

Abstract
Two functional electron transfer (ET) chains, related by a pseudo-C2 symmetry, are present in the reaction center of photosystem I (PSI). Due to slight differences in the environment around the cofactors of the two branches, there are differences in both the kinetics of ET and the proportion of ET that occurs on the two branches. The strongest evidence that this is indeed the case relied on the observation that the oxidation rates of the reduced phylloquinone (PhQ) cofactor differ by an order of magnitude. Site-directed mutagenesis of residues involved in the respective PhQ-binding sites resulted in a specific alteration of the rates of semiquinone oxidation. Here, we show that the PsaA-F689N mutation results in an ∼100-fold decrease in the observed rate of oxidation. This is the largest change of oxidation kinetics observed so far for a single-point mutation, resulting in a lifetime that exceeds that of the terminal electron donor, . This situation allows a second photochemical charge separation event to be initiated before has decayed, thereby mimicking in PSI a situation that occurs in type II reaction centers. The results indicate that the presence of does not impact the overall quantum yield and leads to an almost complete redistribution of the fractional utilization of the two functional ET chains, in favor of the one that does not bear the charged species. The evolutionary implications of these results are also briefly discussed.
Introduction
Photosynthetic reaction centers (RCs) are the most efficient radiation energy converters known. In particular, the quantum efficiency of photochemical charge separation in photosystem I (PSI), a large macromolecular cofactor-protein supercomplex that catalyzes the light-dependent oxidation of plastocyanin and the reduction of ferredoxin, has been estimated to be higher than 0.95 (1–3), with maximal values of ∼0.97–0.99 reported in the literature (4,5). On the other hand, in the other photosystem involved in oxygenic photosynthesis, PSII, the maximal quantum efficiency has been estimated to range from 0.8 to 0.85 (6). Understanding the molecular mechanism that gives rise to such a high quantum efficiency in PSI is of pivotal interest from biological and biophysical perspectives and is important for the development of bio-inspired synthetic compounds that might be implemented in photovoltaic devices.
In contrast to the case of the better-known type II RC (6), in which a single cofactor chain is engaged in an electron transfer (ET) reaction, in PSI two parallel redox chains are active, according to what is commonly referred to as a bidirectional ET mechanism (7,8). The PSI structural models (9,10) clearly identify the ET cofactors, which are organized into two symmetric chains with respect to the pseudo-C2 symmetry axis parallel to the membrane plane. This is a common structural feature of all photosynthetic RCs whose structure is known (i.e., the purple bacterial RC (11,12), PSI (9,10), and PSII (13,14)). The photochemical and redox active cofactors are principally coordinated by the RC subunits PsaA and PsaB, which form a transmembrane heterodimer. The terminal iron-sulfur clusters, FA and FB, are bound to the extrinsic PsaC subunit. Each ET chain (ETC) is primarily, although not exclusively, coordinated by one of the two main RC subunits, and therefore the two chains are referred to here as ETCA and ETCB. Charge separation and successive radical pair stabilization reactions take place in a cluster of six chlorophyll (Chl) a molecules (1–3,7). Two of these Chls form a Chl dimer coordinated at the interface of the PsaA/PsaB heterodimer, which is known as P700. The other four Chls are organized symmetrically into two pairs, referred to here as ec2A/ec3A and ec2B/ec3B. Charge separation has been considered to initiate at the level of the P700 excited state, thereby acting as the primary electron donor (1,7,15,16). More recently, it was shown that charge separation takes place independently within each ec2/ec3 pair (17,18). The ec3 Chls are generally considered to be the primary electron acceptors (also known as A0) (1,7,15–18). Irrespective of the detailed mechanism of charge separation, the radical pair is populated in a few tens of picoseconds (1,3,7,15–18), followed in <100 ps by a reduction of the phylloquinone (PhQ) on the utilized branch (PhQA or PhQB), also known as A1 (7,8,19). The quinone in turn reduces FX, an interpolypeptide [4Fe-4S] cluster where ETCA and ETCB converge.
Whereas the two ETCs present in PSI are functional and are used at a similar level, with possible variations among species (19–27), they are not functionally identical. Different kinetics have been suggested at the level of primary charge separation/stabilization events (18) and, even more obviously, in oxidation of the phyllosemiquinone (PhQ−): the lifetimes attributed to and differ by an order of magnitude i.e., ∼250 ns and ∼20 ns, respectively (7,8,19–21). As the cofactors bound to each ETC are chemically identical, and the intercofactor distances differ only by fractions of an Ångstrom, the distinct kinetics of ET must stem from subtle differences in the interactions of the cofactors with their binding niches that finely tune the physicochemical properties of the redox moieties. This is illustrated by the fact that a single amino acid substitution can cause PhQ− oxidation kinetics to vary over a wide range, either increasing (19–23,27–30) or decreasing (31–33) the oxidation lifetime. A clear example of protein-mediated tuning of redox cofactor properties was provided by mutation analyses of the PhQA binding niche, the oxidation lifetime of which was shown to vary from ∼150 ns to 1.5 μs in response to single residue substitution (7,8,19–23,31–33).
It has also been shown that substitution of single residues involved in the coordination of the ec3A or ec3B cofactors, which are located upstream of the PhQ in ETCA or ETCB, respectively, affects the fraction of electrons transiting through each branch (18,24–28,34,35). Moreover, functional studies of double mutants that combine mutations affecting the coordination of ec3A and PhQA demonstrated that their effects are additive (30). Hence, the functional symmetry of the system appears to be rather plastic, since a few selected amino acid substitutions can change the properties of the two individual ETCs significantly without affecting the functionality of the ensemble. This is particularly interesting when placed in an evolutionary perspective, since type I and type II RCs are thought to share an ancestor that was homodimeric and hence symmetrical (36).
Here, we present the analysis of a mutation in the PhQA binding pocket (PsaA-F689N) in PSI of Chlamydomonas reinhardtii (see Fig. 1 A), which slows down oxidation kinetics by almost two orders of magnitude. This creates an unprecedented situation in which the reduction of is faster than the oxidation of the semiquinone, thereby providing the opportunity to initiate a second photochemical event while is still present in ETCA. We show that the probability for ET to occur down ETCA is dramatically decreased while PhQA is reduced, and that this decrease is compensated for by the increased occurrence of ET down ETCB so that the overall yield of charge separation remains unaffected. This further illustrates the functional plasticity that we propose is inherent to the (co)existence of the two ETCs and may have allowed evolutionary tinkering while preserving the function of the RC.
Figure 1.
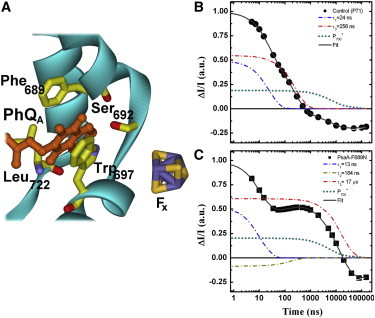
(A) Schematic representation of the binding site for PhQA (orange), highlighting the main cofactor-protein interactions. Also shown is the iron-sulfur center FX (S: yellow, Fe: violet). (B and C) Kinetics of PhQ− oxidation monitored at 395 nm in the control strain (B) or PsaA-F689N mutant (C). Solid symbols are the experimental results and the thick solid line is the fit to a sum of exponential functions. Also shown are the contributions of the decay components ascribed primarily to oxidation of (blue, 24 ns in the WT and 13 ns in the mutant), oxidation of (red, 256 ns in the WT and 17 μs in the mutant), inter-FeS cluster ET (gold, 184 ns in the mutant), and reduction of (the latter is actually the sum of the ∼6 μs and ∼55 μs components). Note that the ∼180 ns component due to inter-FeS cluster ET is not resolvable in the WT. Data are normalized on same total amplitude and same initial intensity. To see this figure in color, go online.
