

Abstract
Prions are proteins that adopt self-propagating aberrant folds. The self-propagating properties of prions are a direct consequence of their distinct structures, making the understanding of these structures and their biophysical interactions fundamental to understanding prions and their related diseases. The insolubility and inherent disorder of prions have made their structures difficult to study, particularly in the case of the infectious form of the mammalian prion protein PrP. Many investigators have therefore preferred to work with peptide fragments of PrP, suggesting that these peptides might serve as structural and functional models for biologically active prions. We have used x-ray fiber diffraction to compare a series of different-sized fragments of PrP, to determine the structural commonalities among the fragments and the biologically active, self-propagating prions. Although all of the peptides studied adopted amyloid conformations, only the larger fragments demonstrated a degree of structural complexity approaching that of PrP. Even these larger fragments did not adopt the prion structure itself with detailed fidelity, and in some cases their structures were radically different from that of pathogenic PrPSc.
Introduction
Prion diseases are diseases caused by prions, proteins having self-propagating aberrant folds. The key term is self-propagating: the distinct prion structures themselves catalyze the transition from the normal structure to the aberrant structure, and thus give rise to heritable characteristics encoded in the protein structure. Prion diseases are thus disorders of abnormal molecular structure and the anomalous transitions between normal and aberrant molecular folds. Self-propagation was originally recognized in prions derived from the mammalian prion protein (PrP); the term prion was coined to describe the PrP-derived, protein-only infectious pathogen found in scrapie, a neurodegenerative disease of sheep and goats (1). PrP prions have since been implicated in related diseases in bovines, cervids, and humans; these diseases are sometimes called the transmissible spongiform encephalopathies.
Amyloid diseases, which include all currently known prion diseases, are diseases associated with abnormal deposits of misfolded proteins in the amyloid structural form. This form, cross-β structure, is a simple motif that consists of β-strands extending roughly perpendicular to the axes of long filaments (fibrils), forming sheets running parallel to the fibril axis. It is now widely held (2–4) that the aberrant protein folds found in many amyloid diseases are self-propagating. Thus the structurally determined, self-propagating properties of PrPSc (the infectious form of PrP (5)) are common to the distinct aberrant folds of other proteins, including the Alzheimer’s disease–associated peptide Aβ, the Parkinson’s disease–associated protein α-synuclein, and the microtubule-associated protein tau, which is associated with a number of amyloid neurodegenerative diseases including Alzheimer’s.
The self-propagating properties of prions are derived from their structures and structural interactions. Structural studies of PrPSc have been limited and extremely difficult, owing to difficulties in purification and a high degree of structural disorder. Limited proteolysis of PrPSc removes ∼65 residues from the N-terminus to form PrP 27–30, which retains prion infectivity (6), and useful information has been obtained from x-ray fiber diffraction (7) and electron microscopy (EM) (8–10) of this form. Many researchers, however, have preferred to use much smaller peptides as structural and functional models for biologically active PrPSc (11–14). Over the past century, many structural studies of amyloids in general have been reported, dating back to the pioneering work of Astbury (15). These have included studies (12,16,17) by fiber diffraction, crystallography, and EM, and more recently solid-state NMR (ssNMR). However, in most cases (a small number of ssNMR studies are exceptions), either the data are poor and the models extremely limited, or the amyloids studied were not shown to be self-propagating in vivo. The self-propagating properties of biologically and pathologically important amyloids are unique consequences of their molecular structures, so studies of generic (but not self-propagating) amyloids tell us little about the biological mechanisms of pathological amyloids. This is not to say that work on simple amyloid structures has not been valuable—it provided an essential starting point. But it leaves many questions unanswered.
Most of the fiber diffraction studies and all of the crystallographic studies of amyloids have targeted small amyloidogenic fragments of biologically significant amyloids (12,13,18–21). These fragments share a generic cross-β architecture found in nonphysiological amyloids derived from a number of complete proteins, but many of those proteins (15) form amyloids only after exposure to nonphysiological conditions such as high temperature or extreme pH, making their formation and possibly their structures substantially different from spontaneously forming biological amyloids. The generic architectures of these fragments and nonphysiological amyloids do not have the level of complexity found in biologically significant amyloids such as the β-solenoids (7,9,22,23) of HET-s and PrPSc or the U-shaped two-β-strand structures found in Aβ (24,25). More significantly, generic amyloid structures have not been shown to share the self-propagating character of the structurally more complex prions.
