

Background: Krüppel-like factor 3 (KLF3) is a transcriptional repressor with multiple biological roles.
Results: Phosphorylation of KLF3 by homeodomain-interacting protein kinase 2 (HIPK2) promotes DNA binding and transcriptional repression.
Conclusion: Signal transduction pathways mediated by HIPK2 control and direct KLF3 activity.
Significance: Determining the pathways that control KLF3 function offers potential for regulating its activity for therapeutic benefit.
Keywords: Gene Expression, Kruppel-like Factor (KLF), Post-translational Modification (PTM), Protein Phosphorylation, Transcription Regulation, C-terminal Binding Protein (CtBP2), Homeodomain-interacting Protein Kinase 2 (HIPK2), Kruppel-like Factor 3 (KLF3/BKLF)
Abstract
Krüppel-like factor 3 (KLF3/BKLF), a member of the Krüppel-like factor (KLF) family of transcription factors, is a widely expressed transcriptional repressor with diverse biological roles. Although there is considerable understanding of the molecular mechanisms that allow KLF3 to silence the activity of its target genes, less is known about the signal transduction pathways and post-translational modifications that modulate KLF3 activity in response to physiological stimuli. We observed that KLF3 is modified in a range of different tissues and found that the serine/threonine kinase homeodomain-interacting protein kinase 2 (HIPK2) can both bind and phosphorylate KLF3. Mass spectrometry identified serine 249 as the primary phosphorylation site. Mutation of this site reduces the ability of KLF3 to bind DNA and repress transcription. Furthermore, we also determined that HIPK2 can phosphorylate the KLF3 co-repressor C-terminal binding protein 2 (CtBP2) at serine 428. Finally, we found that phosphorylation of KLF3 and CtBP2 by HIPK2 strengthens the interaction between these two factors and increases transcriptional repression by KLF3. Taken together, our results indicate that HIPK2 potentiates the activity of KLF3.
Introduction
Krüppel-like factor 3 (KLF3/BKLF) is a member of the Krüppel-like factor (KLF)4 family of transcription factors (1, 2). KLF proteins are characterized by their highly homologous DNA binding domain, consisting of three C2H2 zinc fingers that direct sequence-specific binding to CACCC box motifs and GC-rich sequences in the promoters and enhancers of target genes (3). To date, 18 family members have been identified, with diverse biological roles in many aspects of cellular development and differentiation (4, 5).
KLF3 generally functions as a transcriptional repressor via recruitment of C-terminal binding proteins 1 and 2 (CtBP1/2) (6), which in turn facilitates assembly of a potent silencing complex that drives epigenetic modification of gene regulatory regions (7). KLF3 is widely expressed and has accordingly been shown to have physiological roles in a number of different developmental processes, including erythropoiesis, metabolism, cardiac development, myogenesis, and B lymphopoiesis (8–15). Despite recent progress in both defining the biological functions of KLF3 and in understanding the molecular mechanisms whereby KLF3 regulates expression of its target genes, the signaling pathways and post-translational modifications that control KLF3 activity remain to be fully defined.
To address this and identify novel interacting partners of KLF3, we performed a yeast two-hybrid screen and discovered that KLF3 can bind to homeodomain-interacting protein kinases (HIPKs). The HIPK family consists of four members, defined by the presence of a highly homologous p38-MAPK-like kinase domain (16). The greatest identity is shown between HIPK1 and HIPK2, which exhibit functional overlap, such that mice deficient in both factors display early embryonic lethality due to numerous development defects (17). HIPKs were first discovered as binding partners of NK homeodomain transcription factors, and HIPK-dependent phosphorylation at Ser/Thr-Pro consensus sites has been shown to greatly enhance transcription factor activity (18, 19).
Having identified HIPK proteins as potential binding partners of KLF3 by a yeast two-hybrid assay, we sought to validate this interaction and investigate the effect of HIPK-mediated phosphorylation on KLF3 transcriptional activity. Our investigations confirmed that HIPK2 can directly phosphorylate KLF3 in vitro, with phosphorylation occurring primarily at a single site, serine 249. In vivo, multiple residues are phosphorylated, with serine 249 again identified as a preferential HIPK consensus site. We also examined phosphorylation of the KLF3 cofactor CtBP2 by HIPK2 and identified a single phosphorylation site at serine 428. We found that phosphorylation of KLF3 is important for DNA binding and observed a reduction in binding strength when serine 249 is mutated to prevent phosphorylation. Finally, we determined that phosphorylation of KLF3 by HIPK2 promotes its DNA binding activity, increases the strength of its interaction with CtBP2, and enhances its ability to act as a transcriptional repressor. Our data therefore show that phosphorylation of KLF3 and its corepressor CtBP2 by HIPK2 can potentiate the activity of this repressor complex in certain contexts.
EXPERIMENTAL PROCEDURES
Vectors and Plasmids
For mammalian transfection, KLF3, HIPK2, and CtBP2 sequences were expressed in pcDNA3, pMT2, or pMT3 or in FLAG- or HA-modified versions thereof (2, 20). GST and His fusion proteins were expressed in pGEX-2T and pET-15b vectors, respectively (21, 22). pEGFP-HIPK2 and pEGFP-HIPK2-K221R plasmids were kindly provided by C. Y. Choi (NHLBI, National Institutes of Health (18)). Growth hormone transactivation assays were performed using the previously described reporter pAγ-GH, containing the Aγ-globin promoter (23). For yeast two-hybrid assays, pGBT9-KLF3(1–268) expressed a region of KLF3 containing amino acids 1–268, and pGAD10-HIPK1(615–1057) expressed a region of HIPK1 containing amino acids 615–1057.
λ-Phosphatase Treatment of Nuclear Extracts
15 μg of nuclear protein extract was incubated at 30 °C for 30 min in phosphatase buffer (50 mm Tris-HCl, 0.1 mm Na2EDTA, 5 mm dithiothreitol, 0.01% Brij 35, 2 mm MnCl2) with or without the addition of λ-protein phosphatase (1000 units), prior to Western blot analysis or electrophoretic mobility shift assay (EMSA).
Induction of G1E-ER4 Cells
4-Hydroxytamoxifen induced erythroid differentiation of G1-ER4 cells (24–26).
Yeast Two-hybrid Assays
Assays were performed as described previously (27, 28), using the Clontech two-hybrid system according to the manufacturer's instructions. Briefly, test proteins were expressed in yeast strain HF7c as either Gal4-DNA binding domain or Gal4-activation domain fusions, with transformant colonies being selected on Leu/Trp-deficient plates and patched onto His/Leu/Trp-deficient plates. Growth was scored following 72 h of incubation.
Co-immunoprecipitation Assays
Following transfection, cells were lysed on ice in 500 μl of co-immunoprecipitation buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40 (v/v), 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mm PMSF, or 25 mm Tris, pH 7.9, 10% glycerol (v/v), 0.1% Nonidet P-40 (v/v), 0.5 mm DTT, 5 mm MgCl2, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mm PMSF). Supernatants were collected by centrifugation; two 5-μl aliquots were kept for input, whereas the remaining whole cells extracts were used for immunoprecipitation. Extracts were precleared by adding 20 μl of protein G-agarose slurry (Roche Applied Science) and incubating at 4 °C for 30 min. Beads were removed, and extracts were incubated with 5 μg of antibody (α-FLAG or α-HA) for 1 h at 4 °C. Extracts were transferred to a new tube containing 20 μl of protein G-agarose slurry and incubated for a further 1 h at 4 °C. After incubation, beads were washed with co-immunoprecipitation buffer prior to Western blot analysis.
In Vitro Kinase Assays
5 μg of His-KLF3, KLF3(1–253), and GST-CtBP were each incubated with and without 2.5 μg of HIPK2 in kinase buffer (20 mm HEPES, 20 mm MgCl2, 0.5 mm DTT, 125 mm β-glycerol phosphate, 10 μCi of [γ-32P]ATP, and 25 μm cold ATP) at 30 °C for 30 min. The reactions were stopped by adding 5× SDS-PAGE loading buffer. Samples were resolved by 12% SDS-PAGE and autoradiographed. 25 μm cold ATP alone was used when visualizing proteins by Coomassie staining of SDS-polyacrylamide gels prior to subsequent mass spectrometry analysis.
Trypsin Digest
Bands were excised and destained in 40% (v/v) acetonitrile, 12 mm ammonium bicarbonate for 1 h. Gel slices were desiccated for 30 min and rehydrated in 40 mm ammonium bicarbonate containing 12 ng/μl sequencing grade trypsin (Promega) for 1 h at 4 °C. Excess trypsin was removed, and 40 mm ammonium bicarbonate was added before incubation at 37 °C overnight.
Mass Spectrometry (MS)
0.5 μl of trypsin digest was spotted onto a target plate with an equal volume of matrix solution (10 mg/ml α-cyano-4-hydroxycinnamic acid in 70% (v/v) acetonitrile, 0.1% (v/v) trifluoroacetic acid (TFA)). Where necessary, peptide mixtures were concentrated using Perfect Pure C18 microtips (Eppendorf). Peptide maps were acquired by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) MS using either a QSTAR Elite system (AB Sciex) equipped with MALDI source or a Voyager-DE STR (PerSeptive Biosystems). Data were processed with Analyst and Data Explorer software (Applied Biosystems). Tryptic peptide peak masses were searched against the MASCOT database (Matrix Science) and matches based on MASCOT score and E-value, which were determined by the number of matching peptides and the percentage of sequence coverage of matching peptide masses. Search parameters allowed one missed tryptic cleavage, a 50-ppm error window between observed and theoretical peptide masses, and methionine sulfoxide as a “variable” peptide modification.