Materials and Methods
Creation of the PsaA-Phe689 substitution mutants
Mutant strains were constructed as previously described (37). Briefly, site-directed mutations were constructed by PCR using plasmids designed to reinsert the psaA-3 gene (38). Plasmids bearing mutations in codon 689 of psaA exon 3 (psaA-3) were introduced into strain KRC91-1A (P71 psbAΔ psaA-3Δ) via the bio-ballistic method, followed by selection for resistance to spectinomycin and streptomycin. This strain has low expression of antenna proteins due to the P71 nuclear mutation, and lacks PSII due to the deletion of the psbA gene, which encodes the D1 core polypeptide. All transformants were grown under low continuous illumination (∼10 μE m−2 s−1) in Tris-acetate-phosphate medium (39).
Transient optical spectroscopy
Pump-probe experiment
We monitored the kinetics of ET after a single-turnover laser-flash excitation by time-resolved difference absorption spectroscopy using an in-house-built pump-probe setup previously described in detail (40). In brief, the pump flash (centered at 700 nm) is provided by a dye (LDS 698) laser pumped by the second harmonic of an Nd:YAG laser (Brilliant; Quantel). The tunable probe pulses are obtained from an optical parametric oscillator (Panther; Continuum) pumped by the third harmonic of an Nd:YAG laser (Surelite II; Continuum). In both cases, the pulse duration is ∼5 ns, the intensity of the pump is attenuated so as to excite ∼70% of the RCs, and the probe is attenuated to avoid actinic effects. The pump intensity is sufficiently low to avoid multiple turnovers and nonlinear absorption processes. The pump-probe delays are controlled by a programmable in-house-built pulse sequencer.
Pump-pump-probe experiment
The setup described above was modified to perform pump-pump-probe experiments. The first actinic flash was provided by an optical parametric oscillator (SLOPO; Continuum) pumped by the third harmonic of an Nd:YAG laser (Surelite II; Continuum). The second pump flash and the probe flash were as described above. The delay between the first and second pump pulses was controlled by an electronic delay unit (Stanford) triggered by the same pulse programmer described above.
Data analysis
The kinetics recorded at multiple detection wavelengths were fitted globally by a sum of exponentials as previously described (31,41). The plot of the amplitude as a function of wavelengths yields the decay-associated spectra (DAS). Goodness of fit is judged by the reduced χ2, inspection of residual plots, and cross-correlation analysis. The data presented are the averages of at least four independent data sets.
Kinetic modeling
The kinetics of secondary ET reactions were modeled as previously described (7,29,31) by solving a system of linear differential equations. Further details are provided in the Supporting Material.
Results
Kinetic characterization of the PsaA-F689N mutation
The kinetics of PhQ− oxidation (monitored at 395 nm) in the PsaA-F689N mutant of C. reinhardtii are shown in Fig. 1 C and compared with a control strain harboring wild-type (WT) PSI (Fig. 1 B). Also shown are the fits to the experimental data by a sum of exponential functions and the contribution of each exponential phase to the total decay. These different components were obtained by a global fit of the individual kinetics recorded at several wavelengths, from which the DAS were retrieved (shown in Fig. 2). As some components are markedly slower in the PsaA-F689N mutant compared with the WT, we extended the time range to hundreds of microseconds for both strains. The investigated time window therefore exceeded four orders of magnitude, requiring four kinetic components in the WT and five components in the mutant to obtain good fits.
Figure 2.
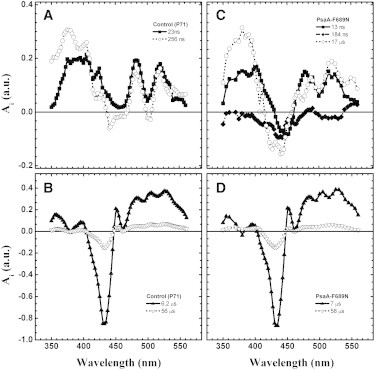
(A–D) DAS retrieved from global analysis in the nanosecond-to-microsecond timescale using control (A and B) or PsaA-F689N (C and D) cells. (A) 24 ns (solid squares) and 256 ns (open circles) components in the WT. (B) 6 μs (solid triangles) and 56 μs (open triangles) components in the WT. (C) 13 ns (solid squares), 184 ns (solid diamonds), and 17 μs (open circles) components in PsaA-F689N. (D) 7 μs (solid triangles) and 58 μs (open triangles) components in PsaA-F689N.
In the WT, two exponential components fall in the submicrosecond time range, with lifetimes of 24 ± 2 ns and 256 ± 6 ns, as previously observed in PSI of C. reinhardtii (7,8,17). These are primarily due to the oxidation of and , respectively. Two exponential decay components were required in the microsecond time window (6.2 ± 0.9 μs and 56 ± 3 μs). Based on their lifetimes and DAS (Fig. 2, B and D), these are assigned to the reduction of by prebound or unbound plastocyanin, respectively (41).
In the PsaA-F689N strain of C. reinhardtii, the kinetics are described by a sum of five exponential decay components with lifetimes of 13 ± 3 ns, 184 ± 4 ns, 7 ± 1 μs, 17 ± 2 μs, and 58 ± 5 μs. The components with lifetimes of 7 and 58 μs are similar in terms of both lifetime and DAS to the corresponding ones obtained with the control strain (Fig. 2 D), and thus are assigned to reduction as well. The three remaining components (13 ns, 184 ns, and 17 μs) are specific to the mutant. The DAS associated with the 13 ns and 17 μs components in PsaA-F689N exhibit significant positive absorption in the near-UV region and their band shapes are similar to the DAS of the 24 ns and 256 ns components, respectively, observed in the WT PSI and assigned to PhQ− oxidation (Fig. 2, A and C).
The lifetime of the fastest component, which is ascribed mainly to the oxidation of by FX, is in the range commonly reported for this reaction (7,8,18–23,28–32), albeit somewhat on the low end of the spread. The DAS of the 17 μs component displays significant amplitude in the near-UV region and a feature corresponding to an electrochromic signal in the 450–550 nm spectral window. Thus, this component is assigned to the oxidation of , making it almost two orders of magnitude slower than in the WT (200–300 ns). Several studies have shown that single mutations in the PhQA binding pocket can slow down the kinetics of PhQA– oxidation (22,23,28,29), but the change rarely exceeded 5-fold (7,8,18–23). Although a remarkable slowing of PhQA– oxidation was also observed in mutants of the PhQ biosynthetic pathway, leading to the replacement of PhQ by plastoquinone (or other exogenous quinones) in the PhQ site of PSI (19), the effect seen in the PsaA-F689N mutant is by far the largest described to date for a single-residue substitution of a RC subunit residue harboring the endogenous PhQ.
In the PsaA-A689N mutant, an additional kinetic component is detected with a lifetime of 184 ± 4 ns. Kinetic components with similar lifetimes, falling in the 150–200 ns range and exhibiting similar DAS, were previously observed in other mutants that slow oxidation and were assigned to ET from FX to FA/FB (28,30). The presence of a kinetic phase with a lifetime intermediate between those ascribed principally to and oxidation was also observed in a temperature-dependence study in the WT PSI (21,41,42). However, this component is not resolved in measurements performed at a single temperature, due to its low relative amplitude and temporal overlap with the ∼250 ns component. Thus, as previously discussed (28,30), the ∼250 ns component in WT PSI will contain spectral changes due to both oxidation and intra-FeS cluster ET.
