

Abstract
We report on Dutch and Iranian families with affected individuals who present with moderate to severe intellectual disability and additional phenotypes including progressive tremor, speech impairment, and behavioral problems in certain individuals. A combination of exome sequencing and homozygosity mapping revealed homozygous mutations c.484G>A (p.Gly162Arg) and c.1898C>G (p.Pro633Arg) in SLC6A17. SLC6A17 is predominantly expressed in the brain, encodes a synaptic vesicular transporter of neutral amino acids and glutamate, and plays an important role in the regulation of glutamatergic synapses. Prediction programs and 3D modeling suggest that the identified mutations are deleterious to protein function. To directly test the functional consequences, we investigated the neuronal subcellular localization of overexpressed wild-type and mutant variants in mouse primary hippocampal neuronal cells. Wild-type protein was present in soma, axons, dendrites, and dendritic spines. p.Pro633Arg altered SLC6A17 was found in soma and proximal dendrites but did not reach spines. p.Gly162Arg altered SLC6A17 showed a normal subcellular distribution but was associated with an abnormal neuronal morphology mainly characterized by the loss of dendritic spines. In summary, our genetic findings implicate homozygous SLC6A17 mutations in autosomal-recessive intellectual disability, and their pathogenic role is strengthened by genetic evidence and in silico and in vitro functional analyses.
Introduction
Intellectual disability (ID) is a neurodevelopmental disorder that has a great impact on the quality of life of affected individuals and their families.1,2 Despite its high prevalence of 2% in the population, so far only approximately 60% of individuals with ID receive a conclusive genetic diagnosis.3,4 The identification of causative mutations is complicated by the enormous genetic heterogeneity, which is observed for all types of inheritance (dominant, recessive, and X-linked). Particularly for non-specific types of ID, the identification of causative mutations has been hampered by the lack of phenotypic features that might guide molecular analyses in both diagnostic and research settings. To date, a handful of genes have been implicated in non-specific forms of autosomal-recessive ID (ARID).5–7 Only recently, the combined application of SNP microarrays for homozygosity mapping and advancement of next-generation sequencing technologies has accelerated the elucidation of causative mutations in ARID, mostly in consanguineous families and specific syndromes.7–9 In addition, exome sequencing has proven to be very powerful in identifying the genetic defects in small outbred families, including those affected by ARID.10 The challenge in such studies is to select the causative variant, because usually multiple homozygous and compound-heterozygous variants need to be considered. Compelling evidence of causality can be obtained by the identification of variants affecting the same gene in unrelated subjects who share the same phenotypic features. It is difficult to recognize phenotypic similarity for genuine non-specific ID; however, it often happens that features associated with an unremarkable ID phenotype only become apparent when clinical comparisons can be carried out for individuals sharing mutations that affect the same gene.
Here, we studied two recessive families by combining homozygosity mapping and exome sequencing. In SLC6A17 (MIM 610299), we identified two missense variants that segregated with the phenotype in each of the families. Further, in silico and in vitro analyses supported the causal effects of the identified mutations.
Material and Methods
Family Ascertainment
Family W10-3099, with three affected females (II-4, II-5, and II-6; Figures 1A and 1B), was ascertained during a cohort study comprising over 250 individuals (both children and adults) with ID of unknown etiology. These individuals were selected from a large cohort of more than 2,000 individuals living in residential settings for people with intellectual disabilities in the eastern part of the Netherlands.4 All of them received a clinical genetic evaluation, including SNP array analysis, phenotype-guided DNA analysis, and a metabolic screen. Individuals without a conclusive diagnosis were candidates for further studies by exome sequencing. The study was approved by the local ethics committee of the Radboud university medical center (Nijmegen). Informed consent was obtained from the parents or legal representatives of the participants.
Figure 1.
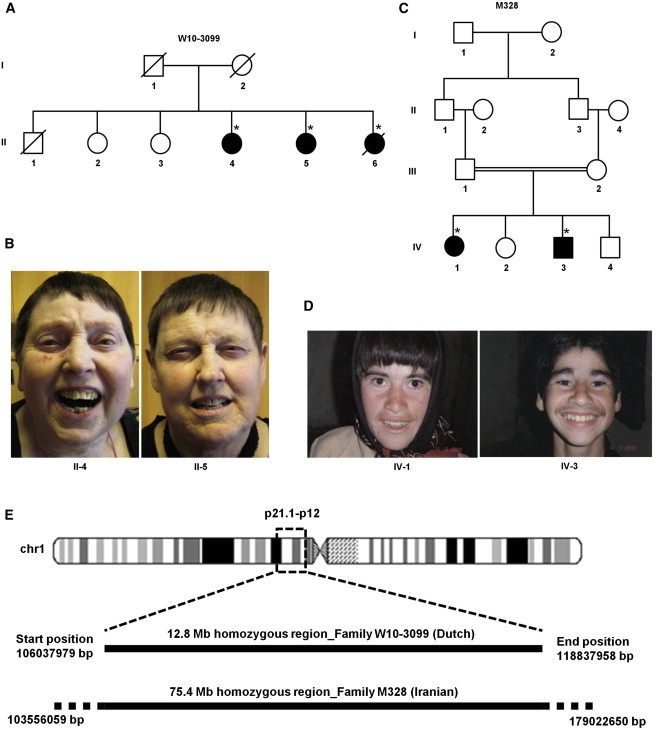
Pedigree Structures of Families W10-3099 and M328, Photographs of Affected Individuals, and Illustration of Homozygosity Mapping
(A) The pedigree structure of family W10-3099. The asterisks indicate the individuals used for homozygosity mapping and CNV analysis.
(B) Photographs of three affected individuals of family W10-3099.
(C) The pedigree structure of family M328. The asterisks indicate the individuals used for homozygosity mapping and CNV analysis.
(D) Photographs of two affected individuals of family M328.
(E) An ideogram of chromosome 1 indicates the location of the homozygous regions at p21.1–p12. Both homozygous regions shared 12.8-Mb interval chr1: 106,037,979–118,837,958 in families W10-3099 and M328.