Because the distinctive biological activity of PrPSc is a direct result of its structure, simpler model systems must share the structural features of biological prions to be maximally useful. In this article, we use x-ray fiber diffraction to characterize a series of different-sized fragments of PrP. Using these structural characterizations, we have determined the structural commonalities among the fragments and compared these structural features with those found in biologically active, self-propagating PrP prions.
Materials and Methods
Peptide purification
Four peptide fragments of PrP, PrP21 (26), PrP55 (27), PrP89, and PrPSc106 (28) (Table 1), as well as a prion protein paralog similar to PrP55, Shadoo (29,30) (Sho), were examined. The amino acid sequences of the fragments corresponded to fragments of human (Hu), mouse (Mo), and bank vole (BV) PrP.
Table 1.
Peptides used for fiber diffraction
HuPrP(106–126) (PrP21) was obtained from Bachem (Torrance, CA), and fibrillized in 20 mM Tris-HCl pH 8.0, following the published protocol (31) as closely as possible.
MoPrP(89–143,P101L) (PrP55), a generous gift from Dr. David Wemmer (University of California, Berkeley), was synthesized as described (32). The lyophilized peptide was resuspended in 20 mM sodium acetate, pH 5.0, 20% in acetonitrile, and 20 mM in sodium chloride. The solution was allowed to fibrillize at room temperature for two to three weeks before fibers were prepared.
Constructs encoding mouse Shadoo (Sho) (residues 25 to 122) or human Sho (residues 25 to 126), both with an N-terminal His tag (MKHHHHHH), were inserted into the pET-11a vector; recombinant Sho was expressed in Rosetta2(DE3) Escherichia coli (EMD Millipore, Billerica, MA). Cells were lysed using Lysonase (EMD Millipore, Billerica, MA) and recombinant Sho was purified by immobilized metal affinity chromatography using TALON resin (Clontech, Mountain View, CA). The N-terminal His tags were not removed before fibril formation. Purified recombinant mouse and human Sho were dialyzed against 20 mM sodium phosphate pH 7.4, 150 mM NaCl (PBS) and concentrated using an Amicon Ultra 3K MW cutoff spin filter (EMD Millipore). Proteins were diluted to 1 mg/ml in PBS and incubated at 37°C with shaking for a period of 5 days. Fibrillization was monitored using Thioflavin T fluorescence. Aliquots of the amyloid preparations were snap-frozen in liquid nitrogen and stored at -80°C.
MoPrP(89–177) (MoPrP89) and BVPrP(90–178) (BVPrP89) were expressed recombinantly. The BVPrP89 used for this study carried the I109 polymorphism. The sequences spanning residues 89–177 of murine PrP and 90–178 of bank vole PrP were cloned into pET11a and expressed in BL21 DE3 CodonPlus-RP (Stratagene). Purification was as described (33), omitting the final oxidation step to form the disulfide bond in recPrP constructs containing the C-terminal residues 178 to 230. MoPrP89 and BVPrP89 lyophilisates were resuspended in 10 M urea at a concentration of 10 mg/ml. For the induction of fibril formation, the recombinant peptides were diluted to 1 mg/ml and adjusted to either 1 M urea (50 mM citrate buffer, pH 3; 0.2 M NaCl) or 4 M urea (50 mM sodium acetate buffer pH 5; 0.2 M NaCl). Samples were incubated for 48 h at 37°C under constant agitation at 900 rpm. After amyloid formation samples were spun down for 30 min at 100,000 g and resuspended in double-distilled water to remove high concentrations of urea and salts.
PrPSc106 was purified from the brains of RML-infected Tg8872 mice expressing PrP(Δ23–88,Δ141–176) (34). The purification was based on the specific precipitation of PrPSc106 by phosphotungstic acid (PTA) and also included a limited digestion with proteinase K. PTA-precipitated PrPSc106 was centrifuged through a sucrose cushion to remove extraneous lipids, pelleted to remove the sucrose, and resuspended in concentrated form in water (7).
The fibrillar structure of the aggregates was confirmed by EM.
Fiber diffraction
Fibers were prepared (35) by suspending a 5 to 10 μl drop of fibril suspension between two glass rods, either tipped with beeswax or silanized and sanded at the tips, ∼1.5 mm apart. The fibers were allowed to dry for a period of hours to days in a closed chamber under high humidity (98% in vapor equilibrium with saturated potassium sulfate or nominal 100% in equilibrium with water). Some fibers were allowed to dry at ∼86% relative humidity in equilibrium with saturated potassium chloride; the effect of this lower humidity on the PrP55 fibers is discussed below.