Kinetics of ET studied by pump-pump-probe experiments
A remarkable consequence of the PsaA-F689N mutation is that it makes the oxidation of slower than the reduction, 85% of which is reduced with a ∼6 μs lifetime by prebound plastocyanin (Fig. 2) (41). This opens up the possibility of exploring the photochemical activity of PSI under conditions where PhQA is present in the semiquinone state. This would mimic what occurs in type II RCs, where the second charge separation occurs in the presence of a semiquinone anion (at the QB site), as two successive photochemical acts are required for its double reduction and protonation (6). To study secondary ET after initiation of photochemistry in the PSI RC containing , we designed a novel (to our knowledge) experimental setup that is based on double laser-flash excitation and hence consists of a pump-pump-probe sequence, as opposed to the classical pump-probe experiment. The first actinic excitation prepares the system in a given redox state and the second actinic excitation induces a second charge separation in the context of that state. One can then assess the consequence(s) of the presence of by comparing the kinetics of the individual reactions measured after one or two actinic flashes.
Fig. 3 shows the kinetics of PhQ− oxidation, monitored at 395 nm in the control (A) and PsaA-F689N mutant (B) strains, obtained after either the pump-probe or the pump-pump-probe sequence with a delay of 15 μs between the two pump flashes. The data were fitted by the function , where τi and τj are the lifetimes; Ai and Aj are the relative amplitudes of each individual component in the pump-probe (i) and pump-pump-probe (j) experiments, respectively; and x is the molar fraction of centers that effectively perform a second charge separation upon the second pump flash. It is worth noting that x is equivalent to the fraction of centers in which P700 is reduced at the moment of the second pump flash, and that it can be retrieved from the pump-probe data set described above. The same is true for the values of Ai and τi, which significantly decreases the number of fit parameters.
Figure 3.
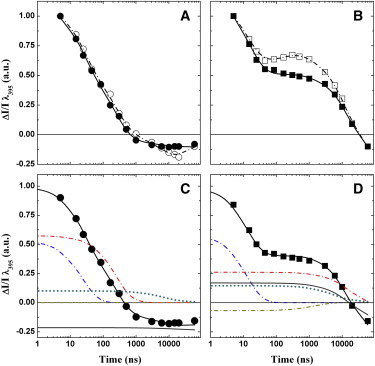
(A and B) Comparison of kinetics monitored at 395 nm in pump-pump probe and pump-probe experiments in the WT (A) and PsaA-F689N (B) strains, using a pump-pump delay (ΔT) of 15 μs. The solid symbols and solid lines are the data and fits, respectively, for the pump-pump probe experiment. The open symbols and dashed lines show the experimental kinetics and fits, respectively, for the classic pump-probe experiment. (C and D) Fit of the pump-pump-probe data at 395 nm for the WT (C) and PsaA-F689N (D) strains. The solid lines are the fits to the data sets; the contributions of different decay phases are also shown. (C) 24 ns (dash-dotted blue line), 258 ns (dash-dotted red line), sum of the 6 μs and 56 μs (dotted green line) phases attributed to reduction, and sum of the decay tail elicited from the first pump pulse in the sequence (thick gray line). (D) 13 ns (dash-dotted blue line), 17 μs (dash-dotted red line), 184 ns (dash-dotted golden line), sum of 7 μs and 58 μs phases of reduction (dotted green line), and sum of the decay tail from the first pump pulse in the sequence (thick gray line). The traces are normalized to the same total amplitude (extrapolated to t0) and the same offset to facilitate comparison. To see this figure in color, go online.
In the case of the control strain, the amplitude-normalized transient absorption kinetics monitored at 395 nm with a pump-pump delay of 15 μs hardly differed from that obtained after a single actinic flash (Fig. 3 A). In contrast, the kinetics obtained by the two protocols differed markedly in the PsaA-F689N mutant (Fig. 3 B) and became significantly faster overall in the pump-pump-probe experiment compared with the pump-probe kinetics. Fitting of the data with the expression described above (Fig. 3 D) indicates that whereas the lifetimes remained unaltered (within the parameter confidence bounds), the amplitude of the 13 ns phase increased significantly with respect to the 17 μs phase, shifting the ratio of fast/slow phases of PhQ− oxidation from 0.41:0.59 to 0.62:0.38. This represents a 50% increase of the relative amplitude of the 13 ns component, with a concomitant 35% decrease of the 17 μs phase, corresponding to a significant overall redistribution in the kinetic components. It is important to consider that for a pump-pump delay of 15 μs, a significant fraction of PSI RCs will have relaxed to a state in which all ET cofactors are in the ground state. Based on the kinetics of reduction and PhQ− oxidation, this relaxed fraction would account for ∼80% of the RCs that are capable of performing stable charge separation (i.e., in which P700 is in its neutral state). The increase of the 13 ns phase from 41% to 62% in the pump-pump-probe kinetics is thus remarkably close to the predicted fraction of photochemically active RCs containing , indicating that the majority of ET leads to the reduction of PhQB (for a more in-depth description, see Fig. S3).
The relative increase of the 13 ns phase with respect to the 17 μs phase was observed at several monitoring wavelengths in the PsaA-F689N mutant in the double-pump experiment. This is shown in Fig. 4, where the relative amplitudes of the fast PhQ− oxidation phase in the pump-probe and pump-pump-probe experiments at selected wavelengths (395, 430, 445, and 480 nm) are compared. The relative increase of the 13 ns phase in PsaA-F689N was similar at all wavelengths tested (Fig. 4 B). This was not the case for WT PSI (Fig. 4 A), where smaller changes of variable sign were observed.
Figure 4.
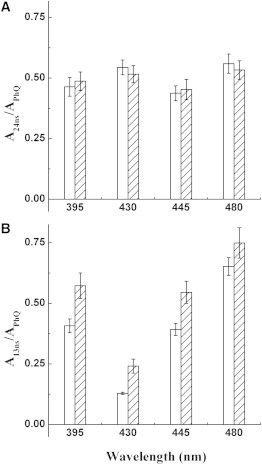
The fractional amplitude of the faster PhQ– oxidation component (24 ns for the WT and 13 ns for PsaA-F689N) to the total amplitude of PhQ– oxidation is shown as a function of wavelength for both the pump-probe (open) and pump-pump-probe (hatched) experiments. (A) WT. (B) PsaA-F689N mutant. Error bars are the propagation of the standard deviation of the mean values.
This consistency of the relative increase of the 13 ns phase is especially significant at 445 nm, as this wavelength is a sensitive indicator of charge recombination of the P700+A0– radical pair (28,43,44). The charge recombination process occurs with a lifetime of ∼20–40 ns and overlaps temporally with the fast PhQ− oxidation phase (7,8,15,28,43). However, the DAS associated with the P700+A0– charge recombination process is markedly different from that of PhQ− oxidation. Therefore, if charge recombination were to occur, it would lead to an alteration of the DAS (and lifetime) of the fastest PhQ− oxidation component, especially at 445 nm, as previously observed in mutants that promote P700+A0– recombination (28,30,44). However, such changes were not observed here (see Fig. S2). On the contrary, the increase of the relative amplitude of the 13 ns component is more pronounced at 395 nm (PhQ– absorption) and 480 nm (electrochromic band shift; Fig. 4 B), both of which are marker wavelengths for processes associated with forward ET rather than charge recombination.
