

Abstract
Verrucous venous malformation (VVM), also called “verrucous hemangioma,” is a non-hereditary, congenital, vascular anomaly comprised of aberrant clusters of malformed dermal venule-like channels underlying hyperkeratotic skin. We tested the hypothesis that VVM lesions arise as a consequence of a somatic mutation. We performed whole-exome sequencing (WES) on VVM tissue from six unrelated individuals and looked for somatic mutations affecting the same gene in specimens from multiple persons. We observed mosaicism for a missense mutation (NM_002401.3, c.1323C>G; NP_002392, p.Iso441Met) in mitogen-activated protein kinase kinase kinase 3 (MAP3K3) in three of six individuals. We confirmed the presence of this mutation via droplet digital PCR (ddPCR) in the three subjects and found the mutation in three additional specimens from another four participants. Mutant allele frequencies ranged from 6% to 19% in affected tissue. We did not observe this mutant allele in unaffected tissue or in affected tissue from individuals with other types of vascular anomalies. Studies using global and conditional Map3k3 knockout mice have previously implicated MAP3K3 in vascular development. MAP3K3 dysfunction probably causes VVM in humans.
Main Text
Verrucous venous malformation (VVM) is a non-hereditary vascular anomaly that is most commonly referred to as verrucous “hemangioma” in the literature.1–5 VVMs occur equally in males and females. The typical VVM lesion is raised, reddish-purple, hyperkeratotic, and extends into the subcutis. VVMs can be single or multiple (Figure 1). 91% of lesions occur in an extremity (usually legs) and 9% involve the trunk.4 Other developmental anomalies have not been reported in persons with VVM. Over time, a VVM lesion might slowly enlarge, become increasingly hyperkeratotic, and exhibit more bleeding episodes. Histologically, affected tissue contains clusters of venule-like channels that immunostain positive for GLUT1 (Figure 2).4 VVMs might represent another example of a genetic lethal mutation that survives as a consequence of somatic mosaicism.6 Therefore, we looked for somatic mutations in VVM lesions via whole-exome sequencing (WES).
Figure 1.
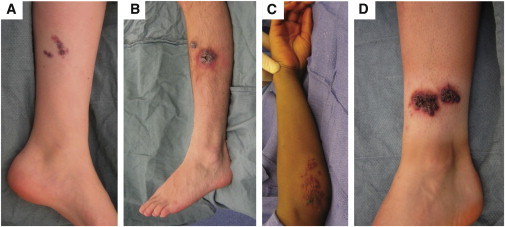
Photographs of Lower and Upper Extremities of Four Participants whose Verrucous Venous Malformations Contain a Somatic Mosaic MAP3K3 Missense Mutation
Shown are participant 4, age 13 years (A); participant 6, age 19 years (B); participant 8, age 9 years (C); and participant 9, age 16 years (D).
Figure 2.
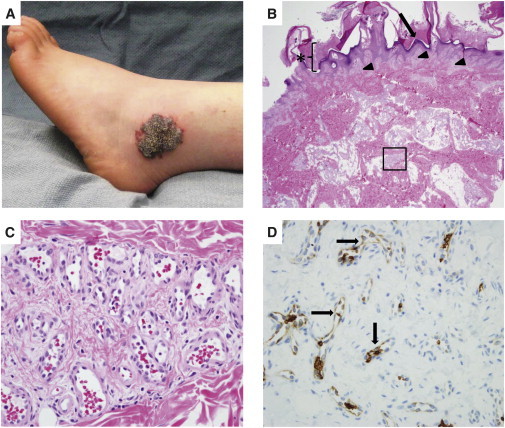
Photograph and Photomicrographs of the Verrucous Venous Malformation from Participant 5
(A) VVM lesion that contains the somatic mosaic MAP3K3 missense mutation.
(B) Hematoxylin-and-eosin-stained section of the excised VVM. Note hyperkeratosis (asterisk), papillomatous epidermis (arrow), and vascular clusters (arrowheads and boxed area) within the papillary dermis and reticular dermis, respectively, that extend into subcutaneous fat.
(C) Higher-magnification photomicrograph of the boxed area in (B) showing a cluster of venule-like channels.
(D) Immunostaining revealing GLUT1 expression in many endothelial cells within the VVM lesion (arrows).
