

Abstract
Many cellular processes are sensitive to levels of cholesterol in specific membranes and show a strongly sigmoidal dependence on membrane composition. The sigmoidal responses of the cholesterol sensors involved in these processes could arise from several mechanisms, including positive cooperativity (protein effects) and limited cholesterol accessibility (membrane effects). Here, we describe a sigmoidal response that arises primarily from membrane effects due to sharp changes in the chemical activity of cholesterol. Our models for eukaryotic membrane-bound cholesterol sensors are soluble bacterial toxins that show an identical switch-like specificity for endoplasmic reticulum membrane cholesterol. We show that truncated versions of these toxins fail to form oligomers but still show sigmoidal binding to cholesterol-containing membranes. The nonlinear response emerges because interactions between bilayer lipids control cholesterol accessibility to toxins in a threshold-like fashion. Around these thresholds, the affinity of toxins for membrane cholesterol varies by >100-fold, generating highly cooperative lipid-dependent responses independently of protein-protein interactions. Such lipid-driven cooperativity may control the sensitivity of many cholesterol-dependent processes.
Introduction
Cholesterol-sensing proteins respond to small changes in the concentration of cholesterol in mammalian cell membranes with a sharp, switch-like sensitivity (1–3). For example, a small increase in endoplasmic reticulum (ER) membrane cholesterol from 5 mol % to 8 mol % of total ER lipids triggers an all-or-none response from Scap, a cholesterol-sensing oligomeric membrane protein that controls the activation of sterol-regulatory element binding proteins (SREBPs), which are transcription factors that stimulate lipid synthesis and uptake (1,4). Another example of such a sensor is perfringolysin O (PFO), a soluble bacterial toxin that specifically binds to cholesterol-containing membranes and forms large oligomeric pores (5). Binding of PFO to purified ER membranes occurs only after the concentration of cholesterol exceeds a threshold of 5 mol %, precisely the same concentration at which Scap is activated (2). Binding of PFO to purified plasma membranes also shows a threshold response, except that the threshold cholesterol concentration is shifted to 35 mol % (3). Binding of PFO to much simpler model membranes composed of just two components, cholesterol and a phospholipid, also occurs only after the cholesterol concentration exceeds a threshold ranging from 20 mol % to 50 mol % depending on the phospholipid headgroup and acyl chain structure (2,6,7).
The molecular basis for these thresholds remains poorly understood. It is not known whether such highly sigmoidal responses arise due to allosteric changes in the binding of cholesterol to Scap or PFO oligomers, or due to properties of the membrane that affect the chemical activity of cholesterol and thus its accessibility to Scap or PFO. Determining the relative contribution of either mechanism is crucial for understanding the sensitivity of cholesterol sensors and guiding their use as probes for cholesterol in the membranes of living cells.
Scap is a polytopic membrane protein, and studying its interaction with membrane cholesterol is technically challenging (8). Unlike Scap, PFO is a soluble protein that does not require detergents for stability and can be easily produced in large quantities. Moreover, there are two remarkable similarities in how Scap and PFO detect membrane cholesterol. The first similarity is their common threshold sensitivity for ER membrane cholesterol. Both Scap and PFO bind to ER cholesterol only after the cholesterol concentration exceeds a threshold of 5 mol % of total lipids (1,2). The second similarity is their identical sterol structural specificity. Both Scap and PFO bind to cholesterol, dihydrocholesterol, desmosterol, and β-sitosterol, but not to epicholesterol, lanosterol, 19-hydroxy-cholesterol, or 25-hydroxy-cholesterol (2,9). Their common ability to distinguish between cholesterol and epicholesterol, a diastereomer that differs only in the orientation of the sterol 3-hydroxyl group, is especially striking. PFO is thus a convenient model for investigating the sensitivity of cholesterol sensors for membrane cholesterol.
PFO is the best-studied member of a large family of cholesterol-dependent cytolysins (CDCs) that are produced by more than 25 bacterial species and share a high degree (>45%) of sequence similarity (5,10). To date, high-resolution crystal structures of the soluble forms of six members of this family—PFO (11), intermedilysin (12), anthrolysin O (ALO) (13), suilysin (14), listeriolysin O (15), and streptolysin O (16)—have been solved. There are no high-resolution crystal structures of the cholesterol-bound or oligomeric forms of any member of the CDC family. Nevertheless, extensive biophysical studies of PFO (6,17–19), combined with a cryo-electron microscopy study of oligomers of another CDC, pneumolysin (20), have revealed many details of CDC pore formation, as illustrated in the schematic diagram in Fig. 1 A. CDCs are elongated proteins that can be divided into four domains (11). The carboxy-terminal domain, referred to as domain 4 (D4) and shaded yellow in Fig. 1 A, is necessary and sufficient for binding to membrane cholesterol (17). This initial binding event is followed by oligomerization into a circular prepore complex. Dramatic restructuring involving all four domains eventually leads to the formation of a transmembrane β-barrel pore comprised of 35–50 monomers with an inner diameter of 250–300 Å. Unfortunately, the elegant studies cited above do not explain PFO’s threshold sensitivity for membrane cholesterol. To date, no studies have decoupled the roles of PFO oligomerization from those of membrane effects in determining PFO’s sharp sigmoidal binding to cholesterol-containing membranes.
Figure 1.
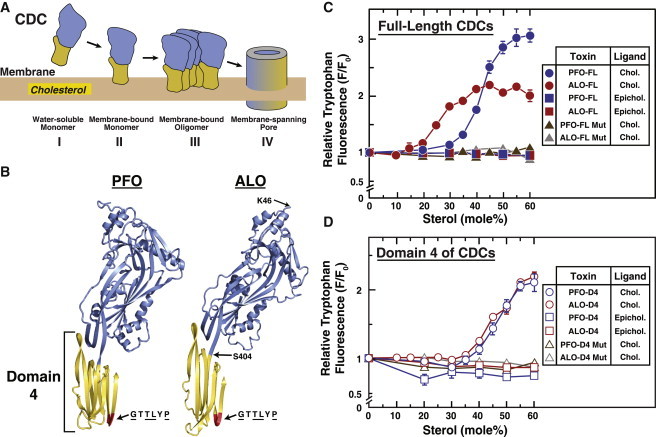
Interaction of PFO and ALO with sterol-containing membranes. (A) General model for the interaction of cholesterol-dependent cytolysins (CDCs) with cholesterol-containing membranes. Soluble monomeric CDCs bind to membrane cholesterol, oligomerize on the membrane surface, and undergo large conformational changes to form a membrane-spanning pore. The cholesterol-binding domain is shaded in yellow. (B) Structures of PFO and ALO. A ribbon representation of the α-carbon backbone of the crystal structures of PFO (11) and ALO (13) is shown with domains 1–3 (amino acids 30–390 in PFO; amino acids 46–403 in ALO) in blue, and domain 4 (D4; amino acids 391–500 in PFO; amino acids 404–512 in ALO) in yellow. A conserved hexapeptide sequence (GTTLYP) is shaded red. Underlined residues of this hexapeptide are identical in 26 related CDCs (10). Also shown are locations of residues in ALO (K46 and S404) that were mutated to cysteines for covalent attachment of fluorescent labels. (C and D) Intrinsic tryptophan fluorescence of PFO and ALO. Recombinant wild-type (WT) and mutant (Mut) versions of full-length (FL) PFO and ALO, as well as D4 of PFO and ALO, were overexpressed and purified as described in Materials and Methods. Each reaction mixture, in a total volume of 200 μL of buffer B, contained 4.4 μM of the indicated protein and 600 μM liposomes composed of DOPC and varying mole fractions of cholesterol (Chol.) or epicholesterol (Epichol.). After incubation for 1 h at room temperature, intrinsic tryptophan fluorescence from the samples was measured (excitation wavelength, 290 nm; emission wavelength, 340 nm). For each version of PFO or ALO, the fluorescence from mixtures of protein with liposomes containing 0% sterol is normalized to 1.
