

Abstract
In the major peripheral plant light-harvesting complex LHCII, excitation energy is transferred between chlorophylls along an energetic cascade before it is transmitted further into the photosynthetic assembly to be converted into chemical energy. The efficiency of these energy transfer processes involves a complicated interplay of pigment-protein structural reorganization and protein dynamic disorder, and the system must stay robust within the fluctuating protein environment. The final, lowest energy site has been proposed to exist within a trimeric excitonically coupled chlorophyll (Chl) cluster, comprising Chls a610-a611-a612. We studied an LHCII monomer with a site-specific mutation resulting in the loss of Chls a611and a612, and find that this mutant exhibits two predominant overlapping fluorescence bands. From a combination of bulk measurements, single-molecule fluorescence characterization, and modeling, we propose the two fluorescence bands originate from differing conditions of exciton delocalization and localization realized in the mutant. Disruption of the excitonically coupled terminal emitter Chl trimer results in an increased sensitivity of the excited state energy landscape to the disorder induced by the protein conformations. Consequently, the mutant demonstrates a loss of energy transfer efficiency. On the contrary, in the wild-type complex, the strong resonance coupling and correspondingly high degree of excitation delocalization within the Chls a610-a611-a612 cluster dampens the influence of the environment and ensures optimal communication with neighboring pigments. These results indicate that the terminal emitter trimer is thus an essential design principle for maintaining the efficient light-harvesting function of LHCII in the presence of protein disorder.
Introduction
Photosynthesis begins with solar light absorption by a light-harvesting antenna, followed by energy transfer to a reaction center where charge separation occurs (1–5). The antennae pigment-protein complexes play a dual role by both funneling excitation energy to the reaction center and protecting the organism from the deleterious effects of excess sunlight (6–9). The interchanging light-harvesting and photoregulation functionalities proceed in cooperation with structural reorganization and dynamic disorder in the pigment protein complexes.
In higher plants and algae, the major antenna is the light-harvesting complex LHCII, which is trimeric in its native form. The high-resolution x-ray structure of the LHCII trimer has been obtained at 2.5 to 2.72 Å (10,11). Each monomeric unit contains 8 Chls a, 6 Chls b, and 4 xanthophylls (one neoxanthin, two luteins, and one violaxanthin or zeaxanthin, depending on light conditions). Protein structure and the composition of the binding sites play a significant role in establishing the energy levels of the various Chls (12,13). They also determine relative orientations between the Chls, thus defining mutual resonance interaction between their excited states. This interaction leads to formation of a complex excitonic manifold (4,14,15). Upon photoexcitation, energy is transferred between the excitonic states along an energetic cascade. Over the years, structural (10,11,16), biochemical (17–21), and spectroscopic (22–27) studies have been combined with modeling to map the energy transfer processes in LHCII (28–32).
The structural studies indicate that the chlorophylls are arranged in two layers within the membrane, at the stromal and lumenal sides. Site-specific mutagenesis of pigment binding residues and subsequent protein reconstitution have further mapped the chlorophyll binding sites and substructural pigment spectral signatures (18–20). Novoderezhkin et al. (28,29) utilized a combined Redfield-Förster approach to simultaneously fit both the linear spectra and the transient absorption spectral evolution using the published structural information and an experimentally derived spectral density. The fit of the data also allowed estimation of the site energies for the Chls. Each monomeric subunit contains a group of three strongly excitonically coupled clusters at the stromal side, the Chls a602-a603, a610-a611-a612, and b608-b609-b601’, where the b601’ pigment is from an adjacent monomeric unit. On the lumenal side there are two clusters consisting of Chls a613-a614 and a604-b605-b606-b607. (Nomenclature is from Liu et al. (10).)
The lowest energy state of LHCII has been assigned to the Chls a610-a611-a612 cluster (18,19,28,33). In the PSII supercomplexes this terminal emitter domain is in direct contact with the Chls of either the core complex or of the minor antenna CP29, and is therefore directly involved in the energy transfer to the core (34,35). The same cluster of Chls has also been purported to play a role in the switching mechanism between light-harvesting and photoprotection functions (36,37). Femtosecond spectroscopy of quenched LHCII shows excited state absorption features reminiscent of a carotenoid S1→Sn transition, which was associated with Lutein 620 in close proximity of the terminal emitter (38). Fluorescence intermittency in single-molecule spectroscopy experiments (SMS) on single LHCII trimers can also be explained by small and reversible protein conformational changes enabling energy dissipation via the lutein S1 state (39). Observed fluorescence bands at and above 700 nm in single complexes were hypothesized to originate from a different site in the vicinity of Lutein 621 (40,41).
The LHCII-A2 mutant, lacking the ligand for Chl a612, (N183L, named A2 according to previous nomenclature (42)), does not bind the Chls a611 and a612, thus altering the terminal emitter domain. Previous work on this mutant demonstrated loss of absorbance on the red side of the Qy band and a blue-shift of the room temperature fluorescence (18–20). Circular dichroism and modeling studies further concluded that the arrangement of the remaining chlorophylls and the interactions between them in the monomeric LHCII-A2 are unperturbed relative to the LHCII-WT (43).
We report in this article further investigation of fluorescence characteristics of the reconstituted LHCII-A2 monomer. We find that this mutant exhibits two overlapping fluorescence bands, which are in turn temperature dependent in relative ratio. The higher energy (bluer) fluorescence peak corresponds to the previously described room temperature (RT) fluorescence emission. A second emission feature appears at the lower energy (redder) edge of the emission band in low temperature bulk fluorescence experiments and is reported in this study, to our knowledge, for the first time. This feature is furthermore consistent with the disorder dependent appearance of red-shifted fluorescence peaks in SMS experiments (44). We model the steady-state fluorescence properties of an LHCII monomer lacking Chls a611 and a612 using established methods (28,45) and find good agreement with our experimental results. Based on this agreement, we propose that the two fluorescence bands originate from differing protein conformations, which are interchangeable at higher temperatures. These conformations furthermore result in varying excitonic energy level populations. The higher energy emission originates primarily from Chls a602-a603. The previously unobserved fluorescence component is assigned to a low energy and relatively low oscillator strength exciton localized on Chl a610.