The Committee on Clinical Investigation at Boston Children’s Hospital approved this study, and informed consent was obtained. VVM tissue specimens were collected from individuals undergoing a clinically indicated surgical resection. VVM was diagnosed based on history, physical examination, and histopathology. Genomic DNA was extracted from frozen specimens via the QIAamp DNA Mini kit (QIAGEN). Participant-specific indexed DNA libraries were made as previously described7 and enriched for exonic sequences via the SureSelect Human All Exon V5 kit (Agilent Technologies). We generated ≥50 million 100-basepair paired-end reads for each DNA library by using the Illumina HiSeq 2500 platform. WES data analysis was performed as previously described,8 with additional processing steps done as recommended by GATK: Best Practices.9 Sequence reads were de-barcoded with Novobarcode (Novocraft Technologies) and aligned to the reference genome (UCSC hg19) by BWA.10 PCR duplicates were removed for each WES dataset via Picard MarkDuplicates. Reads around known insertions and deletions in the genome were re-aligned with GATK software.9,11 Comprehensive variant lists for each sample were populated with VarScan2 software.12 Variants were annotated with ANNOVAR software.13
For the six samples, we averaged 108×–299× coverage across the exome; we obtained ≥30× coverage and ≥60× coverage for 80%–95% and 52%–88% of the exome, respectively (Figure 3). We began filtering the data via several criteria (Table 1). First, we considered only variants that were present in ≥3 independent reads at loci having ≥30× coverage. We did this to reduce false-positive results that can be caused by library preparation, sequencing error, or mismapping. Second, we removed synonymous variants and variants outside of coding regions and splice sites. Third, we removed variants that were present in dbSNP (build 138),14 the Exome Variant Server (ESP6500), and/or the 1000 Genomes Project15 datasets, because they probably represent heritable alleles. Fourth, we further reduced the number of variants under consideration by requiring ≥5 variant reads and by removing variants that exhibited complete strand bias (i.e., only sequence in one direction produced a variant read). Fifth, we excluded variants found in >40% of the reads assuming these too were probably inherited alleles. Sixth, we focused on specific variants or on genes that contained a different variant in ≥2 and then ≥3 specimens. A total of 14 candidates genes, EPPK1 (MIM 607553), IGFN1, KRTAP1-1 (MIM 608819), MAP3K3 (MIM 602539), MUC2 (MIM 158370), MUC6 (MIM 158374), PHGR1, PRSS3 (MIM 613578), RHPN2, RP1L1 (MIM 608581), RPTN (MIM 613259), SPATA3, TMEM163, and ZNF141 (MIM 194648), remained after filtering (Table 1).
Figure 3.
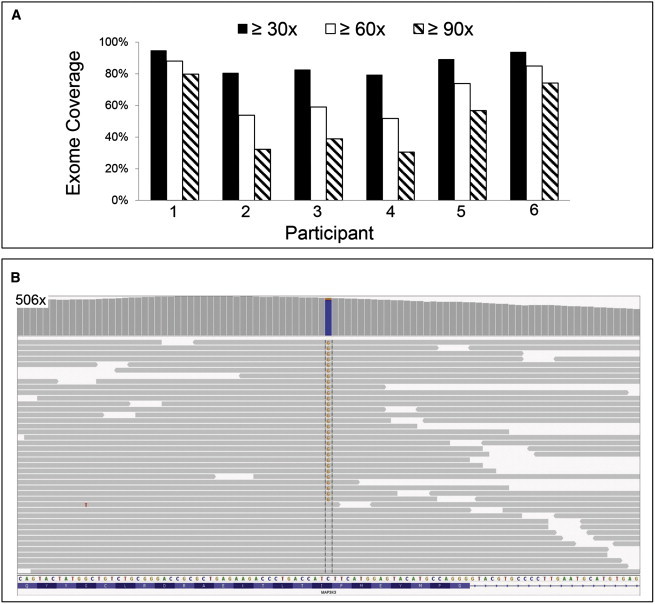
Whole-Exome Sequencing of VVM DNA and Identification of a MAP3K3 Somatic Missense Mutation
(A) Bar graph indicating fold coverages across the exome for the VVM specimens subjected to WES. Specimens from participants 1 and 6 yielded ≥90× coverage for ∼80% of the exome.