In a simultaneous study, the consanguineous family M328 (Figure 1C), with two affected individuals (IV-1 and IV-3; Figure 1D), was recruited among a cohort study comprising 1,000 Iranian families affected by ID with an autosomal-recessive pattern of inheritance. The family was recruited by the Genetics Research Center (Tehran). The affected persons were examined for physical and mental status. Written consent was obtained from their parents. This study was approved by the ethics committee of the University of Social Welfare and Rehabilitation Sciences (Tehran).
Homozygosity Mapping and Analysis of Copy-Number Variation
In order to detect possible copy-number variations (CNVs) in family W10-3099, we performed Affymetrix 250K SNP array analysis and analyzed data with the Copy Number Analyzer for GeneChip (v.2.0)11 (Affymetrix). Genotype calls were generated with Affymetrix Genotyping Console (v.2.1) software. The shared runs of homozygosity (ROHs) were determined with PLINK (v.1.06).12 In each window of 50 SNPs, up to five SNPs with a missing call and a maximum of two heterozygous called SNPs were allowed. We used a cutoff of 1 Mb to identify shared ROHs.13 In order to screen additional individuals, we used the same criteria described above to determine the ROHs in an additional cohort of >5,000 idiopathic ID individuals who were systematically ascertained at our institute. We selected individuals with ROHs ≥ 5 Mb in size.
To detect ROHs in common between the affected individuals in family M328, we mixed genomic DNA from individuals IV-1 and IV-3 in an equimolar concentration. The DNA mixture was hybridized to the Genome-wide Human SNP Array 6.0 (Affymetrix) with a resolution of one million SNPs. Data were analyzed with Partek Genomics Suite software.
Exome Sequencing
Exome enrichment was performed with the Agilent v.2 XT Multiplex (BC7) Enrichment Kit (Agilent) with DNA from individual II-6 of family W10-3099. Emulsion PCR and bead preparation were performed with the EZbead system according to the manufacturer’s (Life Technologies) instructions. We used the SOLiD v.4 platform (Life Technologies). A total of 8.4 Gb of sequence was obtained and mapped to the human reference genome (UCSC Genome Browser hg19) with LifeScope v.2.1 software (Life Technologies).
DNA from individual IV-1 of family M328 was analyzed by exome sequencing as described before.14 In brief, a whole-exome sequencing library was prepared with the SureSelect Human All Exon 50 Mb Kit (Agilent Technologies) in accordance with the manufacturer’s protocol. The captured library was sequenced with a Hi-Seq 2000 sequencing system (Illumina) in accordance with the manufacturer’s protocol.
Sanger Sequencing
Primers for the amplification of exons and exon-intron boundaries were designed with the Primer3 program15 for SLC6A17 as well as for other identified rare variants in their respective genes. PCR products were purified with a NucleoFast 96 PCR plate (Clontech Lab) according to the manufacturer’s protocol. We used the ABI PRISM BigDye Terminator Cycle Sequencing v.3.1 Ready Reaction Kit and the ABI PRISM 3730 DNA Analyzer (Applera Corporation) to perform sequence analysis.
Molecular Modeling
The 3D structure of SLC6A17 has not yet been solved experimentally. Therefore, we used the structure of the leucine transporter (LeuT) as a template to build a homology model of SLC6A17. PDB identifier 3GWU16 contains a sequence of Aquifex aeolicus VF5 LeuT, which has a sequence identity of 24.4% with that of SLC6A17 over a stretch of 447 residues. The (partial) model was built with the automatic modeling script in the YASARA17 and WHAT IF18 Twinset with standard parameters.19
Slc6a17 Neurodevelopmental Expression Analysis in Mouse Brain Tissues
We used mouse neurodevelopmental TissueScanTMqPCR Arrays (OriGene Technologies) to screen for Slc6a17 expression in a total of 48 different brain regions in five neurodevelopmental stages (embryonic days 13, 15, and 18, postnatal day 7, and adult week 5) by using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) gene-expression assays according to the manufacturer’s instructions. These arrays contained pre-standardized amounts of cDNA according to the manufacturer’s specifications. The cDNA was amplified by PCR with Slc6a17-specific primers located in exons 9 and 10 (Table S2). Quantification was carried out with a method derived from the 2−ΔΔCp method.20 A normalized ratio was obtained for each target gene with LC480 software (Roche), qPCR efficiencies were 100%, and Gapdh (glyceraldehyde 3-phosphate dehydrogenase) was used as a reference gene.
Generation of Wild-Type and Mutant Mouse Slc6a17 Constructs for Overexpression Studies
For transient expression, full-length open reading frames of mouse wild-type (WT) and mutant Slc6a17 constructs were amplified from the IMAGE clone 950654248, which contains the mouse Slc6a17 cDNA (GenBank accession number BC171962; Source Bioscience), with primer set Slc6a17-GFPinfus-Fd and Slc6a17-GFPinfus-Rv (Table S2). PCR products were inserted into the pAcGFP1-N In-Fusion Ready Vector (Clontech) with the In-Fusion HD Cloning Kit (Clontech). The clones with mutations c.484G>A (p.Gly162Arg) and c.1898C>G (p.Pro633Arg) were first generated in two PCR fragments including an overlapping region containing the mutation (Table S2). The PCR fragments were then inserted into the pAcGFP1-N In-Fusion Ready Vector with the In-Fusion HD Cloning Kit. Correctness of all sequences was determined by Sanger sequencing. The Slc6a17-WT-GFP, Slc6a17-p.Gly162Arg-GFP, and Slc6a17-p.Pro633Arg-GFP constructs were then used for transfection studies in primary neuronal cultures.
