

Background: The general anesthetic propofol binds many on-pathway and off-pathway proteins.
Results: The deacetylase SIRT2 is a target of propofol, and propofol can inhibit the enzymatic function of the protein.
Conclusion: SIRT2 is a novel, presumably off-pathway protein target of the general anesthetic propofol.
Significance: Propofol might affect cellular events that are regulated by the acetylation state of proteins.
Keywords: Anesthesia, Anesthetic, Myelin, Photoaffinity Labeling, Sirtuin, SIRT2, Azi-propofol, Photolabel, Propofol
Abstract
meta-Azi-propofol (AziPm) is a photoactive analog of the general anesthetic propofol. We photolabeled a myelin-enriched fraction from rat brain with [3H]AziPm and identified the sirtuin deacetylase SIRT2 as a target of the anesthetic. AziPm photolabeled three SIRT2 residues (Tyr139, Phe190, and Met206) that are located in a single allosteric protein site, and propofol inhibited [3H]AziPm photolabeling of this site in myelin SIRT2. Structural modeling and in vitro experiments with recombinant human SIRT2 determined that propofol and [3H]AziPm only bind specifically and competitively to the enzyme when co-equilibrated with other substrates, which suggests that the anesthetic site is either created or stabilized in enzymatic conformations that are induced by substrate binding. In contrast to SIRT2, specific binding of [3H]AziPm or propofol to recombinant human SIRT1 was not observed. Residues that line the propofol binding site on SIRT2 contact the sirtuin co-substrate NAD+ during enzymatic catalysis, and assays that measured SIRT2 deacetylation of acetylated α-tubulin revealed that propofol inhibits enzymatic function. We conclude that propofol inhibits the mammalian deacetylase SIRT2 through a conformation-specific, allosteric protein site that is unique from the previously described binding sites of other inhibitors. This suggests that propofol might influence cellular events that are regulated by protein acetylation state.
Introduction
Propofol is an injectable hypnotic that is used for induction and maintenance of general anesthesia. Proteins involved in neurotransmission are among the pharmacologic targets that are affected by propofol to cause hypnosis (1, 2). Characterizing the molecular interactions between propofol and these proteins should allow the rational design of new hypnotics that specifically target relevant binding sites. Designing ligands with greater selectivity for specific sites should also alleviate side effects caused by propofol binding to off-pathway protein targets that do not contribute to hypnosis. For example, propofol has been reported to cause multiple adverse neurological side effects in mammals (3–6); however, the molecular targets underlying their causes are not clear. Therefore, comprehensive knowledge of propofol substrates in all tissue types is necessary to improve drug action.
To assist in the identification of propofol targets and binding sites, photoactive analogs that retain anesthetic efficacy have been developed by our group and others (7–10). These photolabels can be used to discover novel propofol targets from complex protein mixtures (11–13). In this work, we used meta-azi-propofol (AziPm)2 (see Fig. 1A), a photoactive propofol analog developed in our laboratory (7), to investigate drug binding in rat CNS tissue. We identified sirtuin isoform 2 (SIRT2), a protein deacetylase, as a specific target of propofol and AziPm in mammalian myelin. During enzymatic catalysis, SIRT2 couples deacetylation of acetyl lysine with NAD+ hydrolysis, which results in the formation of nicotinamide and O-acetyl-ADP-ribose. We found that clinical concentrations of propofol inhibit this enzymatic function of SIRT2, and we characterized the allosteric protein site that propofol binds. Possible physiological implications for SIRT2 inhibition by propofol are also discussed.
FIGURE 1.

A, chemical structures of propofol and AziPm. B, Coomassie R-250-stained membrane and corresponding autoradiograph of 50 μg of myelin protein separated with SDS-PAGE after photolabeling with 4 μm [3H]AziPm ± 400 μm propofol (+p) or 180 μm AziPm (+A). Optical density (O.D.) quantification from the autoradiograph lanes is shown in the aligned traces. C, Coomassie G-250-stained gel of 50 μg of myelin protein separated with SDS-PAGE after photolabeling with 4 μm (non-tritiated) AziPm. The boxed gel regions were excised for LC-MS/MS and processed with either trypsin or chymotrypsin digestion as described under “Experimental Procedures.”
EXPERIMENTAL PROCEDURES
Materials
2,6-Diisopropylphenol (propofol) was purchased from Sigma-Aldrich, and AziPm was synthesized according to published methods (7). The applications of [3H]AziPm (11, 14) and epothilone D (15) have been described in previous work, and all other chemicals and reagents were commercially available. Recombinant human SIRT2, representing residues 13–319, and recombinant human SIRT1, representing the full-length protein, were purchased from Sigma-Aldrich, and the identities of the proteins were confirmed with mass spectrometry. For tissue preparation, adult female Sprague-Dawley rats (∼300 g) were used, and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. Electrophoresis apparatuses, gels, molecular weight markers, and PVDF were from Bio-Rad, and 3H-sensitive Amersham Biosciences Hyperfilm MP was used for autoradiography. Developed autoradiographs, Coomassie G-250-stained membranes, and Coomassie G-250-stained gels were scanned with a Bio-Rad GS-800 calibrated densitometer, and the Quantity One software that accompanies the instrument was used for optical density quantification. Western blots were scanned with a Kodak Image Station 4000MM Pro, and band quantification was performed with the accompanying Carestream molecular imaging software. For liquid scintillation counting, MP Biomedicals EcoLite(+) liquid scintillation cocktail was used with a PerkinElmer Life Sciences Tri-Carb 2800TR instrument. A Synergy H1 microplate reader from BioTek was used for fluorescence-based assays.
Preparation of Rat Myelin for Photolabeling
Rats briefly anesthetized with isoflurane were decapitated, and the brains were removed. The brains were washed in ice-cold isolation buffer (0.32 m sucrose, 5 mm Tris, pH 7.4, supplemented with Roche Applied Science complete protease inhibitor cocktail), and then the brains were transferred to fresh isolation buffer. After mincing, tissue was homogenized by hand with a glass and Teflon homogenizer to produce total brain homogenate, and the enriched myelin fraction was prepared without detergents as described elsewhere (16). After a protein assay, 200-μg aliquots were frozen at −80 °C.
