. F., Snyder M. (2005) Global analysis of protein phosphorylation in yeast. Nature 438, 679–684 [DOI] [PubMed] [Google Scholar]
- 9. Ubersax J. A., Woodbury E. L., Quang P. N., Paraz M., Blethrow J. D., Shah K., Shokat K. M., Morgan D. O. (2003) Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864 [DOI] [PubMed] [Google Scholar]
- 10. Pearce L. R., Komander D., Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 11. Sparks J. W., Brautigan D. L. (1986) Molecular basis for substrate specificity of protein kinases and phosphatases. Int. J. Biochem. 18, 497–504 [DOI] [PubMed] [Google Scholar]
- 12. Kennelly P. J., Krebs E. G. (1991) Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 266, 15555–15558 [PubMed] [Google Scholar]
- 13. Knighton D. R., Zheng J. H., Ten Eyck L. F., Xuong N. H., Taylor S. S., Sowadski J. M. (1991) Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 414–420 [DOI] [PubMed] [Google Scholar]
- 14. Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., Brunak S. (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 [DOI] [PubMed] [Google Scholar]
- 15. Miller M. L., Jensen L. J., Diella F., Jørgensen C., Tinti M., Li L., Hsiung M., Parker S. A., Bordeaux J., Sicheritz-Ponten T., Olhovsky M., Pasculescu A., Alexander J., Knapp S., Blom N., Bork P., Li S., Cesareni G., Pawson T., Turk B. E., Yaffe M. B., Brunak S., Linding R. (2008) Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pearson R. B., Kemp B. E. (1991) Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol. 200, 62–81 [DOI] [PubMed] [Google Scholar]
- 17. Songyang Z., Blechner S., Hoagland N., Hoekstra M. F., Piwnica-Worms H., Cantley L. C. (1994) Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 4, 973–982 [DOI] [PubMed] [Google Scholar]
- 18. Endicott J. A., Noble M. E., Johnson L. N. (2012) The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613 [DOI] [PubMed] [Google Scholar]
- 19. Huse M., Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 20. Nolen B., Taylor S., Ghosh G. (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 21. Taylor S. S., Kornev A. P. (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eisenhaber B., Eisenhaber F. (2007) Posttranslational modifications and subcellular localization signals: indicators of sequence regions without inherent 3D structure? Curr. Protein Pept. Sci. 8, 197–203 [DOI] [PubMed] [Google Scholar]
- 23. Fuxreiter M., Tompa P., Simon I. (2007) Local structural disorder imparts plasticity on linear motifs. Bioinformatics 23, 950–956 [DOI] [PubMed] [Google Scholar]
- 24. Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Landry C. R., Levy E. D., Michnick S. W. (2009) Weak functional constraints on phosphoproteomes. Trends Genet. 25, 193–197 [DOI] [PubMed] [Google Scholar]
- 26. Ren S., Uversky V. N., Chen Z., Dunker A. K., Obradovic Z. (2008) Short linear motifs recognized by SH2, SH3 and Ser/Thr kinase domains are conserved in disordered protein regions. BMC Genomics 9, S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tyanova S., Cox J., Olsen J., Mann M., Frishman D. (2013) Phosphorylation variation during the cell cycle scales with structural propensities of proteins. PLoS Comput. Biol. 9, e1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vandermarliere E., Martens L. (2013) Protein structure as a means to triage proposed PTM sites. Proteomics 13, 1028–1035 [DOI] [PubMed] [Google Scholar]
- 29. Hoffman L., Stein R. A., Colbran R. J., Mchaourab H. S. (2011) Conformational changes underlying calcium/calmodulin-dependent protein kinase II activation. EMBO J. 30, 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Masterson L. R., Yu T., Shi L., Wang Y., Gustavsson M., Mueller M. M., Veglia G. (2011) cAMP-dependent protein kinase A selects the excited state of the membrane substrate phospholamban. J. Mol. Biol. 412, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feng Y., Walsh C. A. (2004) The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 6, 1034–1038 [DOI] [PubMed] [Google Scholar]
- 32. Stossel T. P., Condeelis J., Cooley L., Hartwig J. H., Noegel A., Schleicher M., Shapiro S. S. (2001) Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2, 138–145 [DOI] [PubMed] [Google Scholar]