Mouse Primary Neuronal Cultures
All mouse experiments were performed according to protocols approved by the University Francois-Rabelais of Tours and INSERM. Hippocampi or cortices were dissected from embryonic day 17.5 C57BL/6J mouse embryos (Janvier), manually dispersed in cold PBS (Fisher Scientific), and triturated with papain (10 U/ml; Worthington) for 22 min at 37°C. Cells were resuspended in Dulbecco’s modified Eagle’s medium/F12 (Fisher Scientific) with 10% fetal bovine serum (Eurobio) and centrifuged at 250 × g for 3 min, and the final pellet was resuspended in primary neuron growth medium (PNGM; Lonza). Dissociated cells were then plated onto glass coverslips coated in poly-D-lysine (Sigma) and laminin at a density of 400 cells per mm2. The cultures were kept in PNGM, and half of the medium was changed twice a week.
Transfection
Hippocampal neurons were transfected on days 10–12 in vitro with the appropriate plasmids (WT and mutated forms of Slc6a17) with the Magnetofectamine Transfection Kit (OZ Biosciences). In brief, 1 μg of DNA was combined with 2 μl of Lipofectamine 2000. The DNA and Lipofectamine 2000 mixture was immediately added to 1 μl of CombiMag and incubated for 20 min at room temperature. Complexes were added dropwise onto cells growing in serum-containing culture medium. Next, cells were incubated for 15 min on the magnetic plate at room temperature. After incubation, medium was changed, and the magnetic plate was removed. Microscopy analyses were carried out 24–48 hr after transfection.
Immunocytochemistry
Transfected primary neurons were fixed with 4% paraformaldehyde at room temperature for 20 min and permeabilized with PBS (Fisher Scientific), 3% BSA (Interchim), and 0.2% Triton X-100 (Sigma) for 1 hr at room temperature. The following incubations were performed with blocking solution (PBS, 3% BSA, and 0.2% Triton X-100) at room temperature. Cells were first incubated for 1 hr at room temperature with primary antibodies diluted in blocking solution: monoclonal mouse anti-PSD95 (1/100; Fisher Scientific), goat anti-VGLUT1 (1/50; Clinisciences), and mouse anti-synaptophysin (1/50; Abcam). After three washes in PBS for 5 min each, the secondary antibody (FluoProbes FP-594 donkey anti-mouse and anti-goat antibodies, 1/300; Interchim) was diluted in blocking solution and incubated with the neurons for 45 min in dark conditions. No staining was detected when primary antibodies were not added in parallel preparations (data not shown). The cells were mounted with the DAPI ProLong gold antifade solution (Fisher Scientific).
Image Analysis
The neuronal cellular-imaging study was performed at the microscopy research platform of the University of Tours. Sequential acquisitions were made, and high-resolution z stack images of neurons were taken with the 63× plan apochromat objective of a confocal FluoView 500 microscope (Olympus) with optical section separation (z interval) of 0.5–0.7 μm. Maximal projections were made for image production.
Results
Clinical Description
Family W10-3099 was ascertained as an ARID-affected family. This family has three affected females (II-4, II-5, and II-6) and three healthy siblings (Figures 1A and 1B). Pregnancy and birth of the three affected individuals were uncomplicated. In the neonatal period, they cried excessively. Psychomotor development was delayed from birth. Specific timeframes for these childhood milestones were not available because individuals II-4, II-5, and II-6 were seen at 64, 62, and 60 years of age, respectively, and no previous documentation was present. Their developmental age was between 1 and 4 years. They could speak up to three-word sentences. Their behavior was characterized by mood instability, aggression, and self-mutilation. Between 40 and 50 years of age, they developed a progressive essential hand tremor, which manifested as an intentional tremor and slight tremor at rest. This was not present in their healthy siblings. One of the affected sisters (II-5) had a renal anomaly, including a double renal pelvis with double ureter. One of the affected individuals (II-6) had an incidental epileptic insult at 40 years of age. However, no seizures were further reported. Electroencephalography (EEG) was performed in all three affected family members, but no epileptic features were found. In one of the sisters (II-5), a cerebral computed-tomography scan was taken when she was 55 years old. Results were normal. In the other two sisters, cerebral imaging was not performed. Individual II-6 developed melanoma at 48 years of age and breast cancer at 50 years of age. Individual II-5 was diagnosed with endometrium carcinoma at 59 years of age. Tumor characteristics and ages of presentation were not suggestive of hereditary cancer. The three sisters had a normal height (5th–20th percentile) and normal head circumference (20th–50th percentile). Facial dysmorphisms included large ears, a prominent chin, narrow palpebral fissures, a long philtrum, and a slightly high and narrow palate (Figure 1B). In addition, they had small hands (<3rd percentile; Table 1). They had a waddling walking pattern. Metabolic screening in the serum and urine of individual II-6 and in the serum of individual II-4 included quantification of amino acids, creatine biosynthesis, carnitines, very-long-chain fatty acids, transferrin subfractions, mucopolysaccharides, purines and pyrimidines, and organic acids. All the values were found in the normal range. The analysis of FMR1 (MIM 309550) in individual II-4 did not reveal evidence of expansion of the CGG repeat.
Table 1.