Materials and Methods
Samples
LHCII-WT and LHCII-A2 aproproteins were prepared using the Lhcb1.3 gene (AT1G29930) as described in Caffarri et al. (46). The mutant sequence was obtained by site directed mutagenesis replacing the Chl 612 ligand, asparagine 183, with leucine, a residue that cannot coordinate the central magnesium of the Chl. WT and A2 apoproteins were produced by expressing them in Rosetta2DE3. The pigment-protein complexes were reconstituted and purified as described in detail in Natali et al. (47) using purified pigments extracted from plants with a Chl a/b ratio of 3. The pigment composition of the obtained proteins was analyzed by HPLC and fitting of the acetone extract (48).
Characterization
Steady-state absorption was done on a Perkin-Elmer Lambda40 spectrophotometer and steady-state fluorescence was recorded on a Fluorolog-3 spectrofluorometer and the excitation and emission slits widths were 2 nm (HORIBA Jobin-Yvon, Edison, NJ). Temperature dependent measurements were done using an Oxford cryostat (DN1704) and temperature controller. Samples were in 25 mM Hepes buffer (0.06% β-DM, 1 mM MgCl, pH 7.8) with an OD of 0.1 at the Qy absorption band.
Single-molecule spectroscopy
The fluorescence emission of single LHCII monomers was investigated via a home-built confocal microscope as described earlier (44,49). The complexes were excited at 632.8 nm by a CW He-Ne laser (JDS Uniphase, Eindhoven, The Netherlands) with an excitation power of 1 μW corresponding to a diffraction-limited focal irradiance of ∼500 W/cm2. This corresponds to an average absorption rate of one photon per μs per complex. The probability of having two excitations in one complex during one pulse is below 0.01% according to the Poisson distribution of absorbed photons. Therefore, singlet-singlet annihilation effects can be neglected. The high Chl triplet yield of ∼30%, however, leads to the formation of carotenoid triplets with a lifetime of 9 μs and results in a strong singlet-triplet annihilation regime. Assuming a fraction of 70% of the excitations being annihilated with an annihilation time of 35 ps, <5% of the detected photon counts in a fluorescence emission spectrum originate from the annihilation regime.
Near-circular polarized light was obtained by means of a Berek polarization compensator (5540 New Focus, Santa Clara, CA). Thirty consecutive fluorescence spectra, each with an integration time of 1 s, were obtained by dispersing the fluorescence light via a grating (Optometrics LLC, HR830/800nm, Ayer, MA) onto a CCD camera (Roper Scientific, Spec10:100BR, Trenton, NJ). The particles were diluted down to pM concentrations in a 25 mM Hepes buffer (0.03% β-DM, 1 mM MgCl, pH 7.8) and attached to PLL (poly-L-Lysine, Sigma Aldrich) coated microscope cover slips. The immobilized particles were flushed with an oxygen scavenger system containing 750 μg/ml Pyranose Oxidase, 100 μg/ml Catalase, and 7.5 mg/ml Glucose (all from Sigma). The removal of oxygen prevented the formation of singlet oxygen and therefore significantly improved the photostability of complexes. Furthermore, the temperature within the closed sample chamber was controlled and set to 278K, which further prolonged the average survival time of fluorescent particles. The maximum spectral acquisition time was 30 s per complex. All particles with the exception of clearly denatured particles (blue-shifted emission of <670 nm) were fitted with a single-skewed Gaussian in Matlab (The MathWorks, Natick, MA) as described previously (44) and further analyzed in OriginPro. In LHCII-WT 90% of particles could be used for analysis, contrary to 75% in LHCII-A2, which is an indication of the decreased stability of the mutant.
Modeling
The modeling is based on the Hamiltonian and spectral densities used in previously reported work (28,29) with the parameters adjusted to fit simultaneously the spectra of LHCII-WT and LHCII-A2. The fluorescence spectra are calculated from populations Pi of the steady state calculated by the kinetic equations as follows:
where kij are the population transfer rates, Γi are the population relaxation rates, χi(ω) is the absorption spectrum of the i-th exciton, and W(ω) is the spectrum of incident light (50).
These equations can be rigorously derived from the Liouville-von Neumann equation for the reduced density matrix using second-order perturbation theory for the interaction with light, the secular approximation, and the fact that populations vary much slower than optical coherences (time-independent population transfer rates). Under these assumptions these equations have general validity and allow calculation of excitation dynamics.
The excitonic absorption spectrum is given by the transition dipole strength, which can be calculated from the structure and coupling between pigments, and the calculated lineshape. The lineshape and population transfer rates are given by the bath properties, which are expressed by means of the spectral density. The absorption and fluorescence spectra are then calculated as follows (45,51):
Here, |μi0|2 is the transition dipole moment length for the i-th exciton, ωi is the optical transition frequency of the i-th exciton, and is the lineshape function of the i-th exciton (cin are coefficients of the site ↔ exciton basis transformation, and |cin|2 is then the participation of the n-th site in the i-th exciton). The lineshape function of the n-th site is given by , where Cn(t) is the bath correlation function related to the spectral density by Fourier transform (45). We assume the same but uncorrelated bath oscillations on all the different sites here. The reorganization energy of the i-th exciton is given by , where is the reorganization energy of the n-th site and Cn(ω) is the bath spectral density. The dependence of the excitonic reorganization energy on the participation coefficients |cin|2 leads to the difference between reorganization energies assigned to low energy states of the LHCII-WT and LHCII-A2, with increased delocalization leading to smaller reorganization energy.
Based on the crystal structure determined by Liu et al. (10), the excitonic coupling between pigments is calculated in the dipole-dipole approximation. Effective dipole strengths used were 4 D for Chl a and 3.4 D for Chl b (for definition, see Novoderezhkin et al. (52)), and the spectral density was taken from previous work (53). The Chl orientations were taken into account, and the Mg-Mg distances were used for dipole distances. The Chl population lifetime is taken to be 3 ns and the light intensity is, in accordance with the experimental conditions of the ensemble measurements, chosen to be arbitrarily small so as to ensure a nonannihilation regime.
Results
Pigment analysis
The pigment composition of the different samples was measured and is indicated in Table 1. In reconstituted complexes, the Chl a/b ratio depends on the pigment mixture utilized during sample preparation. Therefore, the changes in LHCII-A2 are calculated relative to the LHCII-WT that is reconstituted from the same pigment mixture. Considering that the mutation does not lead to loss of carotenoids, our results suggest the loss of at least two Chls a in LHCII-A2, relative to LHCII-WT, in agreement with previous work (17,18). One of these is assigned to Chl a612, as in the mutant the ligand for this Chl (asparagine) was substituted with an amino acid (leucine) that cannot coordinate the central Mg of the Chl. The second Chl lost is Chl a611, which in the WT interacts strongly with Chl a612 and does not have a protein ligand. For more details on these assignments, see Mozzo et al. and Georgakopolou et al. (17,43). The composition is consistent across different reconstitutions.