Diffraction data were collected at beamline 4-2 at the Stanford Synchrotron Radiation Laboratory (Menlo Park, CA). Some preliminary data and the data from the 98% humidity PrP55 fibers were collected at the BioCAT beamline of the Advanced Photon Source synchrotron, Argonne National Laboratory (Argonne, IL). Fibers were dusted with calcite and specimen-to-detector distances were determined from the 012 calcite diffraction ring at 3.8547 Å resolution and the 104 ring at 3.0355 Å (36); for brain-derived samples, specimen-to-detector distances were taken from dusted samples mounted in the same position. Diffraction patterns were analyzed using the program WCEN (37) to determine experimental parameters and positions of reflections.
Electron microscopy
Samples at concentrations of ∼1 mg/ml or less were applied to continuous carbon film on 400 square mesh copper EM grids (Electron Microscopy Sciences, Hatfield, PA, or Ted Pella, Redding, CA). The grids were stained with a combination of 2% uranyl formate and 1% uranyl acetate (PrP21, PrP55), or with 2% uranyl acetate, and micrographs were collected on an FEI T-12 operating at 100 keV (PrP21) (FEI, Hillsboro, OR), a Philips CM-12 operating at 80 keV (PrP55), or an FEI Tecnai F20 at 80 kV (PrP89, PrPSc106, and Sho).
Results and Discussion
Electron micrographs (Fig. 1) showed that all of the samples under study formed fibrils.
Figure 1.
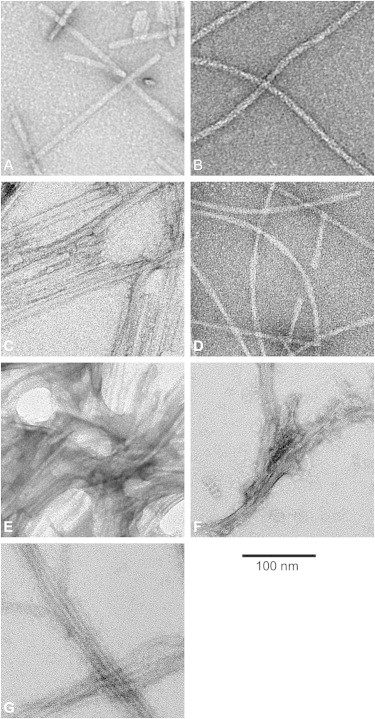
Electron micrographs from amyloid fibrils show that all samples fibrillized. (A) PrP21. (B) PrP55. (C) MoPrP89, refolded in 1 M urea at pH 3. (D) MoPrP89, refolded in 4 M urea at pH 5. (E) BVPrP89, refolded in 1M urea at pH 3. (F) PrPSc106. (G) Sho. Scale bar applies to all panels. Appearances vary because of differences in staining, sources, and electron microscopes, but all samples have clearly fibrillized.
PrP consists of ∼200 amino acids. A four-turn, left-handed β-helix has been proposed (9) as a key element in the structure of PrP 27–30; fiber diffraction data support this model (7). X-ray fiber diffraction patterns from the four peptide fragments of PrP are shown in Fig. 2 and are compared in the following paragraphs with those obtained from the ∼140-amino-acid, brain-derived PrP 27–30 (7).
Figure 2.
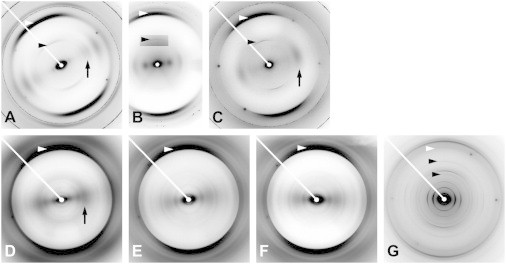
X-ray fiber diffraction patterns from fibers of PrP fragments. (A) PrP21. (B) PrP55 at ∼98% relative humidity. (C) PrP55 at ∼86% relative humidity. (D) MoPrP89 refolded in 1 M urea at pH 3. (E) MoPrP89 refolded in 4 M urea at pH 5. (F) BVPrP89 refolded in 1 M urea at pH 3. (G) PrPSc106. White arrowheads: meridional cross-β diffraction at ∼4.8 Å resolution. Black arrowheads: meridional diffraction at ∼9.6 Å in (A), (B), (C), and (G), and also at ∼6.4 Å in (G). Arrows: equatorial diffraction at 8 to 10 Å in (A), (C), and (D). The inset in (B), which includes the black arrowhead, uses a more intense color table to show the weak meridional intensity at 9.6 Å more clearly. Most panels include sharp crystalline rings from calcite, used as a standard to determine sample-to-detector distance. In (G), diffraction from PrPSc106, the only brain-derived sample, includes many intensities attributed to lipids (7), including an intense meridional intensity at ∼4.2 Å.