Clinical Features of Affected Members of Families W10-3099 and M328
|
Family W10-3099 |
Family M328 |
||||
|---|---|---|---|---|---|
| II-4 | II-5 | II-6 | IV-1 | IV-3 | |
| Age | 64 years | 62 years | 60 years | 22 years | 20 years |
| Consanguinity | no | no | no | yes | yes |
| Height (percentile) | 161 cm (<10%) | 164 cm (15%) | 165 cm (20%) | 150 cm (25%) | 152 cm (25%) |
| Head circumference (percentile) | 56 cm (>50%) | 55.4 cm (25%) | 54 cm (20%) | 54 cm (50%) | 56 cm (50%) |
| Intellectual disability | moderate to severe | moderate to severe | severe | severe (IQ 29) | severe (IQ 30) |
| Tremor | progressive | progressive | progressive | progressive | progressive |
| Behavior | inappropriate laughter, compulsive, mood instability | mood instability | aggressive, compulsive, mood instability | aggressive, poor impulse control | aggressive, poor impulse control |
| Hand length (percentile) | (3%) | small | (3%) | normal (50%) | normal (50%) |
| Other | – | congenital renal anomaly (double renal pelvis with double ureter), endometrium carcinoma at 59 years of age | hypothyroidism, incidental epileptic insult, melanoma at 48 years of age, breast cancer at 50 years of age | no | no |
In the Iranian family (M328), two affected individuals (IV-1 and IV-3; Figures 1C and 1D) and two healthy siblings (IV-2 and IV-4) were born to first-cousin consanguineous parents (Figure 1C). Affected individuals IV-1 and IV-3 (now aged 22 and 20 years, respectively) were initially referred for intellectual disability. Pre-, peri-, and postnatal periods were uneventful. They had severe mental impairment (IQs of 29 and 30 for IV-1 and IV-3, respectively) and no speech development. Progressive essential tremors of the hands developed in both IV-1 and IV-3 at the age of 13 and 16 years, respectively. Progressive tremors were not seen in the healthy family members. Brain imaging and EEG were not performed in the affected individuals. The behavioral problems were mainly characterized by aggressiveness and poor impulse control in both affected persons. They were not able to walk and were unable to take care of themselves. They had normal head circumference (50th percentile) and normal height (25th percentile). They had normal length of hands (50th percentile) (Table 1). Facial features included large ears, a prominent chin, thick eyebrows, and slightly narrow palpebral fissures (Figure 1D). Karyotype analysis by G-banding and tandem mass spectrometry screening for metabolic disorders as well as FMR1 screening were performed for individual IV-3. None of these tests revealed any chromosomal aberrations or mutations that could explain the phenotype.
Genetic Analysis
In family W10-3099, CNV analysis did not reveal any pathogenic aberrations. We performed homozygosity mapping on individuals II-4, II-5, and II-6. A single shared ROH of 12.8 Mb in size was identified in chromosomal region 1p21.1–p12 (chr1: 106,037,979–118,837,958), encompassing about 155 genes (Figure 1E). To identify the genetic defect in this family, we performed exome sequencing. From our exome data, we obtained a median coverage of 103×, and 89% of targets were covered at least 10×. A total of 14,986 variants were identified in coding regions and canonical splice sites. Prioritization was done as described before.21,22 Variants that are known in dbSNP134 and in our in-house database, which consists of 672 samples from healthy and affected individuals with other rare disorders, were excluded. After this filter was applied, the number of variants was reduced to 299. Further prioritization was performed on the basis of an autosomal-recessive model of inheritance, resulting in 21 candidate genes: 18 with potential homozygous variants (>80% variant reads) and three with compound-heterozygous variants (20%–80% variant reads). We analyzed all of these variants by inspecting the raw read alignment files (BAM files) in order to determine the presence and the minimal percentage (20%) of the variant reads, which reduced the number of candidate genes to ten with homozygous variants and two with compound-heterozygous variants. We excluded two genes, TAS1R3 (MIM 605865) and MUC16 (MIM 606154), because these are commonly found in the exome data to carry either homozygous or compound-heterozygous variants. Another gene, CWH43, was excluded because frequent (1.2%) truncating variants are encountered in this gene in the NHLBI Exome Sequencing Project Exome Variant Server (EVS). After this, eight homozygous variants and one compound-heterozygous variant were left (Table S1). All the identified variants were tested by Sanger sequencing. None but a single homozygous variant, chr1:110716634G>A (c.484G>A [p.Gly162Arg]), in SLC6A17 (RefSeq accession number NM_001010898.2) segregated with the phenotype (Figure 2A). This variant is located in SLC6A17 exon 4, which maps to the identified 12.8 Mb homozygous region in 1p21.1–p12 (Figure 1E). It has a phyloP score of 6.3 (Table S1) and a Grantham score of 125. Gly162 was found to be highly conserved down to zebrafish (Figure 2D). The uncharged Gly162, which is replaced by a positively charged arginine residue, is located in the third transmembrane domain of the protein (Figure 2C). We used three prediction programs—PolyPhen2,23 SIFT,24 and SNPs&GO25—to predict the pathogenic nature of the identified missense variant. All three prediction programs classified c.484G>A as “possibly damaging.” A Combined Annotation Dependent Depletion (CADD)26 score of 34 was observed for this variant. The missense mutation was absent from dbSNP138, 1000 Genomes, and the NHLBI EVS.
Figure 2.
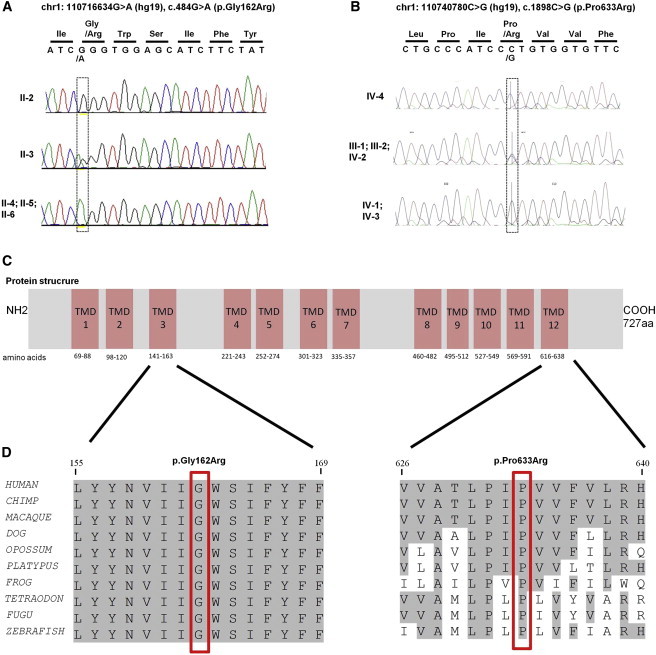
Segregation Analysis and Schematic Representation of the Protein, Including the Location and Conservation of the Identified Amino Acid Changes
(A) Chromatograms revealing the complete segregation of homozygous missense mutation chr1:110716634G>A (c.484G>A [p.Gly162Arg]) with the phenotype in family W10-3099.
(B) Chromatograms show complete segregation of homozygous missense mutation chr1:110740780C>G (c.1898C>G [p.Pro633Arg]) with the phenotype in family M328.