(B) Portion of an Integrative Genomic Viewer screen shot depicting WES coverage at the site of the somatic missense mutation in participant 6. The bar graph indicates the depth of coverage in the interval, which peaked at 506×. Coloring at the site of the mutation indicates the relative proportions of reference (blue) and variant (orange) alleles in the sample. Examples of individual sequencing reads (horizontal gray bars) containing the variant and reference allele are depicted below. Note that the 29 sequencing reads that contain the variant (G) allele are in both directions and have no other variant residues indicative of poor sequence quality or mismapping.
Table 1.
Variant Filtering for Six VVMs Subjected to Whole-Exome Sequencing
|
Participant Specimen |
||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Minimum of 30× coverage and ≥3 variant reads | 106,199 | 51,833 | 60,642 | 50,903 | 68,503 | 86,968 |
| Considering only nonsynonymous variants | 13,222 | 9,986 | 11,148 | 10,092 | 11,823 | 10,932 |
| After excluding common variants (SNVs/indels) | 1,600 (1,183/417) | 1,846 (1,576/270) | 2,597 (2,287/310) | 1,911 (1,609/302) | 2,609 (2,251/358) | 1,540 (1,202/338) |
| After excluding SNVs with ≤5 variant reads | 775 | 238 | 410 | 231 | 566 | 701 |
| After excluding SNVs exhibiting strand bias | 194 | 96 | 131 | 83 | 142 | 119 |
| After excluding SNVs with allele frequencies ≥40% | 87 | 45 | 74 | 39 | 78 | 55 |
| Genes having variants in ≥2 specimens | 30 | |||||
| Genes having variants in ≥3 specimens | 14: EPPK1, IGFN1, KRTAP1-1, MAP3K3, MUC2, MUC6, PHGR1, PRSS3, RHPN2, RP1L1, RPTN, SPATA3, TMEM163, ZNF141 | |||||
Abbreviations are as follows: indel, insertions and deletions; SNV, single-nucleotide variant.
We visually examined and compared variants in each of the 14 candidates with other WES datasets by using Integrative Genomics Viewer.16 Visual examination enabled us to exclude 13 of 14 genes as representing sequencing or mapping artifact, because these variants were also seen in WES data from non-VVM samples. In contrast, we observed a MAP3K3 missense variant (RefSeq accession number NM_002401.3, c.1323C>G; RefSeq NP_002392, p.Iso441Met) in three of six VVM samples and in no other sample (Figure 3). This finding, coupled with published data implicating mouse Map3k3 in endothelial cell differentiation,17,18 led us to explore this mutation in greater depth.
We validated the presence of the mutant MAP3K3 allele by PCR amplifying the implicated sequence from newly extracted affected tissue DNA from one participant who had the variant allele in 29/477 WES reads (∼6% mutant allele frequency). Sanger sequencing of 48 different PCR-amplimer-subclones detected the mutant allele in three clones (∼6% mutant allele frequency). To screen for this mutation more efficiently, we developed a droplet digital PCR (ddPCR) assay19 with forward (5′-TGCAGTACTATGGCTGTCTG-3′) and reverse (5′-GTCTCACATGCATTCAAGG-3′) primers and with fluorescent reference (5′-HEX-CCTGACCATcTTCATGGAGTACA-IBlk-3′) and mutant allele (5′-FAM-CCTGACCATgTTCATGGAGTACA-IBlk-3′) probes (Integrated DNA Technologies). Approximately 30 ng of template DNA were used in each reaction. To validate the sensitivity of our ddPCR assay, we serially diluted genomic DNA that contained the mutant allele with wild-type genomic DNA. The ddPCR detected a mutant allele frequency as low as 1 mutant per 1,693 total alleles (data not shown). We also determined whether ddPCR yielded the same mutant allele frequencies as WES by using the WES libraries as template in the ddPCR assay (data not shown); there was no statistically significant difference in allele frequencies determined by WES and ddPCR (Table 2 and data not shown).
Table 2.