PrP21
Diffraction from PrP21 (Fig. 2 A) is dominated by a very strong intensity on the meridian (the axis parallel to the fibril axis) at 4.75 Å. This intensity is characteristic of cross-β structure and corresponds to the spacing between β-strands. A weaker but clear meridional intensity at 9.5 Å shows that the repeating unit contains not one, but two β-strands along the fibril axis. A series of broad intensities on the equator (the axis through the origin perpendicular to the meridian) includes maxima at 8.5, 7.5, and 5.5 Å. This pattern is strongly indicative of a simple or generic amyloid structure (7,17,23), with β-sheets stacked ∼8 Å apart. The most likely structure that would produce this pattern is that of an antiparallel β-sheet. A parallel sheet would have to include a large meander between two extremely short strands, a very implausible structure, or an equally implausible arrangement in which the repeating unit was two out-of-register parallel strands. Neither of these structures has ever been observed, whereas for short peptides such as PrP21, antiparallel arrangements are not at all uncommon (38). Details notwithstanding, the structure is clearly a stacked β-sheet with a two-strand repeat along the fibrillar axis, and is very different from the solenoidal structure proposed for PrP 27–30 (7).
The amyloid form of PrP21 has been shown to be toxic in cell culture (26), but not self-propagating. Previously published fiber diffraction patterns from PrP21 (13) did not include a meridional 9.5 Å reflection, but fiber diffraction was not a major part of that study, and the use of film for recording diffraction, an in-house sealed tube x-ray generator, and poorly oriented fibers makes it unsurprising that this feature was not seen.
Our data show that this fragment has a stacked-sheet structure with a two-strand repeating unit, differing from an ssNMR model (31) (a single-strand-repeat, parallel in-register stacked sheet). Although we attempted to duplicate the fibrillization conditions of the ssNMR study, it is possible that this polymerization process is so sensitive to solution conditions that different amyloid structures (parallel and antiparallel) could form under very similar conditions. However, in either case, the peptide clearly has a stacked β-sheet structure, and not the structure of PrP 27–30 (7).
PrP55
Diffraction patterns from PrP55 vary in appearance, depending on the conditions of amyloid formation and fiber diffraction specimen preparation, with a particular sensitivity to humidity (Fig. 2, B and C). In all cases, there is strong cross-β meridional diffraction at 4.75 Å, and a weaker reflection at 9.5 Å. The 9.5 Å reflection was seen more clearly in patterns from low-humidity fibers, which is probably a consequence of low background scattering, characteristic of low-humidity patterns. In high-humidity patterns (Fig. 2 B), there are broad equatorial intensity maxima at ∼20 Å and 10.3 Å. In low-humidity patterns (Fig. 2 C), the equator is dominated by a broad, strong maximum at ∼8 Å, with no other significant features. The low-humidity equator is typical of a stacked-sheet amyloid, whereas the high-humidity equator is much more consistent with diffraction from a roughly cylindrical structure such as a β-solenoid. The marked differences between diffraction patterns to be expected from β-solenoidal and stacked-sheet structures have been discussed with calculated and experimental examples elsewhere (7,23,39). The persistence of the meridional 9.5 Å reflection suggests that the high-humidity structure has simply collapsed into a stacked-sheet structure, supporting the hypothesis that the high-humidity structure is indeed a solenoid. These patterns are strikingly analogous to those seen in high and low humidity for the fungal prion HET-s (39), where the solenoidal structure of the high-humidity form has been independently determined by ssNMR (22). The high-humidity patterns, supported by the transition observed at lower humidity, strongly support a two-rung solenoidal model for PrP55. The low-humidity patterns also resemble the patterns obtained from the amyloid form (40) of the PrP paralog Sho (29,30) (Fig. 3), suggesting the possibility that the Sho structure may also include a collapsed two-rung solenoid.
Figure 3.