(C) A schematic representation of SLC6A17 indicates the location of both alterations on the 3rd and 12th transmembrane domains.
(D) Partial amino acid sequence alignment of human SLC6A17 and its homologs in the animal kingdom. The positions of the amino acid substitutions—p.Gly162Arg in family W10-3099 and p.Pro633Arg in family M328—are indicated by red rectangles. Both altered residues are conserved down to zebrafish.
In family M328, CNV analysis did not reveal any pathogenic structural changes. Homozygosity mapping revealed eight shared ROHs (Table S3). We performed whole-exome sequencing in one affected individual (IV-1). Sequencing was carried out in a 101-nt single-end sequencing format and showed 95% coverage and an average depth of 52 reads. To detect single-nucleotide variants, we aligned high-quality reads to the human reference genome (UCSC Genome Browser hg19) with Short Oligonucleotide Analysis Package (SOAP) 2.20. Variant lists were filtered against dbSNP137, whole genomes from 185 healthy individuals studied by the 1000 Genomes Project, exomes from 200 Danish individuals, and more than 6,500 exomes present in the NHLBI EVS. In addition, variants were compared to an in-house database containing more than 200 exomes. Variants were ranked as potential candidates as previously described27 with an improved version of the Medical Re-sequencing Analysis Pipeline (MERAP).28 In addition, we used OMIM and the Human Gene Mutation Database (HGMD) as filters to identify all previously described pathogenic changes.
After filtering the variants by using an improved version of the MERAP algorithm and their annotation with RefSeq, we ranked two homozygous missense variants—SLC6A17 chr1:110740780C>G (c.1898C>G [p.Pro633Arg]) and FAM213B chr1:2519225T>G (c.404T>G [p.Phe135Cys])—as potentially pathogenic. Both are located in identified homozygous regions. Upon co-segregation testing, only the SLC6A17 mutation segregated with the disease in the family (Figure 2B). SLC6A17 is located in the largest homozygous region (Figure 1E; Table S3), and the identified mutation is present in exon 12 of the gene. SLC6A17 c.1898C>G was not reported in dbSNP138, 1000 Genomes, or the NHLBI EVS. Moreover, the variant was absent in our in-house database containing more than 200 exomes. The mutation affects a highly conserved amino acid (Figure 2D). In line with this, the phyloP score was found to be 5.3, and the Grantham score was 103. The uncharged Pro633, which is replaced by a positively charged arginine residue, is located in the 12th transmembrane domain of the protein (Figure 2C). In silico analyses with several pathogenicity prediction programs, including SIFT, PolyPhen-2, and SNPs&Go, predicted the amino acid substitution to be damaging to the protein function. A CADD score of 23.8 was obtained for this variant.
In order to find other possible homozygous mutations, we selected 18 additional unrelated ID subjects (both consanguineous and non-consanguineous) with shared homozygosity of a >5 Mb region encompassing SLC6A17. These individuals were selected from a large cohort of individuals (>5,000) who lacked a molecular diagnosis and were seen at the Department of Human Genetics of Radboud university medical center (Nijmegen). We performed Sanger sequencing in these individuals to sequence all the exons and intron-exon boundaries of SLC6A17. The sequencing analysis did not reveal any potentially pathogenic variants (data not shown).
Molecular Modeling
To collect further evidence of the causality of the identified variants, we performed prediction analysis with the 3D protein structure. Gly162 is located in an alpha helix that also contains residues that form the substrate binding site (Figures S1A and S1B). Gly162 is not in contact with the substrate but instead is located on the opposite side of the helix. The substitution converts this residue into an arginine, a large hydrophilic amino acid whose side chain most likely does not fit at this position and causes a clash with Leu231, located on helix 4. This model suggests that p.Gly162Arg affects protein structure and potentially interferes with the function of the protein. Pro633 is located in one of the helices on the surface of the protein and is far from the ligand binding site (Figures S1A and S1C). This hydrophobic proline is in contact with membrane lipids and could be important for anchoring SLC6A17 in the membrane. Substitution of arginine at this position could interfere with protein function by disturbing the membrane anchoring given that it is a hydrophilic, charged residue.
Slc6a17 Is Expressed in the Central and Peripheral Nervous Systems
To get more insight into the expression pattern of SLC6A17, we performed a quantitative cDNA analysis of the orthologous mouse Slc6a17 in a panel representing 40 distinct mouse tissues. Slc6a17 expression levels increased during embryonic brain development, and the highest levels were observed postnatally. This suggests that an important role for Slc6a17 during brain development starts in the neuritogenesis and synaptogenesis stages. The strongest expression levels in embryonic, postnatal, and adult stages were found in both cortical and hippocampal tissues (Figure S2). Our data are also in accordance with a recent study that described the Slc6a17 mRNA expression pattern in the mouse adult brain.29
Subcellular Localization of WT and Mutant Slc6a17 in Mouse Embryonic Hippocampal Primary Neurons
To assess the functional impact of both variations in SLC6A17, we transfected mature mouse embryonic primary hippocampal neuronal cultures with plasmids, leading to the overexpression of WT or variant forms of SLC6A17-GFP fusion proteins.
Confocal-microscopy analyses indicated a wide subcellular localization of SLC6A17-WT-GFP throughout the cells; compared to GFP-overexpressing neurons, these cells showed no alteration in neuronal morphology (data not shown). SLC6A17-WT-GFP was partly present at dendritic spines (arrows, Figure 3A) and colocalized with PSD95 (an excitatory post-synaptic marker) and was also present at pre-synaptic sites, as indicated by the pre-synaptic markers VGLUT and synaptophysin (Figure 3B). Our data suggest that SLC6A17-WT-GFP is localized at synaptic junctions—at both pre- and post-synaptic sites—particularly in excitatory glutamatergic terminals, in agreement with a recent study by Hägglund et al.,29 who studied the endogenous localization pattern of SLC6A17 in the rat brain.
Figure 3.