Table 1.
Pigment composition of samples as determined by acetone extract fitting
| Chl a/Chl b | Chl/Car | |
|---|---|---|
| LHCII-WT | 1.69 ± 0.05 | 4.73 ± 0.05 |
| LHCII-A2 | 1.31 ± 0.06 | 3.82 ± 0.08 |
Steady-state absorption
The steady-state absorption of LHCII-WT and LHCII-A2 is shown in Fig. 1 and Fig. S1 in the Supporting Material for RT and 77 K conditions, respectively. The calculated difference spectra are also shown. The LHCII-A2 sample differs from the WT most notably in the 470 to 500 nm region and the red edge of the Qy band. The loss of absorption around 680 nm in LHCII-A2 relative to the WT is in accordance with the lack of the Chls a611 and a612 and in agreement with previous results (18). These Chls have site energies red-shifted with respect to the rest of the pigments (28,32,33) and consequently the excitonic states in which they participate absorb in the red edge of the LHCII-WT absorption spectrum (18,20). There is a relative increase at 487 nm in the LHCII-A2 absorption, which is also consistent with previous work. This increase has been attributed to a combination of hypochromism and bathochromic shift in the absorption of the carotenoid Lut 620, because of the loss of excitonic coupling between this pigment and the Chls a611 and a612 (17). Although this region corresponds to the absorption of all of the carotenoids in the complex, the mutation was shown to have a very local effect in the overall pigment arrangement (43). Therefore, the assignment to a Lut 620 absorption shift remains the most likely explanation. The LHCII-A2 relative absorption reduction at 650 nm in the 77K measurements is attributed to the loss of a small amount of Chl b, made clearer because of the band narrowing at low temperature (Fig. S1). The concomitant absorption differences around 472 nm may also be explained by the same reasoning, although since all the pigments have some absorption at this wavelength, it is difficult to assign this change conclusively.
Figure 1.
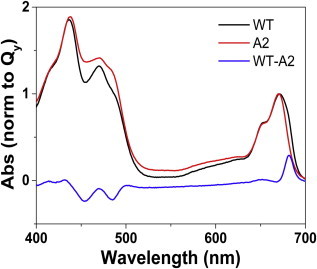
Room temperature steady-state absorption spectra of LHCII-WT, LHCII-A2, and the calculated difference. To see this figure in color, go online.
Steady-state fluorescence
Temperature dependent steady-state emission spectra of LHCII-WT and LHCII-A2 samples of equivalent OD are shown in Figs. S2 and 2, respectively, for λex = 600 nm. The fluorescence emission of the reconstituted LHCII-WT monomer exhibits a peak at 681 nm at 290 K and 679 nm at 77 K, in agreement with literature reports of isolated native complexes (24,54). The emission peak narrows with lowering temperature. LHCII-A2 exhibits a fluorescence peak at 677 nm at 290 K. This 4 nm blue-shift relative to LHCII-WT is in agreement with earlier studies of this mutant at this temperature (18,20). The mutant also shows an increase in intensity of the vibronic band centered at 735 nm, relative to the primary emission. With lowering temperature, the emission of LHCII-A2 broadens, particularly on the redder side of the fluorescence band, differing significantly from the behavior of LHCII-WT. Furthermore, whereas the vibronic band of LHCII-WT narrows with lowering temperature, that of LHCII-A2 does not narrow. The fluorescence quantum yield of LHCII-A2 is quenched with respect to LHCII-WT, and the amount of quenching increases from 30% at RT to 50% at 77 K.
Figure 2.
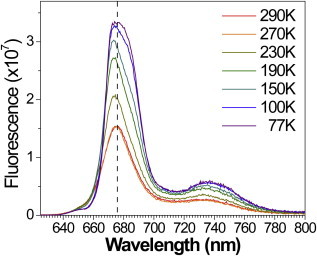
Temperature dependent steady-state emission of LHCII-A2, λex = 600 nm. To see this figure in color, go online.
The fluorescence emission lineshape of LHCII-A2 at 77 K is dependent on excitation wavelength (Fig. 3). Particularly with excitation λex = 475 nm, two emission peaks are observed at 674 and 684 nm. In contrast, LHCII-WT spectra do not show such a change in lineshape under the same conditions (Fig. S3). These steady-state and temperature-dependent measurements suggest that the mutant exhibits two overlapping fluorescence bands in bulk measurements.
Figure 3.
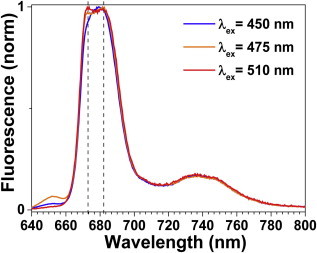
77 K emission spectra of LHCII-A2 at various excitation wavelengths exhibit differing lineshapes. Dashed lines indicate peaks at 674 and 684 nm. To see this figure in color, go online.
Single molecule spectroscopy
SMS of LHCII-A2 and LHCII-WT was conducted to further elucidate the bulk fluorescence characteristics of LHCII-A2. Continuous illumination of surface-attached single monomers at λex = 633 nm allowed measurement of the fluorescence dynamics over tens of seconds. The sample temperature was maintained at 278 K. LHCII-WT particles exhibit a constant narrow (FWHM ≈ 18 nm) emission centered at 681 nm (Fig. S4). The exact peak position varies because of static disorder altering the steady-state exciton composition. These results are consistent with SMS of LHCII-WT trimers isolated from spinach (44). Additionally, ∼3% of the LHCII-WT particles exhibit switching behavior to redder emitting states at and above 700 nm, also in agreement with previous results on isolated LHCII trimers (44). SMS of LHCII-A2 demonstrated lower fluorescence intensity than LHCII-WT, which is consistent with the bulk measurements. The average spectra of all measured single particles furthermore match the bulk steady-state emission spectra for both the LHCII-WT and LHCII-A2 (Fig. S5). The mutant particles exhibited spectral features of four general profiles (Fig. 4). The first category (a) represents particles that give a narrow (FWHM ≈ 18 nm) emission band centered at 678 nm, in agreement with the peak emission of LHCII-A2 in bulk measurements at RT. The second category (b) consists of particles with a broadened spectrum (FWHM up to 27 nm) and a red-shifted peak wavelength up to 684 nm. Accounting for shifts because of temperature, this center wavelength falls between the two emission peaks resolved at 77 K in bulk LHCII-A2 measurements. The third profile (c) is associated with particles that demonstrate switching between profiles (a) and (b). About half of all the measured particles fall into category (a) and the second half of particles with broadened spectra include a fraction of ∼5% of the sampled particles that exhibit the switching feature of category (c). Finally, ∼3% of all particles emit at and above 700 nm, analogous to that seen in the WT sample and depicted in profile (d).