Here, we addressed this problem by conducting a detailed study of the threshold cholesterol sensitivities of two members of the CDC family: PFO and ALO. Using truncated forms of PFO and ALO that do not form oligomers, we isolated the role of membrane cholesterol accessibility in defining the threshold-like sensitivity of these toxins. We found that the primary trigger for their switch-like responses is encoded by the lipid composition of the membrane. Interactions of cholesterol with membrane phospholipids can modulate the affinities of PFO and ALO for membrane cholesterol by more than 100-fold, resulting in sharp, switch-like responses. Toxin oligomerization is not necessary, but can further sensitize this response.
Materials and Methods
Materials and buffers
We obtained 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhyPC) from Avanti Polar Lipids (Alabaster, AL); Dulbecco’s phosphate buffered saline (DPBS) from Corning (Corning, NY); Alexa Fluor 594 C5 maleimide, dimethyl sulfoxide (DMSO), Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (TR-DHPE), Marina Blue 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (MB-DHPE), and isopropyl-1-thio-β-d-galactopyranoside (IPTG) from Invitrogen; cholesterol and tris (2-carboxyethyl) phosphine (TCEP) from Sigma-Aldrich (St. Louis, MO); epicholesterol from Steraloids (Newport, RI) and bovine serum albumin (BSA) from Thermo Fischer Scientific (Waltham, MA). Newborn calf lipoprotein-deficient serum (LPDS, d < 1.215 g/mL) was prepared by ultracentrifugation as described previously (21). Buffer A contained 50 mM Tris-HCl (pH 7.5) and 150 mM NaCl. Buffer B consisted of buffer A supplemented with 1 mM dithiothreitol (DTT). Buffer C was buffer A supplemented with 1 mM TCEP. Buffer D was DPBS supplemented with 2% (v/v) LPDS and 1 mM EDTA.
Expression plasmids
All genetic constructs were cloned into the pRSET B expression vector. A plasmid containing the gene encoding the signal-peptide deficient PFO from Clostridium perfringens (amino acids 29–500) in which the sole cysteine was mutated to alanine (C459A) was a gift from Art Johnson (Texas A&M University). This construct has been described previously (6) and is hereafter referred to as PFO-FL. A plasmid containing the gene encoding a truncated fragment of PFO-FL (amino acids 391–500) was kindly provided to us by Akash Das (University of Texas Southwestern Medical Center). This fragment was defined as the fourth of four distinct structural domains of the soluble form of PFO (11) and was previously shown to bind to cholesterol-containing membranes without causing membrane lysis (22,23). This construct is hereafter referred to as PFO-D4. The gene encoding signal-peptide deficient ALO from Bacillus anthracis (amino acids 35–512) with flanking BamHI and EcoRI restriction sites was synthesized by GenScript (Piscataway, NJ) with a codon sequence optimized for efficient bacterial overexpression, and provided to us in the pUC57 cloning vector. This ALO gene was excised and ligated into the pRSET B expression vector, and this construct is hereafter referred to as ALO-FL. Using ALO-FL as template, a plasmid encoding a truncated fragment of ALO (amino acids 404–512) was generated by site-directed mutagenesis (QuikChange II XL Site-Directed Mutagenesis Kit; Agilent). This fragment of ALO was defined as D4 of ALO in a study comparing the structures of the soluble forms of ALO and PFO (13), and is hereafter referred to as ALO-D4. The PFO-FL, ALO-FL, and ALO-D4 constructs had an NH2-terminal hexahistidine tag followed by an enterokinase cleavage site. The PFO-D4 construct had an NH2-terminal octahistidine tag. Mutations in all four constructs were generated by site-directed mutagenesis. The integrity of each plasmid was verified by DNA sequencing of its entire open reading frame.
Overexpression and purification of recombinant proteins
Expression plasmids were transformed into BL21 (DE3) pLysS Escherichia coli-competent cells (Invitrogen), and protein overexpression was carried out as previously described (6) with the following modifications: PFO-FL and derivatives were induced with 1 mM IPTG at 37°C for 3.5 h; ALO-FL and derivatives were induced with 0.5 mM IPTG at 30°C for 16 h; and PFO-D4, ALO-D4, and derivatives were induced with 1 mM IPTG at 18°C for 16 h. A cell pellet from a 6 L bacterial culture was resuspended in 120 ml of buffer B containing 1 mg/mL lysozyme, 0.4 mg/mL phenylmethanesulfonyl fluoride, and six protease inhibitor cocktail tablets (Complete Mini, EDTA free; Roche, Basil, Switzerland), and incubated at 4°C for 30 min. The lysozyme-disrupted cells were lysed using a dounce homogenizer followed by a tip sonicator (Branson, Danbury, CT) and then subjected to 220,000 × g centrifugation for 1 h. The resulting supernatant was loaded on a column packed with Ni-NTA (nickel-nitrilotriacetic acid) agarose beads (Qiagen, Hilden, Germany). The column was washed with 10 column volumes of buffer B containing 50 mM imidazole, and bound proteins were eluted with either buffer B containing 300 mM imidazole (PFO-FL, ALO-FL, and derivatives) or with buffer B containing a linear gradient of 50–300 mM imidazole (PFO-D4, ALO-D4, and derivatives). The eluted fractions with the desired proteins were pooled and concentrated using an Amicon Ultra centrifugal filter (Millipore, Billerica, MA; 30,000 MWCO for PFO-FL and ALO-FL, and 10,000 MWCO for PFO-D4 and ALO-D4) and further purified by gel filtration chromatography on a Tricon 10/300 Superdex 200 column (GE Healthcare, Uppsala, Sweden) equilibrated with buffer B. Protein-rich fractions were pooled, concentrated to 1–10 mg/mL, and stored at 4°C until use. Protein concentrations were measured using a NanoDrop instrument (Thermo Fisher Scientific) or by using a bicinchoninic acid kit (Pierce).
Labeling of cysteine-substituted proteins with fluorescent groups
Derivatives of ALO containing single cysteines were purified as described above except that the final gel filtration step was carried out on a column equilibrated with buffer C, which contains TCEP instead of DTT. In a typical 200 μL labeling reaction, 20 nmoles of protein (100 μM) was incubated with 200 nmoles of Alexa Fluor 594 C5-maleimide. After incubation for 16 h at 4°C, the reaction was quenched by addition of DTT to a final concentration of 10 mM. Free dye was separated from labeled protein by passing the reaction mixture twice through Zeba spin desalting columns (Pierce, Waltham, MA; 40,000 MWCO for ALO-FL and 7,000 MWCO for ALO-D4) equilibrated in buffer B. Finally, labeled proteins were subjected to gel filtration chromatography as described above to remove any residual free dye and to ensure that no protein aggregation had occurred. Protein-rich fractions were pooled, concentrated, and stored at 4°C until use. The final protein concentration and extent of protein labeling were measured on a Nanodrop instrument using the following parameters: reported ε590 of 100,000 M−1cm−1 for Alexa Fluor 594 (Invitrogen, Waltham. MA) and calculated ε280 of 61,000 and 35,000 M−1cm−1 for ALO-FL and ALO-D4, respectively. The degree of protein labeling ranged from 0.4 to 0.9.