Myelin Photolabeling with [3H]AziPm and SDS-PAGE
After thawing, myelin samples were diluted to 1 mg/ml with isolation buffer. 4 μm [3H]AziPm was added with 180 μm non-radioactive AziPm, the indicated concentrations of propofol, or DMSO vehicle (all contained 0.5% DMSO). After transferring to a quartz cuvette (1-mm path length), samples were photolabeled for 20 min with a Rayonet photochemical reactor with a 350 nm bulb (Southern New England Ultraviolet Co., Branford, CT). Subsequently, the sample was pelleted, and the supernatant was removed. The pellet was gently resuspended in 300 μl of 25 mm Tris, pH 7.4, the sample was centrifuged, and the supernatant was discarded. The pellet was washed again by resuspension in 300 μl of 25 mm Tris, pH 7.4, and after recentrifugation, the supernatant was discarded, and the pellet was dissolved in buffer with detergent (5% glycerol, 1% Triton X-100, 0.5% SDS, and 20 mm Tris, pH 7.6). Detergent-solubilized protein was separated on 4–15% polyacrylamide gels. For autoradiography, protein was transferred to PVDF, and after drying, the membranes were exposed directly to film for 31 days at 4 °C. After the films were developed, the membranes were stained with Coomassie R-250, and the membranes and films were scanned. For scintillation counting, the gels were stained with Coomassie G-250, and after vertically separating the lanes, these were sliced horizontally into 1-mm pieces. The gel slices were dissolved overnight in sealed scintillation vials containing 350 μl of 30% hydrogen peroxide. After cooling, scintillation fluid was added for counting.
Myelin Photolabeling and SDS-PAGE for Mass Spectrometry
Myelin diluted to 1 mg/ml with isolation buffer was photolabeled for 20 min with 4 μm non-radioactive AziPm. After photolabeling, the sample was centrifuged and washed twice, as described above, with 25 mm Tris, pH 7.4, and the final pellet was solubilized. 50 μg of protein was separated by SDS-PAGE in adjacent lanes. The gel was stained with Coomassie G-250, and the three gel pieces indicated in Fig. 1C were excised from each lane for either trypsin or chymotrypsin digestion. After digestion, mass spectrometry analysis was performed by microcapillary reverse-phase ultra performance liquid chromatography with online nanospray tandem mass spectrometry (LC-MS/MS) on a Thermo LTQ-Orbitrap XL mass spectrometer. Raw data were acquired with Xcalibur, and the spectra were searched with SEQUEST against either a full tryptic or a partial chymotryptic rat proteome database for protein identification. Methionine oxidation was permitted, and filters for protein identification included 10-ppm parent ion tolerance, 1 atomic mass unit fragment ion tolerance, Δ CN of 0.05, and two unique peptides. The sequences of the proteins identified from all three bands were then compiled into a new database. With SEQUEST, we searched the spectra against this new database for an AziPm mass modification (216.07620 atomic mass units) on any amino acid of every full tryptic or partial chymotryptic peptide; only three residue modifications, including methionine oxidation, were permitted on each peptide. Filters for identification of modified peptides included 10-ppm parent ion tolerance, 1 atomic mass unit fragment ion tolerance, Δ CN of 0.05, and Xcorr scores of 1, 2, and 3 for +1 ions, +2 ions, and +3 ions, respectively. All spectra were then inspected manually for verification.
Structural Analyses of Sirtuins
For SIRT2 structures, chains A from Protein Data Bank (PDB) codes 3ZGO and 3ZGV were used (17). In the 3ZGO structure, which was re-refined from the dataset of PDB code 1J8F, SIRT2 is bound only to the structural cofactor zinc, and the 3ZGO structure is referred to as “apo-SIRT2” in this work (17, 18). In the 3ZGV structure, SIRT2 is bound to zinc and ADP-ribose, and the 3ZGV structure is referred to as “ADPr-SIRT2”. Both SIRT2 structures were loaded into SWISS-MODEL via the ExPASy web server (19–22), and models of human SIRT2 were rebuilt using the original structures (i.e. 3ZGO and 3ZGV) as the templates. This procedure built missing side chains in the structures while keeping all other atoms in the exact coordinates as the available crystal structures. Similar to SIRT2, a SIRT1 structure bound to zinc was used and is referred to as “apo-SIRT1” (PDB code 4IG9, chain A), and a SIRT1 structure bound to zinc and ADP-ribose was used and is referred to as “ADPr-SIRT1” (PDB code 4XKQ) (23). During crystallization, both ADPr-SIRT2 and ADPr-SIRT1 also engaged neighboring molecules in their respective active sites, effectively simulating acetyl peptide substrate binding. For ADPr-SIRT2, the peptide substrate binding groove of the enzyme was engaged by a loop from a neighboring protein molecule, and a leucine side chain pointed into the acetyl lysine binding site (17); for ADPr-SIRT1, the extended C-terminal tail from an adjacent protein molecule was inserted into the peptide substrate binding groove (23). The structure of human SIRT3 bound to zinc and the inhibitor Ex-527 was from PDB code 4BV3 (24), and the structure of Thermotoga maritima Sir2 (Sir2Tm) bound to zinc and nicotinamide was from PDB code 1YC5 (25). Hydrogen atoms were removed from all sirtuin structures that were used for modeling. For protein cavity analysis, the program fpocket was used (26). Visual molecular dynamics (27) and PyMOL (28) were used for other structural analyses and preparation of structural images, and sequence alignments were performed with ClustalW2 (29–31).
Photolabeling Recombinant Human SIRT2 and SIRT1
At a final concentration of 50 μg/ml, recombinant human SIRT2 or recombinant human SIRT1 was photolabeled with 4 μm [3H]AziPm ± propofol with and without 2 mm ADP-ribose, 100 mm nicotinamide, and 250 ng/μl of core histones purified from HeLa cell chromatin (histones were purchased form Active Motif, Carlsbad, CA). Prior to these experiments, histone acetylation was confirmed by Western blot with an acetyl lysine antibody (Cell Signaling Technology, Danvers, MA). The substrates and ligands for photolabeling were mixed in buffer (10 mm KCl and 10 mm Hepes, pH 7.6) and briefly vortexed, and samples were photolabeled in a 1-mm quartz cuvette with a lamp described elsewhere (11). After photolabeling, the substrates were separated with SDS-PAGE and stained with Coomassie G-250. The protein bands were then excised and dissolved in 350 μl of 30% hydrogen peroxide, and scintillation fluid was added for counting.
Preparation of Soluble Brain Extract
A rat was briefly anesthetized with isoflurane before decapitation. The brain was removed and washed in ice-cold assay buffer (25 mm Tris, pH 7.6, 2 mm MgCl2, 50 mm NaCl, 2 μm trichostatin A (from Sigma-Aldrich), and Roche Applied Science complete protease inhibitor cocktail). The brain was transferred to ∼5 ml of fresh assay buffer and homogenized by hand. The homogenate was centrifuged at 15,000 × g for 15 min, and the supernatant was set aside. The pellet was resuspended in 1 ml of fresh assay buffer, and after another 15,000 × g centrifugation, the supernatants were combined. This was centrifuged at 30,000 × g for 15 min, and the final supernatant was used as the soluble brain extract.