Figure 4.
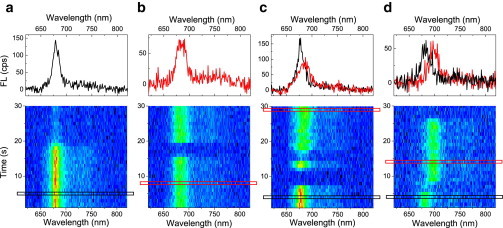
Fluorescence spectral time traces of single LHCII-A2 complexes at 278 K with continuous illumination at λex = 633 nm exhibits four discernible profiles: (a) narrow emission peaking at 678 nm, (b) broad emission at up to 684 nm, (c) switching between the two conditions seen in (a) and (b), and (d) 700 nm emission analogous to that observed in LHCII-WT. Top profiles correspond to the selected 1 s time bins indicated. To see this figure in color, go online.
The measured SMS spectra were each fit with a single-skewed Gaussian after subtracting the vibronic contribution (Fig. S6). The spectral peak width distribution shows the FWHM of these fits versus fluorescence peak position (Fig. 5). This allows us a statistical overview of our data sample. In LHCII-WT, the peak width remains constant, across the emission wavelength. LHCII-A2, however, exhibits narrower emission peaks for bluer peak positions and a trend toward broader peaks at redder wavelengths. There is also no evidence of a narrow emission band from LHCII-A2 at the longer wavelengths. This observation, in addition to the switching behavior observed in profile (c) (Fig. 4), suggests that both of the overlapping emissive bands observed in the bulk LHCII-A2 measurements are originating from single complexes and are thus not due to separate subpopulations of a heterogeneous sample preparation. These two emission bands are furthermore present at all temperatures.
Figure 5.
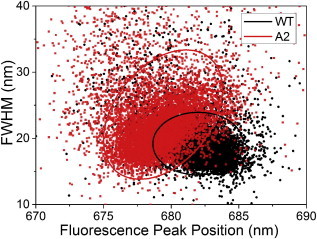
FWHM as derived from single Gaussian fits to SMS spectra versus fluorescence peak position for LHCII-WT (black) and LHCII-A2 (red). Ellipses represent the 68% confidence regions. To see this figure in color, go online.
Discussion
The LHCII-A2 mutant has been previously shown to lose Chls emitting and absorbing near the red edge of the spectrum. These were identified as the Chls a611and a612, members of the strongly excitonically coupled cluster forming the terminal emitter in LHCII. The pigment composition as well as steady-state absorption at RT and 77 K of the LHCII-A2 preparation used in this work match previous reports (17–20,43), and the observed 4 nm blue-shift of the LHCII-A2 fluorescence peak relative to LHCII-WT at RT is also in agreement with earlier studies (18,20). The experiments and simulations presented in this study are fully consistent with the disruption of excitonic states involving Chls a610, a611, and a612 (28,33). The comparison of mutated and WT LHCII systems thus provides us with a framework to study the role of excitonic delocalization in establishing efficient excitation energy transfer pathways within and out of LHCII. The primary goal in this work is to elucidate the nature of the LHCII-A2 monomer emission and in so doing establish the functional role that the excitonic trimer Chl a610-a611-a612 plays within the WT complex.
Our experimental results show that LHCII-A2 exhibits a broader primary fluorescence band than LHCII-WT, an increase in relative intensity of the vibronic band at 735 nm, and a reduced quantum yield of fluorescence. Additionally, at 77 K, the LHCII-A2 emission resolves into a lineshape suggestive of two overlapping fluorescence bands with peaks at 674 and 684 nm. This indicates that the absence of Chls a611 and a612, which has a local effect on the complex structure, has a more drastic effect on the energy distribution in the system.
The samples used in this study were prepared by pigment-protein reconstitution, a well-established biochemical technique for photosynthetic proteins (47,55). The earlier circular dichroism modeling on LHCII-A2 finds that the mutation results in minimal changes to the overall organization of the remaining pigments and consequently of the relevant protein structure (43). Moreover, recent in vivo mutation analysis of minor light-harvesting complexes has shown that reconstitution produces complexes with the same properties as complexes from plants, even in case of mutants (56). However, the reproducibility and homogeneity of samples prepared by this in vitro process should be carefully considered, as it is critical for the reliability of the conclusions we draw from the comparison between LHCII-WT and LHCII-A2. The emission behavior reported in this article was observed in multiple sample preparations, with small differences attributable to slightly differing pigment ratios in the reconstitution. It is thus reasonable to conclude that the observed specific emission features are reproducible and inherent characteristics of the LHCII-A2 monomer.
Bulk measurements provide only averaged information about the energy transfer properties of the studied system. SMS results give further insight into the emissive behavior of individual complexes forming the studied ensemble. The sampling population of type (d) in LHCII-A2 (Fig. 4) matches the red-shifted emission behavior observed in both the LHCII-WT and in isolated LHCII trimers (44). This supports earlier conclusions that this far-red emission is not originating from the terminal emitter trimer. Regarding the emission of LHCII-A2, the sampling population of type (c) demonstrates switching between the narrow blue emission of population type (a) and the broad red emission of population type (b), which indicates that varying fluorescence profiles occur within single particles (Fig. 4). The experimental peak width versus emission wavelength distribution shows no evidence for narrow emission bands from the red edge of the LHCII-A2 fluorescence (Fig. 5). The SMS results thus demonstrate that the overlapping fluorescence bands observed in the bulk measurements of LHCII-A2 are not because of a heterogeneous sample preparation. Varying emission profiles of single light-harvesting complexes have been observed previously in bacterial systems, and were therein attributed to fluctuations in the electron-phonon coupling because of differing protein conformation and concomitantly exciton localization (57). We analogously assign the varying LHCII-A2 emission profiles to a change in the excitonic manifold of the mutant, resulting in altered energy transfer and distribution relative to LHCII-WT. Considering again the temperature dependent bulk measurements, the population of these two bands is furthermore dependent on excitation wavelength at 77 K (Fig. 3). This result, combined with the SMS experiments, suggests either that the system is not fully equilibrated at 77 K, and/or that there is a significant change in absorption cross section of the realizations associated with the two emissive bands.