Preparation of liposomes
All lipids were used without further purification. Mixtures containing the indicated proportions of phospholipids and sterols (from chloroform stock solutions) were evaporated to dryness under a steady stream of nitrogen gas and stored under vacuum for at least 16 h. A trace amount (<0.2 mol %) of a fluorescently labeled phospholipid (TR-DHPE for assays with unlabeled proteins, MB-DHPE for assays with Alexa-594 labeled proteins) was included for detection and quantification of liposomes. The dried lipid mixtures were hydrated by adding 500 μL of buffer A (final lipid concentration, 800 μM), agitated on a vortexer for 1 h, and subjected to three freeze-thaw cycles (one cycle = 60 s in a liquid nitrogen bath, 3 min in a room temperature water bath). The resulting lipid dispersions were placed in a water bath at 37°C and subjected to sonication for 15 min followed by a 15 min pause for cooling (two cycles). Finally, the lipid mixtures were extruded through a polycarbonate filter (100 nm pore size) 11 times to yield homogeneous unilamellar liposomes. Liposomes were stored at 4°C and used within 5 days. Spot checks were carried out to verify cholesterol concentrations in liposomes as described previously (2). In all cases, the measured concentration of cholesterol in liposomes was within 5 mol % of the expected values.
Assays for interaction of purified proteins with liposomes
Reaction mixtures (200 μL) containing 600 μM liposomes (total lipid) and 4.4 μM protein in buffer B were set up in 1.5 mL tubes (Phenix Research Products, Candler, NC). After incubation for 1 h at room temperature, a portion of each reaction mixture (100 μL) was transferred to a 96-well plate (black, flat-bottom, nonbinding; Greiner Bio-One, Monroe, NC) and intrinsic tryptophan fluorescence was measured using a microplate reader (Tecan, Männedorf, Switzerland) (excitation wavelength, 290 nm; emission wavelength, 340 nm; band pass, 5 nm for each). A portion of each reaction mixture (20 μL) was mixed with SDS loading buffer, heated for 10 min at 37°C, and subjected to SDS-PAGE. Proteins were visualized with Coomassie Brilliant Blue R-250 stain (Bio-Rad, Hercules, CA). In assays measuring the affinity of PFO for membrane cholesterol, reaction mixtures (1 mL) containing 100 nM PFO and 0–500 μM liposomes, all in buffer B, were set up in 1.5 mL tubes. After incubation for 2 h at room temperature, 40 μL of Ni-NTA agarose bead slurry that had been washed twice and resuspended in 100 μL of buffer B was added to each reaction. After additional incubation for 1 h at room temperature, the His6-tagged PFO was pelleted by centrifugation at 16,000 × g for 5 min. The supernatant was discarded and the Ni-bound PFO was eluted by the addition of 20 μL of buffer B supplemented with 500 mM imidazole. After addition of 4 μL of 5× SDS loading buffer and heating for 10 min at 37°C, samples were centrifuged at 16,000 × g for 5 min and the resulting supernatant was subjected to SDS-PAGE. Proteins were visualized with Coomassie Brilliant Blue R-250 stain. Densitometry analysis was carried out using ImageJ software (NIH, version 1.36B, Bethesda, MD). For assays in which fluorescently labeled proteins were used, reaction mixtures (120 μL) containing 67 μM liposomes (total lipid), 0.5 μM fluorescently labeled protein, and 0–10 μM unlabeled protein in buffer B were set up in 1.5 mL tubes. After incubation for 1 h at room temperature, a portion of each reaction mixture (100 μL) was transferred to a 96-well plate (Greiner Bio-One), and fluorescence was measured using a microplate reader (excitation wavelength, 590 nm; emission wavelength, 617 nm; band pass, 2.5 nm for each).
Hemolysis assays
For a typical assay, 4 mL of fresh rabbit blood was centrifuged at 120 × g for 10 min and the erythrocyte pellet was resuspended in 4 mL of ice-cold buffer D. After gentle mixing by hand, the mixture was centrifuged at 500 × g for 10 min and the resulting pellet was again resuspended in 4 mL of ice-cold buffer D. After gentle mixing by hand, the mixture was centrifuged at 1000 × g for 20 min and the resulting pellet was resuspended in 36 mL of ice-cold buffer D. Standard hemolysis reaction mixtures (500 μL) containing 450 μL of erythrocytes (washed and diluted as described above) and 50 μL of buffer A containing protein (0–300 nM final concentration) were set up in 1.5 mL tubes. In some hemolysis assays, proteins were preincubated with cholesterol or epicholesterol dissolved in DMSO (4% (v/v) final concentration) for 1 h at room temperature before addition of erythrocytes. After incubation for 10 min at 37°C, the mixtures were centrifuged at 380 × g for 15 min and a portion of the supernatant (100 μL) was transferred to a 96-well plate (clear, flat-bottom; Evergreen Scientific, Los Angeles, CA). The extent of hemolysis was quantified using a microplate reader by measuring the absorbance of released hemoglobin at 540 nm.
Data analysis
The data points in all plots of Figs. 1, 2, 3, 4, and 5 represent the mean of three independent assays (except for Fig. 3 B, which shows the average of two independent assays). Error bars represent the mean ± SE. When not visible, error bars are smaller than the size of the data symbols. In Fig. 4, the binding curves represent a weighted least-squares fit of a sigmoidal function to the data points. The best-fit values of switch-points (cholesterol mole percentages where the normalized Trp fluorescence equals 0.5) for the various protein/phospholipid pairs are 41 mol % for PFO-FL/DOPC; 31 mol % for PFO-FL/DPhyPC; 26 mol % for ALO-FL/DOPC; 17 mol % for ALO-FL/DPhyPC; 45 mol % for PFO-D4/DOPC; 27 mol % for PFO-D4/DPhyPC; 45 mol % for ALO-D4/DOPC; and 27 mol % for ALO-D4/DPhyPC). In Fig. 5, B and D, the curves for PFO-FL oligomerization, and inhibition of PFO-FL and ALO-FL hemolysis by cholesterol represent fits to a one-site receptor-ligand binding model. The best-fit values for half-maximal oligomerization in Fig. 5 B are 11 μM and 16 μM for 40 mol % cholesterol and 50 mol % cholesterol, respectively, and >1000 μM for 20 mol % and 30 mol % cholesterol. The best-fit values for 50% inhibition in Fig. 5 D are 108 nM for ALO-FL and 97 nM for PFO-FL.
Figure 2.
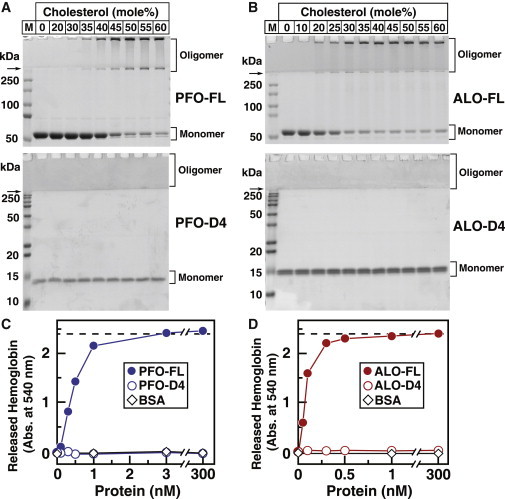
Oligomerization and pore formation by PFO and ALO after binding to cholesterol-containing membranes. (A and B) Coomassie staining. Aliquots (10% of total) of the reaction mixtures from Fig. 1, C and D, containing recombinant FL or D4 of PFO and ALO with membranes containing DOPC and varying mole fractions of cholesterol were subjected to SDS-PAGE. Proteins were visualized with Coomassie Brilliant Blue R-250 stain. The molecular masses of protein standards are indicated. Arrows indicate the interface between stacking and resolving gels. (C and D) Hemolysis assays. Each reaction mixture, in a final volume of 500 μL, contained varying amounts of the indicated version of PFO, ALO, or BSA, and 450 μL rabbit erythrocytes that had been washed and diluted as described in Materials and Methods. After incubation for 10 min at 37°C, the extent of hemolysis was quantified by measuring the release of hemoglobin (absorbance at 540 nm). The dashed line represents the amount of hemoglobin that was released after treatment with 1% (w/v) Triton-X 100 detergent.