MAP3K3 Mutation Detection in Ten Participants Affected with VVM
| Participant | Age | Sex | Location | DNA Source | Whole-Exome Sequencinga | ddPCRb |
|---|---|---|---|---|---|---|
| 1 | 9 months | M | leg | frozen | 0/539 (0%) | 0/3,973 (0%) |
| 2 | 1 year | F | arm | frozen | 0/176 (0%) | 0/1,298 (0%) |
| 3 | 3 years | M | trunk | frozen | 0/228 (0%) | 0/3,826 (0%) |
| 4 | 13 years | F | leg | frozen | 13/167 (7.8%)c | 347/2,627 (10.7%) |
| saliva | – | 0/3,394 (0%) | ||||
| 5 | 18 years | F | leg | frozen | 13/253 (5.1%)c | 239/3,675 (5.5%) |
| saliva | – | 0/3,241 (0%) | ||||
| 6 | 19 years | M | leg | frozen | 29/477 (6.1%)c | 431/3,834 (9.1%) |
| 7 | 2 years | M | arm | FFPE | – | 248/1,228 (16.3%) |
| 8 | 9 years | F | arm | FFPE | – | 247/1,016 (19.3%) |
| saliva | – | 0/2,428 (0%) | ||||
| 9 | 16 years | F | leg | FFPE | – | 207/2,946 (6.1%) |
| saliva | – | 0/4,261 (0%) | ||||
| 10 | 23 years | F | leg | FFPE | – | 0/2,053 (0%) |
Mutant reads/total reads (mutant allele percentage). – indicates test not performed.
Mutant droplets/wild-type droplets (mutant allele percentage calculation considers droplets containing mutant, mutant and wild-type, and wild-type amplimers).
The mutant allele frequency determined by ddPCR using the same template DNA is not statistically different from the frequency determined by WES.
In addition to studying DNA from the first six VVM samples, we analyzed four additional VVM DNA samples from other participants by ddPCR (one frozen tissue, three formalin-fixed paraffin-embedded tissues). We also studied DNA that had been extracted from the skin of healthy individuals (n = 3) and from affected tissues from individuals with other vascular anomalies (infantile hemangioma [n = 3], capillary malformation [n = 3], venous malformation [n = 3], lymphatic malformation [n = 3], arteriovenous malformation [n = 3]). With the ddPCR assay, we confirmed that the MAP3K3 mutation was present in the three VVM specimens where it had been initially found by WES and in three of four new VVM specimens. The mutant allele frequencies in the six VVM tissues in which the mutation was detected ranged from 6% to 19% (Table 2 and Figure 4). We did not detect the mutant allele in healthy skin or affected tissue from individuals with other vascular anomalies (data not shown). Also, we did not detect the mutant allele in saliva DNA from four VVM participants who had the mutant allele in their affected tissue (Table 2).
Figure 4.
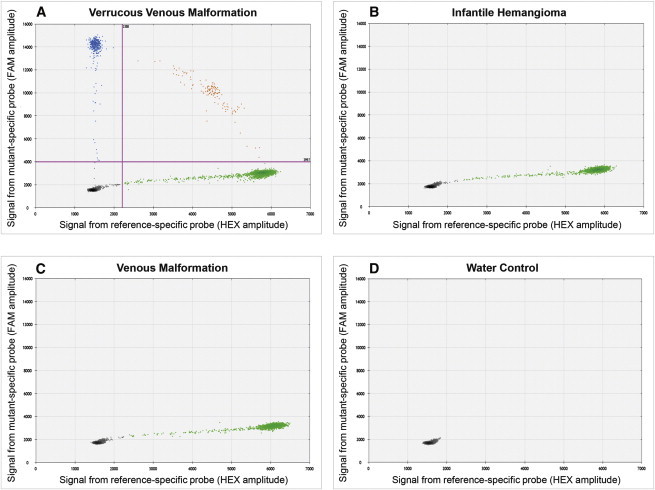
Representative Results for the MAP3K3 c.1323C>G Mutation ddPCR Assay
(A) Graph depicting the amplitudes of FAM fluorescence droplets (blue dots) containing mutant alleles (y axis) and HEX fluorescence droplets (green dots) containing wild-type alleles (x axis) from a ddPCR reaction performed with VVM DNA as template. Cross-hairs indicate the fluorescence cutoffs that we used to call a droplet positive for a wild-type or mutant allele. Note the presence of mutant allele containing droplets in this sample. Some droplets contain mutant and wild-type amplimers (orange dots). The numbers of empty, mutant, mutant and wild-type, and wild-type amplimer containing droplets in this reaction are 12,987, 406, 129, and 4,323, respectively.
(B and C) Similar graphs for ddPCR reactions performed with DNA from infantile hemangioma (B) and venous malformation (C) specimens as template. Note the absence of mutant amplimer containing droplets in each of these two reactions.
(D) Results from a ddPCR reaction performed with no template DNA. All droplets are negative for wild-type and mutant DNA amplimer.