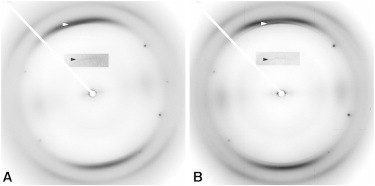
X-ray fiber diffraction patterns from fibers of the nonprion protein Shadoo (Sho), which is a paralog of PrP. (A) Mouse Sho. (B) Human Sho. Meridional intensities at ∼9.6 Å (insets, black arrowhead) show that the repeating unit along the filament axis has the thickness of two β-strands. The inserts, which include the black arrowheads, use a more intense color table to show the weak meridional intensities at 9.6 Å more clearly.
PrP55 shares some of the biological properties of PrPSc, being infectious in transgenic mice expressing PrP(P101L) at a level similar to that of wild-type PrP (27,41). Our PrP55 fiber diffraction data depend on humidity and are similar to but distinctly different from previously published patterns (19), showing that this peptide can be polymorphic. The samples used in earlier work (19) were prepared in 50% acetonitrile, with no humidity control, whereas our samples were in 20% acetonitrile, and we found humidity to be a critical determinant of the diffracting structure. Since the equatorial diffraction from our high-humidity fibers suggests that the structure may be β-solenoidal, PrP55 may be a better model for PrPSc than PrP21, but it still lacks key elements of the PrP 27–30 (and by implication, the PrPSc) structure. For example, our data clearly show that the repeating unit is a two-rung structure rather than the four-rung structure (7,9) proposed for PrP 27–30. PrP55 contains barely enough peptide to make a complete two-rung structure, although a structure might be built using a total of five β-strands instead of six, with highly efficient turns between the strands. Such a model structure would probably not, however, reflect the PrPSc structure in detail; it is quite likely that the infectivity of PrP55 comes from heterogeneous seeding (42–45), that is, seeding between distinct amyloid architectures. PrP55 might be long enough to contain a structured segment that can act as a seed, in contrast to PrP21. Such a seed structure would contain elements of the PrPSc structure, and to that extent would be a partial structural model, but it would still not contain all of the important molecular interactions found in the true structure.
PrP89
MoPrP89 and BVPrP89 (Table 1) represent equivalent sequences of 89 residues from mouse and bank vole PrP respectively. Bank vole PrP has been shown to adopt an infectious conformation spontaneously when expressed in the brains of transgenic mice (46). Refolded recombinant (rec) MoPrP89 and BVPrP89 gave two distinct types of diffraction pattern, depending on the sequence and the conditions of refolding. These two pattern types were strongly reminiscent of those obtained from recombinant and brain-derived PrP (7), from HET-s depending on folding conditions (23,44,45), and from PrP55 as described in the previous section. The pattern from MoPrP89 refolded in 1 M urea at pH 3 (Fig. 2 D) was very similar to that of recSHaPrP (90–231) or recMoPrP (89–230) (7,47), with strong meridional cross-β diffraction and an equator dominated by strong intensities at 14 and 10 Å, strongly indicating a generic stacked-sheet structure. In contrast, patterns from both MoPrP89 refolded in 4 M urea at pH 5 (Fig. 2 E) and BVPrP89 refolded in 1 M urea at pH 3 (Fig. 2 F) include a series of equatorial intensities progressively diminishing as the distance from the origin increases, characteristic of a cylindrical structure such as a β-solenoid (7,23). None of the patterns included meridional diffraction other than the cross-β diffraction at 4.8 Å, but the meridional intensities from the 19.2 Å four-β-strand repeat observed in patterns from brain-derived PrP 27–30 (7) are weak, as would be expected from a four-β-strand structure, and may well have been reinforced by some sort of decoration of the four-strand protein structure by the GPI anchor, brain lipids, or other such molecules absent from the recombinant preparation. The PrP 27–30 patterns (7) include a number of strong intensities indicative of the presence of an ordered lipid domain.
The patterns from MoPrP89 and BVPrP89 folded under suitable conditions are thus consistent with the suggestion that the refolded structure could be that of a multirung solenoid, such as the β-solenoidal structure proposed (7,9) for PrP 27–30. Although this is of necessity a limited conclusion, it is nevertheless an encouraging advance from structural studies (7) of recSHaPrP (90–231) and recMoPrP (89–230); diffraction from those constructs indicated the presence only of stacked-sheet structures, in contrast to diffraction from brain-derived PrP 27–30. This fragment may therefore have some value as a model for PrPSc.