Titanium Dioxide Enrichment of Phosphopeptides for MALDI-TOF MS
Tryptic peptides were diluted in loading buffer (100 mg/ml 2,5-dihydroxybenzoic acid in 80% acetonitrile and 2% TFA) and added to a 3-mm TiO2 column containing 5 μl of TiO2 suspension in 100% acetonitrile. Flow-through (unbound fraction) was collected for MS analysis. The column was washed with 5 μl of loading buffer, followed by 30 μl of wash buffer (80% acetonitrile and 2% TFA). Phosphopeptides were eluted in 0.5% ammonia solution. The eluant was acidified with 100% formic acid (1 μl per 10 μl of eluate, pH 2–3). The elution and unbound fractions were desalted and concentrated using POROS Oligo R3 and POROS 20 R2 columns, respectively (∼5 mg of POROS reversed phase material in 200 μl of 50% acetonitrile). Samples were washed in 30 μl of 0.1% TFA and eluted with 1 μl of R2/R3 elution buffer (20 mg/ml 2,5-dihydroxybenzoic acid in 50% acetonitrile with 1% orthophosphoric acid).
Titanium Dioxide Enrichment of Phosphopeptides for LC-ESI-MS/MS
Peptides were enriched as described above but with 1 m glycolic acid replacing 2,5-dihydroxybenzoic acid in the loading buffer. Samples were concentrated in R3 columns and eluted in 20 μl of 50% acetonitrile, 0.05% TFA. They were then lyophilized, dissolved in 0.5 μl of 100% formic acid, and finally diluted in 50 μl of 20% acetonitrile. Phosphopeptides were separated using an Agilent 1100 liquid chromatography (LC) system (Agilent Technologies). Peptides were bound to a C18 analytical column before being eluted with a 5–65% acetonitrile gradient, directly into a QSTAR Elite instrument equipped with a nano-ESI source for tandem MS (MS/MS). MS/MS spectra of phosphopeptides were manually annotated according to standard nomenclature (29, 30).
Dephosphorylation of Trypsin Digests
10 μl of trypsin digest was incubated with 1 unit of alkaline phosphatase (Roche Applied Science) for 15 min at 37 °C. The reaction was terminated by adding TiO2 loading buffer.
Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were carried out as described previously (2). When detecting KLF3 binding, equal amounts of nuclear extracts (typically 10 μg in a volume of 2 μl) were loaded in a total volume of 30 μl containing 50 μg/ml poly(dI-dC), 4.4 mm dithiothreitol, 100 μg/ml bovine serum albumin, 10 mm HEPES (pH 7.8), 50 mm KCl, 5 mm MgCl2, 1.07 mm EDTA, 6.67% glycerol, 1.33 mm MnCl2, 3.33 mm NaCl, 35 mm Tris-HCl (pH 7.5), 3.33 μm EGTA, 0.007% Brij35, 1 μl of preimmune serum or antibody (as appropriate), and 0.5 ng of 32P-radiolabeled probe. Oligonucleotides used in the synthesis of the β-globin radiolabeled probes were 5′-TAGAGCCACACCCTGGTAAG-3′ and 5′-CTTACCAGGGTGTGGCTCTA-3.
Transient Transactivation Assays
Luciferase reporter assays were performed 48 h after transfection using the Promega Luciferase Assay System according to the manufacturer's instructions.
Expression of GST- and His-tagged Fusion Proteins
Overexpression and purification of fusion constructs were performed as described previously (21, 22).
Statistical Analysis
Data are presented as means ± S.D. or S.E., as indicated. Significance was determined using Student's t tests with p < 0.05 being taken as statistically significant.
RESULTS
KLF3 Is Post-translationally Modified
In preliminary experiments to investigate KLF3 post-translational modification, we examined fibroblast and erythroid cells where KLF3 is known to be expressed (9, 31). We first investigated NIH3T3 fibroblasts and found by Western blot that KLF3 migrates as a doublet, characterized by a more intense slower migrating band (Fig. 1). Similar banding patterns were seen upon examination of erythroid murine erythroleukemia and G1E-ER4 (25) cells, suggesting that post-translationally modified forms of KLF3 are also present in these lines. We then treated nuclear extracts with λ-phosphatase prior to Western blot and consistently observed altered migration patterns across all lines, indicating that although various modifications may occur, phosphorylation plays a detectable role in the migration of KLF3 (Fig. 1).
FIGURE 1.
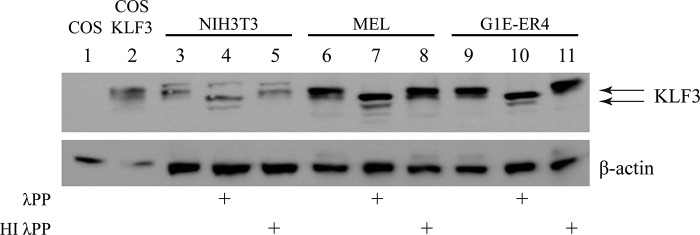
KLF3 is modified by post-translational phosphorylation. Nuclear extracts were purified from NIH3T3, murine erythroleukemia (MEL), and induced G1E-ER4 cells (25). Untreated extracts (lanes 3, 6, and 9), extracts treated with λ-phosphatase (λPP; lanes 4, 7, and 10), and extracts treated with heat-inactivated λ-phosphatase (HI λPP; lanes 5, 8, and 11) were then Western blotted with an anti-KLF3 antibody. The blots were then stripped and incubated with an anti-β-actin antibody to provide a loading control. Lane 1, nuclear extract from COS cells; lane 2, nuclear extract from COS cells transfected with KLF3.
KLF3 Interacts with HIPKs
We used a candidate yeast two-hybrid approach to initially assess potential kinases that may bind and modify KLF3. We detected a strong interaction between KLF3 and the serine/threonine kinase HIPK1 (Fig. 2A). Following this observation, we next examined whether KLF3 can bind to the related and better studied kinase, HIPK2 (16, 32, 33). We found that HIPK2 can be co-immunoprecipitated with KLF3 from transfected COS nuclear extracts (Fig. 2B, lane 3). Because HIPK2 is known to bind the KLF3 partner protein CtBP (34), we asked whether the interaction between KLF3 and HIPK2 is dependent on CtBP2. However, we found that HIPK2 interacted efficiently with a mutant version of KLF3 that is unable to recruit CtBP2, suggesting a direct association between HIPK2 and KLF3 independent of CtBP2 (Fig. 2B, lane 6). Thus, these findings suggest that HIPK2 binds to both CtBP2 and KLF3.
FIGURE 2.
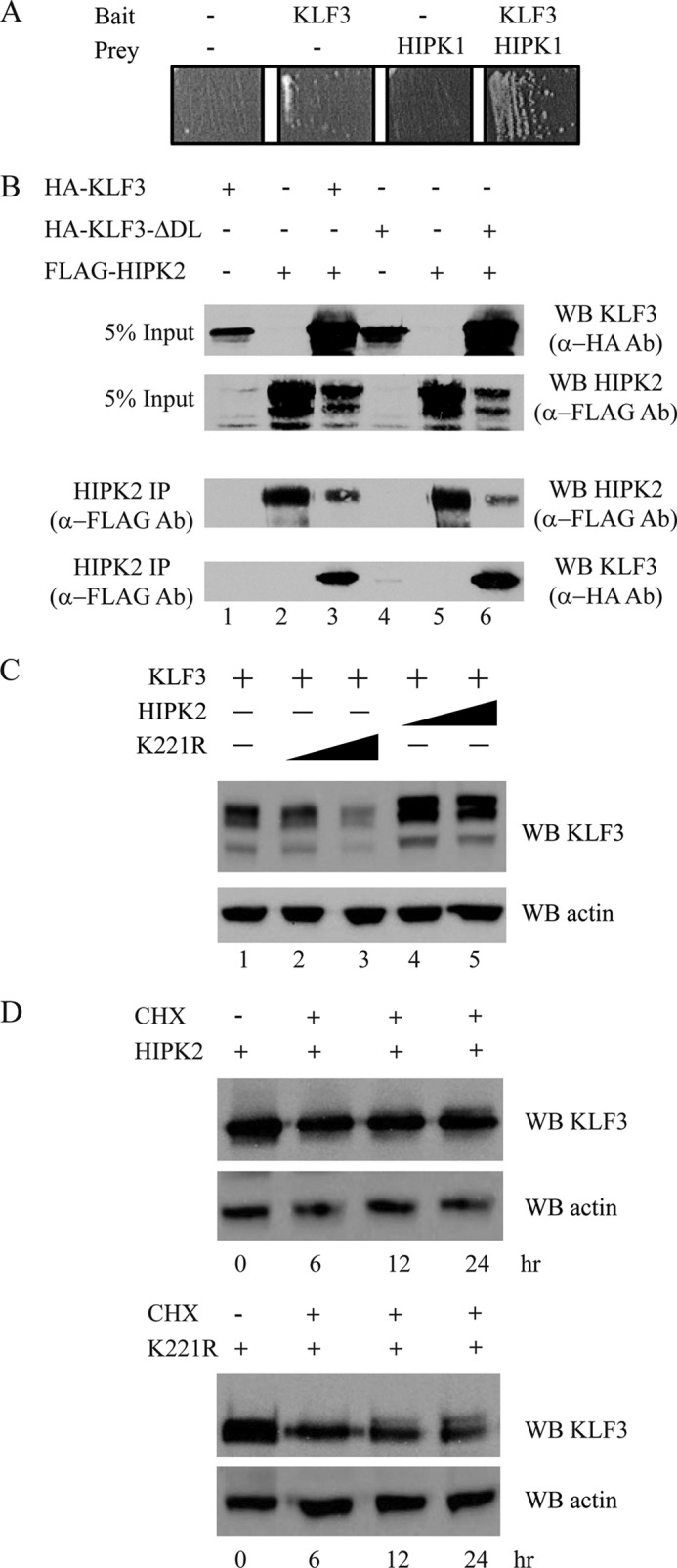
KLF3 interacts with HIPKs. A, HIPK1 and KLF3 associate in yeast two-hybrid assays. Transfection of pGBT9-KLF3(1–268) and pGAD10-HIPK1(617–901) into yeast resulted in growth on selective media. B, KLF3 and HIPK2 co-immunoprecipitate. COS-7 cells were transfected with 2 μg of pMT3-HA.KLF3 (lane 1), 2 μg of pMT3-FLAG.HIPK2 (lane 2), or 2 μg of pMT3-HA.KLF3 and 2 μg of pMT3-FLAG.HIPK2 (lane 3). Lanes 4–6 are identical to lanes 1–3, except pMT3-HA.KLF3 was replaced with pMT3-HA.KLF3-ΔDL, which expresses a mutant form of KLF3 unable to bind the cofactor CtBP (6). 48 h after transfection, cells were harvested, whole cell extracts were prepared, and HIPK2 was immunoprecipitated (IP) using an anti-FLAG antibody. Input (top two panels) and immunoprecipitate (bottom two panels) were Western blotted (WB) with anti-FLAG and anti-HA antibodies for detection of HIPK2 and KLF3, respectively. C, HIPK2 alters the migration of KLF3 in SDS-PAGE. COS-7 cells were transfected with KLF3 and either wild type HIPK2 or a kinase-inactive mutant HIPK2-K221R (18). Nuclear extracts were prepared and Western blotted with an anti-KLF3 antibody. Lane 1, 1 μg of pcDNA3-KLF3; lane 2, 1 μg of pcDNA3-KLF3 and 0.5 μg of pEGFP-Hipk2-K221R; lane 3, 1 μg of pcDNA3-KLF3 and 2 μg of pEGFP-Hipk2-K221R; lanes 4 and 5 are identical to lanes 2 and 3, respectively, except kinase-inactive pEGFP-Hipk2-K221R was replaced with wild type pEGFP-Hipk2. Bottom, the blot was stripped and probed with an anti-β-actin antibody to confirm equal loading of nuclear extracts. D, HIPK2 stabilizes KLF3 protein. COS-7 cells were transfected with KLF3 and either wild type or kinase-inactive HIPK2. Cells were treated with cycloheximide (CHX) 24 h after transfection, and nuclear extracts were harvested at the indicated time points for Western blotting with an anti-KLF3 antibody. Bottom, the blot was stripped and probed with an anti-β-actin antibody to confirm equal loading of nuclear extracts.
