

Abstract
The image-mean square displacement technique applies the calculation of the mean square displacement commonly used in single-molecule tracking to images without resolving single particles. The image-mean square displacement plot obtained is similar to the mean square displacement plot obtained using the single-particle tracking technique. This plot is then used to reconstruct the protein diffusion law and to identify whether the labeled molecules are undergoing pure isotropic, restricted, corralled, transiently confined, or directed diffusion. In our study total internal reflection fluorescence microscopy images were taken of Cholera toxin subunit B (CtxB) membrane-labeled NIH 3T3 mouse fibroblasts and MDA 231 MB cells. We found a population of CTxB undergoing purely isotropic diffusion and one displaying restricted diffusion with corral sizes ranging from 150 to ∼1800 nm. We show that the diffusion rate of CTxB bound to GM1 is independent of the size of the confinement, suggesting that the mechanism of confinement is different from the mechanism controlling the diffusion rate of CtxB. We highlight the potential effect of continuous illumination on the diffusion mode of CTxB. We also show that aggregation of CTxB/GM1 in large complexes occurs and that these aggregates tend to have slower diffusion rates.
Introduction
In recent years, several technological developments (fluorescence recovery after photobleaching (FRAP), fluorescence resonance energy transfer (FRET), single-particle tracking (SPT), spot variation fluorescence correlation spectroscopy (svFCS), stimulation emission depletion-FCS (STED-FCS), and photoactivation localization microscopy-SPT) have been used to study the lateral diffusion and segregation of lipids and proteins in the cell membrane (for review see (1)). Lateral segregation of lipids in the membrane is thought to be responsible for the formation of nanodomains (often called rafts when enriched in cholesterol) that can mediate signal transduction across the membrane (2). Some proteins such as the Cholera Toxin subunit B (CTxB) have the ability to cross-link lipids and induce phase separation in a model membrane system (3) as well as in giant plasma membrane vesicles produced by blebbing (4). This ability to cross-link membrane glycolipids has been linked to the toxin uptake into cells (5). CTxB binding to five GM1 molecules promote the stabilization of lipid rafts and facilitate their coalescence into larger physiologically relevant structures resulting in toxin trafficking (6). A recent study by Day and Kenworthy (7) investigated the factors responsible for the surprisingly slow diffusion of CTxB/GM1 complex on the cell membrane at physiological temperature. These authors used confocal FRAP microscopy and found that the diffusion of CTxB was significantly increased following treatment with Latrunculin A but was not affected by the presence of caveolae and suggested that the diffusion of GM1/CTxB is slowed directly or indirectly as a result of its interaction with the actin cytoskeleton. Contrasting with these results, the diffusion of labeled GM1 studied by svFCS was found to be unaffected by Latrunculin treatment (8), whereas STED nanoscopy showed an increased trapping of GM1 following treatment (9). These apparently conflicting results could arise from a modification of the diffusion behavior of GM1 induced by CTxB clustering or could be inherent to the experimental modality used to study their diffusion. Indeed, FRAP is essentially an ensemble or bulk measurement and subpopulations cannot be easily identified. FRAP probes macroscopic areas of the cells, whereas both STED and svFCS probe very small observation volumes. Therefore, FRAP is less sensitive to short-range processes. In addition, svFCS does not measure processes below the diffraction limit but extrapolate the data to infer dynamical organization of cell-membrane components below the diffraction limit. The image-mean square displacement technique (iMSD) is based on the calculation of the spatiotemporal image correlation function but it introduces a quantity similar to the mean square displacement (MSD) used in SPT while analyzing the entire image and without separation of the particles. The iMSD versus time plot obtained is then used to reconstruct the protein diffusion law (10). From this plot one can directly identify and determine the number of molecules under observation and identify whether the labeled molecules are undergoing pure isotropic, confined (or corralled), transiently confined, or directed diffusion (Fig. 1). Given that the size of the corrals can be determined with nanometer resolution, regions of confined barriers can be mapped in the cell. An advantage of the iMSD method with respect to the traditional MSD analysis obtained by the SPT method is that iMSD does not require the acquisition of a large number of trajectories so that it can be applied to the entire cell under analysis. Therefore, iMSD analysis provides considerably more information than can be obtained by other current methods such has single point FCS, STED-FCS, and svFCS, which provide very good temporal resolution at a single point but very limited in mapping the diffusional behavior of molecules in the entire cell (1). Raster image correlation spectroscopy (RICS) (11) and STED-RICS (12) techniques provide some spatial resolution but are limited in temporal resolution due to the scanning process and they do not provide diffusion laws. Spatiotemporal image correlation spectroscopy (STICS) gives information on directed diffusion but does not report on faster diffusion processes. Therefore, for analysis of the diffusion of molecules in the entire cell the iMSD method has great potentials.
Figure 1.