- 33. van der Flier A., Sonnenberg A. (2001) Structural and functional aspects of filamins. Biochim. Biophys. Acta 1538, 99–117 [DOI] [PubMed] [Google Scholar]
- 34. Ithychanda S. S., Hsu D., Li H., Yan L., Liu D. D., Liu D., Das M., Plow E. F., Qin J. (2009) Identification and characterization of multiple similar ligand-binding repeats in filamin: implication on filamin-mediated receptor clustering and cross-talk. J. Biol. Chem. 284, 35113–35121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li M., Bermak J. C., Wang Z. W., Zhou Q. Y. (2000) Modulation of dopamine D2 receptor signaling by actin-binding protein (ABP-280). Mol. Pharmacol. 57, 446–452 [DOI] [PubMed] [Google Scholar]
- 36. Simon E. J., Onoprishvili I. (2010) The interaction between the μ opioid receptor and filamin A. Neurochem. Res. 35, 1859–1866 [DOI] [PubMed] [Google Scholar]
- 37. Jay D., García E. J., de la Luz Ibarra M. (2004) In situ determination of a PKA phosphorylation site in the C-terminal region of filamin. Mol. Cell. Biochem. 260, 49–53 [DOI] [PubMed] [Google Scholar]
- 38. Bedolla R. G., Wang Y., Asuncion A., Chamie K., Siddiqui S., Mudryj M. M., Prihoda T. J., Siddiqui J., Chinnaiyan A. M., Mehra R., de Vere White R. W., Ghosh P. M. (2009) Nuclear versus cytoplasmic localization of filamin A in prostate cancer: immunohistochemical correlation with metastases. Clin. Cancer Res. 15, 788–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carroll R. C., Gerrard J. M. (1982) Phosphorylation of platelet actin-binding protein during platelet activation. Blood 59, 466–471 [PubMed] [Google Scholar]
- 40. Glogauer M., Arora P., Chou D., Janmey P. A., Downey G. P., McCulloch C. A. (1998) The role of actin-binding protein 280 in integrin-dependent mechanoprotection. J. Biol. Chem. 273, 1689–1698 [DOI] [PubMed] [Google Scholar]
- 41. Hastie L. E., Patton W. F., Hechtman H. B., Shepro D. (1997) H2O2-induced filamin redistribution in endothelial cells is modulated by the cyclic AMP-dependent protein kinase pathway. J. Cell. Physiol. 172, 373–381 [DOI] [PubMed] [Google Scholar]
- 42. Zhang J., Neal J., Lian G., Shi B., Ferland R. J., Sheen V. (2012) Brefeldin A-inhibited guanine exchange factor 2 regulates filamin A phosphorylation and neuronal migration. J. Neurosci. 32, 12619–12629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lad Y., Kiema T., Jiang P., Pentikäinen O. T., Coles C. H., Campbell I. D., Calderwood D. A., Ylänne J. (2007) Structure of three tandem filamin domains reveals auto-inhibition of ligand binding. EMBO J. 26, 3993–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rust H. L., Thompson P. R. (2011) Kinase consensus sequences: a breeding ground for crosstalk. ACS Chem. Biol. 6, 881–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 46. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 47. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 48. Fischer H., de Oliveira Neto M., Napolitano H. B., Polikarpov I., Craievich A. F. (2010) Determination of the molecular weight of proteins in solution from a single small-angle x-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43, 101–109 [Google Scholar]
- 49. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Svergun D., Barberato C., Koch M. H. (1995) CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 51. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kozin M. B., Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 54. Ithychanda S. S., Qin J. (2011) Evidence for multisite ligand binding and stretching of filamin by integrin and migfilin. Biochemistry 50, 4229–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nakamura F., Pudas R., Heikkinen O., Permi P., Kilpeläinen I., Munday A. D., Hartwig J. H., Stossel T. P., Ylänne J. (2006) The structure of the GPIb-filamin A complex. Blood 107, 1925–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ithychanda S. S., Das M., Ma Y. Q., Ding K., Wang X., Gupta S., Wu C., Plow E. F., Qin J. (2009) Migfilin, a molecular switch in regulation of integrin activation. J. Biol. Chem. 284, 4713–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nakamura F., Stossel T. P., Hartwig J. H. (2011) The filamins: organizers of cell structure and function. Cell Adh. Migr. 5, 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Heikkinen O., Permi P., Koskela H., Ylänne J., Kilpeläinen I. (2009) 1H, 13C and 15N resonance assignments of the human filamin A tandem immunoglobulin-like domains 16–17 and 18–19. Biomol. NMR Assign. 3, 53–56 [DOI] [PubMed] [Google Scholar]