PrPSc106
Diffraction patterns from brain-derived PrPSc106 reflect a considerable degree of disorder, and in some cases even the cross-β reflection is difficult to discern, but in favorable cases diffraction patterns bearing distinct similarities to those of brain-derived PrP 27–30 (7) can be obtained (Fig. 2 F). Like the PrP 27–30 patterns, they include numerous intensities attributable to lipids or lipid-detergent assemblies, but also meridional maxima at 9.6, 6.4, and 4.8 Å, corresponding to the second, third, and fourth orders, respectively, of a 19.2 Å repeat, the size of a four-stranded β-sheet. The breadth and weakness of the 4.8 Å cross-β reflection are strongly reminiscent of the corresponding reflection in the PrP 27–30 patterns. Although the lipid diffraction makes equatorial measurements impossible in these patterns, the 19.2 Å repeat is an encouraging indication of structural similarities between PrPSc106 and PrP 27–30.
PrPSc106 has been shown to support prion propagation in transgenic mice (34) and shares the 19.2 Å repeat of PrP 27–30. To that extent, it may be the best peptide model for PrPSc. Its most obvious limitations are that it exhibits disorder comparable with, and at times greater than that of PrP 27–30, and is consequently difficult to obtain high-quality diffraction data from. Nevertheless, comparisons of PrPSc106, PrP 27–30, and PrPSc may provide useful insights.
Conclusions
Although all of the peptides studied adopted amyloid conformations, as do many other peptide fragments of PrP (12,18–21), only the larger fragments have a degree of structural complexity approaching that of PrP 27–30. In particular, only the larger fragments appear to have any type of β-solenoidal structure, and even PrP55 is too small to adopt the four-rung PrP 27–30 structure. Fragments of increasing length show increasing similarity to PrP 27–30 (and by implication to PrPSc), but from the point of view of understanding structure none has a real advantage over purified PrPSc or PrP 27–30. In particular, the shorter peptides (PrP21 and by implication anything shorter, and perhaps even peptides as large as PrP55), although useful in providing insight into the generic stacked-sheet structure that underlies amyloid structure in general, tell us little or nothing about the structural and functional basis of self-propagation in prions.
Although we interpret the PrP 27–30 data in terms of a solenoidal structure, our conclusions do not depend on the accuracy of that interpretation. Regardless of the structural details, there are clearly at least two quite different structures that can be adopted by these peptides. The shorter peptides have never been seen to adopt structures similar to that of PrP 27–30, the self-propagating prion structure. This is most probably because the short lengths are not sufficient for the formation of complex amyloid structures. Although the longer fragments may adopt variations of the prion structure (the two-turn solenoid, for example), they do not adopt the prion structure itself with high fidelity, and often do not adopt any structure close to it.
It appears that the definitive property of prions, self-propagation, is inextricably tied to a requisite level of structural complexity. We have shown that this structural complexity is not found in overly simple model systems. Longer models that recapitulate the structural features found in prions tend to have similar levels of structural disorder, indicating that simple solutions for understanding the biophysical properties of prions remain elusive.
Author Contributions
W.W., H.W., S.B.P, and G.S. designed the research; W.W., H.W., J.S., A.K., W.B., M.M., J.C.W., and G.S. performed research and analysis; W.B. and S.T. contributed research methods; W.W., H.W., J.S., A.K., J.C.W., S.B.P., and G.S. wrote the article; all authors contributed to the final article.
Acknowledgments
We thank the staff of beamline 4-2 at the Stanford Synchrotron Radiation Lightsource (SSRL) and BioCAT at the Advanced Photon Source (APS). We also thank Lillian Bloch and Aleksandra Kijac for assistance preparing diffraction samples.
This work was supported by U.S. National Institutes of Health (NIH) grants AG002132, AG021601, AG010770, and F31-AG040947. The SSRL is a national user facility operated by Stanford University on behalf of the U.S. Department of Energy (DOE), Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the DOE and NIH. The APS is supported by the DOE. BioCAT is supported by NIH grant 9 P41 GM103622. Some negative stain electron micrographs were collected in the VUMC Cell Imaging Shared Resource, supported by NIH grants.
Footnotes
William Wan’s present address is Structural and Computational Biology Unit, European Molecular Biology Laboratory, Heidelberg, Germany.
Holger Wille’s present address is Department of Biochemistry and Centre for Prions and Protein Folding Diseases, University of Alberta, Edmonton, Alberta, Canada.
Joel Watts’s present address is Tanz Centre for Research in Neurodegenerative Diseases, University of Toronto, Toronto, Ontario, Canada.
References
- 1.Prusiner S.B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Holmes B.B., Diamond M.I. Cellular mechanisms of protein aggregate propagation. Curr. Opin. Neurol. 2012;25:721–726. doi: 10.1097/WCO.0b013e32835a3ee0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prusiner S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336:1511–1513. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jucker M., Walker L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prusiner, S. B. 2007. Prions. In Fields Virology. D. M. Knipe, P. M. Howley, …, S. E. Straus, editors. Lippincott Williams & Wilkins, Philadelphia, pp. 3059–3092.