Therefore, the best explanation for the increase in the relative amplitude of the 13 ns component in pump-pump-probe experiments is an increase in the relative yield of the photochemical reduction of when is present in the ETCA chain. We further tested this hypothesis by measuring the relative amplitude of the PhQ− oxidation phases as a function of the delay between the two pump flashes. In this framework, it is expected that 1) the total number of open centers that can perform charge separation will increase as plastocyanin reduces produced by the first flash (i.e., with time constants of ∼6 μs and ∼55 μs), and 2) the ratio of the fast and slow phases of PhQ– oxidation after the second flash will decrease as is oxidized by FX (i.e., with a time constant of ∼17 μs). The experimental kinetics at 390 nm at pump-pump delays (ΔT) are shown in Fig. 5 A, while Fig. 5 B shows the dependence of the relative amplitude of the fast phase (Afast) and the total signal (Atot) as a function of ΔT. As expected, Atot increased as a function of the pump-pump delay, and its time dependency can be described by the function , with τB ∼11 μs. This value is close to the average lifetime of reduction, when the contributions of the ∼6 μs and ∼55 μs components are weighted by their fractional amplitudes. In fact, a closer match to the experimental data is obtained by a biexponential fit (Fig. 5 B), yielding lifetimes of τB,1 = 6 ± 1 μs (72%) and τB,2 = 62 ± 4 μs (28%), which is fully consistent with Atot representing the fraction of PSI with neutral P700 (i.e., open) when the second pump pulse excites the RC.
Figure 5.
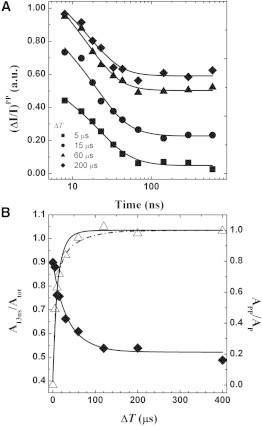
(A) Initial decay of the absorption difference signal at 390 nm in pump-pump-probe experiments in the PsaA-F689N mutant, using different values of ΔT: 5 μs (squares), 15 μs (circles), 60 μs (triangles), and 200 μs (diamonds). The symbols and lines are the data and fits, respectively. (B) Dependence of the total signal amplitude (open triangles, left y axis) and the ratio between the fast phase of PhQ− oxidation and the total signal amplitude (solid diamonds, right y axis) as a function of ΔT. The increase of total signal amplitude is described by either a monoexponential recovery with τB = 11 ± 1 μs (solid line) or a biexponential recovery with τB,1 = 6 ± 1 μs and τB,2 = 62 ± 4 μs (dash-dot line). The Afast/Atot(ΔT) ratio is described by an exponential decay with a lifetime of 20 ± 3 μs.
The fractional amplitude of the ∼13 ns phase of PhQ− oxidation (Afast/Atot) decreases monotonically as a function of pump-pump delay ΔT, approaching a final value similar to that seen in the simple pump-probe experiment. Its time dependency can be described by a single exponential decay with a lifetime of ∼20 μs (Fig. 5 B), which is close to the 17 μs lifetime of oxidation measured in the PsaA-F689N mutant by the pump-probe experiment. Since it is possible to exclude the notion that the increase in the relative amplitude of the 13 ns phase is due to charge recombination of P700+A0A–, it must therefore reflect the redistribution of the statistical utilization of ETCA and ETCB, from a ratio of ∼0.4:0.6, under neutral conditions, to larger than 0.9:0.1 under conditions of charge separation occurring in the presence of .
Simulation of the pump-pump kinetics in PsaA-F689N
To gain further insights into the redistribution of utilization of ETCA and ETCB caused by the presence of , we performed a simulation of the kinetics based on a few simple rationales. The simulations consider that when the second pump pulse excites the sample at any delay time (ΔT) longer than ∼100 ns after the first actinic flash (i.e., after complete oxidation of ), PSI can be present in four possible redox states: 1) the fraction in which the primary donor (P700) is oxidized and the secondary acceptor (PhQA) is reduced is denoted as , 2) the fraction in which both P700 and PhQA are oxidized is denoted as , 3) the fraction in which P700 is reduced and PhQA is oxidized is denoted , and 4) the fraction in which both P700 and PhQA are reduced is denoted . The probability that a particular PSI RC will be in any of these states depends on the kinetics with which each of the cofactors returns to its neutral state after photochemical oxidation/reduction. The molar fractions of the various redox states described above are then equivalent to the combinatorial probability of the population evolution of the individual cofactors (presented in Fig. S3).
The pump-pump-probe kinetics are modeled according to the following assumptions (for a more in-depth discussion, see Supporting Material).
-
1.
When the electron donor is oxidized (i.e., ), the system cannot undergo charge separation (i.e., it is closed). Therefore, fractions and will not contribute to the light-induced signal.
-
2.
In the fraction of centers, the system is in its relaxed state and the evolution of the state produced by the second pump flash is identical to that observed with a single flash in the submicrosecond timescale, with the exception of the reduction kinetics. The ratio of the fast (6 μs) and slow (55 μs) phases of reduction slightly differs from that observed after the first flash, because the dissociation of oxidized plastocyanin and association of reduced plastocyanin occur within the microsecond timescale (see Supporting Material for more details).
-
3.
In the fraction, the presence of will modify the energetics of charge separation in ETCA (e.g., by Coulombic interactions), causing a complete redistribution in favor of ETCB. It is assumed (for the sake of simplicity) that the ET rates through the B-branch and to the FeS clusters will not be significantly different due to the presence of .
Part of the absorption difference signal detected at long delays in whole cells is due to late electron transport events and is described in the analysis by a nondecaying component (e.g., Fig. 1, B and C). Since the intensity of the nondecaying signal in a pump-pump-probe experiment is not predictable in a straightforward manner, it was allowed to vary in the simulation to enable a closer match to the experimental data, as it does not affect the kinetics in the investigated time window (further details are given in Fig. S4 and the Supporting Material). The results of the simulation of the kinetics in the PsaA-F689N mutant made under these assumptions are shown in Fig. 6, where it can be seen that they satisfyingly agree with the experimental results.
Figure 6.
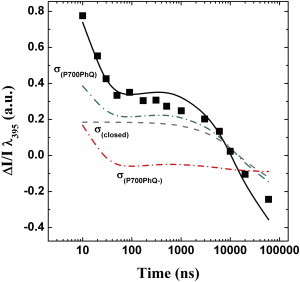
Simulation of pump-pump-probe kinetics in the PsaA-F689N mutant. The simulations (black line) are based on the fit of the pump-probe data (Fig. 1) and are compared with the experimental results (squares; as in Fig. 3, but without amplitude normalization). Also shown is the deconvolution of the contributions to the pump-pump-probe absorption difference kinetics from the various redox state fractions of the RC at the time of the second pump pulse: (dash-dotted red line), (dash-dotted blue-green line), and σclosed (dashed gray line). To see this figure in color, go online.
Based on the same rationale, it is possible to simulate the ΔT dependence of the pump-pump-probe signal (Fig. S5). There are two implications to the striking agreement between the simulations and the experimental results. First, it confirms that P700+A0– charge recombination is not taking place to a significant extent during the pump-pump-probe experiments in the PsaA-F689N mutant. Such a good agreement between simulated and experimental kinetics would be possible only in the case that at 395 nm, and a comparison of the DAS attributed to and PhQ− (i.e., Fig. 2) indicates that this is not the case. Second, it strongly suggests that the presence of at the moment that charge separation takes place leads to an almost complete redistribution of primary charge separation probabilities between the two active ETCs of PSI, favoring initial photochemistry in ETCB.