To explore this further, we have modeled the steady-state spectral properties of LHCII-A2. Previous work reported a model of the excitonic manifold, including site energies and energy transfer pathways, in LHCII trimers and monomers based on a quantitative fit of linear and transient absorption spectra using structural information from crystallography studies (28,29). We have applied this model to this study. The initial set of site energies used was from the work of Novoderezhkin et al. (29). The temperature dependence was incorporated only through the spectral density, i.e., through the lineshapes and energy transfer rates. LHCII-A2 is considered to have the same structure as LHCII-WT, but lacking the Chls a611 and a612, as supported by the previously discussed experimental evidence (43). As we wanted the simplest model that explained our results, we chose not to alter the LHCII-A2 site energies relative to LHCII-WT. This allows for a simultaneous fit of the absorption and fluorescence spectra of both LHCII-WT and LHCII-A2 at RT and 77 K with the same set of parameters. All simulations are averaged over 110 cm−1 FWHM Gaussian disorder on all the site energies, and the fluorescence spectra are calculated for 650 nm illumination. Since the reconstituted samples are in the monomeric state, the Chl b601’ is not associated with the b608-b609 as in the trimer model. Further details on the theory and input parameters are presented in the Materials and Methods section.
Both the LHCII-WT and LHCII-A2 spectra were fitted and, from the diagonalization of the Hamiltonian with the resulting site energies (Table S1), excitonic contributions to the spectra were calculated at 290 and 77 K. These are shown in Fig. 6 for LHCII-A2 and Fig. S7 for LHCII-WT. The steady-state spectral lineshapes of LHCII-A2 and LHCII-WT are both reasonably well reproduced. The origins of the absorption and emission lineshapes can be elucidated by considering the energies of the excitonic states and the participation of the Chls in these states, shown for the lowest six energy levels in Table S2. The differences between the excitonic energy levels of the two samples manifest predominantly in the lowest six levels. This is reasonable as these correspond as well to the excitonic levels with altered participation of Chl a610, the only remaining pigment of the disrupted terminal emitter trimer in LHCII-A2. Fig. 7 illustrates the differences between the lowest energy states of the LHCII-A2 and LHCII-WT excitonic manifolds. In LHCII-WT the primary contribution to the fluorescence, at both RT and 77 K, is from the lowest excitonic state of Chl a610-a611-a612 terminal emitter trimer, which furthermore has a high oscillator strength because of oscillator strength borrowing facilitated by the large exciton delocalization (an effect known as superradiance). In LHCII-A2 we find that the lowest two excitonic levels contribute strongly to the fluorescence. The bluer of these two, denoted A2-02 (see Table S2 for the exciton notation) is composed mostly of Chl a602-a603, and also has high oscillator strength, agreeing with its analog in LHCII-WT, exciton WT-03. The redder emission (A2-01) originates primarily from Chl a610, which is now less coupled to other Chls and consequently demonstrates a much reduced oscillator strength because of the loss of superradiance. From the modeling, the fluorescence quantum yield of LHCII-A2 is reduced 23% at room temperature and 36% at 77 K relative to LHCII-WT, supporting the experimentally observed fluorescence quenching of the mutant. Furthermore, the A2-01 emission is redder than that observed for WT-01. In LHCII-A2, A2-01 is localized on Chl a610, resulting in an increased reorganization energy with respect to the delocalized WT-01 exciton, and subsequently a greater Stokes shift. The modeling also suggests that the broader 735 nm band results from overlapping vibronic contributions from A2-01 and A2-02. Additionally, the localized a610 state exhibits increased electron-phonon coupling as well as larger inhomogeneous broadening. This effect is the straightforward consequence of the change in delocalization as demonstrated by the equations for the reorganization energy of a delocalized excitonic state in the Materials and Methods section. These factors also contribute to the higher relative intensity and broadened lineshape of the 735 nm vibronic band in LHCII-A2 as compared with the analogous ratio in the LHCII-WT spectrum.
Figure 6.
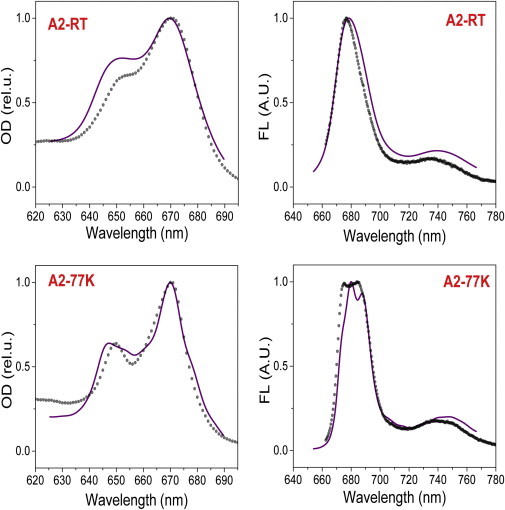
Modeling (solid line) as compared with experimental results (dotted line) of LHCII-A2 absorption and emission at RT and 77K. Fluorescence traces shown are for λex = 650 nm. To see this figure in color, go online.
Figure 7.
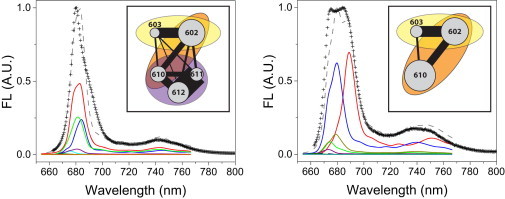
Illustration of the change in the excitonic manifold between (left) LHCII-WT and (right) LHCII-A2. Excitonic contributions to the modeled fluorescence spectra are shown for the lowest energy levels, as well as a depiction of the pigment connectivity. The thickness of the black connecting line demonstrates the tendency of the excitation to be delocalized over these two pigments: , the size of the circle depicts the contribution of that pigment to the emission spectrum: , and the semi-transparent ovals are to distinguish the tendency of the contained pigments to be in an excitonically coupled dimer or trimer. For notation see the section on modeling in the Materials and Methods. To see this figure in color, go online.