Figure 3.
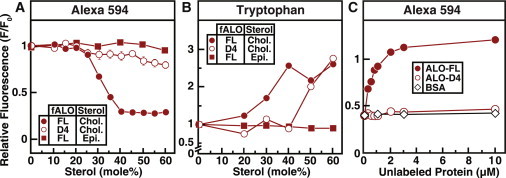
Interaction of fluorescently labeled ALO with sterol-containing membranes. Recombinant ALO-FL and ALO-D4 were overexpressed and purified, and fluorescently labeled versions (fALO-FL and fALO-D4) were generated as described in Materials and Methods. (A and C) Alexa 594 fluorescence of the labeled proteins. Reaction mixtures, in a final volume of 120 μL buffer B, contained 0.5 μM of the indicated fluorescently labeled protein and 67 μM liposomes comprised of DOPC and varying amounts of cholesterol (Chol.) or epicholesterol (Epi.) (A) or 0.5 μM fALO-FL, 67 μM liposomes comprised of 50 mol % DOPC and 50 mol % cholesterol, and varying amounts of the indicated unlabeled protein (C). (B) Intrinsic tryptophan fluorescence of the labeled proteins. Reaction mixtures, in a volume of 100 μL buffer B, contained 3.6 μM of the indicated protein and 600 μM liposomes composed of DOPC and varying mole fractions of cholesterol or epicholesterol. After incubation for 1 h at room temperature, Alexa Fluor 594 fluorescence (A and C) (excitation wavelength, 590 nm; emission wavelength, 617 nm; band pass, 2.5 nm for each) or intrinsic tryptophan fluorescence (B) (excitation wavelength, 290 nm; emission wavelength, 340 nm) was measured. For each protein, fluorescence from mixtures of protein with liposomes containing 0% cholesterol is normalized to 1.
Figure 4.
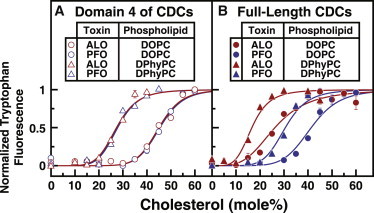
Cholesterol thresholds for ALO and PFO are determined by membrane phospholipids. (A and B) Recombinant FL and D4 of PFO and ALO were overexpressed and purified as described in Materials and Methods. Each reaction mixture, in a total volume of 200 μL of buffer B, contained 4.4 μM of the indicated protein and 600 μM liposomes composed of DOPC or DPhyPC and varying mole fractions of cholesterol. After incubation for 1 h at room temperature, the intrinsic tryptophan fluorescence of the samples was measured (excitation wavelength, 290 nm; emission wavelength, 340 nm). For each combination of protein and phospholipid, fluorescence values were normalized to range from 0 to 1.
Figure 5.
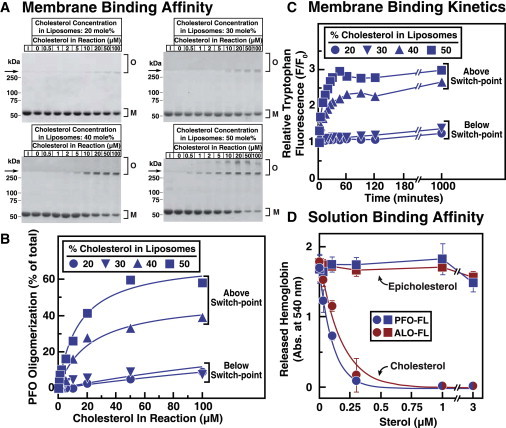
Affinity of PFO and ALO for cholesterol is determined by membrane phospholipids. Recombinant PFO-FL and ALO-FL were overexpressed and purified as described in Materials and Methods. (A and B) Affinity of PFO-FL for membrane cholesterol. Each reaction mixture, in a total volume of 1 mL of buffer B, contained 100 nM PFO-FL (5.7 μg) and varying amounts of liposomes composed of DOPC and the indicated amounts of cholesterol. (A) After incubation for 2 h at room temperature, PFO-FL was concentrated to a volume of 20 μL as described in Materials and Methods, and the entire amount of protein was subjected to SDS-PAGE. Lane 1 (I) in each gel contains 5.7 μg of PFO-FL (input amount) as a reference to judge the efficiency of PFO-FL concentration by Ni beads. Proteins were visualized with Coomassie Brilliant Blue R-250 stain. The molecular masses of protein standards are shown. Arrows indicate the interface between stacking and resolving gels. O, membrane-bound oligomeric form of PFO; M, free monomer form of PFO. (B) Gels were scanned and densitometric analysis was carried out to determine the percentage of the oligomeric, membrane-bound form of PFO relative to the total (membrane-bound oligomer plus free monomer). (C) Binding kinetics. Each reaction mixture, in a total volume of 200 μL of buffer B, contained 4.4 μM of PFO-FL and 600 μM liposomes composed of DOPC and the indicated amounts of cholesterol. After incubation at room temperature for the indicated times, intrinsic tryptophan fluorescence from the samples was measured (excitation wavelength, 290 nm; emission wavelength, 340 nm). The fluorescence from mixtures of PFO-FL with liposomes containing 0% sterol is normalized to 1. (D) Binding of DMSO-solubilized sterols to PFO-FL and ALO-FL. Each reaction mixture, in a final volume of 50 μL, contained either ALO-FL (1 nM) or PFO-FL (3 nM), and varying amounts of cholesterol or epicholesterol dissolved in DMSO (4% (v/v) final concentration). After incubation for 1 h at room temperature, 450 μL of rabbit erythrocytes (washed and diluted as described in Materials and Methods) was added to each reaction mixture. After incubation for 10 min at 37°C, the extent of hemolysis was quantified by measuring the release of hemoglobin (absorbance at 540 nm).
Results and Discussion
Cholesterol switch-points of PFO and ALO: identical for D4s, but different for full-length versions
The crystal structures of PFO and ALO monomers are shown in Fig. 1 B with their carboxy-terminal D4s shaded yellow. These proteins share ∼70% sequence identity and a common elongated, β-sheet-rich architecture. Previous studies showed that D4s from PFO and ALO were sufficient for binding to membrane cholesterol, but were unable to form oligomers or membrane pores (23,24). These earlier studies suggested that a comparative study of full-length (FL) versus D4 fragments of CDCs might shed light on the role of oligomerization in determining the threshold cholesterol sensitivity of CDCs. To this end, we expressed FL and D4 versions of both PFO and ALO, all with amino-terminal His-tags, in bacteria. We purified the resulting recombinant proteins (hereafter referred to as PFO-FL, PFO-D4, ALO-FL, and ALO-D4) to homogeneity using nickel chromatography followed by gel filtration chromatography. We then adapted many of the assays established by the Johnson and Tweten groups for PFO-FL (5,6,17–19) and extended them to study the various domains of PFO and ALO.
PFO-FL contains seven Trp residues, six of which are located in its D4, and binding to membranes results in a 2- to 3-fold increase in its Trp fluorescence (2,25,26). Since ALO-FL contains six Trp residues (five in its D4) at similar locations compared with PFO-FL, we monitored the intrinsic Trp fluorescence of both proteins to measure their binding to cholesterol-containing membranes. Initially, we used model membranes composed of binary mixtures of DOPC and cholesterol, and varied the mole fraction of cholesterol while keeping the total amount of membranes constant in each reaction. In this approach, changes in membrane cholesterol content are accompanied by an opposite change in the content of DOPC.