Encouraged by an inspection of the amino acid sequence of KLF3 that revealed the presence of 12 potential HIPK phosphorylation sites, conforming to the consensus of (S/T)P (19), we next examined whether HIPK2 can phosphorylate KLF3. We observed altered migration of KLF3 in gel electrophoresis of nuclear extracts purified from COS cells cotransfected with KLF3 and HIPK2 (Fig. 2C, lanes 4 and 5). This effect was not observed when KLF3 was cotransfected with a mutant version of HIPK2, HIPK2-K221R, in which the active site of the kinase domain has been inactivated by a lysine to arginine substitution (18) (Fig. 2C, lanes 2 and 3). Furthermore, it appeared that KLF3 levels were increased in the presence of wild type HIPK2, which could be explained by either altered Klf3 gene expression or protein stability (Fig. 2C, compare lane 2 with lane 4 and compare lane 3 with lane 5).
To further investigate the effect of HIPK2 on KLF3 protein stability, we cotransfected COS cells with KLF3 and either wild-type or kinase-inactive HIPK2 and then treated cells with cycloheximide to inhibit de novo protein synthesis (Fig. 2D). In the 24-h period following treatment, we noticed considerably more degradation of KLF3 when cells were cotransfected with the kinase-inactive form of HIPK2, in line with the hypothesis that phosphorylation of KLF3 by HIPK2 directly increases its stability.
KLF3 Is Directly Phosphorylated by HIPK2
Having established that HIPK2 can bind KLF3 and cause its modification in cellular assays, we wished to confirm that this was a direct effect. To do this, we carried out in vitro kinase assays using bacterially expressed KLF3 and HIPK2 and radiolabeled ATP (Fig. 3, A and B). We investigated both full-length KLF3 and a truncated form of the protein (amino acids 1–253), which lacks the zinc-finger DNA binding domain, a region that does not contain any canonical HIPK2 phosphorylation sites. We found that preincubation of both forms of KLF3 with HIPK2 resulted in the appearance of an intense radiolabeled band corresponding to the expected size of expressed KLF3, whereas no phosphorylated protein was detected in the absence of HIPK2. In our assays, we observed greater radiolabeling of the truncated form of KLF3, suggesting that it is a better substrate for phosphorylation and that the DNA-binding domain may influence HIPK2 activity (Fig. 3B).
FIGURE 3.

KLF3 is directly phosphorylated by HIPK2. Full-length His-tagged KLF3 (A) or a truncated form of KLF3 (amino acids 1–253) lacking the DNA binding domain (B) was expressed in bacteria and incubated with radiolabeled ATP in the absence (lanes 1 and 3) or presence of HIPK2 (lanes 2 and 4). Samples were resolved by SDS-PAGE (left) and autoradiographed (right). C, amino acid sequence of KLF3 with serine and threonine residues conforming to the HIPK phosphorylation site consensus underlined. The phosphorylated peptide identified by mass spectrometry is boxed. D–G, MALDI-TOF MS spectra of TiO2-bound tryptic peptides from bacterially expressed KLF3. Shown are the spectrum of His-tagged full-length KLF3 (D) and His-tagged KLF3 incubated with HIPK2 (E). Spectrum from truncated KLF3 containing amino acids 1–253 (KLF3(1–253)) (F). G, spectrum from KLF3(1–253) incubated with HIPK2. The peptide sequences shown in E and G correspond to the major peaks and have a potential HIPK2 phosphorylation site at Ser-249 underlined.
KLF3 contains 12 potential phosphorylation sites that conform to the HIPK2 consensus of Ser/Thr-Pro (19) (Fig. 3C). To determine which of these are directly phosphorylated in vitro by HIPK2, we excised the major radiolabeled band and performed MALDI-TOF MS, following tryptic digest and TiO2 enrichment for phosphopeptides (Fig. 3, D–G). In the case of both full-length and truncated KLF3, we observed a single TiO2-purified phosphopeptide with an observed mass suggesting phosphorylation of a serine residue at amino acid 249 (Fig. 3, E and G) (threonine 252 phosphorylation could also generate this mass, although this is not a HIPK2 consensus site). This phosphopeptide was not observed in the absence of HIPK2 (Fig. 3, D and F). MALDI-TOF MS/MS of the phosphopeptide from full-length KLF3 confirmed phosphorylation at Ser-249 (Fig. 4A), the most C-terminal HIPK2 consensus site, which is located close to the zinc finger DNA-binding domain of KLF3 (Fig. 4B). Mutation of Ser-249 to an alanine residue resulted in a noticeable reduction in the level of HIPK2-mediated in vitro phosphorylation of KLF3 (Fig. 4C). The observation of residual phosphorylation of this mutant form of KLF3 indicates that although Ser-249 is the major target, one or more additional sites can also be phosphorylated by HIPK2 in vitro, albeit to a noticeably lesser extent.
FIGURE 4.

HIPK2 phosphorylates KLF3 at serine 249 in vitro. A, full-length KLF3 was incubated with HIPK2 and subjected to MALDI-TOF MS/MS following tryptic digest and TiO2 enrichment of phosphopeptides. Shown is a representative spectrum. Based on the y ion series (y1, y3–5, y9) and the b ion series (b1–3, b6–11), the sequence of the parent ion with mass 1474.8 was determined to be RPLPVESPDTQR, corresponding to KLF3 residues 243–254. Within this peptide, phosphorylation of Ser-249 was determined by signals b6, b7-H2O, and b8. The neutral loss of phosphoric acid (H3PO4, 98 Da) is indicated. B, schematic diagram of KLF3, indicating the position of potential phosphorylation sites conforming to the HIPK consensus of (S/T)P. The in vitro phosphorylation site at serine 249 is in boldface type and underlined. C, wild type KLF3 (lanes 1 and 2) and a mutant form of KLF3 in which serine 249 has been mutated to an alanine residue (lanes 3 and 4) were subject to in vitro kinase assays in the presence or absence of HIPK2.
KLF3 Is Phosphorylated at Multiple Sites in Vivo
To assess in vivo phosphorylation, we transfected COS cells either with KLF3 alone or with KLF3 and HIPK2 and performed mass spectrometry on TiO2-enriched KLF3 phosphopeptides (Fig. 5 and Table 1). We observed phosphorylation at Ser-249 of KLF3 but also observed signals consistent with predicted HIPK2 consensus phosphorylation at Ser-71, Ser-78, Ser-91, Ser-100, Ser-215, and Ser-223. Because the action of other kinases may also contribute to phosphopeptides via both HIPK2 and alternative kinase consensus motifs, we employed cotransfection of HIPK2 with KLF3 to determine whether elevated HIPK2 had a notable effect on phosphorylation events at these and other potential sites. The region containing amino acids 70–83 has two potential phosphorylation sites at Ser-71 and Ser-78. In cells transfected with KLF3 alone, we identified peptides where one or both of these sites were phosphorylated, whereas in the presence of transfected HIPK2, we could detect only a single peak, consistent with phosphorylation at both sites (Fig. 5, A and B). We saw the same effect with the peptide containing Ser-215 and Ser-223 (Fig. 5, C and D). Additionally, we observed new phosphopeptides with masses spanning amino acids 105–116, 106–116, 105–132, and 106–132 in the presence of HIPK2 (Fig. 5, compare A with B and C with D). These suggest HIPK2-driven phosphorylation at Ser-107 and Ser-110, which we confirmed by MS/MS of the 1463.02 m/z ion (data not shown). We also found that the level of phosphorylation at Ser-249 is elevated in the presence of transfected HIPK2, as shown by the increase in intensity of a phosphopeptide peak covering amino acids 243–254 (Fig. 5, A and B). We were unable to detect peaks corresponding to phosphopeptides containing Thr-40 and Thr-49 in TiO2-enriched fractions, both in the presence and absence of transfected HIPK2, suggesting that these sites may not be phosphorylated in vivo (Fig. 5, A, B, E, and F) (data not shown). Our analysis of the TiO2-unbound samples also revealed peaks corresponding to unphosphorylated forms of Ser-91, Ser-100, and Ser-249 (Fig. 5, E and F). The phosphorylation status of Ser-137 remains unclear because we were unable to detect a peak for the peptide containing this site in any of our fractions.