We also compared the modeling results with that of the SMS experiments. The timescale of the switching observed in LHCII-A2 is seconds, which means that the protein-pigment conformation responsible for switching process can be regarded static for the duration of the absorption-emission cycle (few ns). The modeling of the SMS experiment is therefore performed with a static disorder in pigment site energies. In this view the observed switching behavior corresponds to changing from one realization of the static disorder to another. Single realizations of the modeled exciton composition of LHCII-A2 reproduce the increasing FWHM versus peak wavelength correlation observed in SMS (Fig. S8). This trend supports the interpretation of static disorder controlling the extent of exciton localization.
Although spectroscopic experiments allow us to measure energy transfer rates between different spectral regions only, providing thus a limited assignment for the spatial energy transfer whenever delocalization takes place, modeling allows us to translate these results into the rates for the populations of excited states of particular Chls. This helps us to illustrate the role of excitonic coupling in the energy transfer efficiency and distribution in LHCII. In particular, modeling results allow us to comment on the biological design principle of the excitonically coupled chlorophyll trimer constituting the terminal emitter of native LHCII. The major light-harvesting antenna, LHCII must be very efficient in transporting solar excitation energy into the core of the PSII supercomplex. This requires fast and robust energy transfer between pigments to the terminal emitter, from where it can be transferred to a neighboring antenna. Our modeling results show that the average transfer rate from Chl a602 to a610, i.e., to the terminal emitter site, slows down by 35% in LHCII-A2, relative to LHCII-WT. Single realizations of the disorder with a highly localized Chl a610 show even slower rates (<10 ps−1). These slower rates result in part from fewer pathways to Chl a610, because of the absence of Chl a611 and a612. Another contributing factor to the slower excitation energy transfer rates in LHCII-A2 is the increased energy gap between the lowest exciton levels. In LHCII-WT, the three lowest exciton levels, all within the Chls a610-a611-a612, are on average ∼50 cm−1 apart. In contrast, in LHCII-A2, the two lowest excitons (A2-01 and A2-02) have an energy gap of 200 cm−1. The terminal emitter trimer in the WT thus ensures good connectivity to neighboring pigments, as well as stabilized and optimized energy levels because of the reduced influence of static disorder, as evidenced by the simulations of single realizations of disorder. The increased sensitivity to the dynamic protein disorder in LHCII-A2 highlights another important design principle in the native complex. In the mutant, the site energy of the decoupled Chl a610 varies because of the disorder, resulting in unfavorable situations wherein excitations populate mainly Chl a602. Our modeling results show that this happens roughly one-third of the time. In the WT, the lowest exciton, and thus final population, is almost always on the Chl a610-a611-a612 trimer. Additionally, in the native complex, the terminal emitter must be able to back transfer excitation energy in the event that a suitable neighboring antenna is not available to accept transfer out of the LHCII. This requires that the Chls a610-a611-a612 cluster is well connected to the Chls a602-a603 cluster. In LHCII-A2, however, for some realizations of the disorder, excitations that populate Chl a610 will become trapped because of the above-mentioned increased energy gap between the lowest exciton levels in the mutant. The stabilized energetic structure of the terminal emitter trimer in the WT is thus essential for the necessary energy migration within and between the monomeric subunits of an LHCII trimer.
In this work we have demonstrated that disrupting the terminal emitter trimer in LHCII-A2 results in emissive features consistent with a loss of energy transfer robustness and the average efficiency because of an increased sensitivity of the excited state energy landscape to the disorder induced by the protein conformations. In light of these results, the existence of the terminal emitter trimer with strong resonance coupling and a correspondingly high degree of excitation delocalization is found to be an essential design principle of LHCII functional structure.
Author Contributions
R.C. and R.v.G. designed the research. C.R., J.M.G., and M.N. performed the experiments. C.R., J.M.G., and T.P.J.K. analyzed the data. V.N., T.M. and P.M. designed the modeling, and P.M. carried out the modeling calculations.
Acknowledgments
C.R., J.M.G., and R.v.G. were supported by the VU University and by an Advanced Investigator grant from the European Research Council (No. 267333, PHOTPROT) to R.v.G. R.v.G. was further supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek, Council of Chemical Sciences (NWO-CW) via a TOP-grant (700.58.305), and by the EU FP7 project PAPETS (GA 323901). R.v.G. gratefully acknowledges his Academy Professor grant from the Netherlands Royal Academy of Sciences (KNAW). M.N. was supported by the Earth and Life Sciences Council of the NWO (NWO-ALW) through a Vici grant to R.C. R.C. is also supported by a Consolidator Investigator grant from the European Research Council (No. 281341 ASAP). T.M. acknowledges financial support from the Czech Science Foundation (GACR, No. 14-25752S) and an NWO visitor grant 040.11.423. V.N. was supported by the NWO visitor grant 040.11.428.
Footnotes
Charusheela Ramanan and J. Michael Gruber contributed equally to this work.
Contributor Information
Charusheela Ramanan, Email: c.ramanan@vu.nl.
Rienk van Grondelle, Email: r.van.grondelle@vu.nl.