Tubulin Deacetylase Assays
For each deacetylase assay, 30 μg of the soluble brain extract protein was used, and the final assay volume was 30 μl. All components were dissolved in the assay buffer described above, and the assays were performed in 0.2-ml PCR tubes. When added to the reaction, final concentrations of 1 mm NAD+, 100 mm nicotinamide, and 3 μg of recombinant human SIRT2 were used. After briefly vortexing the assay mixture, the tubes were incubated at 37 °C for 3 h with brief mixing every 30 min. The reactions were terminated by adding Laemmli buffer and then placing the tubes in boiling water. SDS-PAGE and Western blots for acetylated α-tubulin were typically performed with both 1 μg and 3 μg of soluble extract protein.
Acetyl Peptide Deacetylase Assays
Two different microplate- and fluorescence-based sirtuin assay kits were purchased and used. 1) The SIRT2 inhibitor screening assay kit (catalog number EPI010 from Sigma-Aldrich), which is designed for IC50 determination, was used with the manufacturer-recommended dilutions of SIRT2 and the provided acetyl peptide substrate, and with variable concentrations of NAD+. Enzyme reactions were performed at 37 °C for 1 h. In this assay, recombinant human SIRT2 deacetylates an acetyl peptide that is conjugated to a fluorescent group. Upon peptide deacetylation, the fluorescent group is released and provides a detectable signal (λEX/λEM ≈ 395/541 (where EX and EM designate excitation and emission)) that should theoretically increase or decrease in the presence of an activator or inhibitor, respectively. As a positive control with this kit, we measured that 10 mm nicotinamide inhibited SIRT2 activity by 91 ± 2%. 2) The SIRTainty class III histone deacetylase assay kit (catalog number 17-10090 from EMD Millipore) was used with recombinant human SIRT2. In this assay, SIRT2 activity is measured through the formation of the enzymatic product nicotinamide, which is converted to nicotinic acid and NH3+ by the enzyme nicotinamidase (co-incubated in the enzyme assay); NH3+ is subsequently reacted with a developing reagent to provide a fluorescent signal (λEX/λEM ≈ 420/460) that should also theoretically increase or decrease in the presence of an activator or inhibitor, respectively. These experiments were performed with 1 μm SIRT2, 0.5 mm NAD+, 40 μm acetylated peptide (provided in the SIRTainty kit), and the recommended dilution of nicotinamidase. Enzyme reactions were performed at 37 °C for 4 min. Based on the mol of nicotinamide produced, ∼15% of the NAD+ was consumed under these conditions during control experiments.
Western Blotting
Detergent-solubilized protein was separated by SDS-PAGE and then transferred to PVDF. Membranes were blocked with 1.5% bovine serum albumin in TBS-T (Tris-buffered saline with 0.1% Tween 20). A 1:1000 dilution of primary antibody was applied overnight in TBS-T at 4 °C on a shaker. The anti-SIRT2 antibody was from Abcam (Cambridge, England), and the immunogen was the synthetic peptide LEDLVRREHANI corresponding to amino acids 341–352 of rat SIRT2; the anti-acetylated α-tubulin antibody, clone 6-11B-1, was from Sigma-Aldrich and recognizes acetyl lysine 40. After removing the primary antibody, the membrane was washed with TBS-T, and a secondary antibody conjugated to horseradish peroxidase was applied for 1 h. After washing with TBS-T, the blots were developed with Amersham Biosciences ECL Select reagent and scanned. The blots were then washed briefly in TBS-T followed by water before staining with Coomassie R-250 and drying before scanning.
Data Analysis and Figure Preparation
Where applicable, mean values with standard error are shown unless noted otherwise. For the Western blot standard curves, the net intensities from acetylated α-tubulin blots of known amounts of soluble brain extract were plotted and curve-fit according to a one-phase, exponential decay, and the resulting coefficients of determination (R2) were equal to 0.9999. To estimate the concentration of propofol that inhibits SIRT2 by 50%, we first plotted relative Western blot intensity versus absolute deacetylase activity using experimental values (SIRT2 assays with 0, 0.3, and 3 μm propofol, and the no activity baseline); R2 of the linear fit was 0.9906. From the regression equation, we calculated absolute deacetylase activities for assays with 30 and 300 μm propofol using experimentally determined relative intensities. The absolute activities were plotted as a percentage of control, and a sigmoidal curve with a variable slope was fit with the minimum and maximum constrained to 0 and 100, respectively. All curve fits and data analyses were performed within the GraphPad Prism version 6.0e software.
RESULTS
Myelin Photolabeling
We isolated a myelin-enriched fraction from rat brain for photolabeling with 4 μm [3H]AziPm, a concentration that approximates the EC99 dose for anesthetizing tadpoles (7, 11). To measure the specificity of anesthetic binding, we also photolabeled with 4 μm [3H]AziPm while co-equilibrating with 400 μm propofol or 180 μm (non-radioactive) AziPm, which are concentrations that approach their maximum aqueous solubility. The protein was separated by SDS-PAGE and transferred to a membrane for autoradiography. Based on optical density from the radioactivity (Fig. 1B), propofol and AziPm inhibited [3H]AziPm binding to myelin proteins an average of 41 and 53%, respectively, throughout the lanes. [3H]AziPm incorporation was concentrated in four bands shown as peaks at: 1) >250 kDa, 2) 80–95 kDa, 3) 55–70 kDa, and 4) 35–40 kDa, and propofol decreased [3H]AziPm labeling of these peak regions by 50–69%.
The >250-kDa band did not enter the resolving gel and was likely aggregated protein; however, the high selectivity of photolabeling in the remaining bands suggested strong binding to few proteins. To identify these proteins, we photolabeled myelin with 4 μm AziPm and excised the bands from a separate SDS-PAGE gel (Fig. 1C). We then employed a mass spectrometry-based approach whereby we first identified all the proteins in the bands and then searched for an AziPm mass adduct on those proteins. Six unique peptides from trypsin- and chymotrypsin-digested samples of the 35–40-kDa band were identified as photolabeled by AziPm (Table 1 and supplemental Fig. S1), and these peptides were all assigned to SIRT2. With this approach, we did not identify adducts on proteins from the other bands and therefore pursued the relevance of propofol binding to SIRT2.
TABLE 1.