- 6.McKinley M.P., Meyer R.K., Prusiner S.B. Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J. Virol. 1991;65:1340–1351. doi: 10.1128/jvi.65.3.1340-1351.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wille H., Bian W., Stubbs G. Natural and synthetic prion structure from X-ray fiber diffraction. Proc. Natl. Acad. Sci. USA. 2009;106:16990–16995. doi: 10.1073/pnas.0909006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wille H., Michelitsch M.D., Prusiner S.B. Structural studies of the scrapie prion protein by electron crystallography. Proc. Natl. Acad. Sci. USA. 2002;99:3563–3568. doi: 10.1073/pnas.052703499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Govaerts C., Wille H., Cohen F.E. Evidence for assembly of prions with left-handed β-helices into trimers. Proc. Natl. Acad. Sci. USA. 2004;101:8342–8347. doi: 10.1073/pnas.0402254101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wille H., Govaerts C., Prusiner S.B. Electron crystallography of the scrapie prion protein complexed with heavy metals. Arch. Biochem. Biophys. 2007;467:239–248. doi: 10.1016/j.abb.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inouye H., Kirschner D.A. X-ray fiber and powder diffraction of PrP prion peptides. Adv. Protein Chem. 2006;73:181–215. doi: 10.1016/S0065-3233(06)73006-6. [DOI] [PubMed] [Google Scholar]
- 12.Sawaya M.R., Sambashivan S., Eisenberg D. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 13.Tagliavini F., Prelli F., Forloni G. Synthetic peptides homologous to prion protein residues 106-147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA. 1993;90:9678–9682. doi: 10.1073/pnas.90.20.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helmus J.J., Surewicz K., Jaroniec C.P. Conformational flexibility of Y145Stop human prion protein amyloid fibrils probed by solid-state nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 2010;132:2393–2403. doi: 10.1021/ja909827v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Astbury W.T., Dickinson S., Bailey K. The x-ray interpretation of denaturation and the structure of the seed globulins. Biochem. J. 1935;29:2351–2360.1. doi: 10.1042/bj0292351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wasmer C., Lange A., Meier B.H. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 17.Jahn T.R., Makin O.S., Serpell L.C. The common architecture of cross-beta amyloid. J. Mol. Biol. 2010;395:717–727. doi: 10.1016/j.jmb.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen J.T., Inouye H., Kirschner D.A. X-ray diffraction of scrapie prion rods and PrP peptides. J. Mol. Biol. 1995;252:412–422. doi: 10.1006/jmbi.1995.0507. [DOI] [PubMed] [Google Scholar]
- 19.Inouye H., Bond J., Kirschner D.A. Structural changes in a hydrophobic domain of the prion protein induced by hydration and by Ala—>Val and Pro—>Leu substitutions. J. Mol. Biol. 2000;300:1283–1296. doi: 10.1006/jmbi.2000.3926. [DOI] [PubMed] [Google Scholar]
- 20.Salmona M., Morbin M., Tagliavini F. Structural properties of Gerstmann-Straussler-Scheinker disease amyloid protein. J. Biol. Chem. 2003;278:48146–48153. doi: 10.1074/jbc.M307295200. [DOI] [PubMed] [Google Scholar]
- 21.Apostol M.I., Wiltzius J.J., Eisenberg D. Atomic structures suggest determinants of transmission barriers in mammalian prion disease. Biochemistry. 2011;50:2456–2463. doi: 10.1021/bi101803k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Melckebeke H., Wasmer C., Meier B.H. Atomic-resolution three-dimensional structure of HET-s(218-289) amyloid fibrils by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010;132:13765–13775. doi: 10.1021/ja104213j. [DOI] [PubMed] [Google Scholar]
- 23.Wan W., Wille H., Stubbs G. Degradation of fungal prion HET-s(218-289) induces formation of a generic amyloid fold. Biophys. J. 2012;102:2339–2344. doi: 10.1016/j.bpj.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald M., Box H., Stubbs G. Fiber diffraction data indicate a hollow core for the Alzheimer’s aβ 3-fold symmetric fibril. J. Mol. Biol. 2012;423:454–461. doi: 10.1016/j.jmb.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu J.X., Qiang W., Tycko R. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forloni G., Angeretti N., Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- 27.Kaneko K., Ball H.L., Cohen F.E. A synthetic peptide initiates Gerstmann-Sträussler-Scheinker (GSS) disease in transgenic mice. J. Mol. Biol. 2000;295:997–1007. doi: 10.1006/jmbi.1999.3386. [DOI] [PubMed] [Google Scholar]