Supporting Material
References
- 1.Blankenship R.E. Wiley-Blackwell; Oxford, UK: 2002. Molecular Mechanisms of Photosynthesis. [Google Scholar]
- 2.Fleming G.R., van Grondelle R. The primary steps of photosynthesis. Phys. Today. 1994;47:48–55. [Google Scholar]
- 3.Van Grondelle R., Dekker J.P., Sundstrom V. Energy transfer and trapping in photosynthesis. Biochim. Biophys. Acta Bioenerg. 1994;1187:1–65. [Google Scholar]
- 4.van Grondelle R., Novoderezhkin V.I. Energy transfer in photosynthesis: experimental insights and quantitative models. Phys. Chem. Chem. Phys. 2006;8:793–807. doi: 10.1039/b514032c. [DOI] [PubMed] [Google Scholar]
- 5.Novoderezhkin V.I., van Grondelle R. Physical origins and models of energy transfer in photosynthetic light-harvesting. Phys. Chem. Chem. Phys. 2010;12:7352–7365. doi: 10.1039/c003025b. [DOI] [PubMed] [Google Scholar]
- 6.Niyogi K.K., Truong T.B. Evolution of flexible non-photochemical quenching mechanisms that regulate light harvesting in oxygenic photosynthesis. Curr. Opin. Plant Biol. 2013;16:307–314. doi: 10.1016/j.pbi.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Ruban A.V., Johnson M.P., Duffy C.D.P. The photoprotective molecular switch in the photosystem II antenna. Biochim. Biophys. Acta. 2012;1817:167–181. doi: 10.1016/j.bbabio.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Müller P., Li X.P., Niyogi K.K. Non-photochemical quenching. A response to excess light energy. Plant Physiol. 2001;125:1558–1566. doi: 10.1104/pp.125.4.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horton P., Ruban A.V., Walters R.G. Regulation of light harvesting in green plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996;47:655–684. doi: 10.1146/annurev.arplant.47.1.655. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z., Yan H., Chang W. Crystal structure of spinach major light-harvesting complex at 2.72 A resolution. Nature. 2004;428:287–292. doi: 10.1038/nature02373. [DOI] [PubMed] [Google Scholar]
- 11.Standfuss J., Terwisscha van Scheltinga A.C., Kühlbrandt W. Mechanisms of photoprotection and nonphotochemical quenching in pea light-harvesting complex at 2.5 A resolution. EMBO J. 2005;24:919–928. doi: 10.1038/sj.emboj.7600585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adolphs J., Renger T. How proteins trigger excitation energy transfer in the FMO complex of green sulfur bacteria. Biophys. J. 2006;91:2778–2797. doi: 10.1529/biophysj.105.079483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croce R., van Amerongen H. Natural strategies for photosynthetic light harvesting. Nat. Chem. Biol. 2014;10:492–501. doi: 10.1038/nchembio.1555. [DOI] [PubMed] [Google Scholar]
- 14.Croce R., van Amerongen H. Light-harvesting and structural organization of Photosystem II: from individual complexes to thylakoid membrane. J. Photochem. Photobiol. B Biology. 2011;104:142–153. doi: 10.1016/j.jphotobiol.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 15.Van Amerongen H., van Grondelle R. Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J. Phys. Chem. B. 2001;105:604–617. [Google Scholar]
- 16.Kühlbrandt W. Structure and function of the plant light-harvesting complex, LHC-II. Curr. Opin. Struct. Biol. 1994;4:519–528. [Google Scholar]
- 17.Mozzo M., Passarini F., Croce R. Photoprotection in higher plants: the putative quenching site is conserved in all outer light-harvesting complexes of Photosystem II. Biochim. Biophys. Acta Bioenerg. 2008;1777:1263–1267. doi: 10.1016/j.bbabio.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 18.Remelli R., Varotto C., Bassi R. Chlorophyll binding to monomeric light-harvesting complex. A mutation analysis of chromophore-binding residues. J. Biol. Chem. 1999;274:33510–33521. doi: 10.1074/jbc.274.47.33510. [DOI] [PubMed] [Google Scholar]
- 19.Rogl H., Schödel R., Schubert A. Assignment of spectral substructures to pigment-binding sites in higher plant light-harvesting complex LHC-II. Biochemistry. 2002;41:2281–2287. doi: 10.1021/bi015875k. [DOI] [PubMed] [Google Scholar]
- 20.Rogl H., Kühlbrandt W. Mutant trimers of light-harvesting complex II exhibit altered pigment content and spectroscopic features. Biochemistry. 1999;38:16214–16222. doi: 10.1021/bi990739p. [DOI] [PubMed] [Google Scholar]
- 21.Yang C., Kosemund K., Paulsen H. Exchange of pigment-binding amino acids in light-harvesting chlorophyll a/b protein. Biochemistry. 1999;38:16205–16213. doi: 10.1021/bi990738x. [DOI] [PubMed] [Google Scholar]
- 22.Fuciman M., Enriquez M.M., Frank H.A. Role of xanthophylls in light harvesting in green plants: a spectroscopic investigation of mutant LHCII and Lhcb pigment-protein complexes. J. Phys. Chem. B. 2012;116:3834–3849. doi: 10.1021/jp210042z. [DOI] [PubMed] [Google Scholar]
- 23.Calhoun T.R., Ginsberg N.S., Fleming G.R. Quantum coherence enabled determination of the energy landscape in light-harvesting complex II. J. Phys. Chem. B. 2009;113:16291–16295. doi: 10.1021/jp908300c. [DOI] [PubMed] [Google Scholar]
- 24.Palacios M.A., de Weerd F.L., van Amerongen H. Superradiance and exciton (de)localization in light-harvesting complex II from green plants? J. Phys. Chem. B. 2002;106:5782–5787. [Google Scholar]
- 25.Gradinaru C.C., Özdemir S., van Amerongen H. The flow of excitation energy in LHCII monomers: implications for the structural model of the major plant antenna. Biophys. J. 1998;75:3064–3077. doi: 10.1016/S0006-3495(98)77747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleima F.J., Gradinaru C.C., van Amerongen H. Energy transfer in LHCII monomers at 77K studied by sub-picosecond transient absorption spectroscopy. Biochemistry. 1997;36:15262–15268. doi: 10.1021/bi9716480. [DOI] [PubMed] [Google Scholar]
- 27.Bittner T., Irrgang K.-D., Wasielewski M.R. Ultrafast excitation energy transfer and exciton-exciton annihilation processes in isolated light harvesting complexes of photosystem II (LHC II) from spinach. J. Phys. Chem. 1994;98:11821–11826. [Google Scholar]
- 28.Novoderezhkin V.I., Palacios M.A., van Grondelle R. Excitation dynamics in the LHCII complex of higher plants: modeling based on the 2.72 Angstrom crystal structure. J. Phys. Chem. B. 2005;109:10493–10504. doi: 10.1021/jp044082f. [DOI] [PubMed] [Google Scholar]
- 29.Novoderezhkin V., Marin A., van Grondelle R. Intra- and inter-monomeric transfers in the light harvesting LHCII complex: the Redfield-Förster picture. Phys. Chem. Chem. Phys. 2011;13:17093–17103. doi: 10.1039/c1cp21079c. [DOI] [PubMed] [Google Scholar]