SIRT2 peptides identified with LC-MS/MS as photolabeled by AziPm
| Photolabeled peptidea | Peptide charge | Observed MH+ | Calculated MH+ | Enzyme usedb |
|---|---|---|---|---|
| 131F.FALAKELY#PGQF.K144 | +2 | 1599.8101 | 1599.8069 | Chymo |
| 132F.ALAKELY#PGQF.K144 | +2 | 1452.7406 | 1452.7385 | Chymo |
| 134L.AKELY#PGQF.K144 | +2 | 1268.6200 | 1268.6173 | Chymo |
| 136K.ELY#PGQFK.P145 | +1 | 1197.5819 | 1197.5802 | Trypsin |
| 172L.ERVAGLEPQDLVEAHGTF#Y.T192 | +3 | 2347.1260 | 2347.1216 | Chymo |
| 202K.EYTM#SWMK.E211 | +1 | 1291.5378 | 1291.5349 | Trypsin |
a The photolabeled peptide is shown between periods. # indicates that a 216.0762-Da modification was detected. Amino acid numbering is according to full-length rat SIRT2.
b Chymo indicates chymotrypsin.
Conformation-specific and Isoform-selective Binding
After photolabeling separate myelin samples, we confirmed that propofol concentration-dependently inhibited 4 μm [3H]AziPm photolabeling of the ∼37-kDa SIRT2 band (Fig. 2A). We then photolabeled recombinant human SIRT2 with 4 μm [3H]AziPm ± propofol to test the specificity of binding in solution. Human and rat SIRT2 are highly conserved and share 88% sequence identity, and thus it was surprising when our initial attempts to inhibit [3H]AziPm photolabeling of human SIRT2 with propofol were unsuccessful (Fig. 2B).
FIGURE 2.
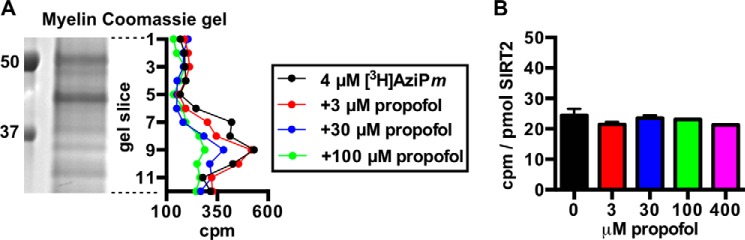
A, representative Coomassie G-250-stained gel lane of myelin protein separated with SDS-PAGE after photolabeling with 4 μm [3H]AziPm ± 3, 30, or 100 μm propofol. Each gel lane was cut horizontally into 1-mm pieces, and the radioactivity content in each slice is shown in the aligned graph. B, shown are the levels of covalent [3H]AziPm binding to human SIRT2 in solution. In this experiment, SIRT2 was photolabeled with 4 μm [3H]AziPm ± the indicated concentrations of propofol, and no additional substrates were added. Each mean was derived from 3–6 values from separate photolabeling experiments. Mean values are shown with S.E.
To investigate these contrasting results, we analyzed the AziPm binding site on high-resolution structures of SIRT2, which were also derived from recombinant human protein. Crystal structures of human SIRT2 represent two enzymatic conformations that are dictated by the presence or absence of bound substrates. In one structure, referred to here as apo-SIRT2, the enzyme is bound only to its structural, non-catalytic cofactor zinc (17, 18, 23). The second structure, referred to here as ADPr-SIRT2, represents an enzyme bound to zinc, a pseudo-peptide substrate, and ADP-ribose, which binds in the cleft occupied by NAD+ during enzymatic catalysis (17). In ADPr-SIRT2, with substrate bound, the helical domain containing the zinc subdomain is rotated over the NAD+ cleft, hinging on loops that connect to the Rossmann fold domain (17); similar conformational transitions have been observed with other mammalian sirtuins (23, 32). AziPm photolabeled Tyr139, Phe190, and Met206 on the helical domain of rat SIRT2, and these residues converge in the folded protein at a single site (Fig. 3, A and B).
FIGURE 3.

A, top, the positions of residues that were photolabeled by AziPm are shown in the apo-SIRT2 structure. The Rossmann fold of SIRT2 is colored dark blue, zinc is colored orange, and the indicated photolabeled residues are shown as red sticks outlined by a transparent surface topology. Bottom, enlarged view of the photolabeled residues on the apo-SIRT2 structure, but with the solid surface topology of the protein shown. B, identical views as in A, but with the structure of ADPr-SIRT2. ADP-ribose is colored yellow. C, the cavity in the ADPr-SIRT2 structure is shown with a black surface representation, which has a volume of 364 Å3. D, segments of the human and rat SIRT2 sequences, which were derived from the indicated UniProt codes, are aligned. Identical residues are indicated by the asterisks, residues photolabeled by AziPm are bolded and red, residues lining the anesthetic cavity in ADPr-SIRT2 are highlighted yellow, and boxed residues form the protein C-pocket.
Although the photolabeled residues are similarly positioned in both SIRT2 conformations, the protein topology revealed that these amino acids surround a cavity that is present in ADPr-SIRT2, but that is absent in apo-SIRT2 (Fig. 3, A–C). The volume of this cavity is ∼360 Å3 and should therefore accommodate propofol and AziPm, which have van der Waals volumes of 192 and 197 Å3, respectively. Residues that line this cavity are identical in rat and human SIRT2, with the exception of rat Met206, which is a leucine in the human protein (Fig. 3D).
This suggested that [3H]AziPm and propofol might bind selectively to SIRT2 conformations that can be induced and/or stabilized by substrate binding. To test this experimentally, we photolabeled recombinant human SIRT2 while co-incubating with >10-fold concentration (mol to mol) of acetylated human histones, which are protein substrates of SIRT2, and excess ADP-ribose and nicotinamide, which together mimic the co-substrate NAD+ (Fig. 4A). Without allowing for enzymatic catalysis, simultaneous binding of these substrates should increase the equilibrium fraction of the enzyme that assumes a conformation more similar to ADPr-SIRT2 than apo-SIRT2. As predicted with modeling, [3H]AziPm preferentially photolabeled SIRT2 when the enzyme was co-equilibrated with these substrates, and propofol readily displaced [3H]AziPm from the SIRT2 site under these conditions (Fig. 4B). In the absence of propofol, we also measured ∼20-fold greater [3H]AziPm binding per mol of SIRT2 relative to histones, despite the excess amounts of the latter, and co-equilibration with propofol had no effect on histone photolabeling.
FIGURE 4.