Discussion
Effect of the PsaA-F689N mutation on forward ET kinetics
The PsaA-F689N substitution causes a significant increase of the lifetime of oxidation, from ∼250 ns in the WT to 17 μs in the mutant (Fig. 1). This effect is more than one order of magnitude greater than those previously observed as a result of a point mutation affecting the protein environment of the PhQA-binding site (7,8,18–23). Although there remains some disagreement concerning the effective midpoint of PhQs in PSI (7,15,19,21,45–49), it has been shown that both the WT kinetics and the effect of mutations in the PhQA binding site can be rationalized on a semiquantitative basis by a model that considers the oxidation of to be energetically uphill and that of to be downhill (7,21,27,29,31). The lifetime observed in the PsaA-F689N mutant can be qualitatively explained, with all other energetic parameters being unchanged from those of the WT, by a ∼100-fold increase of the equilibrium constant, corresponding to an upshift by ∼125 mV of the midpoint potential of the couple (Fig. S1; Table S1). This description reproduces the experimental lifetimes of both the slower component (17 μs) and the smaller time constant for the faster decay component. However, the amplitude associated with the slow phase is significantly overestimated in the simulations for the PsaA-F689N mutant (ratio of ∼0.96:0.04; Table S1) compared with the relatively modest fractional increase of this component observed experimentally, which is 0.75:0.25 on average, using the kinetics monitored in the 360–400 nm range (Fig. 2).
Even though the kinetics simulations should be considered only a semiquantitative description of the system, previous descriptions of PhQA-binding-site mutants that considered the variation of the redox potential as the sole mutation-induced perturbation to the energetic parameters that control ET yielded a rather satisfactory description of both the experimental lifetimes and associated amplitudes (7,21,27,29,31). Thus, the discrepancy between simulations and experimental results for the PsaA-F689N substitution suggests that the mutation does not solely affect the redox potential. Among the other parameters that would affect the ET rates, one may consider an increase in the (total) reorganization energy (λtot) from a value of 0.7 to 1 eV for . These values fall in the range commonly reported for ET proteins, including PSI (7,15,42,50–52). This, combined with an upshift of the midpoint potential of 105 mV, results in a good prediction of the lifetimes, as well as the fractional amplitude of the slow/fast phase of PhQ− oxidation (∼0.81:0.19; Table S1).
Although the parameters retrieved from kinetic modeling should be considered only as semiquantitative, because of the unavoidable approximations required in the calculations, the simulations nonetheless indicate that a shift of the potential by at least ∼100 mV is induced by the PsaA-F689N mutation. Therefore, irrespective of the midpoint potential of considered for the WT, which has been reported as being in the range of ±120 mV that of FX (7,15,19,21,45–49), the mutation-induced perturbation would render the reaction in PsaA-F689N as energetically either barely downhill or significantly uphill.
Modulation of ET directionality within PSI by the presence of
The amplitude of the fast PhQ− oxidation phase, which, as established by previous studies (18,53), can be taken as a reliable measure of ETCB utilization, is strongly increased if is present when charge separation is initiated. We have already discussed the assignment of this component to the oxidation of rather than to charge recombination of P700+A0–. Notably, our normalization procedure uses the total amplitudes obtained after the first pump flash as an internal standard. Although this does not represent an absolute estimate of the photochemical quantum efficiency, which for PSI of higher plants approaches unity (4,5), it allows us to rule out any significant change in quantum efficiency in pump-pump-probe experiments performed in the PsaA-F689N mutant. Consistent with this, the data can be simulated assuming that the redistribution in favor of ETCB is close to 100% (Fig. 6; Supporting Material).
Charge recombination from the so-called stabilized radical pair P700+A0– occurs in ∼20–40 ns and leads to the repopulation of the RC excited state, as well as the ground and triplet states (3P700). Whereas the excited state can be retrapped with high probability by the ETCB cofactors, thereby promoting a redistribution of the chain utilization without a significant reduction of the quantum yield, repopulation of the ground state or the population of 3P700 would instead be photochemically unproductive, thereby lowering the quantum efficiency by some significant extent. Since, as discussed above, we did not observe such a decrease, redistribution is unlikely to involve repopulation of the excited state from a relatively long-lived radical pair (e.g., P700+ec3A–) that would have been formed on the A branch.
The most straightforward hypothesis to explain our results is that the charge borne by modifies the standard free-energy difference (ΔG0) between the initial radical pair produced by charge separation and the RC excited state, disfavoring its formation (and/or favoring the backward repopulation of the RC excited state). Such an electrostatic effect would not be unprecedented, since similar conclusions were reached regarding the energetics of charge separation in PSII in the presence of (54). In addition, this interpretation is consistent with the proposal advanced by Müller et al. (18) that the statistical utilization of the two ETCs is determined by the competition for conversion of the excited state into either ec2A+ec3A– or ec2B+ec3B–, which are considered to be the primary radical pair states in their model. The effects of mutations that alter the redox properties of ec3A or ec3B by breaking a hydrogen bond (H-bond) to the 131 keto oxygen of the targeted Chl, leading to a partial redistribution of the statistical utilization of ETCA/ETCB (18,53), were described in terms of a decrease of ΔG0 of initial charge separation from –90 meV in the WT to approximately –70 meV in the mutants (18). The apparent effect on directionality observed in the PsaA-F689N mutant when photochemistry was performed in the presence of seems to be significantly larger than that observed in these point mutants of the ec3 Chl-binding sites in PSI of C. reinhardtii (18,28,53). Thus, the change in ΔG0 associated with primary radical pair formation in the presence of would need to be larger than the change caused by the H-bond mutants. This is reasonable in light of the facts that the center-to-center distance between PhQΑ and ec3A is ∼9 Å, and the dielectric constant associated with membrane proteins is typically considered to be low (in the 4–20 range), leading to fairly large electrostatic interactions (i.e., ∼80–400 meV). These are larger perturbations than would be expected from the disruption of a single H-bond to the 131-keto group of a Chl a (18,30,53,55). Hence, although the mechanisms of primary charge separation and the associated energetics in PSI are still under debate (1,7,17,18,49,56), Coulombic interactions in the range of 80–400 meV would likely affect the charge separation kinetics on ETCA even when one considers larger values of ΔG0 for charge separation (under normal conditions) than those estimated by Müller et al. (18). Therefore, regardless of the exact mechanism considered, electrostatic repulsion would decrease the driving force for charge reparation on ETCA significantly, thereby favoring trapping by ETCB, in agreement with the experimental observations reported here. This represents a novel (to our knowledge) mechanism of directionality control in photosynthetic RCs.
Insights into the evolution of photosynthetic RCs
These results provide insights into the evolution of photosynthetic RCs, from a bioenergetics perspective. In type II RCs, initial charge separation is fully asymmetric (unidirectional) and the lifetime of the QB semiquinone form is such that it is compatible with the double reduction (and protonation) of the terminal quinone through the reaction, which occurs after a second charge separation event. If indeed, as is commonly agreed upon, type I and type II RCs evolved from a common ancestor that was homodimeric, there should be a reasonable scenario describing their progressive evolution. Gene duplication represents a decisive initial step, as it made possible the divergent evolution of the two subunits that make the heterodimer constituting the core of all known type II RCs.