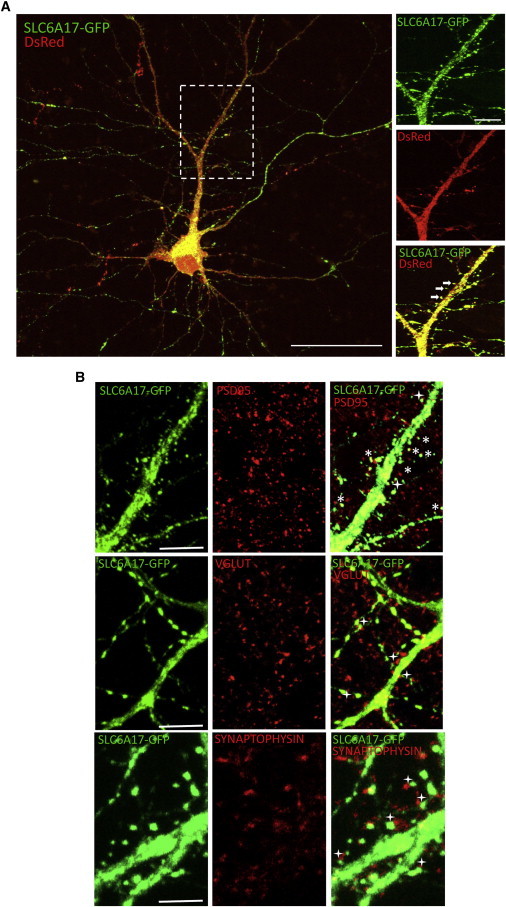
Subcellular Neuronal Localization of SLC6A17-GFP
(A) Mature hippocampal neurons (11–19 days in vitro) were co-transfected with Slc6a17-GFP and DsRed. A zoomed part of the cell (white rectangle) provides higher resolution regarding the localization in dendritic spines (arrows) (n = 5 transfections, 38 cells analyzed). Scale bars represent 50 μm.
(B) Mature hippocampal neurons (11–19 days in vitro) were transfected with Slc6a17-GFP and then analyzed by immunocytochemistry against PSD95, VGLUT, or synaptophysin. (n = 3 transfections, 12 cells analyzed). Scale bars represent 10 μm.
Asterisks indicate colocalization of SLC6A17-GFP and a synaptic marker; plus signs indicate juxtaposition of SLC6A17-GFP and a synaptic marker.
By contrast, the analysis of the subcellular localization of p.Gly162Arg and p.Pro633Arg SLC6A17 variants revealed different findings. Whereas SLC6A17-p.Gly162Arg-GFP displayed a wide distribution within the cell, the morphology of transfected mature neurons was altered and characterized by a systematic loss of dendritic spines (Figure 4A). In addition, SLC6A17-p.Pro633Arg-GFP was observed in soma and proximal dendrites, but not in spinal structures, contrary to the WT form (Figure 4B). The overall neuronal morphology was not affected in a comparison of SLC6A17-WT-GFP and GFP-overexpressing neurons.
Figure 4.
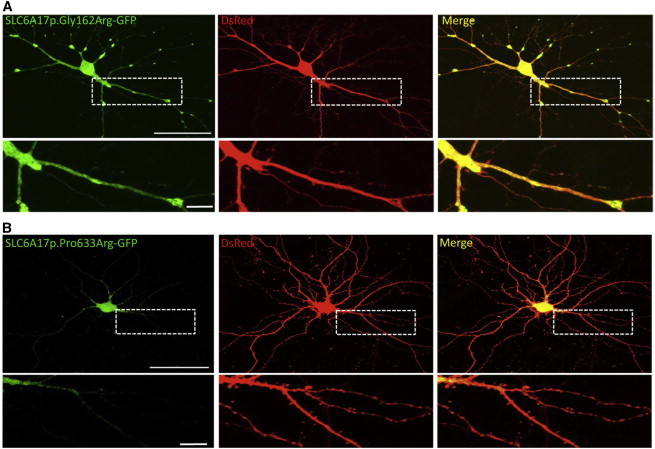
Functional Analysis of SLC6A17 Alterations in Mature Primary Hippocampal Neurons
(A) Primary neuronal cultures were co-transfected at 14 days in vitro with Slc6a17-p.Gly162Arg-GFP and pDsRedmonomer (for whole-cell labeling) and visualized 48 hr later on confocal microscopy under living condition (n = 7 transfections, 46 cells analyzed). Scale bars represent 50 μm (neuron) and 10 μm (zoomed image).
(B) Confocal images of mature hippocampal neurons (16 days in vitro) transfected with Slc6a17-p.Pro633Arg-GFP and pDsRedmonomer (n = 3 transfections, 6 cells analyzed). Scale bars represent 50 μm (neuron) and 10 μm (zoomed image).
Discussion
In this study, we have identified SLC6A17 mutations in individuals with an ARID phenotype. Two independent mutations were identified in two families with different ethnic backgrounds. In silico analysis based on PolyPhen-2, SIFT, and SNPs&GO predicted “damaging” effects of the identified missense variants on the protein function. Moreover, 3D modeling revealed that identified amino acid substitutions are predicted to result in a conformational change influencing normal SLC6A17 function. To further test this notion, we performed functional analyses by using primary hippocampal neuronal cells from mouse embryos. Our in vitro data obtained from transfected primary neurons indicate that the p.Gly162Arg and p.Pro633Arg amino acid substitutions in SLC6A17 affect either its dendritic localization or the neuronal morphology and suggest a specific pathophysiological mechanism possibly linked to the protein domains including the Gly162 or the Pro633 amino acids. It should, however, be noted that our in vitro data were obtained from overexpression experiments of either WT or altered SLC6A17-GFP and thus might not perfectly reflect the in vivo conditions.
On the basis of the clinical features from the two families, the phenotypic spectrum that could be caused by SL6A17 mutations comprises moderate to severe ID with hardly any speech development, progressive tremors, and behavioral problems. Motor development might also be severely impaired, possibly inhibiting independent walking. There is no distinct facial phenotype. Remarkably, all affected individuals from family W10-3099 had small hands, whereas this was not present in the affected members of the Iranian family (M328).