- 59. Heikkinen O. K., Ruskamo S., Konarev P. V., Svergun D. I., Iivanainen T., Heikkinen S. M., Permi P., Koskela H., Kilpeläinen I., Ylänne J. (2009) Atomic structures of two novel immunoglobulin-like domain pairs in the actin cross-linking protein filamin. J. Biol. Chem. 284, 25450–25458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Madhusudan, Trafny E. A., Xuong N. H., Adams J. A., Ten Eyck L. F., Taylor S. S., Sowadski J. M. (1994) cAMP-dependent protein kinase: crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 3, 176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu J., Janknecht R. (2002) Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase A. J. Biol. Chem. 277, 42669–42679 [DOI] [PubMed] [Google Scholar]
- 62. Lu P. J., Zhou X. Z., Liou Y. C., Noel J. P., Lu K. P. (2002) Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J. Biol. Chem. 277, 2381–2384 [DOI] [PubMed] [Google Scholar]
- 63. Rochette-Egly C., Oulad-Abdelghani M., Staub A., Pfister V., Scheuer I., Chambon P., Gaub M. P. (1995) Phosphorylation of the retinoic acid receptor-alpha by protein kinase A. Mol. Endocrinol. 9, 860–871 [DOI] [PubMed] [Google Scholar]
- 64. Steichen J. M., Kuchinskas M., Keshwani M. M., Yang J., Adams J. A., Taylor S. S. (2012) Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J. Biol. Chem. 287, 14672–14680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hou S., Guan H., Ricciardi R. P. (2003) Phosphorylation of serine 337 of NF-κB p50 is critical for DNA binding. J. Biol. Chem. 278, 45994–45998 [DOI] [PubMed] [Google Scholar]
- 66. Zhong H., Voll R. E., Ghosh S. (1998) Phosphorylation of NF-κ B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1, 661–671 [DOI] [PubMed] [Google Scholar]
- 67. Aksaas A. K., Eikvar S., Akusjärvi G., Skålhegg B. S., Kvissel A. K. (2011) Protein kinase a-dependent phosphorylation of serine 119 in the proto-oncogenic serine/arginine-rich splicing factor 1 modulates its activity as a splicing enhancer protein. Genes Cancer 2, 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Evans G. J., Wilkinson M. C., Graham M. E., Turner K. M., Chamberlain L. H., Burgoyne R. D., Morgan A. (2001) Phosphorylation of cysteine string protein by protein kinase A. Implications for the modulation of exocytosis. J. Biol. Chem. 276, 47877–47885 [DOI] [PubMed] [Google Scholar]
- 69. Chen D., Pace P. E., Coombes R. C., Ali S. (1999) Phosphorylation of human estrogen receptor α by protein kinase A regulates dimerization. Mol. Cell. Biol. 19, 1002–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vang T., Torgersen K. M., Sundvold V., Saxena M., Levy F. O., Skålhegg B. S., Hansson V., Mustelin T., Taskén K. (2001) Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J. Exp. Med. 193, 497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Marazita M. C., Ogara M. F., Sonzogni S. V., Martí M., Dusetti N. J., Pignataro O. P., Cánepa E. T. (2012) CDK2 and PKA mediated-sequential phosphorylation is critical for p19INK4d function in the DNA damage response. PLoS One 7, e35638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Koshland D. E. (1958) Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. U.S.A. 44, 98–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Changeux J. P., Edelstein S. (2011) Conformational selection or induced fit? 50 years of debate resolved. F1000 Biol. Rep. 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Monod J., Wyman J., Changeux J. P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 [DOI] [PubMed] [Google Scholar]
- 75. Hammes G. G., Benkovic S. J., Hammes-Schiffer S. (2011) Flexibility, diversity, and cooperativity: pillars of enzyme catalysis. Biochemistry 50, 10422–10430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Csermely P., Palotai R., Nussinov R. (2010) Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 35, 539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]