As shown in previous studies (2,6,7) and Fig. 1 C, when PFO-FL was incubated with liposomes containing increasing mole fractions of cholesterol, its Trp fluorescence increased by ∼3-fold in a sharp, sigmoidal fashion, with a half-maximum at 41 mol % cholesterol. Hereafter, we use the term “switch-point” to refer to the cholesterol mole fraction corresponding to the midpoint of the switch-like increase in Trp fluorescence, an indicator of CDC binding to membranes (see Materials and Methods for details on curve fitting). When ALO-FL was incubated with DOPC-cholesterol membranes, its Trp fluorescence increased by ∼2-fold in a sharp, sigmoidal fashion as well, but its switch-point was shifted to 26 mol % cholesterol. PFO-FL and ALO-FL showed no binding to membranes containing epicholesterol, even when the mole fraction of this diastereomer of cholesterol reached 60 mol %. Binding was also abolished when a conserved hexapeptide sequence, GTTLYP (shaded red in Fig. 1 B), was mutated to GTAAYP in PFO-FL and to AAAAAA in ALO-FL. The threonine-leucine pair within this hexapeptide region was previously shown to be critical for the binding of PFO and two other CDCs (streptolysin and pneumolysin) to cholesterol-containing membranes (17). Mutation of just the TL pair in ALO-FL to AA only partially affected ALO-FL’s ability to bind to cholesterol-containing membranes.
We then measured the interaction of truncated D4 fragments of PFO and ALO with DOPC-cholesterol membranes. As shown in Fig. 1 D, the Trp fluorescence of PFO-D4 increased by ∼2.5-fold in a sigmoidal fashion, with a switch-point at 45 mol % cholesterol, which is slightly shifted from the 41 mol % switch-point observed for PFO-FL. The Trp fluorescence of ALO-D4 also increased by ∼2.5-fold in a sigmoidal fashion, with a switch-point at 45 mol % cholesterol, which is significantly shifted from the 26 mol % switch-point observed for ALO-FL. Remarkably, although the switch-points for PFO-FL and ALO-FL in Fig. 1 C differed dramatically (41 mol % cholesterol versus 26 mol % cholesterol), the switch-points for PFO-D4 and ALO-D4 were identical (45 mol % cholesterol). Like their FL counterparts, PFO-D4 and ALO-D4 also showed no binding to membranes containing epicholesterol. Introducing the same mutations described above for PFO-FL and ALO-FL into PFO-D4 and ALO-D4 also abolished binding.
D4s of PFO and ALO do not form oligomers
We next sought to determine whether the dramatic difference in cholesterol sensitivity between these proteins was related to differences in their ability to form oligomers. To assay for CDC oligomerization, we took advantage of a previous observation that CDC oligomers are resistant to denaturation by SDS and can be distinguished from CDC monomers by their slower electrophoretic mobility during SDS-PAGE (2,6). As shown in Fig. 2 A (top gel), when mixtures of PFO-FL and DOPC-cholesterol liposomes were subjected to SDS-PAGE, PFO-FL migrated primarily as an ∼50 kDa species (calculated molecular mass: 57 kDa) for cholesterol mole fractions up to 40 mol %. At higher mole fractions of cholesterol, the ∼50 kDa monomeric form of PFO-FL was significantly diminished and the majority of PFO-FL was found as a slower-migrating species (>250 kDa), consistent with a large oligomer. Fig. 2 B (top gel) shows similar results for mixtures of ALO-FL and DOPC-cholesterol liposomes. A major fraction of ALO-FL electrophoresed as a slow-migrating oligomer (>250 kDa) rather than an ∼50 kDa monomer (calculated molecular mass: 56 kDa), only when membrane cholesterol exceeded 25 mol %. The cholesterol mole fractions at which oligomeric forms became dominant (40–45 mol % for PFO-FL; 25–30 mol % for ALO-FL) exactly matched the switch-points observed using the Trp fluorescence assay (Fig. 1 C). In contrast, when we subjected mixtures of PFO-D4 or ALO-D4 and DOPC-cholesterol liposomes to SDS-PAGE, we observed only their monomeric forms at ∼15 kDa (calculated molecular masses: PFO-D4, 14 kDa; ALO-D4, 16 kDa), even at the highest cholesterol mole fraction of 60 mol % (Fig. 2, A and B, bottom gels).
Oligomerization of CDCs on the surface of cholesterol-containing membranes eventually leads to formation of a membrane-spanning pore. We used a hemolysis assay to test the ability of FL and D4 fragments of PFO and ALO to form pores in rabbit erythrocytes. As shown in Fig. 2 C, PFO-FL caused complete hemolysis at a concentration of 3 nM, whereas PFO-D4 or a control protein (BSA) caused no hemolysis, even when added at a concentration of 300 nM. Similarly, as shown in Fig. 2 D, ALO-FL caused complete hemolysis at a concentration of 1 nM, whereas ALO-D4 or BSA did not lyse red cells, even when added at a concentration of 300 nM.
Taken together, the tryptophan fluorescence, gel-shift, and hemolysis assays show that when membrane cholesterol exceeds a threshold concentration, the FL versions of PFO and ALO undergo a transition from a soluble to a membrane-bound form (state I to state II; Fig. 1 A), and then oligomerize to finally form a membrane-spanning pore (states III and IV; Fig. 1 A). The D4 versions of PFO and ALO also bind to cholesterol-rich membranes, but do not form oligomers or pores. The gel-shift assays used to assess oligomerization are not conclusive, since it is possible that D4 oligomers are broken down more readily than FL oligomers by the denaturing conditions of SDS-PAGE. To study the oligomeric properties of CDCs using a different approach, we developed a fluorescence-quenching assay for oligomerization of ALO-FL and ALO-D4. We mutated the sole cysteine in ALO (C472) to alanine and introduced single cysteines near the NH2-terminii of cysteine-less ALO-FL (K46C) and ALO-D4 (S404C). We then covalently attached fluorophores (Alexa Fluor 594) to the sulfhydryl groups of these cysteine residues. These fluorescently labeled proteins are hereafter referred to as fALO-FL and fALO-D4. By placing fluorescent reporters near the NH2-terminus, far from the COOH-terminal tip that is involved in cholesterol binding, we hoped to gain insight into postbinding conformational changes involving oligomerization.
As shown in Fig. 3 A, when fALO-FL was incubated with DOPC-cholesterol liposomes, its Alexa 594 fluorescence was constant until membrane cholesterol reached 25 mol %. At higher cholesterol mole fractions, Alexa 594 fluorescence decreased by >75% in a sharp, sigmoidal fashion. No such reduction in Alexa 594 fluorescence was observed when fALO-FL was incubated with liposomes containing epicholesterol. In contrast to the dramatic, cholesterol-specific quenching observed for fALO-FL, no significant change in Alexa 594 fluorescence was observed when fALO-D4 was incubated with DOPC-cholesterol liposomes, even when the membranes contained 60 mol % cholesterol. The Trp fluorescence assays in Fig. 3 B show that fALO-D4 binds to DOPC-cholesterol membranes with a switch-point of ∼45 mol % cholesterol, similar to what was observed for unlabeled ALO-D4 (Fig. 1 D), thus confirming the activity of this fluorescently labeled protein. The Trp fluorescence assays also show that fALO-FL binds to DOPC-cholesterol membranes with a switch-point of ∼30 mol % cholesterol and does not bind to membranes containing epicholesterol, similar to what was observed for unlabeled ALO-FL (Fig. 1 C). The correlation between changes in Trp and Alexa 594 fluorescence of fALO-FL, but not of fALO-D4, is consistent with oligomerization of membrane-bound fALO-FL, but not of membrane-bound fALO-D4.