A, chemical structures of NAD+, ADP-ribose, and nicotinamide. B, the levels of covalent [3H]AziPm binding to human SIRT2 in solution are shown, as in Fig. 2B. However, in this experiment, SIRT2 was photolabeled with 4 μm [3H]AziPm ± propofol and also ± substrates (acetylated histones, ADP-ribose, and nicotinamide). Each mean was derived from 3–6 values from separate experiments. Two-way analysis of variance determined a significant effect of propofol and substrates on the means and their interaction (***, p < 0.001 for all comparisons). Bonferroni's post hoc tests comparing the indicated means determined that the substrates significantly increased [3H]AziPm binding (p < 0.001), and that this binding was inhibited by propofol (p < 0.001). ns, not significant. C, the levels of covalent [3H]AziPm binding to human SIRT1 in solution are shown. This experiment was performed identically to that shown in B, but with SIRT1 instead of SIRT2, and the means were from 3–5 values. Two-way analysis of variance did not determine a significant effect of propofol or substrates on the means or their interaction (p > 0.05). D, top, on the apo-SIRT1 structure, residues Tyr317, Phe366, and Cys380, which are homologous to the SIRT2 residues that were photolabeled by AziPm, are shown as red sticks outlined by a transparent surface topology; zinc is colored orange. Bottom, enlarged view of the residues, but with the solid surface topology of the protein shown. E, identical views as in D, but with the structure of ADPr-SIRT1. Mean values are shown with S.E.
Of the 15 residues that surround the propofol cavity on SIRT2, 10 residues were identical in SIRT1, which was the highest similarity among the seven human sirtuin isoforms. We therefore tested whether the anesthetics bind to a conserved site on recombinant human SIRT1 using an identical in vitro photolabeling approach. Levels of [3H]AziPm binding per mol of SIRT1 were lower than SIRT2, and propofol did not inhibit photolabeling whether or not the enzyme was co-equilibrated with the acetylated histones, ADP-ribose, and nicotinamide substrates (Fig. 4C). SIRT1 also lacks a discernable cavity at this location in the apo-SIRT1 and ADPr-SIRT1 crystal structures (Fig. 4, D and E), which also represent different conformations due to substrate binding (23); in the ADPr-SIRT1 structure, the residues around the cavity remain compact (Fig. 4E), as opposed to the spreading of residues seen in ADPr-SIRT2 (Fig. 3B). Together, our data suggest that propofol and AziPm show selectivity for binding sirtuin isoform 2.
SIRT2 Functional Assays
To test whether propofol affects SIRT2 enzymatic activity, we measured SIRT2 deacetylation of acetylated α-tubulin that was derived from mammalian tissue (33, 34). For this, we prepared a soluble extract from rat brain that contained only a small amount of native SIRT2 (Fig. 5A). Deacetylation of α-tubulin in this soluble extract was accelerated by the addition of recombinant human SIRT2 and 1 mm NAD+, and enzymatic activity was prevented by the sirtuin inhibitor nicotinamide (Fig. 5B). The inability of other sirtuins to deacetylate α-tubulin (33), and the addition of the histone deacetylase inhibitor trichostatin A to all reactions, ensured that deacetylation was SIRT2-dependent.
FIGURE 5.

A, Western blot showing relative amounts of SIRT2 in rat brain fractions: SE, soluble brain extract; b, brain homogenate; my, myelin. The Coomassie R-250-stained membrane is shown for loading. B, Western blot demonstrating deacetylation of acetylated α-tubulin (Acet-tubulin) from rat brain by recombinant human SIRT2 (hSIRT2). In this assay, SIRT2 activity was pronounced with the addition of 3 μg of SIRT2 and 1 mm NAD+, and deacetylase activity was inhibited by the SIRT2 inhibitor nicotinamide (nicot.); the histone deacetylase inhibitor trichostatin A was added to all reactions. 3 μg of soluble extract protein from an assay was loaded in each lane for this blot. C, propofol concentration-dependently inhibited SIRT2 deacetylation of acetylated α-tubulin. Assays were performed similar to B with the indicated substrates ± propofol. D, representative standard curve from a Western blot used to determine absolute levels of acetylated α-tubulin deacetylation by SIRT2 in the absence and presence of propofol. For this, increasing amounts of soluble brain extract protein were separated via SDS-PAGE, and alongside this, 1 μg of protein from SIRT2 deacetylase assays that contained no inhibitor, 0.3 μm propofol, or 3 μm propofol was separated. Densitometry from the standards allowed generation of the standard curve, from which absolute levels of deacetylase activity in the assay samples were determined. In this assay, 0.3 and 3 μm propofol inhibited SIRT2 activity by 9 ± 1 and 33 ± 4%, respectively. E, from the tubulin deacetylase assays, the absolute levels of SIRT2 inhibition at each propofol concentration are shown and were used to fit the sigmoidal curve drawn in red; 25 ± 2 μm propofol was necessary to inhibit the enzyme by 50%, and the slope of the curve was equal to −0.5 ± 0.1. Mean values are shown with S.E.
With this assay, we observed concentration-dependent inhibition of SIRT2 activity by propofol (Fig. 5C), including at the anesthetic concentration of 3 μm (7, 11). To quantify the absolute inhibition of SIRT2 by propofol, we accounted for the potential non-linearity of the Western blot chemiluminescent signal intensity. We generated internal standard curves by loading increasing amounts of soluble extract protein on a gel, and alongside this, we loaded the soluble extract from enzyme assays containing 0–3 μm propofol (Fig. 5D). In the absence of an inhibitor, 70 ± 2% of total acetylated α-tubulin was deacetylated in the assay, and propofol concentrations of 0.3 and 3 μm inhibited SIRT2 deacetylase activity by 9 ± 1 and 33 ± 4%, respectively. The data from these experiments enabled us to extrapolate the absolute inhibition of SIRT2 by 30 and 300 μm propofol, and a curve fit to the data estimated that 50% inhibition of SIRT2 should occur with 25 ± 2 μm propofol (Fig. 5E).
Because general anesthetics can affect tubulin polymerization (15), and because acetylation of α-tubulin is used as a surrogate for microtubule stability, we also confirmed that propofol inhibition of SIRT2 was independent of the polymerization state of tubulin. The microtubule-stabilizing agent epothilone D (15, 35) was added to separate assays at a concentration of 2 μm, which is sufficient to increase microtubule stability in vivo (15). Epothilone D did not affect SIRT2 deacetylation of α-tubulin, nor did it affect propofol inhibition of SIRT2 (Fig. 6). This is consistent with observations that SIRT2 deacetylation of α-tubulin is not affected by co-incubation with taxol (33).
FIGURE 6.