FIGURE 5.

KLF3 is phosphorylated at multiple sites in vivo. FLAG-tagged KLF3 or FLAG-tagged KLF3 and HIPK2 were transfected into COS-7 cells. Expressed KLF3 was then purified from nuclear extracts by anti-FLAG immunoprecipitation and subjected to SDS-PAGE and tryptic digest. Following enrichment of phosphopeptides on TiO2 columns, fragments were analyzed by MALDI-TOF MS. The masses of ion signals that align to KLF3 peptides are given along with the sequences of the mapped peptides, which are shown on the right (see also Table 1). Potential phosphorylation sites in these peptides are denoted by SP or S(P), where more than one potential site is present and it is unclear which is phosphorylated. Arrows, HIPK2-driven phosphorylation events. Mox, oxidation of a methionine residue (+16 Da). A and B, representative MALDI-TOF MS spectra of TiO2-enriched KLF3 peptides within the m/z range 1200–1900 following COS cell transfection of KLF3 alone (A) or KLF3 and HIPK2 (B). C and D, representative spectra of TiO2-enriched peptides within the m/z range 2000–4500. E and F, representative spectra of the TiO2-unbound fraction.
TABLE 1.
Assignment of KLF3 MALDI-TOF MS ion peaks post-TiO2 enrichment of phosphopeptides
Transfected KLF3 was purified from COS-7 cells and subject to tryptic digest. Samples were then enriched for phosphopeptides and subjected to MALDI-TOF MS analysis. Shown are peptide sequences assigned to ion signals generated in the presence and absence of cotransfected HIPK2, revealing sites that can be phosphorylated by endogenous kinases and/or expressed HIPK2. HIPK2 consensus phosphorylation sites in TiO2-enriched phosphopeptides are indicated by SP, or S(P) (where phosphosite assignment has not been confirmed). Theoretical mass represents the mass of predicted tryptic peptides without modifications. Mox represents potential oxidation of a methionine residue (+16 Da). ▾, missed trypsin cleavage site; ▴, nonspecific tryptic cleavage.
| Observed mass | Assigned residues | Assigned sequence | Theoretical mass | Potential phosphorylation site(s) |
|---|---|---|---|---|
| 1352.6 | 34–45 | YGVIYSTPLPDK | 1352.6 | Thr-40 |
| 2570.2 | 46–68 | FFQTPEGLTHGIQVEPVDLTVNK | 2570.2 | Thr-49 |
| 2727.4 | 46–69 | FFQTPEGLTHGIQVEPVDLTVNK▾R | 2727.4 | Thr-49 |
| 1292.6 | 70–83 | GS(P)PPAAGGS(P)PSSLK | 1212.6 | Ser-71, Ser-78 |
| 1372.6 | 70–83 | GSPPPAAGGSPPSSLK | 1212.6 | Ser-71, Ser-78 |
| 1528.6 | 69–83 | R▾GSPPPAAGGSPPSSLK | 1368.6 | Ser-71, Ser-78 |
| 1787.7 | 89–104 | R▾ASPPGLSMoxPSSSPPPIK | 1611.7 | Ser-91, Ser-100 |
| 1631.6 | 90–104 | ASPPGLSMoxPSSSPPPIK | 1455.7 | Ser-91, Ser-100 |
| 1463.0 | 105–116 | KYSPPPSPPGVQPF▴ | 1303.0 | Ser-107, Ser-110 |
| 1335.5 | 106–116 | YSPPPSPPGVQPF▴ | 1175.5 | Ser-107, Ser-110 |
| 2995.3 | 105–132 | KYS(P)PPS(P)PGVQPFGVPLSMoxPPVMoxAAALSR | 2899.3 | Ser-107, Ser-110 |
| 3075.3 | 105–132 | KYSPPPSPPGVQPFGVPLSMoxPPVMoxAAALSR | 2899.3 | Ser-107, Ser-110 |
| 2867.3 | 106–132 | YS(P)PPS(P)PGVQPFGVPLSMoxPPVMoxAAALSR | 2771.3 | Ser-107, Ser-110 |
| 2947.3 | 106–132 | YSPPPSPPGVQPFGVPLSMoxPPVMoxAAALSR | 2771.3 | Ser-107, Ser-110 |
| 4117.8 | 207–242 | TDYYPEEMoxS(P)PPLMoxNPVS(P)PPQALLQENHPSVIVQPGK | 4005.8 | Ser-215, Ser-223 |
| 4197.8 | 207–242 | TDYYPEEMoxSPPPLMoxNPVSPPPQALLQENHPSVIVQPGK | 4005.8 | Ser-215, Ser-223 |
| 1474.7 | 243–254 | RPLPVESPPDTQR | 1394.7 | Ser-249 |
The results of these experiments (Figs. 4 and 5) and the migration patterns shown in Fig. 1 suggest that KLF3 is subject to complex phosphorylation in vivo. Ser-249 appears to be directly phosphorylated by HIPK2, but other sites are also modified either by HIPK2 or by other kinases that may be activated by HIPK2 or indeed by alternative independent kinases.
CtBP2 Is Also Phosphorylated by HIPK2
Given that KLF3 regulates target gene expression via recruitment of the repressive cofactor CtBP2 (6) and that the activity of CtBP1 is regulated by HIPK2-mediated phosphorylation (34, 35), we were interested to see whether HIPK2 could also directly phosphorylate CtBP2. We performed in vitro kinase assays with bacterially expressed GST-CtBP2 and HIPK2. We observed noticeable phosphorylation of CtBP2 in the presence of HIPK2 (Fig. 6A). In addition to a band migrating at the expected molecular weight for GST-CtBP2, we also observed a faster moving band corresponding to the expected weight of CtBP2 (Fig. 6A, lanes 1 and 2). This band was excised, digested with trypsin, and subjected to MALDI-TOF MS, following TiO2 enrichment of phosphopeptides (Fig. 6, B–E). We were unable to detect any CtBP2 phosphopeptides in the absence of HIPK2 (Fig. 6D). In the TiO2-unbound fractions, we observed identical ion peaks in both the absence and presence of HIPK2 (Fig. 6, B and C). However, when CtBP2 was incubated with HIPK2, a shift was seen in a peak corresponding to a peptide covering amino acids 387–434 (Fig. 6, C, E, and F). This shift reflects an increase in mass of 80 Da and is consistent with HIPK2-mediated phosphorylation of CtBP2 at Ser-428 and is further supported by alkaline phosphatase treatment, which resulted in the disappearance of this peak from the TiO2-enriched fraction (Fig. 6G).
FIGURE 6.

CtBP2 is directly phosphorylated by HIPK2. A, GST-CtBP2 was expressed in bacteria and incubated with radiolabeled ATP in the absence (lanes 1 and 3) or presence of HIPK2 (lanes 2 and 4). Samples were resolved by SDS-PAGE (left) and autoradiographed (right). B–E, MALDI-TOF MS spectra of TiO2-unbound and -bound tryptic peptides from bacterially expressed GST-CtBP2 incubated without (B and D) and with HIPK2 (C and E). F, masses of ion signals that align to CtBP2 peptides and sequences of the mapped peptides. An 80-Da shift in the peak at m/z 4747 to a peak at m/z 4827 is consistent with phosphorylation at Ser-428 (boldface type and underlined). This shift is shown by a dotted line extending from C to E. G, alkaline phosphatase treatment results in the loss of the 4827 peak in the HIPK2-treated sample. Mox, oxidation of a methionine residue (+16 Da).
CtBP2 Is Phosphorylated by HIPK2 at Serine Residue 428 in Vivo
To further investigate the potential phosphorylation of CtBP2 at Ser-428 in vivo, we cotransfected COS cells with CtBP2 in the presence and absence of HIPK2. Transfected CtBP2 was purified from these cells and subjected to MALDI-TOF MS analysis, following tryptic digest and enrichment of phosphopeptides on TiO2 columns (Fig. 7). Several ion peaks were detected and assigned to CtBP2 peptides (Fig. 7, A and B), although no phosphorylated peptides could be detected in the absence of cotransfected HIPK2 (Fig. 7C). In the presence of HIPK2, two peaks at m/z 2065 and m/z 4827 were apparent in the TiO2-enriched fraction that mapped to phosphorylation of CtBP2 at Ser-428 (Fig. 7D and Table 2). LC-ESI-MS/MS of a triply charged precursor ion at m/z 688 was used to generate sequence fragment ions of the 2065 phosphopeptide, which confirmed the HIPK2-mediated phosphorylation of this site (Fig. 8).
FIGURE 7.

CtBP2 is phosphorylated by HIPK2 in vivo. FLAG-tagged CtBP2 was expressed in COS-7 cells in the absence (A and C) and presence (B and D) of transfected HIPK2. FLAG-CtBP2 was immunoprecipitated and subjected to SDS-PAGE and tryptic digest. Phosphopeptides were enriched on TiO2 columns (C and D), and samples were analyzed by MALDI-TOF MS. The masses of ion peaks are indicated, and assigned peptides are given in Table 2.
TABLE 2.