If quenching of fALO-FL fluorescence was due to the close proximity of Alexa 594 fluorophores in membrane-bound oligomers, the addition of unlabeled ALO-FL during the reaction would be expected to result in mixed oligomers and relieve proximity-based quenching. As shown in Fig. 3 C, when fALO-FL was incubated with DOPC-cholesterol liposomes containing 50 mol % cholesterol, its Alexa 594 fluorescence was quenched by ∼60% relative to when it was incubated with liposomes containing no cholesterol. As increasing amounts of unlabeled ALO-FL were added to the reaction, the Alexa 594 fluorescence gradually increased, and complete recovery to unquenched levels occurred when unlabeled ALO-FL concentrations were four times that of fALO-FL. In contrast, no recovery of quenched Alexa 594 fluorescence was observed when unlabeled ALO-D4 or BSA was added to the reaction. These results suggest that ALO-D4 cannot form self-oligomers after binding to membrane cholesterol or be incorporated efficiently into oligomers of ALO-FL. The fluorescence of a labeled version of PFO-FL (D30C) is also quenched in a sharp sigmoidal fashion when incubated with DOPC-cholesterol liposomes containing >40 mol % cholesterol. So far, however, we have been unable to generate a stable fluorescently labeled version of PFO-D4 to study its oligomeric properties using this quenching assay. Single cysteine residues were introduced at 10 locations in cysteine-less PFO-D4 (N395, K417, E418, Y432, Q433, D469, S472, Y474, D475, and N481), but in all cases the purified recombinant protein precipitated and was unusable. Nonetheless, the fluorescence quenching studies with ALO support the gel-based results in Fig. 2, which show that FL versions of CDCs form oligomers after binding cholesterol-containing membranes, whereas D4 fragments do not.
Cholesterol switch-points for D4s of PFO and ALO depend on the bilayer phospholipid
Although they did not form oligomers, PFO-D4 and ALO-D4 showed a sharp, sigmoidal cholesterol dependence in binding to DOPC-cholesterol membranes, with identical switch-points at 45 mol % cholesterol (Fig. 1 D). To test whether this common switch-point reflects phospholipid-dependent accessibility of cholesterol in membranes, we changed the bulk phospholipid from DOPC (Tm = −2°C) to DPhyPC (Tm < −120°C). The Tm is a convenient measure of the ordering tendency of phospholipid acyl chains: the lower the Tm, the lower the affinity for cholesterol (27). As shown by the Trp fluorescence assays in Fig. 4 A, both PFO-D4 and ALO-D4 bound to DPhyPC-cholesterol membranes in a sharp, sigmoidal fashion. Remarkably, their switch-points for DPhyPC-cholesterol membranes were also identical, but at 27 mol % cholesterol, which is significantly lower than the identical 45 mol % switch-point observed for their binding to DOPC-cholesterol membranes (data for DOPC are replotted here from Fig. 1 D in a normalized form). As shown in Fig. 4 B, when we studied the interaction of PFO-FL and ALO-FL with DPhyPC-cholesterol membranes, we found that the binding of these proteins also showed shifts in switch-points to lower cholesterol mole fractions (31 mol % for PFO-FL, 17 mol % for ALO-FL; data for DOPC are replotted here for reference from Fig. 1 C). Whereas the binding curves for PFO-D4 and ALO-D4 to cholesterol-containing membranes collapse into phospholipid-specific groups, the binding curves for PFO-FL and ALO-FL show a wider distribution of switch-points because oligomerization enhances their cholesterol sensitivity. The variation in switch-points (by <10 mol % cholesterol) observed for mutant versions of PFO-FL (28) is likely due to differences in their oligomerization.
Apparent affinities of PFO and ALO for cholesterol are determined by membrane composition
The experiments described so far show that CDC binding to membrane cholesterol is sensitive to the mole fraction or surface density of cholesterol in membranes. To further understand the affinity of CDCs for membrane cholesterol, we designed a set of experiments in which we fixed the surface density (mole fraction) of cholesterol in membranes and varied the amount of cholesterol in the reaction solution by changing the total amount of membranes. In these experiments, we used the gel-shift assay of Fig. 2 A to monitor the interaction of PFO-FL with DOPC membranes containing cholesterol at levels both above and below the 41 mol % switch-point for this protein-phospholipid pair (Figs. 1 C and 2 A). The extent of oligomer formation in the Coomassie-stained gels shown in Fig. 5 A was quantified by densitometry and plotted in Fig. 5 B as a function of total cholesterol concentration in the reaction.
When we subjected mixtures of PFO-FL and increasing amounts of liposomes containing 20 mol % cholesterol to SDS-PAGE, we observed that most of the PFO-FL (90% of total) migrated as a monomeric species, even at the highest total concentration of cholesterol (100 μM). Similar behavior was observed when the liposomes contained 30 mol % cholesterol. When the mole fraction of cholesterol in liposomes increased to 40 mol % and 50 mol %, significant oligomer formation of PFO-FL occurred when the cholesterol concentration rose above 5 μM. At cholesterol concentrations of 100 μM, 30–60% of PFO-FL was found in its slower-migrating oligomeric form. These data show that the apparent affinity of PFO-FL for DOPC-cholesterol membranes depends on the lipid composition, ranging from very little affinity (>1000 μM) when the mole fraction of cholesterol is 20 mol % and 30 mol % to an affinity of 11 μM and 16 μM when the mole fraction of cholesterol is 40 mol % and 50 mol %, respectively. To test whether this context-dependent affinity arose due to kinetic differences, we used the Trp fluorescence assay to measure the time course of PFO-FL binding to DOPC-cholesterol membranes. As shown in Fig. 5 C, PFO-FL rapidly bound to membranes containing 40 mol % and 50 mol % cholesterol with half-maximal binding at ∼15 min. However, no binding of PFO-FL was observed when it was incubated with membranes containing 20 mol % and 30 mol % cholesterol, even when the reaction time exceeded 1000 min. Similar kinetics were observed for the binding of PFO-FL to membranes composed of palmitoyl-oleoyl phosphatidylcholine and cholesterol (26).
Our studies so far highlight the role of the lipid bilayer membrane in determining the sensitivity of CDCs for membrane cholesterol. To study the affinity of CDCs for cholesterol without competing interactions from phospholipids, we designed a solution-binding assay in which cholesterol was not immersed in a lipid bilayer. We took advantage of early observations that hemolysis by CDCs can be inhibited by preincubating the toxin with cholesterol in organic solvents (5). As shown in Fig. 2, C and D, incubation of rabbit erythrocytes with PFO-FL (3 nM) or ALO-FL (1 nM) resulted in complete hemolysis. We preincubated PFO-FL and ALO-FL with cholesterol dissolved in DMSO before addition to rabbit erythrocytes, and measured the subsequent inhibition of hemolysis. As shown in Fig. 5 D, hemolysis was completely inhibited by preincubation of PFO-FL and ALO-FL with 250 nM cholesterol. No inhibition of hemolysis was observed after preincubation with epicholesterol, even at the highest concentration of 3 μM. Half-maximal inhibition of hemolysis, which can be related to a solution affinity of PFO and ALO for cholesterol, occurred at similar concentrations of 97 nM and 108 nM, respectively. These affinities are only a rough estimate, since a large fraction of cholesterol added to the aqueous phase likely becomes rapidly insoluble and inaccessible. The varied threshold-like cholesterol sensitivities observed in Fig. 4 due to differences in cholesterol sequestration by membrane phospholipids, or to differences in PFO and ALO oligomerization on membranes, were no longer observed when the affinity of PFO and ALO for cholesterol was measured in this nonmembrane context.
Conclusions
We have shown that the switch-like sensitivity of cholesterol-sensing bacterial toxins for membrane cholesterol arises primarily due to properties of the lipid bilayer. Interactions with membrane phospholipids control the accessibility of membrane cholesterol to soluble sensors such as PFO and ALO. After binding to accessible cholesterol at the surface of membranes, the membrane-bound toxins are stabilized by large-scale oligomerization. This complex interplay among lipid-lipid, lipid-protein, and protein-protein interactions serves to fine-tune the final varied sensitivities of PFO and ALO for cholesterol in membranes.