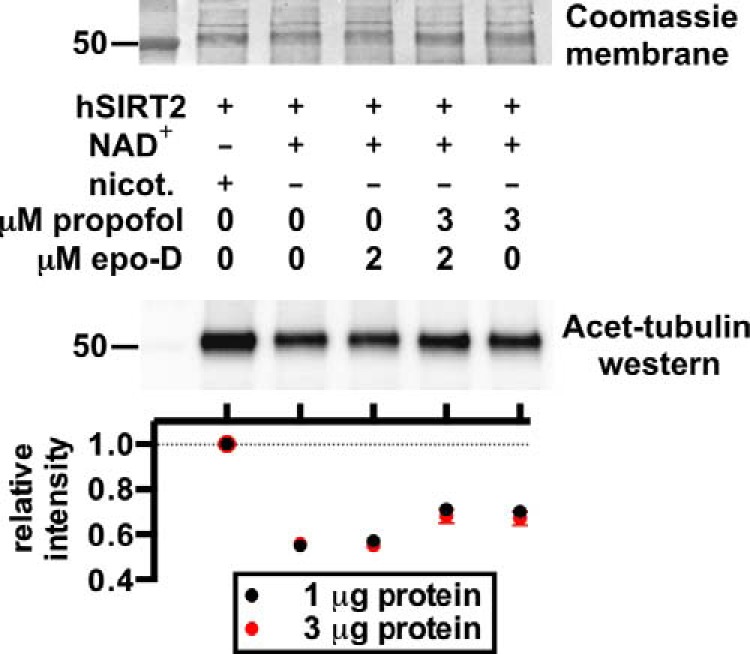
SIRT2 deacetylase assay Western blot and quantification, as in Fig. 5C, but with the microtubule-stabilizing agent epothilone D (epo-D) added to some reactions. Epothilone D did not affect SIRT2 deacetylase activity or propofol inhibition of SIRT2. hSIRT2, human SIRT2; nicot., nicotinamide; Acet-tubulin, acetylated α-tubulin.
Finally, in addition to the experiments using brain extract, we tested whether propofol affected SIRT2 function using enzyme assay kits that were commercially available. Both assays measured SIRT2 deacetylase activity in the absence of the soluble brain milieu. Interestingly, propofol concentrations as high as 300–400 μm had no effect on SIRT2 deacetylase activity in these in vitro assays, which used small acetylated peptides as substrates (Fig. 7, A–C).
FIGURE 7.
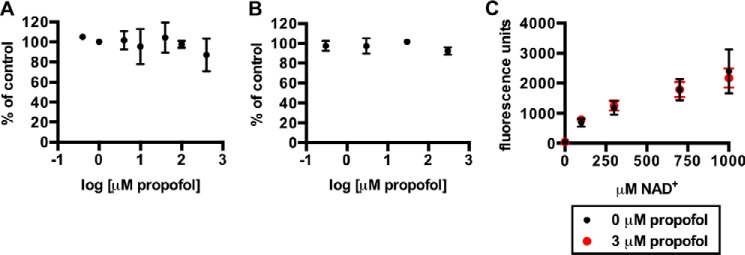
A, SIRT2 deacetylase activity in the presence of propofol, shown as the percentage of control, using the SIRTainty class III histone deacetylase assay kit (EMD Millipore) and the included acetyl peptide substrate. Experimental conditions are described under “Experimental Procedures.” B, SIRT2 activity in the presence of propofol using the SIRT2 inhibitor screening assay (Sigma-Aldrich) and the included fluorophore-conjugated acetyl peptide substrate. Experiments were performed according to the manufacturer's instructions. C, SIRT2 activity using the same kit as in B, with the fluorophore-conjugated acetyl peptide substrate, but with varying concentrations of NAD+. For assays in A–C, mean values are shown with S.D.
DISCUSSION
In this work, we identified mammalian SIRT2 as a target of propofol and showed that this anesthetic can inhibit the tubulin deacetylase activity of the enzyme. Propofol binds to an allosteric cavity that has not been described previously as an inhibitory site. Strong agreement between structural modeling and photolabeling experiments suggested that the propofol site is either created or stabilized when SIRT2 is bound to substrate(s) and has presumably undergone a conformational transition.
Despite the allosteric nature of this site, three residues that line the propofol site (Ile169, Asp170, and Thr171) also line the protein “C-pocket” that is highly conserved across sirtuins (25, 36) (Fig. 3D). Ile169 and Asp170 specifically contact the nicotinamide moiety of NAD+ during enzymatic reactions (25, 36), and mutagenesis of Asp170 reduces enzymatic activity (18). These residues are also adjacent to the acetyl lysine substrate site (17, 37). However, propofol does not bind within the C-pocket itself, in contrast to other sirtuin inhibitors such as nicotinamide (25) and Ex-527 (24, 38). The uniqueness of the propofol site was first demonstrated by the lack of competition between AziPm and nicotinamide for SIRT2 binding (Fig. 4B). This can also be modeled by aligning the SIRT2 C-pocket to those of sirtuins co-crystallized with nicotinamide and Ex-527 (Fig. 8). The residues photolabeled by AziPm are ∼7–14 Å from nicotinamide and Ex-527, and the ligands are separated from these residues by a tunnel that constricts to ∼4 Å in the SIRT2 protein (17).
FIGURE 8.

In A and B, the ADPr-SIRT2 structure is shown in cyan, zinc is colored orange, ADP-ribose is colored yellow, and residues photolabeled by AziPm are shown as red sticks. The propofol binding cavity is filled with the black surface representation. A, the C-pocket residues of SIRT2 were aligned to the C-pocket residues of Sir2Tm (PDB code 1YC5). The alignment root mean square was 0.75 Å, and the Sir2Tm C-pocket residues are traced in the transparent magenta. In 1YC5, Sir2Tm is bound to nicotinamide, and the position of this inhibitor, which binds to a separate site than propofol, is shown as green sticks. B, the C-pocket residues of SIRT2 were aligned to those of SIRT3 (PDB code 4BV3). The alignment root mean square was 0.26 Å, and the SIRT3 C-pocket residues are traced in the transparent magenta. In 4BV3, SIRT3 is bound to the inhibitor Ex-527, which binds to the same site as nicotinamide and is shown as green sticks.
AziPm photolabeling of multiple SIRT2 residues suggests that the ligand is somewhat mobile in the site. The portion of the cavity containing Tyr139 is also dynamic, being part of the flexible loop that connects the Rossmann fold domain to the helical domain. Along with these ligand and site dynamics, specific binding of propofol only to substrate-bound SIRT2 in solution (Fig. 4B) raises intriguing questions about the conformation of the enzyme in myelin. In order for [3H]AziPm to bind strongly and specifically to myelin SIRT2 (Figs. 1B and 2A), there must be a significant fraction of SIRT2 in adult myelin that is complexed with substrate or is otherwise induced into a conformation more similar to ADPr-SIRT2 than apo-SIRT2. The components that mediate this ligand specificity were likely present in the tubulin deacetylase assays that were performed with the soluble brain extract.