Assignment of CtBP2 MALDI-TOF MS ion peaks post-TiO2 enrichment of phosphopeptides
Transfected CtBP was purified from COS-7 cells and subject to tryptic digest. Samples were then enriched for phosphopeptides and subjected to MALDI-MS analysis. Shown are peptide sequences assigned to ion signals generated in the presence and absence of cotransfected HIPK2. HIPK2 consensus phosphorylation sites are indicated by SP. Theoretical mass represents the mass of predicted tryptic peptides without modifications. Mox represents potential oxidation of a methionine residue (+16 Da). ▾, missed trypsin cleavage site.
| Observed mass | Assigned residues | Assigned sequence | Theoretical mass | Potential phosphorylation site(s) |
|---|---|---|---|---|
| 2053.8 | 72–89 | VLNEAVGAMoxMoxYHTITLTR | 2021.8 | |
| 2101.3 | 292–311 | GAALDVHESEPFSFAQGPLK | 2101.3 | |
| 2238.3 | 201–219 | AFGFSVIFYDPYLQDGIER | 2238.3 | |
| 3079.0 | 360–386 | EFFVTSAPWSVIDQQAIHPELNGATYR | 3079.0 | |
| 3402.4 | 115–147 | AAGELGIAVCNIPSAAVEET ADSTVCHILNLYR | 3402.4 | |
| 4043.0 | 226–259 | VYTLQDLLYQSDCVSLHCNLNEHNHHLINDFTIK | 4043.0 | |
| 2065.3 | 416–434 | ▾NLPTVAHPSQAPSPPNQPTK | 1985.3 | Ser-428 |
| 4747.8 | 387–434 | YPPGIVGVAPGGLPPAMoxEGIIPGGIPVTHNLPTVAHPSQAPSPNQPTK | 4731.8 | Ser-428 |
| 4827.2 | 387–434 | YPPGIVGVAPGGLPPAMoxEGIIPGGIPVTHNLPTVAHPSQAPSPPNQPTK | 4731.2 | Ser-428 |
FIGURE 8.

CtBP2 is phosphorylated by HIPK2 at serine 428 in vivo. LC-ESI-MS/MS analysis of the 2065.3 m/z peptide peak detected following cotransfection of CtBP2 and HIPK2. This peptide gave rise to a doubly (m/z 1032.6) and a triply charged ion (m/z 688.7) (inset), which was selected for fragmentation. From the y ion series, (y3, y6, y7, y8, y12, y17) and the a2 and b2 ions, the sequence of this peptide was determined to be NLPTVAHPSQAPSPPNQPTK, which corresponds to residues 416–434 of CtBP2. Ser-428, indicated by SP, was determined to be phosphorylated based on the signals y6, y7, and y8-H2O. Neutral losses of phosphoric acid (H3PO4, 98 Da) are indicated.
HIPK2 Potentiates KLF3-mediated Transcriptional Repression
The ability of KLF3 to silence gene expression is dependent upon interaction with its corepressor CtBP (6). We investigated whether modification by HIPK2 influenced the interaction between KLF3 and CtBP2. Co-immunoprecipitation experiments indicated that the interaction between KLF3 and CtBP is strengthened by the presence of HIPK2 (Fig. 9A). We also found that mutation of the CtBP binding domain of KLF3 prevented co-immunoprecipitation in both the presence and absence of HIPK2, suggesting that, in these experiments at least, the kinase cannot act as a bridging molecule between the two proteins (Fig. 9B).
FIGURE 9.
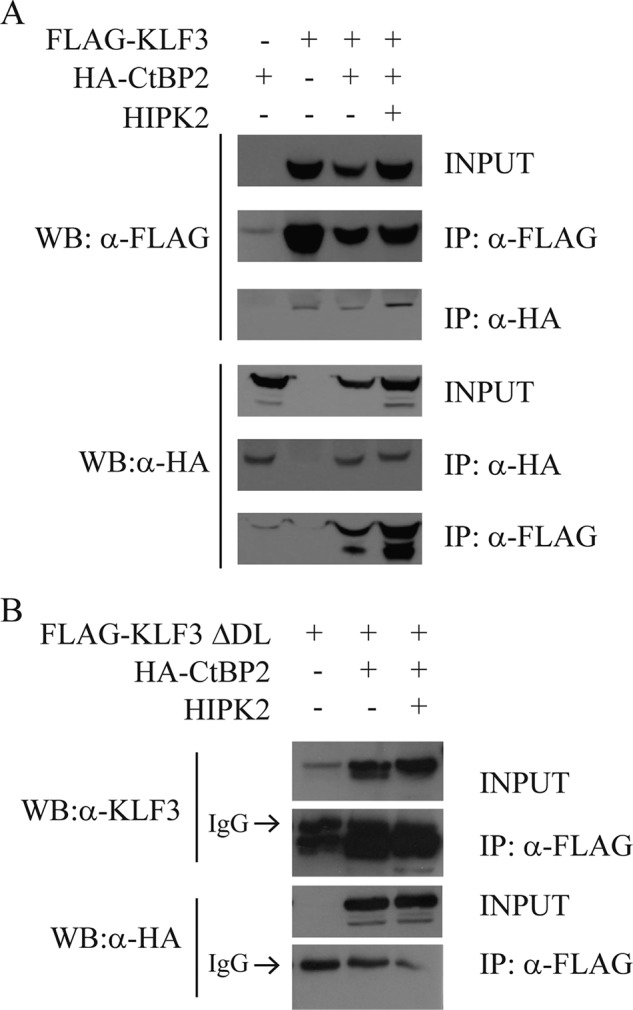
HIPK2 enhances the interaction between KLF3 and its corepressor CtBP2. Co-immunoprecipitation (IP) assays show increased interaction between KLF3 and CtBP2 in the presence of HIPK2. COS-7 cells were transfected with FLAG-KLF3, a mutant form of FLAG-KLF3 unable to bind CtBP (ΔDL), HA-CtBP2, and HIPK2 as indicated. Western blots (WB) for FLAG-KLF3, HA-CtBP2, and FLAG-KLF3-ΔDL were carried out following immunoprecipitation of FLAG-KLF3 and HA-CtBP2 (A) and FLAG-KLF3-ΔDL (B). In B, closely migrating IgG bands are labeled.
To further explore the molecular significance of the interaction between HIPK2 and KLF3, we next investigated whether phosphorylation influences KLF3 DNA binding in EMSAs. We found that treatment of COS-expressed KLF3 with λ-phosphatase resulted in altered electrophoretic mobility and a significant reduction in the ability of KLF3 to interact with DNA (Fig. 10), which was not observed in samples treated with heat-inactivated phosphatase.
FIGURE 10.
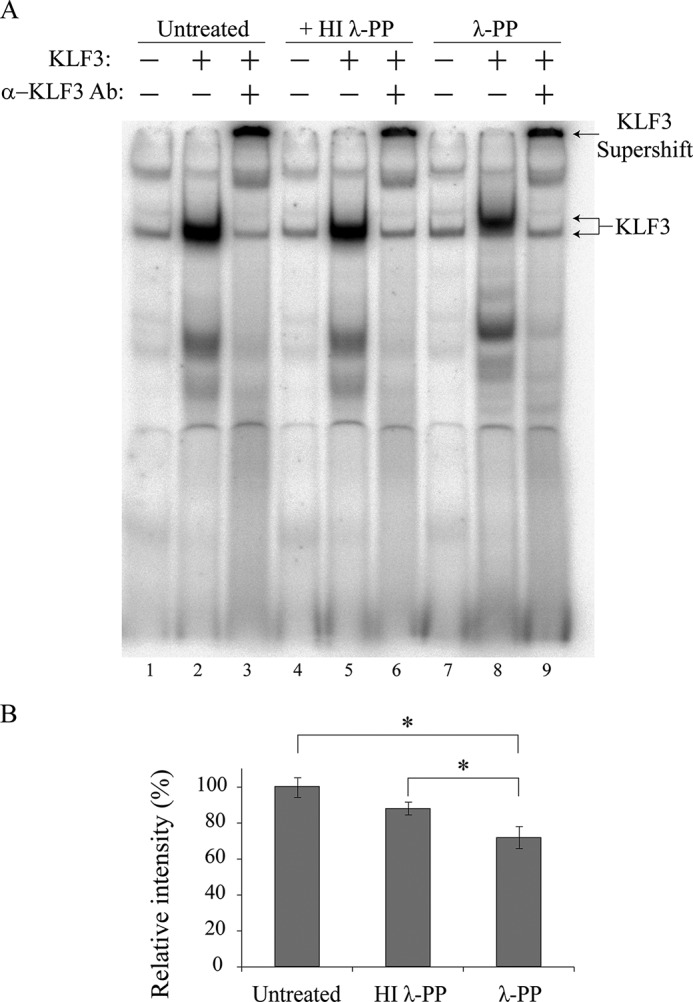
Phosphorylation of KLF3 enhances DNA binding. A, nuclear extracts were prepared from COS-7 cells transfected with pMT3-KLF3. Binding of KLF3 to a β-globin CACCC probe was assessed by EMSA (lane 2) and compared with binding by samples treated with either heat-inactivated λ-phosphatase (HI λ-PP; lane 5) or λ-phosphatase (λ-PP; lane 8). Lanes 1, 4, and 7, untransfected COS nuclear extract controls; α-KLF3 Ab indicates supershift with an anti-KLF3 antibody (lanes 3, 6, and 9). B, relative binding of untreated KLF3, KLF3 incubated with heat-inactivated λ-phosphatase, and KLF3 incubated with λ-phosphatase was quantified using ImageJ software. Error bars, S.E., with binding by untreated KLF3 set to 100% (*, p < 0.05 for a one-tailed Student's t test, n = 3).
To confirm that HIPK2-mediated phosphorylation of KLF3 at Ser-249 can influence DNA binding, HIPK2, KLF3, and KLF3-S249A were expressed in bacteria and purified for EMSA (Fig. 11). In agreement with previous results, incubation of KLF3 with HIPK2 resulted in enhanced DNA binding and a shift in SDS-PAGE migration (compare lanes 3 and 4). These effects are consistent with phosphorylation, which we confirmed by the inclusion of λ-phosphatase (lane 5). In the case of the KLF3-S249A mutant, the presence of HIPK2 and λ-phosphatase treatment had no discernable effect on DNA binding or gel migration (lanes 6–8). This experiment confirms that Ser-249 does have a role in modulating KLF3 activity, but taken together with previous results, it is probable that other phosphorylation events also influence DNA binding by KLF3.