We were able to elucidate the initial sensing of membrane cholesterol by PFO and ALO without the complications of protein oligomerization by engineering nonoligomerizing, truncated versions of these proteins: PFO-D4 and ALO-D4. Figs. 1, 2, 3, and 4 show that although they did not form oligomers, PFO-D4 and ALO-D4 bound to cholesterol-containing membranes in a sharp, sigmoidal manner. The concentration of cholesterol at which this switch-like transition in binding occurs is determined by the phospholipid structure (2,6,7). The absolute specificity for cholesterol over its diastereomer epicholesterol suggests to us a specific binding site in the ∼110 amino acid D4 fragments of PFO and ALO. Positive cooperativity between multiple cholesterol binding sites in these nonoligomerizing D4 fragments could be a source of this sigmoidal behavior; however, we think this explanation is unlikely, as mutation of just a threonine-leucine pair in PFO-D4 to alanines completely abolishes membrane binding (see Fig. 1). Instead, we propose that the sigmoidal binding of PFO-D4 and ALO-D4 to cholesterol-containing membranes is determined by the accessibility of cholesterol at the surface of membranes.
The binding reaction of PFO-D4 or ALO-D4 to membrane cholesterol can be conceptualized as a two-step reaction. The first step involves an equilibrium between cholesterol dissolved in the lipid bilayer membrane and cholesterol in the water layer at the surface of a lipid bilayer. Since cholesterol is virtually insoluble in water, this interfacial cholesterol could partially project into the bilayer-associated water layer without fully escaping the bilayer. The fraction of cholesterol molecules that make excursions into this juxtamembranous water layer is controlled by underlying interactions with membrane phospholipids, and is a measure of the chemical activity of cholesterol in the membrane. This first step is purely a feature of the lipid bilayer. The second step occurs in the aqueous phase and involves the binding of water-soluble PFO-D4 or ALO-D4 to cholesterol in the water-layer at the membrane periphery. In this model, the toxin molecules can be considered to be in a competitive binding equilibrium with the phospholipids for bilayer cholesterol (2). In aqueous solution, akin to the second step of this reaction scheme, the apparent binding affinity of PFO for cholesterol in the water phase is high (∼100 nM, as shown in Fig. 5 D). However, as shown in Fig. 5 B, when cholesterol-phospholipid interactions in bilayers are included, the apparent binding affinity of PFO is much weaker (∼10 μM). In the extreme case where membrane cholesterol is below the switch-point concentration, interactions with phospholipids dominate the reaction and there is very little apparent affinity of PFO for membrane cholesterol (>1000 μM). The sigmoidal dependence for PFO and ALO binding to membranes clearly involves the chemical activity of cholesterol, and this could dominate the reaction even if the binding of toxins to membranes is not reversible and/or involves toxin oligomerization.
The chemical activity of cholesterol, which controls its surface accessibility to sensor proteins, generally increases as the concentration of cholesterol increases, but it can be severely suppressed at lower concentrations in a sigmoidal fashion due to interactions with bilayer phospholipids (complex formation) (2,29,30). Theoretical studies have shown that the cooperative formation of oligomers of such complexes can further sharpen the sigmoidal change in chemical activity, making it more threshold-like (2,30). This simple, intuitive model of complex formation has been extremely useful in accounting for many physical chemical properties of membranes (2,29–33); however, such complexes have not been isolated. This may not be surprising, because molecular complexes in liquids have been described with relatively well-defined structures but very short lifetimes (<10 ps) (34). A well-defined specific structure for phospholipid-cholesterol complexes may be unlikely, since sharp changes in chemical activities are observed for a wide variety of phospholipid and sterol structures (2,31,35). Other models that consider nonrandom arrangements of cholesterol in the bilayer could also result in sharp changes in its chemical activity (36,37). Phase separations provide another possible mechanism for triggering sharp changes in the chemical activity of membrane cholesterol; however, no liquid phase separations have been observed in the simple DOPC-cholesterol and DPhyPC-cholesterol membranes used here (38,39). Regardless of the mechanism that modulates the chemical activity of cholesterol in membranes, it is clear that a property of the lipid bilayer itself can be a key regulator of cholesterol-sensing proteins.
Disappointingly, no high-resolution structures of PFO or ALO bound to cholesterol are available to test our proposed reaction scheme. However, it is worth examining the structure of cholesterol-bound Osh4, a soluble protein from yeast that is related to the family of mammalian oxysterol-binding protein (OSBP)-related proteins (ORPs) that have been implicated in cholesterol homeostasis (40). The structure of cholesterol-Osh4 shows no direct hydrogen bonds between Osh4 amino acid side chains and the hydroxyl group or any other part of cholesterol; instead, the cholesterol is bound through water-mediated interactions. In contrast, the crystal structure of cholesterol-bound N-terminal domain of human Niemann Pick C1 (NPC1), a soluble protein that is involved in cholesterol transport from lysosomes, shows a snug binding pocket and direct close contacts between NPC1 amino acid side chains and the hydroxyl group and tetracyclic steroid nucleus of cholesterol (41). It remains to be seen whether cholesterol sensors such as ALO and PFO employ either one of these strategies for binding to membrane cholesterol.
The chemical activity of membrane cholesterol likely controls its surface accessibility to other soluble molecules such as cholesterol oxidase and cyclodextrin (30,31,42). Of particular interest is a recent report that the cholesterol-binding site in Scap, the mammalian cholesterol sensor, is located not in its transmembrane region but in a membrane-associated loop that projects into the lumen of the ER (43). As noted in the Introduction, the binding of PFO to purified ER membranes and the activation of Scap by ER cholesterol both occur at a common threshold concentration of 5 mol % cholesterol. Based on our current understanding of ALO and PFO, it is tempting to speculate that Scap may be binding to a pool of ER cholesterol that exceeds the sequestration capacity of ER phospholipids and projects out of the ER bilayer. Despite the involvement of many proteins, a property of the ER lipid bilayer alone may be a key regulatory element of cholesterol homeostasis (2,44). Future studies with purified or reconstituted ER membranes and the tools developed here promise to clarify this issue.
Acknowledgments
We thank Harden McConnell, Mike Rosen, Mike Brown, and Joe Goldstein for many helpful discussions; Cameron Fazeli for invaluable technical assistance; Akash Das and Arthur Johnson for providing us with PFO plasmids; and Feiran Lu for her help during an early phase of this work.
This work was supported by the NIH (HL-20948), Welch Foundation (I-1793), and American Heart Association (12SDG12040267). A.G. received a Cell and Molecular Biology Training Grant from the NIH (5T32-GM008203).