The efficacy of propofol as a SIRT2 inhibitor was revealed in the tubulin deacetylase assays, yet our in vitro experiments using acetylated peptides as substrates suggest a complex allosteric pharmacology. It is possible that the same components that promote propofol binding to SIRT2 in myelin also underlie the efficacy of propofol as an inhibitor and that propofol itself does not affect the function of the apo-enzyme in solution. An alternative explanation is that propofol inhibition of SIRT2 is specific to certain substrates, including full-length tubulin; however, the mechanism by which the enzyme could be sensitized to inhibition through the identified allosteric site in a substrate-specific manner is unclear.
There are many potential physiological implications of SIRT2 binding and enzymatic inhibition in myelin and elsewhere. Because SIRT2 is a soluble protein that is not known to directly affect neuronal excitability, it seems unlikely that propofol-SIRT2 interactions would contribute to hypnosis. However, although we used acetylated α-tubulin as a model substrate in our assays, there are numerous other proteins and cellular processes that are regulated by SIRT2. For example, as a major deacetylase in oligodendrocytes (39–41), SIRT2 activity regulates the development of myelin and also remyelination of axons after crush injury (42). In other cell types, SIRT2 deacetylates transcription factors (43) and histones (44, 45) to regulate transcription and chromatin structure, and SIRT2 regulates metabolic enzyme function (46). The relevance of a SIRT2-propofol interaction is therefore likely as an off-pathway target of the drug. Future studies that test the effects of propofol on these processes, specifically in the context of SIRT2 inhibition, could help to improve the optimal administration of propofol and other clinically used general anesthetics.
Finally, in addition to SIRT2, there remain other proteins in myelin and other CNS tissue fractions that bind to propofol and have not yet been identified (Fig. 1B). In this work, the identification of photolabeled protein was contingent on identifying AziPm mass adducts on peptides. The advantage of this strategy is that most proteins are retained in the SDS-PAGE gel and can potentially be identified, which is in contrast to other approaches such as two-dimensional gel electrophoresis that aim to purify photolabeled protein. However, adduct identification is also contingent on extensive sequencing of proteins. Soluble peptides are favored with LC-MS/MS sequencing, hindering detection of more hydrophobic propofol protein sites, and extensive sequencing becomes more challenging for larger proteins. Therefore, although our approach was successfully implemented for SIRT2 identification, this work also demonstrates the challenges associated with unbiased identification of unknown anesthetic protein targets. Alternative proteomic and chemical strategies using anesthetic analogs should continue to uncover additional propofol targets that might contribute to both off-pathway and on-pathway pharmacologic mechanisms.
Supplementary Material
Acknowledgments
We thank Bill Dailey for synthesizing meta-azi-propofol, and we thank the members of The Wistar Institute Proteomics Facility for assistance with mass spectrometry experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants P01-GM055876 (to R. G. E.) and F31-NS080519 (to B. P. W.).

This article contains supplemental Fig. S1.
- AziPm
- meta-azi-propofol
- propofol
- 2,6-diisopropylphenol
- ADPr
- ADP-ribose
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Jurd R., Arras M., Lambert S., Drexler B., Siegwart R., Crestani F., Zaugg M., Vogt K. E., Ledermann B., Antkowiak B., Rudolph U. (2003) General anesthetic actions in vivo strongly attenuated by a point mutation in the GABAA receptor β3 subunit. FASEB J. 17, 250–252 [DOI] [PubMed] [Google Scholar]
- 2. Kretschmannova K., Hines R. M., Revilla-Sanchez R., Terunuma M., Tretter V., Jurd R., Kelz M. B., Moss S. J., Davies P. A. (2013) Enhanced tonic inhibition influences the hypnotic and amnestic actions of the intravenous anesthetics etomidate and propofol. J. Neurosci. 33, 7264–7273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang X., Liu Y., Feng C., Yang S., Wang Y., Wu A.-s, Yue Y. (2009) Proteomic profiling of the insoluble fractions in the rat hippocampus post-propofol anesthesia. Neurosci. Lett. 465, 165–170 [DOI] [PubMed] [Google Scholar]
- 4. Creeley C., Dikranian K., Dissen G., Martin L., Olney J., Brambrink A. (2013) Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. Br. J. Anaesth. 110, Suppl 1, i29–i38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patel S., Wohlfeil E. R., Rademacher D. J., Carrier E. J., Perry L. J., Kundu A., Falck J. R., Nithipatikom K., Campbell W. B., Hillard C. J. (2003) The general anesthetic propofol increases brain N-arachidonylethanolamine (anandamide) content and inhibits fatty acid amide hydrolase. Br. J. Pharmacol. 139, 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whittington R. A., Virág L., Marcouiller F., Papon M.-A., El Khoury N. B., Julien C., Morin F., Emala C. W., Planel E. (2011) Propofol directly increases tau phosphorylation. PloS One. 6, e16648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hall M. A., Xi J., Lor C., Dai S., Pearce R., Dailey W. P., Eckenhoff R. G. (2010) m-Azipropofol (AziPm) a photoactive analogue of the intravenous general anesthetic propofol. J. Med. Chem. 53, 5667–5675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stewart D. S., Savechenkov P. Y., Dostalova Z., Chiara D. C., Ge R., Raines D. E., Cohen J. B., Forman S. A., Bruzik K. S., Miller K. W. (2011) p-(4-Azipentyl)propofol: a potent photoreactive general anesthetic derivative of propofol. J. Med. Chem. 54, 8124–8135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yip G. M. S., Chen Z.-W., Edge C. J., Smith E. H., Dickinson R., Hohenester E., Townsend R. R., Fuchs K., Sieghart W., Evers A. S., Franks N. P. (2013) A propofol binding site on mammalian GABAA receptors identified by photolabeling. Nat. Chem. Biol. 9, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weiser B. P., Woll K. A., Dailey W. P., Eckenhoff R. G. (2014) Mechanisms revealed through general anesthetic photolabeling. Curr. Anesthesiol. Rep. 4, 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weiser B. P., Kelz M. B., Eckenhoff R. G. (2013) In vivo activation of azipropofol prolongs anesthesia and reveals synaptic targets. J. Biol. Chem. 288, 1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xi J., Liu R., Asbury G. R., Eckenhoff M. F., Eckenhoff R. G. (2004) Inhalational anesthetic-binding proteins in rat neuronal membranes. J. Biol. Chem. 279, 19628–19633 [DOI] [PubMed] [Google Scholar]