SLC6 family genes encode structurally related Na+-dependent transporter proteins. They are responsible for the re-uptake of many neurotransmitters.30 SLC6A17 encodes a vesicular transporter protein, also known as XT1 or NTT4.31 This protein is predominantly localized in the nervous system, specifically in glutamatergic and some GABAergic neurons.32,33 SLC6A17 functions as a selective transporter of four amino acids (proline, glycine, leucine, and alanine), as well as glutamate.34,35 Recently, it has been shown that it is involved in regulating glutamatergic synapses.29 Proline has a role in producing electrophysiological actions in the brain.36,37 Moreover, it has been demonstrated that physiologically increased levels of L-proline are localized in many neurons,38 but understanding the specific role of proline in synaptic transmission will require further studies.34 Glycine acts both as an inhibitory neurotransmitter and as an activator of ionotropic glutamate N-methyl-D-aspartate (NMDA) receptors. NMDA receptors are distributed all over the brain and wire different functions such as synaptic plasticity and development of learning and memory. NMDA receptor activation requires both glycine and glutamate. It is known that fluctuation in the extracellular concentration of glycine can alter the functions of NMDA receptors. Several independent studies have shown that such NMDA alterations are involved in neurodevelopmental and neuropsychiatric disorders such as cognitive impairment and schizophrenia.39,40 The role of leucine and alanine in synaptic transmission is not known.34 It would have been very instructive to obtain the amino acid profile in cerebrospinal fluid of the affected individuals, but none of the individuals or guardians wished to participate in such invasive investigations.
Another SLC6 Na+-dependent transporter protein, the creatine transporter SLC6A8, has been implicated in cerebral creatine deficiency syndrome 1 (MIM 300352), which includes ID as a main feature.41 It is proposed that dysfunction of the inhibitory neurotransmitters GABA and glycine plays a role in the development of essential tremor.42–44 The impairment of the GABAergic system is also involved in fragile X syndrome (MIM 300624) and fragile X tremor/ataxia syndrome (FXTAS [MIM 300623]).45
In conclusion, we have identified SLC6A17 mutations that are associated with ARID. Our data are supported by genetic recurrence and in silico and in vitro analyses. Further testing of SLC6A17 in individuals with similar phenotypes or unbiased ARID screening might elucidate the involvement of this gene in cognitive function as well as establish the phenotypic spectrum.
Acknowledgments
We are thankful to all the participating family members in this study. Special thanks go to Annette Schenck, Bonnie Nijhof, and Misa Fenckova for discussions on different experiments. This work was supported by the European Union’s Seventh Framework Programme under grant agreement number 241995 (project GENCODYS to H.v.B., F.L., L.M., H.H.R., and H.H.), the Max-Planck Society, the Dutch Brain Foundation under grant number 2010(1)-30 to A.P.M.d.B., the consortium “Stronger on your own feet” to T.K. and M.H.W., and the Netherlands Organisation for Health Research and Development (ZonMW) under grant number 907-00-365 to T.K. Z.I. was supported by the Higher Education Commission of Islamabad (Pakistan).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/genbank
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer3, http://primer3.ut.ee/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Leonard H., Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 2.Roeleveld N., Zielhuis G.A., Gabreëls F. The prevalence of mental retardation: a critical review of recent literature. Dev. Med. Child Neurol. 1997;39:125–132. doi: 10.1111/j.1469-8749.1997.tb07395.x. [DOI] [PubMed] [Google Scholar]
- 3.Gilissen C., Hehir-Kwa J.Y., Thung D.T., van de Vorst M., van Bon B.W., Willemsen M.H., Kwint M., Janssen I.M., Hoischen A., Schenck A. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–347. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 4.Willemsen M.H., Kleefstra T. Making headway with genetic diagnostics of intellectual disabilities. Clin. Genet. 2014;85:101–110. doi: 10.1111/cge.12244. [DOI] [PubMed] [Google Scholar]
- 5.Ropers H.H. Genetics of early onset cognitive impairment. Annu. Rev. Genomics Hum. Genet. 2010;11:161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 6.van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011;45:81–104. doi: 10.1146/annurev-genet-110410-132512. [DOI] [PubMed] [Google Scholar]
- 7.Iqbal Z., van Bokhoven H. Identifying genes responsible for intellectual disability in consanguineous families. Hum. Hered. 2014;77:150–160. doi: 10.1159/000360539. [DOI] [PubMed] [Google Scholar]
- 8.Ellison J.W., Rosenfeld J.A., Shaffer L.G. Genetic basis of intellectual disability. Annu. Rev. Med. 2013;64:441–450. doi: 10.1146/annurev-med-042711-140053. [DOI] [PubMed] [Google Scholar]
- 9.Musante L., Ropers H.H. Genetics of recessive cognitive disorders. Trends Genet. 2014;30:32–39. doi: 10.1016/j.tig.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Schuurs-Hoeijmakers J.H., Vulto-van Silfhout A.T., Vissers L.E., van de Vondervoort I.I., van Bon B.W., de Ligt J., Gilissen C., Hehir-Kwa J.Y., Neveling K., del Rosario M. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J. Med. Genet. 2013;50:802–811. doi: 10.1136/jmedgenet-2013-101644. [DOI] [PubMed] [Google Scholar]