- 30.Renger T., Madjet M.E., Müh F. How the molecular structure determines the flow of excitation energy in plant light-harvesting complex II. J. Plant Physiol. 2011;168:1497–1509. doi: 10.1016/j.jplph.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Müh F., Renger T. Refined structure-based simulation of plant light-harvesting complex II: Linear optical spectra of trimers and aggregates. Biochim. Biophys. Acta Bioenerg. 2012;1817:1446–1460. doi: 10.1016/j.bbabio.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 32.Müh F., Madjet Mel.-A., Renger T. Structure-based identification of energy sinks in plant light-harvesting complex II. J. Phys. Chem. B. 2010;114:13517–13535. doi: 10.1021/jp106323e. [DOI] [PubMed] [Google Scholar]
- 33.Schlau-Cohen G.S., Calhoun T.R., Fleming G.R. Pathways of energy flow in LHCII from two-dimensional electronic spectroscopy. J. Phys. Chem. B. 2009;113:15352–15363. doi: 10.1021/jp9066586. [DOI] [PubMed] [Google Scholar]
- 34.Bennett D.I.G., Amarnath K., Fleming G.R. A structure-based model of energy transfer reveals the principles of light harvesting in photosystem II supercomplexes. J. Am. Chem. Soc. 2013;135:9164–9173. doi: 10.1021/ja403685a. [DOI] [PubMed] [Google Scholar]
- 35.Caffarri S., Broess K., van Amerongen H. Excitation energy transfer and trapping in higher plant Photosystem II complexes with different antenna sizes. Biophys. J. 2011;100:2094–2103. doi: 10.1016/j.bpj.2011.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duffy C.D.P., Chmeliov J., Ruban A.V. Modeling of fluorescence quenching by lutein in the plant light-harvesting complex LHCII. J. Phys. Chem. B. 2013;117:10974–10986. doi: 10.1021/jp3110997. [DOI] [PubMed] [Google Scholar]
- 37.Wentworth M., Ruban A.V., Horton P. Thermodynamic investigation into the mechanism of the chlorophyll fluorescence quenching in isolated photosystem II light-harvesting complexes. J. Biol. Chem. 2003;278:21845–21850. doi: 10.1074/jbc.M302586200. [DOI] [PubMed] [Google Scholar]
- 38.Ruban A.V., Berera R., van Grondelle R. Identification of a mechanism of photoprotective energy dissipation in higher plants. Nature. 2007;450:575–578. doi: 10.1038/nature06262. [DOI] [PubMed] [Google Scholar]
- 39.Valkunas L., Chmeliov J., van Grondelle R. How photosynthetic proteins switch. J. Phys. Chem. Lett. 2012;3:2779–2784. [Google Scholar]
- 40.Krüger T.P.J., Ilioaia C., van Grondelle R. Disentangling the low-energy states of the major light-harvesting complex of plants and their role in photoprotection. Biochim. Biophys. Acta. 2014;1837:1027–1038. doi: 10.1016/j.bbabio.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 41.Krüger T.P.J., Wientjes E., van Grondelle R. Conformational switching explains the intrinsic multifunctionality of plant light-harvesting complexes. Proc. Natl. Acad. Sci. USA. 2011;108:13516–13521. doi: 10.1073/pnas.1105411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kühlbrandt W., Wang D.N., Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 43.Georgakopoulou S., van der Zwan G., Croce R. Understanding the changes in the circular dichroism of light harvesting complex II upon varying its pigment composition and organization. Biochemistry. 2007;46:4745–4754. doi: 10.1021/bi062031y. [DOI] [PubMed] [Google Scholar]
- 44.Krüger T.P.J., Novoderezhkin V.I., van Grondelle R. Fluorescence spectral dynamics of single LHCII trimers. Biophys. J. 2010;98:3093–3101. doi: 10.1016/j.bpj.2010.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valkunas L., Abramavicius D., Mančal T. Wiley-VCH Verlag; Weinheim, Germany: 2013. Molecular Excitation Dynamics and Relaxation. [Google Scholar]
- 46.Caffarri S., Passarini F., Croce R. A specific binding site for neoxanthin in the monomeric antenna proteins CP26 and CP29 of Photosystem II. FEBS Lett. 2007;581:4704–4710. doi: 10.1016/j.febslet.2007.08.066. [DOI] [PubMed] [Google Scholar]
- 47.Natali A., Roy L.M., Croce R. In vitro reconstitution of light-harvesting complexes of plants and green algae. J. Vis. Exp. 2014:e51852. doi: 10.3791/51852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Croce R., Canino G., Bassi R. Chromophore organization in the higher-plant photosystem II antenna protein CP26. Biochemistry. 2002;41:7334–7343. doi: 10.1021/bi0257437. [DOI] [PubMed] [Google Scholar]
- 49.Rutkauskas D., Novoderezkhin V., van Grondelle R. Fluorescence spectral fluctuations of single LH2 complexes from Rhodopseudomonas acidophila strain 10050. Biochemistry. 2004;43:4431–4438. doi: 10.1021/bi0497648. [DOI] [PubMed] [Google Scholar]
- 50.Chenu A., Malý P., Mančal T. Dynamic coherence in excitonic molecular complexes under various excitation conditions. Chem. Phys. 2014;439:100–110. [Google Scholar]
- 51.Renger T., Marcus R.A. On the relation of protein dynamics and exciton relaxation in pigment–protein complexes: an estimation of the spectral density and a theory for the calculation of optical spectra. J. Chem. Phys. 2002;116:9997–10019. [Google Scholar]
- 52.Novoderezhkin V., Salverda J.M., van Grondelle R. Exciton modeling of energy-transfer dynamics in the LHCII complex of higher plants: a Redfield theory approach. J. Phys. Chem. B. 2003;107:1893–1912. [Google Scholar]
- 53.Novoderezhkin V.I., Palacios M.A., van Grondelle R. Energy-transfer dynamics in the LHCII complex of higher plants: modified Redfield approach. J. Phys. Chem. B. 2004;108:10363–10375. doi: 10.1021/jp044082f. [DOI] [PubMed] [Google Scholar]
- 54.Peterman E.J.G., Hobe S., van Amerongen H. Low-temperature spectroscopy of monomeric and trimeric forms of reconstituted light-harvesting chlorophyll ab complex. Biochim. Biophys. Acta Bioenerg. 1996;1273:171–174. [Google Scholar]
- 55.Plumley F.G., Schmidt G.W. Reconstitution of chlorophyll a/b light-harvesting complexes: Xanthophyll-dependent assembly and energy transfer. Proc. Natl. Acad. Sci. USA. 1987;84:146–150. doi: 10.1073/pnas.84.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Passarini F., Xu P., Croce R. Towards in vivo mutation analysis: knock-out of specific chlorophylls bound to the light-harvesting complexes of Arabidopsis thaliana—the case of CP24 (Lhcb6) Biochim. Biophys. Acta. 2014;1837:1500–1506. doi: 10.1016/j.bbabio.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 57.Kunz R., Timpmann K., Köhler J. Fluctuations in the electron-phonon coupling of a single chromoprotein. Angew. Chem. Int. Ed. Engl. 2013;52:8726–8730. doi: 10.1002/anie.201303231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.