- 28.Muramoto T., Scott M., Prusiner S.B. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc. Natl. Acad. Sci. USA. 1996;93:15457–15462. doi: 10.1073/pnas.93.26.15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Premzl M., Sangiorgio L., Gready J.E. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene. 2003;314:89–102. doi: 10.1016/s0378-1119(03)00707-8. [DOI] [PubMed] [Google Scholar]
- 30.Watts J.C., Drisaldi B., Westaway D. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J. 2007;26:4038–4050. doi: 10.1038/sj.emboj.7601830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walsh P., Simonetti K., Sharpe S. Core structure of amyloid fibrils formed by residues 106-126 of the human prion protein. Structure. 2009;17:417–426. doi: 10.1016/j.str.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 32.Lim K.H., Nguyen T.N., Wemmer D.E. Solid-state NMR structural studies of the fibril form of a mutant mouse prion peptide PrP89-143(P101L) Solid State Nucl. Magn. Reson. 2006;29:183–190. doi: 10.1016/j.ssnmr.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 33.Mehlhorn I., Groth D., Prusiner S.B. High-level expression and characterization of a purified 142-residue polypeptide of the prion protein. Biochemistry. 1996;35:5528–5537. doi: 10.1021/bi952965e. [DOI] [PubMed] [Google Scholar]
- 34.Supattapone S., Bosque P., Scott M. Prion protein of 106 residues creates an artificial transmission barrier for prion replication in transgenic mice. Cell. 1999;96:869–878. doi: 10.1016/s0092-8674(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 35.McDonald M., Kendall A., Stubbs G. Enclosed chambers for humidity control and sample containment in fiber diffraction. J. Appl. Cryst. 2008;41:206–209. [Google Scholar]
- 36.Effenberger H.K., Mereiter K., Zemann J. Crystal structure refinements of magnesite, calcite, rhodochrosite, siderite, smithonite, and dolomite, with discussion of some aspects of the stereochemistry of calcite type carbonates. Z. Kristallogr. 1981;156:233–243. [Google Scholar]
- 37.Bian W., Wang H., Stubbs G. WCEN: a computer program for initial processing of fiber diffraction patterns. J. Appl. Cryst. 2006;39:752–756. [Google Scholar]
- 38.Serpell L.C., Blake C.C.F., Fraser P.E. Molecular structure of a fibrillar Alzheimer’s A β fragment. Biochemistry. 2000;39:13269–13275. doi: 10.1021/bi000637v. [DOI] [PubMed] [Google Scholar]
- 39.Wan W., Stubbs G. Fiber diffraction of the prion-forming domain HET-s(218-289) shows dehydration-induced deformation of a complex amyloid structure. Biochemistry. 2014;53:2366–2370. doi: 10.1021/bi5002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daude N., Ng V., Westaway D. Wild-type Shadoo proteins convert to amyloid-like forms under native conditions. J. Neurochem. 2010;113:92–104. doi: 10.1111/j.1471-4159.2010.06575.x. [DOI] [PubMed] [Google Scholar]
- 41.Tremblay P., Ball H.L., Safar J.G. Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J. Virol. 2004;78:2088–2099. doi: 10.1128/JVI.78.4.2088-2099.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makarava N., Kovacs G.G., Baskakov I.V. Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 2011;7:e1002419. doi: 10.1371/journal.ppat.1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Makarava N., Kovacs G.G., Baskakov I.V. A new mechanism for transmissible prion diseases. J. Neurosci. 2012;32:7345–7355. doi: 10.1523/JNEUROSCI.6351-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wan W., Bian W., Stubbs G. Heterogeneous seeding of a prion structure by a generic amyloid form of the fungal prion-forming domain HET-s(218-289) J. Biol. Chem. 2013;288:29604–29612. doi: 10.1074/jbc.M113.505511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan W., Stubbs G. Heterogeneous seeding of HET-s(218-289) and the mutability of prion structures. Prion. 2014;8:1–5. doi: 10.4161/pri.28126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watts J.C., Giles K., Prusiner S.B. Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc. Natl. Acad. Sci. USA. 2012;109:3498–3503. doi: 10.1073/pnas.1121556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Legname G., Baskakov I.V., Prusiner S.B. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]