With evidence that a specific somatic MAP3K3 missense variant (c.1323C>G [p.Iso441Met]) is present in six of ten VVMs, we reviewed MAP3K3 WES data in the three specimens that did not have this mutation. The average WES coverage across MAP3K3 in these specimens ranged from 68× to 187×, and in one VVM sample the entire MAP3K3 coding sequence was covered at ≥100× coverage. We did not find a suspicious MAP3K3 mutation (i.e., ≥3 variant reads) in any of these three specimens. It remains possible that the specimens have mutant MAP3K3 alleles, but at frequencies too low to detect. Alternatively, VVM might also be associated with somatic mutation affecting another gene. Although all ten specimens were typical of VVM, there exists similarity between VVM and other vascular malformations, including angiokeratoma, capillary-lymphatic malformation, cutaneous lesions associated with cerebral cavernous malformations (CCM [MIM 116860]), and glomuvenous malformation (GVM [MIM 138000]). Therefore, we examined WES data, but did not find evidence for mutation in PIK3CA (MIM 171834), TIE2 (TEK [MIM 600221]), GLMN (MIM 601749), KRIT1 (MIM 604214), CCM2 (MIM 607929), or PDCD10 (MIM 609118), which are the genes responsible for these other disorders.20,21
Our data strongly support a somatic missense mutation in MAP3K3 as a cause of VVM. The isoleucine residue (Iso441) is fully conserved across evolution. MAP3K3 is one of 21 members of the MAP kinase kinase kinase (MAP3K) family of serine/threonine phosphorylases.22 Studies on mice with global and conditional knockout alleles of Map3k3 have previously implicated this gene in vascular development. Mice lacking MAP3K3 die at embryonic day 10.5 because of abnormalities in extra-embryonic vasculature of the yolk sac, the embryonic vasculature, and the heart.18 Conditional ablation of Map3k3 placed it downstream of the angiopoietin-1 (ANG1) and tyrosine kinase with immunoglobulin and EFG-like domains-2 (TIE2) signaling pathway.17 TIE2 is a known vascular malformation locus, with gain-of-function mutations segregating in families with dominant venous malformation phenotypes23 and somatic gain-of-function mutations detected in sporadically occurring venous malformations.24 Because mice lacking one copy of Map3k3 do not develop vascular malformations and mice lacking two copies fail to form vasculature, it seems likely that the somatic mutation we identified in MAP3K3 creates a neomorphic or hypermorphic function.
Imperial and Helwig1 suggested that VVM was a “vascular malformation,” but instead labeled the lesion “verrucous hemangioma” probably because, at that time, most vascular anomalies were called hemangioma.25,26 We have preferred the term verrucous venous malformation for this lesion because it most closely resembles a hyperkeratotic venous malformation based on its clinical, radiologic, and pathologic features.27,28 Our finding that VVM contains a MAP3K3 mutation supports our impression that this lesion is a venous anomaly. No somatic mutation affecting Iso441 has been reported in COSMIC, the catalog of somatic mutation in cancer,29 which is also consistent with VVM being a malformation rather than a tumor. Identifying molecular etiologies for VVM should facilitate the search for better therapies.
Acknowledgments
The authors thank their participants for contributing in this study. This work was supported by NIH grants R01-AR064231 (to M.L.W.) and R21-HD081004 (to A.K.G.).