- 11.Nannya Y., Sanada M., Nakazaki K., Hosoya N., Wang L., Hangaishi A., Kurokawa M., Chiba S., Bailey D.K., Kennedy G.C., Ogawa S. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 12.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuurs-Hoeijmakers J.H., Hehir-Kwa J.Y., Pfundt R., van Bon B.W., de Leeuw N., Kleefstra T., Willemsen M.A., van Kessel A.G., Brunner H.G., Veltman J.A. Homozygosity mapping in outbred families with mental retardation. Eur. J. Hum. Genet. 2011;19:597–601. doi: 10.1038/ejhg.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bainbridge M.N., Hu H., Muzny D.M., Musante L., Lupski J.R., Graham B.H., Chen W., Gripp K.W., Jenny K., Wienker T.F. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med. 2013;5:11. doi: 10.1186/gm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Z., Zhen J., Karpowich N.K., Law C.J., Reith M.E., Wang D.N. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat. Struct. Mol. Biol. 2009;16:652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krieger E., Koraimann G., Vriend G. Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 18.Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. 29. [DOI] [PubMed] [Google Scholar]
- 19.Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77(9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Hoischen A., Gilissen C., Arts P., Wieskamp N., van der Vliet W., Vermeer S., Steehouwer M., de Vries P., Meijer R., Seiqueros J. Massively parallel sequencing of ataxia genes after array-based enrichment. Hum. Mutat. 2010;31:494–499. doi: 10.1002/humu.21221. [DOI] [PubMed] [Google Scholar]
- 22.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 23.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calabrese R., Capriotti E., Fariselli P., Martelli P.L., Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 26.Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 28.Hu H., Wienker T.F., Musante L., Kalscheuer V.M., Kahrizi K., Najmabadi H., Ropers H.H. Integrated sequence analysis pipeline provides one-stop solution for identifying disease-causing mutations. Hum. Mutat. 2014;35:1427–1435. doi: 10.1002/humu.22695. [DOI] [PubMed] [Google Scholar]
- 29.Hägglund M.G., Hellsten S.V., Bagchi S., Ljungdahl A., Nilsson V.C., Winnergren S., Stephansson O., Rumaks J., Svirskis S., Klusa V. Characterization of the transporterB0AT3 (Slc6a17) in the rodent central nervous system. BMC Neurosci. 2013;14:54. doi: 10.1186/1471-2202-14-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kristensen A.S., Andersen J., Jørgensen T.N., Sørensen L., Eriksen J., Loland C.J., Strømgaard K., Gether U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- 31.Chen N.H., Reith M.E., Quick M.W. Synaptic uptake and beyond: the sodium- and chloride-dependent neurotransmitter transporter family SLC6. Pflugers Arch. 2004;447:519–531. doi: 10.1007/s00424-003-1064-5. [DOI] [PubMed] [Google Scholar]
- 32.Bröer S. The SLC6 orphans are forming a family of amino acid transporters. Neurochem. Int. 2006;48:559–567. doi: 10.1016/j.neuint.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 33.Liu Q.R., Mandiyan S., López-Corcuera B., Nelson H., Nelson N. A rat brain cDNA encoding the neurotransmitter transporter with an unusual structure. FEBS Lett. 1993;315:114–118. doi: 10.1016/0014-5793(93)81145-p. [DOI] [PubMed] [Google Scholar]
- 34.Parra L.A., Baust T., El Mestikawy S., Quiroz M., Hoffman B., Haflett J.M., Yao J.K., Torres G.E. The orphan transporter Rxt1/NTT4 (SLC6A17) functions as a synaptic vesicle amino acid transporter selective for proline, glycine, leucine, and alanine. Mol. Pharmacol. 2008;74:1521–1532. doi: 10.1124/mol.108.050005. [DOI] [PubMed] [Google Scholar]
- 35.Zaia K.A., Reimer R.J. Synaptic vesicle protein NTT4/XT1 (SLC6A17) catalyzes Na+-coupled neutral amino acid transport. J. Biol. Chem. 2009;284:8439–8448. doi: 10.1074/jbc.M806407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felix D., Künzle H. The role of proline in nervous transmission. Adv. Biochem. Psychopharmacol. 1976;15:165–173. [PubMed] [Google Scholar]
- 37.Yoneda Y., Roberts E. A new synaptosomal biosynthetic pathway of proline from ornithine and its negative feedback inhibition by proline. Brain Res. 1982;239:479–488. doi: 10.1016/0006-8993(82)90523-6. [DOI] [PubMed] [Google Scholar]
- 38.Takemoto Y., Semba R. Immunohistochemical evidence for the localization of neurons containing the putative transmitter L-proline in rat brain. Brain Res. 2006;1073-1074:311–315. doi: 10.1016/j.brainres.2005.12.064. [DOI] [PubMed] [Google Scholar]
- 39.Harsing L.G., Jr., Matyus P. Mechanisms of glycine release, which build up synaptic and extrasynaptic glycine levels: the role of synaptic and non-synaptic glycine transporters. Brain Res. Bull. 2013;93:110–119. doi: 10.1016/j.brainresbull.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Newcomer J.W., Farber N.B., Jevtovic-Todorovic V., Selke G., Melson A.K., Hershey T., Craft S., Olney J.W. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- 41.Salomons G.S., van Dooren S.J., Verhoeven N.M., Cecil K.M., Ball W.S., Degrauw T.J., Jakobs C. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am. J. Hum. Genet. 2001;68:1497–1500. doi: 10.1086/320595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louis E.D. A new twist for stopping the shakes? Revisiting GABAergic therapy for essential tremor. Arch. Neurol. 1999;56:807–808. doi: 10.1001/archneur.56.7.807. [DOI] [PubMed] [Google Scholar]
- 43.Málly J., Baranyi M., Vizi E.S. Change in the concentrations of amino acids in CSF and serum of patients with essential tremor. J. Neural Transm. 1996;103:555–560. doi: 10.1007/BF01273153. [DOI] [PubMed] [Google Scholar]
- 44.Kralic J.E., Criswell H.E., Osterman J.L., O’Buckley T.K., Wilkie M.E., Matthews D.B., Hamre K., Breese G.R., Homanics G.E., Morrow A.L. Genetic essential tremor in gamma-aminobutyric acidA receptor alpha1 subunit knockout mice. J. Clin. Invest. 2005;115:774–779. doi: 10.1172/JCI23625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Hulst C., Heulens I., Brouwer J.R., Willemsen R., De Geest N., Reeve S.P., De Deyn P.P., Hassan B.A., Kooy R.F. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS) Brain Res. 2009;1253:176–183. doi: 10.1016/j.brainres.2008.11.075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.