In concert with previous work, the results presented here provide some rationales on which the evolutionary scenario may have relied to convert a type I-like RC into a type II-like RC, involving relatively few single amino acid substitutions. First, as previously demonstrated (7,8,18–23), a single amino acid substitution can lead to a significant increase in the semiquinone lifetime; here, we show that an increase of 2 orders of magnitude can be achieved from a single amino acid substitution (∼17 μs in PsaA-F689N compared with ∼250 ns in the WT). A lifetime in the hundreds of microseconds time range, as in the case of in type II RCs (6), would not require much additional change. We interpret the increased lifetime of resulting from the PsaA-F689N substitution as (predominantly) upshifting the reduction potential of the couple. This also implies a large difference in the E0 value between and couples, and thus a large driving force for interquinone ET mediated by FX, according to the reaction scheme , which has been proposed to occur in WT PSI (21,31). The oxidation of is thus thermodynamically driven by coupling with the large exergonic oxidation of by FA/FB(7,15,19). Therefore, in the absence of FA/FB, would represent the most stable state in the PsaA-F689N mutant PSI, and to a smaller extent in the case of WT PSI. The absence of the FA and FB clusters would thus further stabilize , if such a large change did not greatly affect the potentials of the other cofactors (57).
The core of the photosynthetic RC of heliobacteria is composed of a homodimer of the PshA polypeptide, and the soluble PshB polypeptide was originally thought to act analogously to PsaC (58). However, PshB appears to be weakly bound to the PshA homodimer (59) and the same is true of the PscB polypeptide of the chlorobial RC (60), suggesting that this is a general property of homodimeric type I RCs. Thus, the homodimeric ancestor of the type I RCs probably harbored an FX cluster but lacked a tightly bound ferredoxin-like FA/FB-binding subunit. In this context, a mutation that sufficiently delayed oxidation would result in an RC with characteristics of both RC families, i.e., a quinone-reducing acceptor side in a system performing bidirectional ET. Based on the results obtained in this study, one could argue that this might be sufficient to confer a type II-like function on a type I RC. The first photochemical event would result in the formation of the analog of with a quantum yield close to one, and the second photochemical act would produce equally efficiently, as shown here. Subsequent ET from to via FX would result in a double reduction of the quinone, to quinol, assuming that there existed a pathway for efficient protonation of the semiquinone and/or quinolate form(s). (We deliberately omit in this discussion the need to couple proton transfer to the reduction of the quinone, as well as issues regarding binding of the quinone and release of the quinol. These issues are undoubtedly important and deserve careful discussion, but this would go beyond the scope of this article.) In this respect, it is worth noting that PSI is capable of a double reduction of plastoquinone (which has a higher redox potential than PhQ) when it is bound in the PhQ site (44). Although this reaction occurs with low quantum efficiency in PSI (44), the same may not be true of the type I RCs of the anoxygenic phototrophs.
Author Contributions
S.S., F.R., and K.E.R. designed the experiment. B.B. made the mutant and did all genetic work. K.R., F.R., and S.S. participated in data collection and analysis and wrote the manuscript.
Acknowledgments
We acknowledge the involvement of Dr. A. Jasaitis in the initial stages of this study.
This work was supported by grants from the Department of Energy (DE-FG02-08ER15989 to K.R., for the initial creation and characterization of the PsaA-F689N mutant) and the National Science Foundation (MCB-1052573 for the use of this mutant to study the effect of on the directionality of ET within PSI). F.R. received financial support from the CNRS and the Initiative d’Excellence Program of the French state (DYNAMO, ANR-11-LABX-0011-01).
Contributor Information
Fabrice Rappaport, Email: fabrice.rappaport@ibpc.fr.
Kevin E. Redding, Email: kevin.redding@asu.edu.
Supporting Material
Supporting Citations
References (61–68) appear in the Supporting Material.
References
- 1.Gobets B., van Grondelle R. Energy transfer and trapping in photosystem I. Biochim. Biophys. Acta. 2001;1507:80–99. doi: 10.1016/s0005-2728(01)00203-1. [DOI] [PubMed] [Google Scholar]
- 2.Croce R., van Amerongen H. Light-harvesting in photosystem I. Photosynth. Res. 2013;116:153–166. doi: 10.1007/s11120-013-9838-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caffarri S., Tibiletti T., Santabarbara S. A comparison between plant photosystem I and photosystem II architecture and functioning. Curr. Protein Pept. Sci. 2014;15:296–331. doi: 10.2174/1389203715666140327102218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jennings R.C., Zucchelli G., Santabarbara S. Photochemical trapping heterogeneity as a function of wavelength, in plant photosystem I (PSI-LHCI) Biochim. Biophys. Acta. 2013;1827:779–785. doi: 10.1016/j.bbabio.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Galka P., Santabarbara S., Caffarri S. Functional analyses of the plant photosystem I-light-harvesting complex II supercomplex reveal that light-harvesting complex II loosely bound to photosystem II is a very efficient antenna for photosystem I in state II. Plant Cell. 2012;24:2963–2978. doi: 10.1105/tpc.112.100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diner B.A., Rappaport F. Structure, dynamics, and energetics of the primary photochemistry of photosystem II of oxygenic photosynthesis. Annu. Rev. Plant Biol. 2002;53:551–580. doi: 10.1146/annurev.arplant.53.100301.135238. [DOI] [PubMed] [Google Scholar]
- 7.Santabarbara S., Heathcote P., Evans M.C.W. Modelling of the electron transfer reactions in Photosystem I by electron tunnelling theory: the phylloquinones bound to the PsaA and the PsaB reaction center subunits of PSI are almost isoenergetic to the iron-sulfur cluster FX. Biochim. Biophys. Acta. 2005;1708:283–310. doi: 10.1016/j.bbabio.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Rappaport F., Diner B.A., Redding K.E. Optical measurements of secondary electron transfer in photosystem I. In: Golbeck J.H., editor. Photosystem I: The Light-Driven Plastocyanin:Ferredoxin Oxidoreductase. Kluwer Academic Publishers; Dordrecht: 2006. pp. 223–244. [Google Scholar]
- 9.Jordan P., Fromme P., Krauss N. Three dimensional structure of cyanobacterial Photosystem I at 2.5 Å resolution. Nature. 2001;411:909–917. doi: 10.1038/35082000. [DOI] [PubMed] [Google Scholar]
- 10.Ben–Shem A., Frolow F., Nelson N. Crystal structure of plant photosystem I. Nature. 2003;426:630–635. doi: 10.1038/nature02200. [DOI] [PubMed] [Google Scholar]