FIGURE 11.
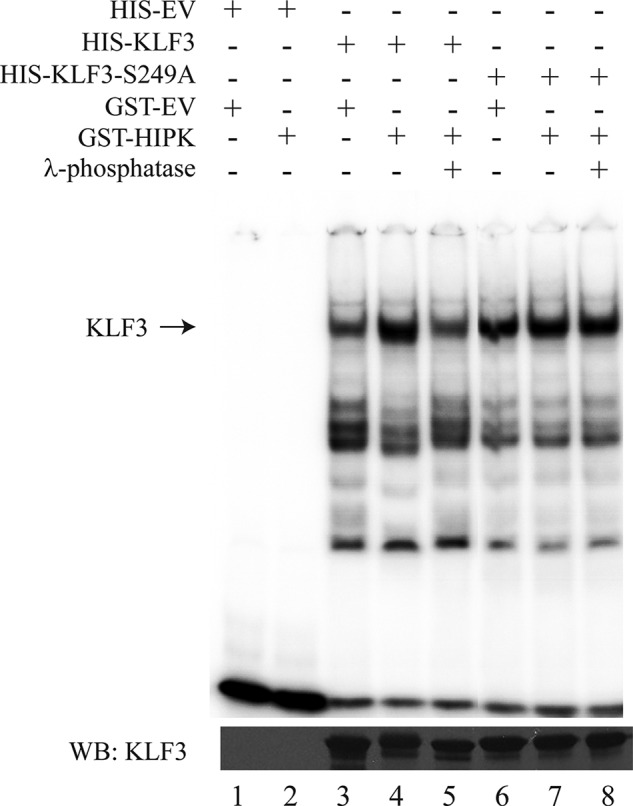
Phosphorylation of KLF3 at serine 249 influences DNA binding. Top, His-KLF3, His-KLF3-S249A, and GST-HIPK2 were expressed in bacteria and purified for EMSA. The binding of KLF3 (lanes 3 and 4) and KLF3-S249A (lanes 6 and 7) to a β-globin CACCC probe was assessed in the presence and absence of HIPK2. The effect of λ-phosphatase treatment on DNA affinity was also examined for both constructs (lanes 5 and 8). HIS-EV, empty pET15b expression vector; GST-EV, empty pGEX-2T expression vector. Bottom, Western blot (WB) of expressed KLF3 and KLF3-S249A. Lanes are as for the top.
Finally, we examined how HIPKs influence the ability of KLF3 to silence expression of Klf8, a well characterized in vivo KLF3 target gene (9, 31, 36), first in a series of cell-based reporter assays and second by examining the effect of ablation of HIPK1 and HIIPK2 on Klf8 promoter activity (Fig. 12). In the reporter experiments (Fig. 12A), we found that increased expression of HIPK2 boosts silencing of the Klf8 promoter in a dose-dependent manner. In further support of a physiological role for HIPKs in potentiating KLF3 activity, we observed a significant increase in Klf8 expression in both HIPK1 knock-out and HIPK2 knock-out murine embryonic fibroblasts, with loss of HIPK2 resulting in the more notable derepression of this well characterized KLF3 target gene (36).
FIGURE 12.
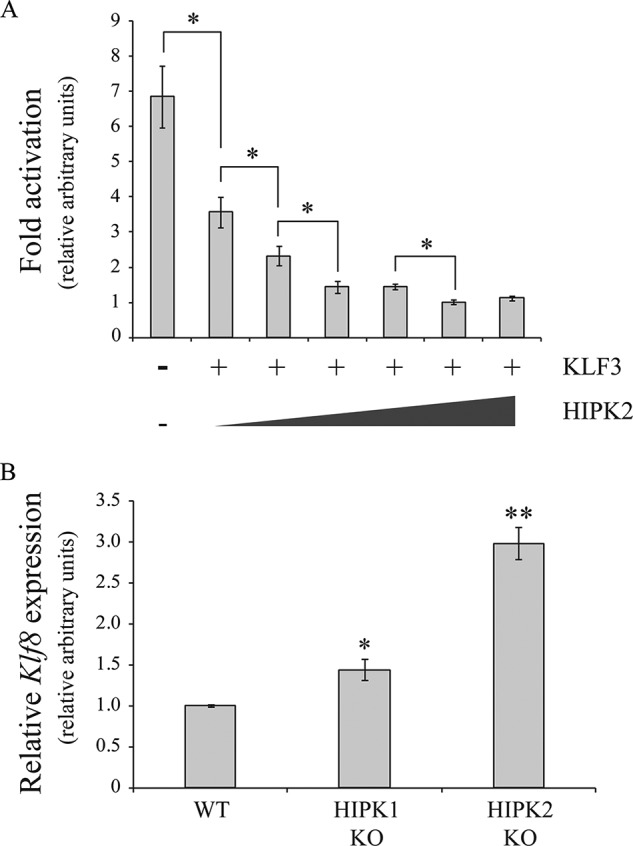
HIPK2 potentiates KLF3 mediated gene repression in vivo. A, COS-7 cells were transfected with 1.0 μg of the KLF3-responsive reporter plasmid pGL4.10-KLF8 (36), 50 ng of pcDNA3-KLF3, and increasing amounts of pEGFP-HIPK2 (0, 25, 50, 100, 250, and 500 ng), as indicated. Shown is relative -fold activation in arbitrary units; error bars, S.E. for n = 3. *, p < 0.05, one-tailed Student's t test for increased repression of the Klf8 promoter. B, expression levels of the KLF3 target gene Klf8 were determined in wild type (WT), HIPK1 knock-out (HIPK1 KO), and HIPK2 knock-out (HIPK2 KO) murine embryonic fibroblasts by real-time RT-PCR. For analysis of real-time data, expression levels were normalized to 18 S rRNA and are shown relative to the WT value, which was set to 1.0. Error bars, S.E. *, p < 0.05; **, p < 0.005, one tailed Student's t test for an increase in Klf8 expression relative to WT control.
DISCUSSION
Recent advances in our understanding of the in vivo biology of KLF3 have revealed widespread physiological roles in diverse tissues, highlighting the importance of this transcriptional repressor in normal cellular development and differentiation (8–15, 37). These studies have also provided some insight into the molecular mechanisms whereby KLF3 regulates expression of target genes. It is clear that recruitment of the corepressor CtBP influences both gene silencing and in vivo specificity (6, 31). However, less is known about the signaling pathways and post-translational modifications that modulate KLF3 activity, although previous work has demonstrated a role for sumoylation in enhancing gene repression (20). In screening for binding partners of KLF3, we have discovered a novel interaction with HIPK, a serine/threonine kinase known to regulate transcription factor activity to control cell survival, proliferation, and differentiation.
KLF3 contains a number of HIPK consensus sites, and we have found that several of these are phosphorylation targets. In vivo, the pattern of phosphorylation appears to be complex, but our results suggest that HIPK2 potentiates the activity of KLF3 through multiple mechanisms. HIPK2 enhances the stability of KLF3, it increases its binding to the co-repressor CtBP2, and it enhances its affinity for DNA. Moreover, HIPK2 also phosphorylates CtBP2, consistent with a view that it is a key kinase in the regulation of KLF3/CtBP2-mediated repression.
HIPK2 has been identified as a transcriptional cofactor in a number of developmental processes and also as a tumor suppressor capable of influencing cellular proliferation and apoptosis (33, 38). The tumor suppressor function of HIPK2 is mediated via both p53-dependent and p53-independent mechanisms, leading to considerable interest in modulating HIPK activity in cancer therapy (39). The p53-dependent pathway is well described; following DNA damage, activation of HIPK leads to site-specific phosphorylation of p53 and up-regulation of downstream targets that promote cell arrest and death (40–42). In contrast, the p53-independent pathway acts via phosphorylation of factors such as CtBP (34, 35).
HIPK2 partners with a number of transcription factors to regulate development in a variety of contexts, in which phosphorylation can result in either protein stabilization and enhanced transcriptional activity or gene silencing due to targeting of the transcription factor for proteosomal degradation. For example, HIPK2-mediated phosphorylation of c-Myb, in combination with a second kinase, Nemo-like kinase, results in phosphorylation at multiple sites, ubiquitination, and proteosomal degradation, with the associated loss of c-Myb activity driving hematopoietic differentiation (43). In contrast, phosphorylation of the developmental regulator Pax6 enhances its association with p300 and leads to significantly elevated target gene activity (44).
Although the effects and in vivo patterns of HIPK2 phosphorylation of KLF3 remain to be fully defined, in the various cellular contexts examined, we have identified a role for Ser-249, which is also the primary target of HIPK2 in vitro. We have shown that mutation of Ser-249 reduces the affinity of KLF3 for DNA. This site is located close to the C-terminal DNA binding domain, and it is possible that phosphorylation results in conformational changes that allow improved DNA access for the zinc fingers of KLF3. In support of this, we have observed that deletion of the DNA binding domain results in noticeably increased phosphorylation of Ser-249 (Fig. 3). This implies an autoinhibitory role for the Ser-249 region; similar inhibitory domains, where steric hindrance of DNA binding is relieved by phosphorylation, are found in the related factor KLF1 (45, 46) and also in p53 (47).
Phosphorylation at two additional sites, Ser-71 and Ser-78, may also influence KLF3 activity. These sites, which lie close to the PVDLT CtBP2 interaction domain (6), appear to show a reduction in phosphorylation intensity in the presence of HIPK2 (Fig. 5, compare A and B). The binding pocket of CtBP is a hydrophobic region (48), with interaction at this site likely to be affected by phosphorylation. It is hence possible that HIPK2 preferentially directs phosphorylation elsewhere as part of a mechanism to promote KLF3 and CtBP2 binding affinity.