References
- 1.Radhakrishnan A., Goldstein J.L., Brown M.S. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 2008;8:512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sokolov A., Radhakrishnan A. Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J. Biol. Chem. 2010;285:29480–29490. doi: 10.1074/jbc.M110.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Das A., Goldstein J.L., Radhakrishnan A. Use of mutant 125I-perfringolysin O to probe transport and organization of cholesterol in membranes of animal cells. Proc. Natl. Acad. Sci. USA. 2013;110:10580–10585. doi: 10.1073/pnas.1309273110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown M.S., Goldstein J.L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 5.Tweten R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flanagan J.J., Tweten R.K., Heuck A.P. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry. 2009;48:3977–3987. doi: 10.1021/bi9002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson L.D., Johnson A.E., London E. How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: insights into the origin of perfringolysin O-lipid raft interaction. J. Biol. Chem. 2008;283:4632–4642. doi: 10.1074/jbc.M709483200. [DOI] [PubMed] [Google Scholar]
- 8.Radhakrishnan A., Sun L.P., Goldstein J.L. Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol. Cell. 2004;15:259–268. doi: 10.1016/j.molcel.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 9.Radhakrishnan A., Ikeda Y., Goldstein J.L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heuck A.P., Moe P.C., Johnson B.B. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell. Biochem. 2010;51:551–577. doi: 10.1007/978-90-481-8622-8_20. [DOI] [PubMed] [Google Scholar]
- 11.Rossjohn J., Feil S.C., Parker M.W. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell. 1997;89:685–692. doi: 10.1016/s0092-8674(00)80251-2. [DOI] [PubMed] [Google Scholar]
- 12.Polekhina G., Giddings K.S., Parker M.W. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc. Natl. Acad. Sci. USA. 2005;102:600–605. doi: 10.1073/pnas.0403229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bourdeau R.W., Malito E., Tang W.J. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J. Biol. Chem. 2009;284:14645–14656. doi: 10.1074/jbc.M807631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu L., Huang B., Du H., Zhang X.C., Xu J., Li X., Rao Z. Crystal structure of cytotoxin protein suilysin from Streptococcus suis. Protein Cell. 2010;1:96–105. doi: 10.1007/s13238-010-0012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Köster S., Hudel M., Yildiz Ö. Crystallization and X-ray crystallographic analysis of the cholesterol-dependent cytolysin listeriolysin O from Listeria monocytogenes. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013;69:1212–1215. doi: 10.1107/S1744309113025761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feil S.C., Ascher D.B., Parker M.W. Structural studies of Streptococcus pyogenes streptolysin O provide insights into the early steps of membrane penetration. J. Mol. Biol. 2014;426:785–792. doi: 10.1016/j.jmb.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrand A.J., LaChapelle S., Tweten R.K. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. USA. 2010;107:4341–4346. doi: 10.1073/pnas.0911581107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramachandran R., Tweten R.K., Johnson A.E. The domains of a cholesterol-dependent cytolysin undergo a major FRET-detected rearrangement during pore formation. Proc. Natl. Acad. Sci. USA. 2005;102:7139–7144. doi: 10.1073/pnas.0500556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato T.K., Tweten R.K., Johnson A.E. Disulfide-bond scanning reveals assembly state and β-strand tilt angle of the PFO β-barrel. Nat. Chem. Biol. 2013;9:383–389. doi: 10.1038/nchembio.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tilley S.J., Orlova E.V., Saibil H.R. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell. 2005;121:247–256. doi: 10.1016/j.cell.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 21.Goldstein J.L., Basu S.K., Brown M.S. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 22.Ishitsuka R., Saito T., Kobayashi T. Fluorescence image screening for chemical compounds modifying cholesterol metabolism and distribution. J. Lipid Res. 2011;52:2084–2094. doi: 10.1194/jlr.D018184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimada Y., Maruya M., Iwashita S., Ohno-Iwashita Y. The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Eur. J. Biochem. 2002;269:6195–6203. doi: 10.1046/j.1432-1033.2002.03338.x. [DOI] [PubMed] [Google Scholar]
- 24.Cocklin S., Jost M., Chaiken I.M. Real-time monitoring of the membrane-binding and insertion properties of the cholesterol-dependent cytolysin anthrolysin O from Bacillus anthracis. J. Mol. Recognit. 2006;19:354–362. doi: 10.1002/jmr.784. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura M., Sekino N., Ohno-Iwashita Y. Interaction of theta-toxin (perfringolysin O), a cholesterol-binding cytolysin, with liposomal membranes: change in the aromatic side chains upon binding and insertion. Biochemistry. 1995;34:6513–6520. doi: 10.1021/bi00019a032. [DOI] [PubMed] [Google Scholar]
- 26.Heuck A.P., Hotze E.M., Johnson A.E. Mechanism of membrane insertion of a multimeric beta-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Mol. Cell. 2000;6:1233–1242. doi: 10.1016/s1097-2765(00)00119-2. [DOI] [PubMed] [Google Scholar]
- 27.Keller S.L., Radhakrishnan A., McConnell H.M. Saturated phospholipids with high melting temperatures form complexes with cholesterol in monolayers. J. Phys. Chem. B. 2000;104:7522–7527. [Google Scholar]
- 28.Johnson B.B., Moe P.C., Heuck A.P. Modifications in perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry. 2012;51:3373–3382. doi: 10.1021/bi3003132. [DOI] [PubMed] [Google Scholar]
- 29.Radhakrishnan A., McConnell H. Condensed complexes in vesicles containing cholesterol and phospholipids. Proc. Natl. Acad. Sci. USA. 2005;102:12662–12666. doi: 10.1073/pnas.0506043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radhakrishnan A., McConnell H.M. Chemical activity of cholesterol in membranes. Biochemistry. 2000;39:8119–8124. doi: 10.1021/bi0005097. [DOI] [PubMed] [Google Scholar]
- 31.Lange Y., Tabei S.M., Steck T.L. Stability and stoichiometry of bilayer phospholipid-cholesterol complexes: relationship to cellular sterol distribution and homeostasis. Biochemistry. 2013;52:6950–6959. doi: 10.1021/bi400862q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McConnell H., Radhakrishnan A. Theory of the deuterium NMR of sterol-phospholipid membranes. Proc. Natl. Acad. Sci. USA. 2006;103:1184–1189. doi: 10.1073/pnas.0510514103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ratajczak M.K., Chi E.Y., Kjaer K. Ordered nanoclusters in lipid-cholesterol membranes. Phys. Rev. Lett. 2009;103:028103. doi: 10.1103/PhysRevLett.103.028103. [DOI] [PubMed] [Google Scholar]
- 34.Zheng J., Kwak K., Fayer M.D. Ultrafast dynamics of solute-solvent complexation observed at thermal equilibrium in real time. Science. 2005;309:1338–1343. doi: 10.1126/science.1116213. [DOI] [PubMed] [Google Scholar]
- 35.Okonogi T.M., Radhakrishnan A., McConnell H.M. Two fatty acids can replace one phospholipid in condensed complexes with cholesterol. Biochim. Biophys. Acta. 2002;1564:1–4. doi: 10.1016/s0005-2736(02)00451-0. [DOI] [PubMed] [Google Scholar]
- 36.Ali M.R., Cheng K.H., Huang J. Assess the nature of cholesterol-lipid interactions through the chemical potential of cholesterol in phosphatidylcholine bilayers. Proc. Natl. Acad. Sci. USA. 2007;104:5372–5377. doi: 10.1073/pnas.0611450104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olsen B.N., Bielska A.A., Ory D.S. The structural basis of cholesterol accessibility in membranes. Biophys. J. 2013;105:1838–1847. doi: 10.1016/j.bpj.2013.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veatch S.L., Gawrisch K., Keller S.L. Closed-loop miscibility gap and quantitative tie-lines in ternary membranes containing diphytanoyl PC. Biophys. J. 2006;90:4428–4436. doi: 10.1529/biophysj.105.080283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veatch S.L., Polozov I.V., Keller S.L. Liquid domains in vesicles investigated by NMR and fluorescence microscopy. Biophys. J. 2004;86:2910–2922. doi: 10.1016/S0006-3495(04)74342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Im Y.J., Raychaudhuri S., Hurley J.H. Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature. 2005;437:154–158. doi: 10.1038/nature03923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwon H.J., Abi-Mosleh L., Infante R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohvo H., Slotte J.P. Cyclodextrin-mediated removal of sterols from monolayers: effects of sterol structure and phospholipids on desorption rate. Biochemistry. 1996;35:8018–8024. doi: 10.1021/bi9528816. [DOI] [PubMed] [Google Scholar]
- 43.Motamed M., Zhang Y., Brown M.S. Identification of luminal Loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. J. Biol. Chem. 2011;286:18002–18012. doi: 10.1074/jbc.M111.238311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lange Y., Steck T.L. Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog. Lipid Res. 2008;47:319–332. doi: 10.1016/j.plipres.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]