- 11.Deisenhofer O., Epp K., Michel H. Structure of the protein subunits in the photosynthetic reaction center of Rhodopseudomonas viridis at 3Å resolution. Nature. 1985;318:618–624. doi: 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- 12.Allen J.P., Feher G., Huber R. Structural homology of reaction centers from Rhodopseudomonas sphaeroides and Rhodopseudomonas viridis as determined by x-ray diffraction. Proc. Natl. Acad. Sci. USA. 1986;83:8589–8593. doi: 10.1073/pnas.83.22.8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zouni A., Witt H.T., Orth P. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]
- 14.Umena Y., Kawakami K., Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 15.Brettel K. Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim. Biophys. Acta. 1997;1318:322–373. [Google Scholar]
- 16.Melkozernov A.N., Blankenship R.E. Structural and functional organization of the peripheral light-harvesting system in photosystem I. Photosynth. Res. 2005;85:33–50. doi: 10.1007/s11120-004-6474-5. [DOI] [PubMed] [Google Scholar]
- 17.Müller M.G., Niklas J., Holzwarth A.R. Ultrafast transient absorption studies on Photosystem I reaction centers from Chlamydomonas reinhardtii. 1. A new interpretation of the energy trapping and early electron transfer stePSIn Photosystem I. Biophys. J. 2003;85:3899–3922. doi: 10.1016/S0006-3495(03)74804-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Müller M.G., Slavov C., Holzwarth A.R. Independent initiation of primary electron transfer in the two branches of the photosystem I reaction center. Proc. Natl. Acad. Sci. USA. 2010;107:4123–4128. doi: 10.1073/pnas.0905407107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srinivasan N., Golbeck J.H. Protein-cofactor interactions in bioenergetic complexes: the role of the A1A and A1B phylloquinones in Photosystem I. Biochim. Biophys. Acta. 2009;1787:1057–1088. doi: 10.1016/j.bbabio.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Redding K.E., van der Est A. The directionality of electron transfer in photosystem I. In: Golbeck J.H., editor. Photosystem I: The Plastocyanin:Ferredoxin Oxidoreductase in Photosynthesis. Kluwer Academic Publishers; Dordrecht: 2006. pp. 413–437. [Google Scholar]
- 21.Santabarbara S., Galuppini L., Casazza A.P. Bidirectional electron transfer in the reaction centre of photosystem I. J. Integr. Plant Biol. 2010;52:735–749. doi: 10.1111/j.1744-7909.2010.00977.x. [DOI] [PubMed] [Google Scholar]
- 22.Guergova-Kuras M., Boudreaux B., Redding K.E. Evidence for two active branches for electron transfer in photosystem I. Proc. Natl. Acad. Sci. USA. 2001;98:4437–4442. doi: 10.1073/pnas.081078898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu W., Chitnis P., Golbeck J.H. Electron transfer in cyanobacterial photosystem I: II. Determination of forward electron transfer rates of site-directed mutants in a putative electron transfer pathway from A0 through A1 to FX. J. Biol. Chem. 2003;278:27876–27887. doi: 10.1074/jbc.M302965200. [DOI] [PubMed] [Google Scholar]
- 24.Santabarbara S., Kuprov I., Evans M.C.W. Bidirectional electron transfer in photosystem I: determination of two distances between P700+ and A1− in spin-correlated radical pairs. Biochemistry. 2005;44:2119–2128. doi: 10.1021/bi048445d. [DOI] [PubMed] [Google Scholar]
- 25.Santabarbara S., Kuprov I., Evans M.C.W. Analysis of the spin-polarized electron spin echo of the [P700+A1−] radical pair of photosystem I indicates that both reaction center subunits are competent in electron transfer in cyanobacteria, green algae, and higher plants. Biochemistry. 2006;45:7389–7403. doi: 10.1021/bi060330h. [DOI] [PubMed] [Google Scholar]
- 26.Berthold T., von Gromoff E.D., Kothe G. Exploring the electron transfer pathways in photosystem I by high-time-resolution electron paramagnetic resonance: observation of the B-side radical pair P700(+)A1B(−) in whole cells of the deuterated green alga Chlamydomonas reinhardtii at cryogenic temperatures. J. Am. Chem. Soc. 2012;134:5563–5576. doi: 10.1021/ja208806g. [DOI] [PubMed] [Google Scholar]
- 27.Santabarbara S., Kuprov I., Evans M.C.W. Directionality of electron-transfer reactions in photosystem I of prokaryotes: universality of the bidirectional electron-transfer model. J. Phys. Chem. B. 2010;114:15158–15171. doi: 10.1021/jp1044018. [DOI] [PubMed] [Google Scholar]
- 28.Byrdin M., Santabarbara S., Rappaport F. Assignment of a kinetic component to electron transfer between iron-sulfur clusters FX and FA/B of Photosystem I. Biochim. Biophys. Acta. 2006;1757:1529–1538. doi: 10.1016/j.bbabio.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 29.Ali K., Santabarbara S., Heathcote P., Evans M.C.W., Purton S. Bidirectional electron transfer in photosystem I: replacement of the symmetry-breaking tryptophan close to the PsaB-bound phylloquinone A1B with a glycine residue alters the redox properties of A1B and blocks forward electron transfer at cryogenic temperatures. Biochim. Biophys. Acta. 2006;1757:1623–1633. doi: 10.1016/j.bbabio.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Santabarbara S., Jasaitis A., Redding K.E. Additive effect of mutations affecting the rate of phylloquinone reoxidation and directionality of electron transfer within photosystem I. Photochem. Photobiol. 2008;84:1381–1387. doi: 10.1111/j.1751-1097.2008.00458.x. [DOI] [PubMed] [Google Scholar]
- 31.Santabarbara S., Reifschneider K., Redding K.E. Interquinone electron transfer in photosystem I as evidenced by altering the hydrogen bond strength to the phylloquinone(s) J. Phys. Chem. B. 2010;114:9300–9312. doi: 10.1021/jp1038656. [DOI] [PubMed] [Google Scholar]
- 32.Srinivasan N., Santabarbara S., Golbeck J.H. Alteration of the H-bond to the A1A phylloquinone in Photosystem I: influence on the kinetics and energetics of electron transfer. J. Phys. Chem. B. 2011;115:1751–1759. doi: 10.1021/jp109531b. [DOI] [PubMed] [Google Scholar]
- 33.Mula S., McConnell M.D., van der Est A. Introduction of a hydrogen bond between phylloquinone PhQA and a threonine side-chain OH group in photosystem I. J. Phys. Chem. B. 2012;116:14008–14016. doi: 10.1021/jp309410w. [DOI] [PubMed] [Google Scholar]
- 34.Cohen R.O., Shen G., Stehlik D. Evidence for asymmetric electron transfer in cyanobacterial photosystem I: analysis of a methionine-to-leucine mutation of the ligand to the primary electron acceptor A0. Biochemistry. 2004;43:4741–4754. doi: 10.1021/bi035633f. [DOI] [PubMed] [Google Scholar]
- 35.Giera W., Gibasiewicz K., Webber A. Electron transfer from A0 to A1 in Photosystem I from Chlamydomonas reinhardtii occurs in both the A and B branch with 25–30-ps lifetime. Phys. Chem. Chem. Phys. 2009;11:5186–5191. doi: 10.1039/b822938d. [DOI] [PubMed] [Google Scholar]
- 36.Baymann F., Brugna M., Muhlenhoff U., Nitschke W. Daddy, where did (PS)I come from? Biochim. Biophys. Acta. 2001;1507:291–310. doi: 10.1016/s0005-2728(01)00209-2. [DOI] [PubMed] [Google Scholar]
- 37.Li Y., Lucas M.G., Redding K.E. Mutation of the putative hydrogen-bond donor to P700 of Photosystem I. Biochemistry. 2004;43:12634–12647. doi: 10.1021/bi036329p. [DOI] [PubMed] [Google Scholar]