It has previously been shown that HIPK2 phosphorylates CtBP1 at serine 422 (35), and in line with this, we have demonstrated phosphorylation of CtBP2 at serine 428 in vivo. Whereas HIPK-mediated phosphorylation results in CtBP1 degradation, we have found that HIPK2 stabilizes the interaction between CtBP2 and KLF3. CtBP1 degradation occurs specifically in response to DNA damage, and it is probable that the physiological mechanisms and consequences of HIPK phosphorylation are context-dependent, as evidenced by the complex phenotype of HIPK1/HIPK2-deficient mice (17). Differences in post-translational modification are also seen in the case of p53, where UV dose influences phosphorylation patterns and hence regulation of downstream target genes, resulting in either cell cycle arrest or apoptosis in stressed cells (49). However, although repression of shared targets with p53 and HIPK, such as the anti-apoptotic gene galectin-3 (Lgals3), imply a role for KLF3 in the DNA damage response (9, 31, 50), it remains to be determined whether KLF3 influences either cell survival or apoptosis.
Although this study has focused on HIPK2 phosphorylation of KLF3 and CtBP2, a number of other kinases may also have in vivo roles in regulating their activity. These include MAPK and related family members, including c-Jun N-terminal kinase (JNK) and Nemo-like kinase. JNK and Nemo-like kinase can themselves be phosphorylated by HIPK (43, 51), and it is possible that such kinases function in a regulatory pathway or network to regulate KLF3 activity.
Finally, although the majority of research has focused on the DNA damage response, it is apparent that HIPKs also influence many aspects of physiology, and in support of their role as activators of KLF3, similarities are emerging in the analysis of phenotypes resulting from reduced expression or ablation of either HIPK or KLF3. These experiments have identified significant and overlapping roles for KLF3 and HIPK in areas such as metabolism and hematopoiesis. For example, detailed studies of the metabolic phenotypes of Klf3−/− and Hipk2−/− mice have revealed that both have smaller adipocytes and reduced adiposity. Furthermore, the absence of either KLF3 or HIPK2 results in improved insulin sensitivity and resistance to diet-induced obesity, despite an energy intake similar to that of wild type counterparts (11, 14, 52).
In erythropoiesis, KLF3, HIPK1, and HIPK2 exhibit nearly identical expression patterns, with levels increasing noticeably as TER119-negative erythroid progenitors transition into TER119-positive cells (9, 53). Erythroid cells that lack KLF3 or have reduced HIPK2 both fail to mature correctly, and it has been shown that KLF3- and HIPK1-deficient mice each have enlarged spleens, most likely as a consequence of compensatory stress erythropoiesis (9, 54).
There is also evidence that KLF3 and HIPK are components of the signaling pathways lying downstream of the B cell receptor that influence B cell development and activity (12, 13, 54). Hipk1−/− mice have notably reduced total splenic B cell numbers, and Klf3−/− hematopoietic progenitors show a distinct disadvantage to wild type cells in re-establishing splenic B cells compartments during competitive reconstitution experiments. HIPK1 and KLF3 both have a significant impact on the B cell lineage decisions that control the follicular to marginal zone (MZ) B cell ratio in the spleen. Here any potential interplay between HIPK and KLF3 is complex and likely to be dependent upon other downstream targets because loss of HIPK1 promotes MZ B cell development, whereas abrogation of KLF3 results in a deficiency of MZ B cells. Despite the increase in MZ B cell number in Hipk1−/− mice, these cells show an impaired immune response and proliferative capacity, a phenotype similar to that observed in Klf3−/− mice.
In conclusion, these data suggest complex overlapping roles for KLF3 and HIPK in a number of physiological processes and indicate that disruption of their expression patterns and activity has a profound and related effect on normal cell development and function. Furthermore, given the established role of KLF3 in diverse biological settings, including metabolism, cardiovascular development, and hematopoiesis, the identification of HIPK as a regulator of KLF3 has potential in directing future therapeutic design.
Acknowledgments
We thank C. Y. Choi for generously providing pEGFP-HIPK2 and pEGFP-HIPK2-K221R plasmids and Haruhiko Koseki for generously providing HIPK knock-out tissue.
Note Added in Proof
In the version of this article that was published as a Paper in Press on February 6, 2015, Beeke Wienert's name was misspelled. The correct spelling is now shown.
This work was supported by grants from the Australian National Health and Medical Research Council (NHMRC) and the Australian Research Council (ARC).
- KLF
- Krüppel-like factor
- HIPK
- homeodomain-interacting protein kinase
- CtBP
- C-terminal binding protein
- ESI
- electrospray ionization
- MZ
- marginal zone.
REFERENCES
- 1. Pearson R. C., Funnell A. P., Crossley M. (2011) The mammalian zinc finger transcription factor Kruppel-like factor 3 (KLF3/BKLF). IUBMB Life 63, 86–93 [DOI] [PubMed] [Google Scholar]
- 2. Crossley M., Whitelaw E., Perkins A., Williams G., Fujiwara Y., Orkin S. H. (1996) Isolation and characterization of the cDNA encoding BKLF/TEF-2, a major CACCC-box-binding protein in erythroid cells and selected other cells. Mol. Cell. Biol. 16, 1695–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pearson R., Fleetwood J., Eaton S., Crossley M., Bao S. (2008) Kruppel-like transcription factors: a functional family. Int. J. Biochem. Cell Biol. 40, 1996–2001 [DOI] [PubMed] [Google Scholar]
- 4. Pei J., Grishin N. V. (2013) A new family of predicted Kruppel-like factor genes and pseudogenes in placental mammals. PLoS One 8, e81109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McConnell B. B., Yang V. W. (2010) Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 90, 1337–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Turner J., Crossley M. (1998) Cloning and characterization of mCtBP2, a co-repressor that associates with basic Kruppel-like factor and other mammalian transcriptional regulators. EMBO J. 17, 5129–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi Y., Sawada J., Sui G., Affar el B., Whetstine J. R., Lan F., Ogawa H., Luke M. P., Nakatani Y., Shi Y. (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738 [DOI] [PubMed] [Google Scholar]
- 8. Kelsey L., Flenniken A. M., Qu D., Funnell A. P., Pearson R., Zhou Y. Q., Voronina I., Berberovic Z., Wood G., Newbigging S., Weiss E. S., Wong M., Quach I., Yeh S. Y., Deshwar A. R., Scott I. C., McKerlie C., Henkelman M., Backx P., Simpson J., Osborne L., Rossant J., Crossley M., Bruneau B., Adamson S. L. (2013) ENU-induced mutation in the DNA-binding domain of KLF3 reveals important roles for KLF3 in cardiovascular development and function in mice. PLoS Genet. 9, e1003612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Funnell A. P., Norton L. J., Mak K. S., Burdach J., Artuz C. M., Twine N. A., Wilkins M. R., Power C. A., Hung T. T., Perdomo J., Koh P., Bell-Anderson K. S., Orkin S. H., Fraser S. T., Perkins A. C., Pearson R. C., Crossley M. (2012) The CACCC-binding protein KLF3/BKLF represses a subset of KLF1/EKLF target genes and is required for proper erythroid maturation in vivo. Mol. Cell. Biol. 32, 3281–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Funnell A. P., Mak K. S., Twine N. A., Pelka G. J., Norton L. J., Radziewic T., Power M., Wilkins M. R., Bell-Anderson K. S., Fraser S. T., Perkins A. C., Tam P. P., Pearson R. C., Crossley M. (2013) Generation of mice deficient in both KLF3/BKLF and KLF8 reveals a genetic interaction and a role for these factors in embryonic globin gene silencing. Mol. Cell. Biol. 33, 2976–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sue N., Jack B. H., Eaton S. A., Pearson R. C., Funnell A. P., Turner J., Czolij R., Denyer G., Bao S., Molero-Navajas J. C., Perkins A., Fujiwara Y., Orkin S. H., Bell-Anderson K., Crossley M. (2008) Targeted disruption of the basic Kruppel-like factor gene (Klf3) reveals a role in adipogenesis. Mol. Cell. Biol. 28, 3967–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vu T. T., Gatto D., Turner V., Funnell A. P., Mak K. S., Norton L. J., Kaplan W., Cowley M. J., Agenès F., Kirberg J., Brink R., Pearson R. C., Crossley M. (2011) Impaired B cell development in the absence of Kruppel-like factor 3. J. Immunol. 187, 5032–5042 [DOI] [PubMed] [Google Scholar]
- 13. Turchinovich G., Vu T. T., Frommer F., Kranich J., Schmid S., Alles M., Loubert J. B., Goulet J. P., Zimber-Strobl U., Schneider P., Bachl J., Pearson R., Crossley M., Agenès F., Kirberg J. (2011) Programming of marginal zone B-cell fate by basic Kruppel-like factor (BKLF/KLF3). Blood 117, 3780–3792 [DOI] [PubMed] [Google Scholar]
- 14. Bell-Anderson K. S., Funnell A. P., Williams H., Mat Jusoh H., Scully T., Lim W. F., Burdach J. G., Mak K. S., Knights A. J., Hoy A. J., Nicholas H. R., Sainsbury A., Turner N., Pearson R. C., Crossley M. (2013) Loss of Kruppel-like factor 3 (KLF3/BKLF) leads to upregulation of the insulin-sensitizing factor adipolin (FAM132A/CTRP12/C1qdc2). Diabetes 62, 2728–2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Himeda C. L., Ranish J. A., Pearson R. C., Crossley M., Hauschka S. D. (2010) KLF3 regulates muscle-specific gene expression and synergizes with serum response factor on KLF binding sites. Mol. Cell. Biol. 30, 3430–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rinaldo C., Siepi F., Prodosmo A., Soddu S. (2008) HIPKs: Jack of all trades in basic nuclear activities. Biochim. Biophys. Acta 1783, 2124–2129 [DOI] [PubMed] [Google Scholar]