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
COSMIC, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/
dbSNP, build 138, http://www.ncbi.nlm.nih.gov/projects/SNP/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
Picard, http://picard.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Imperial R., Helwig E.B. Verrucous hemangioma. A clinicopathologic study of 21 cases. Arch. Dermatol. 1967;96:247–253. doi: 10.1001/archderm.96.3.247. [DOI] [PubMed] [Google Scholar]
- 2.Chan J.K.C., Tsang W.Y.W., Calonje E., Fletcher C.D.M. Verroucous hemangioma: A distinct but neglected variant of cutaneous hemangioma. Int. J. Surg. Pathol. 1995;2:171–176. [Google Scholar]
- 3.Mankani M.H., Dufresne C.R. Verrucous malformations: their presentation and management. Ann. Plast. Surg. 2000;45:31–36. doi: 10.1097/00000637-200045010-00006. [DOI] [PubMed] [Google Scholar]
- 4.Tennant L.B., Mulliken J.B., Perez-Atayde A.R., Kozakewich H.P. Verrucous hemangioma revisited. Pediatr. Dermatol. 2006;23:208–215. doi: 10.1111/j.1525-1470.2006.00219.x. [DOI] [PubMed] [Google Scholar]
- 5.Wang L., Gao T., Wang G. Verrucous hemangioma: a clinicopathological and immunohistochemical analysis of 74 cases. J. Cutan. Pathol. 2014;41:823–830. doi: 10.1111/cup.12385. [DOI] [PubMed] [Google Scholar]
- 6.Happle R. Cutaneous manifestation of lethal genes. Hum. Genet. 1986;72:280. doi: 10.1007/BF00291899. [DOI] [PubMed] [Google Scholar]
- 7.Bowen M.E., Boyden E.D., Holm I.A., Campos-Xavier B., Bonafé L., Superti-Furga A., Ikegawa S., Cormier-Daire V., Bovée J.V., Pansuriya T.C. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet. 2011;7:e1002050. doi: 10.1371/journal.pgen.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyden E.D., Campos-Xavier A.B., Kalamajski S., Cameron T.L., Suarez P., Tanackovic G., Andria G., Ballhausen D., Briggs M.D., Hartley C. Recurrent dominant mutations affecting two adjacent residues in the motor domain of the monomeric kinesin KIF22 result in skeletal dysplasia and joint laxity. Am. J. Hum. Genet. 2011;89:767–772. doi: 10.1016/j.ajhg.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. From FastQ data to high-confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;11:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koboldt D.C., Chen K., Wylie T., Larson D.E., McLellan M.D., Mardis E.R., Weinstock G.M., Wilson R.K., Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng Y., Yang J., McCarty M., Su B. MEKK3 is required for endothelium function but is not essential for tumor growth and angiogenesis. Am. J. Physiol. Cell Physiol. 2007;293:C1404–C1411. doi: 10.1152/ajpcell.00058.2007. [DOI] [PubMed] [Google Scholar]
- 18.Yang J., Boerm M., McCarty M., Bucana C., Fidler I.J., Zhuang Y., Su B. Mekk3 is essential for early embryonic cardiovascular development. Nat. Genet. 2000;24:309–313. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 19.Hindson B.J., Ness K.D., Masquelier D.A., Belgrader P., Heredia N.J., Makarewicz A.J., Bright I.J., Lucero M.Y., Hiddessen A.L., Legler T.C. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blatt J., Powell C.M., Burkhart C.N., Stavas J., Aylsworth A.S. Genetics of hemangiomas, vascular malformations, and primary lymphedema. J. Pediatr. Hematol. Oncol. 2014;36:587–593. doi: 10.1097/MPH.0000000000000260. [DOI] [PubMed] [Google Scholar]
- 21.Kurek K.C., Luks V.L., Ayturk U.M., Alomari A.I., Fishman S.J., Spencer S.A., Mulliken J.B., Bowen M.E., Yamamoto G.L., Kozakewich H.P., Warman M.L. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am. J. Hum. Genet. 2012;90:1108–1115. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Craig E.A., Stevens M.V., Vaillancourt R.R., Camenisch T.D. MAP3Ks as central regulators of cell fate during development. Dev. Dyn. 2008;237:3102–3114. doi: 10.1002/dvdy.21750. [DOI] [PubMed] [Google Scholar]
- 23.Vikkula M., Boon L.M., Carraway K.L., 3rd, Calvert J.T., Diamonti A.J., Goumnerov B., Pasyk K.A., Marchuk D.A., Warman M.L., Cantley L.C. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 24.Limaye N., Wouters V., Uebelhoer M., Tuominen M., Wirkkala R., Mulliken J.B., Eklund L., Boon L.M., Vikkula M. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat. Genet. 2009;41:118–124. doi: 10.1038/ng.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulliken J.B., Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast. Reconstr. Surg. 1982;69:412–422. doi: 10.1097/00006534-198203000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Mulliken J.B. Oxford University Press; Oxford: 2013. Classification of Vascular Anomalies. [Google Scholar]
- 27.Greene A.K., Mulliken J.B. Elsevier Saunders; London: 2013. Vascular Anomalies. [Google Scholar]
- 28.Greene A.K. Quality Medical Publishing; St. Louis: 2013. Venous Malformation. [Google Scholar]
- 29.Forbes S.A., Bindal N., Bamford S., Cole C., Kok C.Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]