- 38.Redding K., MacMillan F., Rochaix J.-D. A survey of conserved histidines in the photosystem I: methodology and analysis of the PsaB-H656L mutant. In: Garab G., editor. Vol. 1. Kluwer Academic Publishers; Dordrecht: 1998. pp. 591–594. (Photosynthesis: Mechanisms and Effects). [Google Scholar]
- 39.Harris E.H. Academic Press; San Diego: 1989. The Chlamydomonas Sourcebook. A Comprehensive Guide to Biology and Laboratory Use. [DOI] [PubMed] [Google Scholar]
- 40.Béal D., Rappaport F., Joliot P. A new high-sensitivity 10-ns time-resolution spectrophotometric technique adapted to in vivo analysis of the photosynthetic apparatus. Rev. Sci. Instrum. 1999;70:202–207. [Google Scholar]
- 41.Santabarbara S., Redding K.E., Rappaport F. Temperature dependence of the reduction of P700+ by tightly bound plastocyanin in vivo. Biochemistry. 2009;48:10457–10466. doi: 10.1021/bi901052c. [DOI] [PubMed] [Google Scholar]
- 42.Agalarov R., Brettel K. Temperature dependence of biphasic forward electron transfer from the phylloquinone(s) A1 in photosystem I: only the slower phase is activated. Biochim. Biophys. Acta. 2003;1604:7–12. doi: 10.1016/s0005-2728(03)00024-0. [DOI] [PubMed] [Google Scholar]
- 43.Vos M.H., van Gorkom H.J. Thermodynamics of electron transfer in Photosystem I studied by electric field-stimulated charge recombination. Biochim. Biophys. Acta. 1988;934:293–302. [Google Scholar]
- 44.McConnell M.D., Cowgill J.B., Redding K.E. Double reduction of plastoquinone to plastoquinol in photosystem I. Biochemistry. 2011;50:11034–11046. doi: 10.1021/bi201131r. [DOI] [PubMed] [Google Scholar]
- 45.Iwaki M., Kumazaki S., Itoh S. ΔG0 dependence of the electron transfer rate in the photosynthetic reaction center of plant photosystem I: natural optimization of reaction between chlorophyll a A0 and quinone. J. Phys. Chem. 1996;100:10802–10809. [Google Scholar]
- 46.Munge B., Das S.K., Rusling J.F. Electron transfer reactions of redox cofactors in spinach photosystem I reaction center protein in lipid films on electrodes. J. Am. Chem. Soc. 2003;125:12457–12463. doi: 10.1021/ja036671p. [DOI] [PubMed] [Google Scholar]
- 47.Ishikita H., Knapp E.W. Redox potential of quinones in both electron transfer branches of photosystem I. J. Biol. Chem. 2003;278:52002–52011. doi: 10.1074/jbc.M306434200. [DOI] [PubMed] [Google Scholar]
- 48.Karyagina I., Pushkar Y., Golbeck J.H. Contributions of the protein environment to the midpoint potentials of the A1 phylloquinones and the Fx iron-sulfur cluster in photosystem I. Biochemistry. 2007;46:10804–10816. doi: 10.1021/bi700846z. [DOI] [PubMed] [Google Scholar]
- 49.Ptushenko V.V., Cherepanov D.A., Krishtalik L.I., Semenov A.Y. Semi-continuum electrostatic calculations of redox potentials in photosystem I. Photosynth. Res. 2008;97:55–74. doi: 10.1007/s11120-008-9309-y. [DOI] [PubMed] [Google Scholar]
- 50.Moser C.C., Dutton P.L. Engineering protein structure for electron transfer function in photosynthetic reaction centers. Biochim. Biophys. Acta. 1992;1101:171–176. [PubMed] [Google Scholar]
- 51.Page C.C., Moser C.C., Dutton P.L. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 52.Schlodder E., Falkenberg K., Brettel K. Temperature dependence of forward and reverse electron transfer from A1–, the reduced secondary electron acceptor in photosystem I. Biochemistry. 1998;37:9466–9476. doi: 10.1021/bi973182r. [DOI] [PubMed] [Google Scholar]
- 53.Li Y., van der Est A., Redding K.E. Directing electron transfer within Photosystem I by breaking H-bonds in the cofactor branches. Proc. Natl. Acad. Sci. USA. 2006;103:2144–2149. doi: 10.1073/pnas.0506537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gibasiewicz K., Dobek A., Leibl W. Modulation of primary radical pair kinetics and energetics in photosystem II by the redox state of the quinone electron acceptor QA. Biophys. J. 2001;80:1617–1630. doi: 10.1016/S0006-3495(01)76134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rappaport F., Guergova-Kuras M., Lavergne J. Kinetics and pathways of charge recombination in photosystem II. Biochemistry. 2002;41:8518–8527. doi: 10.1021/bi025725p. [DOI] [PubMed] [Google Scholar]
- 56.Shelaev I.V., Gostev F.E., Semenov A.Y. Femtosecond primary charge separation in Synechocystis sp. PCC 6803 photosystem I. Biochim. Biophys. Acta. 2010;1797:1410–1420. doi: 10.1016/j.bbabio.2010.02.026. [DOI] [PubMed] [Google Scholar]
- 57.Ishikita H., Stehlik D., Knapp E.W. Electrostatic influence of PsaC protein binding to the PsaA/PsaB heterodimer in photosystem I. Biophys. J. 2006;90:1081–1089. doi: 10.1529/biophysj.105.069781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinnickel M., Shen G., Golbeck J.H. Identification and characterization of PshB, the dicluster ferredoxin that harbors the terminal electron acceptors FA and FB in Heliobacterium modesticaldum. Biochemistry. 2007;46:2530–2536. doi: 10.1021/bi0622165. [DOI] [PubMed] [Google Scholar]
- 59.Romberger S.P., Golbeck J.H. The FX iron-sulfur cluster serves as the terminal bound electron acceptor in heliobacterial reaction centers. Photosynth. Res. 2012;111:285–290. doi: 10.1007/s11120-012-9723-z. [DOI] [PubMed] [Google Scholar]
- 60.Jagannathan B., Golbeck J.H. Unifying principles in homodimeric type I photosynthetic reaction centers: properties of PscB and the FA, FB and FX iron-sulfur clusters in green sulfur bacteria. Biochim. Biophys. Acta. 2008;1777:1535–1544. doi: 10.1016/j.bbabio.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 61.Marcus R.A., Sutin N. Electron transfer in chemistry and biology. Biochim. Biophys. Acta. 1985;811:265–322. [Google Scholar]
- 62.Hopfield J.J. Electron transfer between biological molecules by thermally activated tunnelling. Proc. Natl. Acad. Sci. USA. 1974;71:3640–3644. doi: 10.1073/pnas.71.9.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Devault D. Cambridge University Press; Cambridge: 1980. Quantum Mechanical Tunnelling in Biological Systems. [DOI] [PubMed] [Google Scholar]
- 64.Jortner J. Temperature dependent activation energy for electron transfer between biological molecules. J. Chem. Phys. 1976;64:4860–4868. [Google Scholar]
- 65.Giera W., Ramesh V.M., Gibasiewicz K. Effect of the P700 pre-oxidation and point mutations near A0 on the reversibility of the primary charge separation in Photosystem I from Chlamydomonas reinhardtii. Biochim. Biophys. Acta. 2010;1797:106–112. doi: 10.1016/j.bbabio.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 66.Lefebvre-Legendre L., Rappaport F., Rochaix J.D. Loss of phylloquinone in Chlamydomonas affects plastoquinone pool size and photosystem II synthesis. J. Biol. Chem. 2007;282:13250–13263. doi: 10.1074/jbc.M610249200. [DOI] [PubMed] [Google Scholar]
- 67.Hervás M., Navarro J.A., De La Rosa M.A. Electron transfer between membrane complexes and soluble proteins in photosynthesis. Acc. Chem. Res. 2003;36:798–805. doi: 10.1021/ar020084b. [DOI] [PubMed] [Google Scholar]
- 68.Hope A.B. Electron transfers amongst cytochrome f, plastocyanin and photosystem I: kinetics and mechanisms. Biochim. Biophys. Acta. 2000;1456:5–26. doi: 10.1016/s0005-2728(99)00101-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.