- 13. Darbandi-Tonkabon R., Hastings W. R., Zeng C.-M., Akk G., Manion B. D., Bracamontes J. R., Steinbach J. H., Mennerick S. J., Covey D. F., Evers A. S. (2003) Photoaffinity labeling with a neuroactive steroid analogue. 6-azi-pregnanolone labels voltage-dependent anion channel-1 in rat brain. J. Biol. Chem. 278, 13196–13206 [DOI] [PubMed] [Google Scholar]
- 14. Weiser B. P., Bu W., Wong D., Eckenhoff R. G. (2014) Sites and functional consequence of VDAC-alkylphenol anesthetic interactions. FEBS Lett. 588, 4398–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emerson D. J., Weiser B. P., Psonis J., Liao Z., Taratula O., Fiamengo A., Wang X., Sugasawa K., Smith A. B., 3rd, Eckenhoff R. G., Dmochowski I. J. (2013) Direct modulation of microtubule stability contributes to anthracene general anesthesia. J. Am. Chem. Soc. 135, 5389–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sims N. R., Anderson M. F. (2008) Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nat. Protoc. 3, 1228–1239 [DOI] [PubMed] [Google Scholar]
- 17. Moniot S., Schutkowski M., Steegborn C. (2013) Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J. Struct. Biol. 182, 136–143 [DOI] [PubMed] [Google Scholar]
- 18. Finnin M. S., Donigian J. R., Pavletich N. P. (2001) Structure of the histone deacetylase SIRT2. Nat. Struct. Biol. 8, 621–625 [DOI] [PubMed] [Google Scholar]
- 19. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., Bordoli L., Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 21. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 37, D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guex N., Peitsch M. C., Schwede T. (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 30, S162–S173 [DOI] [PubMed] [Google Scholar]
- 23. Davenport A. M., Huber F. M., Hoelz A. (2014) Structural and functional analysis of human SIRT1. J. Mol. Biol. 426, 526–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gertz M., Fischer F., Nguyen G. T. T., Lakshminarasimhan M., Schutkowski M., Weyand M., Steegborn C. (2013) Ex-527 inhibits sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, E2772–E2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Avalos J. L., Bever K. M., Wolberger C. (2005) Mechanism of sirtuin inhibition by nicotinamide: altering the NAD+ cosubstrate specificity of a Sir2 enzyme. Mol. Cell 17, 855–868 [DOI] [PubMed] [Google Scholar]
- 26. Le Guilloux V., Schmidtke P., Tuffery P. (2009) Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 28. DeLano W. L. (2012) The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC, New York [Google Scholar]
- 29. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 30. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McWilliam H., Li W., Uludag M., Squizzato S., Park Y. M., Buso N., Cowley A. P., Lopez R. (2013) Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 41, W597–W600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jin L., Wei W., Jiang Y., Peng H., Cai J., Mao C., Dai H., Choy W., Bemis J. E., Jirousek M. R., Milne J. C., Westphal C. H., Perni R. B. (2009) Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 284, 24394–24405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. North B. J., Marshall B. L., Borra M. T., Denu J. M., Verdin E. (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 11, 437–444 [DOI] [PubMed] [Google Scholar]
- 34. Borra M. T., Langer M. R., Slama J. T., Denu J. M. (2004) Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry 43, 9877–9887 [DOI] [PubMed] [Google Scholar]
- 35. Ballatore C., Brunden K. R., Huryn D. M., Trojanowski J. Q., Lee V. M.-Y., Smith A. B., 3rd (2012) Microtubule stabilizing agents as potential treatment for Alzheimer's disease and related neurodegenerative tauopathies. J. Med. Chem. 55, 8979–8996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Avalos J. L., Boeke J. D., Wolberger C. (2004) Structural basis for the mechanism and regulation of Sir2 enzymes. Mol. Cell 13, 639–648 [DOI] [PubMed] [Google Scholar]
- 37. Avalos J. L., Celic I., Muhammad S., Cosgrove M. S., Boeke J. D., Wolberger C. (2002) Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol. Cell 10, 523–535 [DOI] [PubMed] [Google Scholar]
- 38. Napper A. D., Hixon J., McDonagh T., Keavey K., Pons J.-F., Barker J., Yau W. T., Amouzegh P., Flegg A., Hamelin E., Thomas R. J., Kates M., Jones S., Navia M. A., Saunders J. O., DiStefano P. S., Curtis R. (2005) Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 48, 8045–8054 [DOI] [PubMed] [Google Scholar]
- 39. Southwood C. M., Peppi M., Dryden S., Tainsky M. A., Gow A. (2007) Microtubule deacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 32, 187–195 [DOI] [PubMed] [Google Scholar]
- 40. Li W., Zhang B., Tang J., Cao Q., Wu Y., Wu C., Guo J., Ling E.-A., Liang F. (2007) Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating α-tubulin. J. Neurosci. 27, 2606–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Werner H. B., Kuhlmann K., Shen S., Uecker M., Schardt A., Dimova K., Orfaniotou F., Dhaunchak A., Brinkmann B. G., Möbius W., Guarente L., Casaccia-Bonnefil P., Jahn O., Nave K.-A. (2007) Proteolipid protein is required for transport of sirtuin 2 into CNS myelin. J. Neurosci. 27, 7717–7730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beirowski B., Gustin J., Armour S. M., Yamamoto H., Viader A., North B. J., Michán S., Baloh R. H., Golden J. P., Schmidt R. E., Sinclair D. A., Auwerx J., Milbrandt J. (2011) Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc. Natl. Acad. Sci. U.S.A. 108, E952-E961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu L., Arun A., Ellis L., Peritore C., Donmez G. (2012) Sirtuin 2 (SIRT2) enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigrostriatal damage via deacetylating forkhead box O3a (Foxo3a) and activating Bim protein. J. Biol. Chem. 287, 32307–32311 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44. Vaquero A., Scher M. B., Lee D. H., Sutton A., Cheng H.-L., Alt F. W., Serrano L., Sternglanz R., Reinberg D. (2006) SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vempati R. K., Jayani R. S., Notani D., Sengupta A., Galande S., Haldar D. (2010) p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J. Biol. Chem. 285, 28553–28564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang W., Wang S., Xiao M., Lin Y., Zhou L., Lei Q., Xiong Y., Guan K.-L., Zhao S. (2011) Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol. Cell 43, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.