- 17. Isono K., Nemoto K., Li Y., Takada Y., Suzuki R., Katsuki M., Nakagawara A., Koseki H. (2006) Overlapping roles for homeodomain-interacting protein kinases hipk1 and hipk2 in the mediation of cell growth in response to morphogenetic and genotoxic signals. Mol. Cell. Biol. 26, 2758–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim Y. H., Choi C. Y., Lee S. J., Conti M. A., Kim Y. (1998) Homeodomain-interacting protein kinases, a novel family of co-repressors for homeodomain transcription factors. J. Biol. Chem. 273, 25875–25879 [DOI] [PubMed] [Google Scholar]
- 19. Hanks S. K., Hunter T. (1995) Protein kinases 6: the eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576–596 [PubMed] [Google Scholar]
- 20. Perdomo J., Verger A., Turner J., Crossley M. (2005) Role for SUMO modification in facilitating transcriptional repression by BKLF. Mol. Cell. Biol. 25, 1549–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matthews J. M., Kowalski K., Liew C. K., Sharpe B. K., Fox A. H., Crossley M., MacKay J. P. (2000) A class of zinc fingers involved in protein-protein interactions biophysical characterization of CCHC fingers from fog and U-shaped. Eur. J. Biochem. 267, 1030–1038 [DOI] [PubMed] [Google Scholar]
- 22. Sunde M., McGrath K. C., Young L., Matthews J. M., Chua E. L., Mackay J. P., Death A. K. (2004) TC-1 is a novel tumorigenic and natively disordered protein associated with thyroid cancer. Cancer Res. 64, 2766–2773 [DOI] [PubMed] [Google Scholar]
- 23. Turner J., Nicholas H., Bishop D., Matthews J. M., Crossley M. (2003) The LIM protein FHL3 binds basic Krüppel-like factor/Kruppel-like factor 3 and its co-repressor C-terminal-binding protein 2. J. Biol. Chem. 278, 12786–12795 [DOI] [PubMed] [Google Scholar]
- 24. Tsang A. P., Visvader J. E., Turner C. A., Fujiwara Y., Yu C., Weiss M. J., Crossley M., Orkin S. H. (1997) FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell 90, 109–119 [DOI] [PubMed] [Google Scholar]
- 25. Weiss M. J., Yu C., Orkin S. H. (1997) Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol. Cell. Biol. 17, 1642–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hancock D., Funnell A., Jack B., Johnston J. (2010) Introducing undergraduate students to real-time PCR. Biochem. Mol. Biol. Educ. 38, 309–316 [DOI] [PubMed] [Google Scholar]
- 27. Quinlan K. G., Verger A., Kwok A., Lee S. H., Perdomo J., Nardini M., Bolognesi M., Crossley M. (2006) Role of the C-terminal binding protein PXDLS motif binding cleft in protein interactions and transcriptional repression. Mol. Cell. Biol. 26, 8202–8213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quinlan K. G., Nardini M., Verger A., Francescato P., Yaswen P., Corda D., Bolognesi M., Crossley M. (2006) Specific recognition of ZNF217 and other zinc finger proteins at a surface groove of C-terminal binding proteins. Mol. Cell. Biol. 26, 8159–8172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Biemann K. (1992) Mass spectrometry of peptides and proteins. Annu. Rev. Biochem. 61, 977–1010 [DOI] [PubMed] [Google Scholar]
- 30. Roepstorff P., Fohlman J. (1984) Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601. [DOI] [PubMed] [Google Scholar]
- 31. Burdach J., Funnell A. P., Mak K. S., Artuz C. M., Wienert B., Lim W. F., Tan L. Y., Pearson R. C., Crossley M. (2014) Regions outside the DNA-binding domain are critical for proper in vivo specificity of an archetypal zinc finger transcription factor. Nucleic Acids Res. 42, 276–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rinaldo C., Prodosmo A., Siepi F., Soddu S. (2007) HIPK2: a multitalented partner for transcription factors in DNA damage response and development. Biochem. Cell Biol. 85, 411–418 [DOI] [PubMed] [Google Scholar]
- 33. Calzado M. A., Renner F., Roscic A., Schmitz M. L. (2007) HIPK2: a versatile switchboard regulating the transcription machinery and cell death. Cell Cycle 6, 139–143 [DOI] [PubMed] [Google Scholar]
- 34. Zhang Q., Yoshimatsu Y., Hildebrand J., Frisch S. M., Goodman R. H. (2003) Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell 115, 177–186 [DOI] [PubMed] [Google Scholar]
- 35. Zhang Q., Nottke A., Goodman R. H. (2005) Homeodomain-interacting protein kinase-2 mediates CtBP phosphorylation and degradation in UV-triggered apoptosis. Proc. Natl. Acad. Sci. U.S.A. 102, 2802–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eaton S. A., Funnell A. P., Sue N., Nicholas H., Pearson R. C., Crossley M. (2008) A network of Kruppel-like factors (Klfs): Klf8 is repressed by Klf3 and activated by Klf1 in vivo. J. Biol. Chem. 283, 26937–26947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mak K. S., Burdach J., Norton L. J., Pearson R. C. M., Crossley M., Funnell A. P. W. (2014) Repression of chimeric transcripts emanating from endogenous retrotransposons by a sequence-specific transcription factor. Genome Biol. 15, R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sombroek D., Hofmann T. G. (2009) How cells switch HIPK2 on and off. Cell Death Differ. 16, 187–194 [DOI] [PubMed] [Google Scholar]
- 39. Puca R., Nardinocchi L., Givol D., D'Orazi G. (2010) Regulation of p53 activity by HIPK2: molecular mechanisms and therapeutical implications in human cancer cells. Oncogene 29, 4378–4387 [DOI] [PubMed] [Google Scholar]
- 40. Hofmann T. G., Möller A., Sirma H., Zentgraf H., Taya Y., Dröge W., Will H., Schmitz M. L. (2002) Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4, 1–10 [DOI] [PubMed] [Google Scholar]
- 41. Dauth I., Krüger J., Hofmann T. G. (2007) Homeodomain-interacting protein kinase 2 is the ionizing radiation-activated p53 serine 46 kinase and is regulated by ATM. Cancer Res. 67, 2274–2279 [DOI] [PubMed] [Google Scholar]
- 42. D'Orazi G., Cecchinelli B., Bruno T., Manni I., Higashimoto Y., Saito S., Gostissa M., Coen S., Marchetti A., Del Sal G., Piaggio G., Fanciulli M., Appella E., Soddu S. (2002) Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 4, 11–19 [DOI] [PubMed] [Google Scholar]
- 43. Kanei-Ishii C., Ninomiya-Tsuji J., Tanikawa J., Nomura T., Ishitani T., Kishida S., Kokura K., Kurahashi T., Ichikawa-Iwata E., Kim Y., Matsumoto K., Ishii S. (2004) Wnt-1 signal induces phosphorylation and degradation of c-Myb protein via TAK1, HIPK2, and NLK. Genes Dev. 18, 816–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim E. A., Noh Y. T., Ryu M. J., Kim H. T., Lee S. E., Kim C. H., Lee C., Kim Y. H., Choi C. Y. (2006) Phosphorylation and transactivation of Pax6 by homeodomain-interacting protein kinase 2. J. Biol. Chem. 281, 7489–7497 [DOI] [PubMed] [Google Scholar]
- 45. Chen X., Bieker J. J. (1996) Erythroid Kruppel-like factor (EKLF) contains a multifunctional transcriptional activation domain important for inter- and intramolecular interactions. EMBO J. 15, 5888–5896 [PMC free article] [PubMed] [Google Scholar]
- 46. Ouyang L., Chen X., Bieker J. J. (1998) Regulation of erythroid Kruppel-like factor (EKLF) transcriptional activity by phosphorylation of a protein kinase casein kinase II site within its interaction domain. J. Biol. Chem. 273, 23019–23025 [DOI] [PubMed] [Google Scholar]
- 47. Hupp T. R., Meek D. W., Midgley C. A., Lane D. P. (1992) Regulation of the specific DNA binding function of p53. Cell 71, 875–886 [DOI] [PubMed] [Google Scholar]
- 48. Nardini M., Spanò S., Cericola C., Pesce A., Massaro A., Millo E., Luini A., Corda D., Bolognesi M. (2003) CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. EMBO J. 22, 3122–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Latonen L., Taya Y., Laiho M. (2001) UV-radiation induces dose-dependent regulation of p53 response and modulates p53-HDM2 interaction in human fibroblasts. Oncogene 20, 6784–6793 [DOI] [PubMed] [Google Scholar]
- 50. Cecchinelli B., Lavra L., Rinaldo C., Iacovelli S., Gurtner A., Gasbarri A., Ulivieri A., Del Prete F., Trovato M., Piaggio G., Bartolazzi A., Soddu S., Sciacchitano S. (2006) Repression of the antiapoptotic molecule galectin-3 by homeodomain-interacting protein kinase 2-activated p53 is required for p53-induced apoptosis. Mol. Cell. Biol. 26, 4746–4757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hofmann T. G., Stollberg N., Schmitz M. L., Will H. (2003) HIPK2 regulates transforming growth factor-β-induced c-Jun NH2-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res. 63, 8271–8277 [PubMed] [Google Scholar]
- 52. Sjölund J., Pelorosso F. G., Quigley D. A., DelRosario R., Balmain A. (2014) Identification of Hipk2 as an essential regulator of white fat development. Proc. Natl. Acad. Sci. U.S.A. 111, 7373–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hattangadi S. M., Burke K. A., Lodish H. F. (2010) Homeodomain-interacting protein kinase 2 plays an important role in normal terminal erythroid differentiation. Blood 115, 4853–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guerra F. M., Gommerman J. L., Corfe S. A., Paige C. J., Rottapel R. (2012) Homeodomain-interacting protein kinase (HIPK)-1 is required for splenic B cell homeostasis and optimal T-independent type 2 humoral response. PLoS One 7, e35533. [DOI] [PMC free article] [PubMed] [Google Scholar]