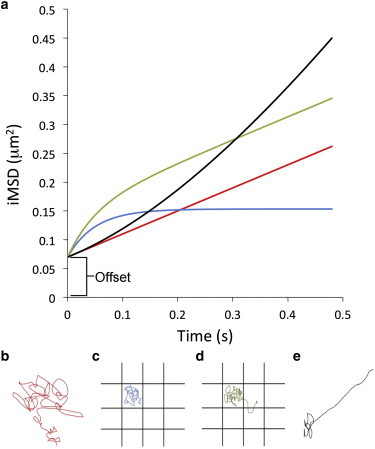
Theoretical curves and different modes of diffusion. (a) iMSD curve for the four different modes of diffusion: isotropic (red curve), confined (blue curve), transiently confined (green curve), and directed diffusion or flow, which could happen when active transport is involved or the cell is moving (black curve). The intercept of the iMSD curve with the y axis at time 0 is the offset that is due to the convolution of the size of the particles and the PSF. (b) Schematic drawing of a molecule trajectory for a long-range isotropic diffusion corresponding to the red iMSD curve in (a). The value obtained from this curve is the rate of diffusion Dmacro and the offset. (c) This time, the molecule diffusion is confined within boundaries represented by the black squares and cannot cross these barriers. This type of diffusion will produce the iMSD curve represented by the blue line in (a). In addition to the offset, the values obtained from the iMSD analysis are the faster local diffusion of the confined molecule Dmicro and the linear size of the confinement or corral. (d) The confined molecules are able to escape the confinement in this model and will therefore also display a long-range diffusion component and is represented by the green curve in (a). The offset, Dmacro, Dmicro, and linear size of the confinement or corral can be obtained from these data (see equations in the Materials and Methods section). (e) Schematic drawing of a molecule undergoing directed diffusion or flow corresponding to the black curve in (a). To see this figure in color, go online.
In this work, we map the modes of CTxB diffusion when bound to GM1 in live cell membranes. We also combined the iMSD analysis with number and brightness analysis to evaluate the influence of cluster formation on the diffusion of CTxB/GM1. We found different types of diffusion across the cell including isotropic, confined, and transiently confined. We show that continuous illumination of the same area of the cell leads to a transition of diffusion type from confined to isotropic diffusion mode, highlighting a potential artifact that can be produced by performing FRAP experiments to investigate diffusion processes at the cell membrane. We also found that CTxB/GM1 are binding to immobile structures or are incorporated into areas of extremely slow diffusion in the cell membrane and that CTxB/GM1 are dynamically partitioning in immobile membrane nanodomains smaller than the diffraction limit. We show that the formation of clusters favors isotropic diffusion and reduces the rate of diffusion. We also demonstrate that in confined diffusion regime we observe a large range of diffusion rates demonstrating that the size of the confinement does not determine the diffusion rates.
Materials and Methods
Cell culture and labeling and total internal reflection fluorescence microscopy are described in the Supporting Material.
Analysis of the iMSD curve
The iMSD data obtained from SimFCS (Laboratory for Fluorescence Dynamics, Irvine, CA) were exported into SigmaPlot (Systat Software, San Jose, CA) for fitting. Each iMSD curve was fitted to each of the equations for the different modes of diffusion described in Di Rienzo et al. (10) and the Supporting Material.
Brightness analysis
Brightness analysis was performed on the same stacks of images as the iMSD analysis with SimFCS (Laboratory for Fluorescence Dynamics) using G1 for fast camera as described in Unruh and Gratton (13). The G1 analysis is equivalent to measuring the first point of the autocorrelation function instead of the zeroth point (G0) and has the advantage of removing the noise from the camera system without the need for additional measurements. The brightness was calculated using identical parameters for each stack of images so that the number of pixels having similar brightness could be compared between the different images.
Simulations of apparent negative iMSD
We used simulations of particles to understand the conditions giving rise to negative iMSD curves. The simulations were obtained using SimFCS (Laboratory for Fluorescence Dynamics). We simulated the diffusion of 1000 particles, for diffusion rates ranging from 0.001 μm2/s to 0.04 μm2/s. The point spread function (PSF) was set to 280 nm and the image size was selected to be 128 × 128 pixels. The frame time was set to 10 ms, the camera mode was used for scanning and 1000 frames were simulated for two-dimensional diffusion. The image stack acquired was then analyzed using 49 overlapping region of interest (ROI) of 32 × 32 pixels and the iMSD curves fitted and processed as described previously. When a negative iMSD was obtained, we assigned it to the ROI and calculated the fractions of ROI in the image that displayed negative iMSD curves. Three simulations were performed for each condition and the average ± SD values were calculated over all ROIs of the images.
Results
Types of diffusion and statistics
The iMSD data were fitted to three models of diffusion; isotropic or free diffusion (Eq. S6 in the Supporting Material), confined diffusion (Eq. S7), and transiently confined diffusion (Eq. S8).
We analyzed the diffusion in a minimum of two regions in 25 NIH3T3 cells and 30 MDA 231 MB cells and found that the variations between areas of the same cells were of the same order as the variations between cells with similar diffusion modes or diffusion rates between the cells analyzed (see Fig. S1 for the variations between cells and insert, see Figs. 4 and 9 for the variations within a single cell). Therefore, rather than presenting diffusion data of isolated cell regions; we focused our analysis on the variation of diffusion across an entire single cell.
Figure 4.
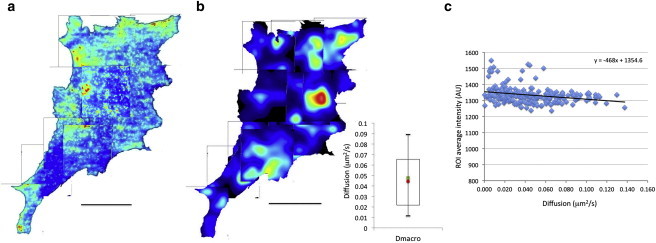
Intensity and isotropic diffusion maps of the NIH3T3 cell. (a) Intensity image of the cell represented in pseudocolors. (b) Isotropic diffusion (Dmacro) rates mapped on the cell in pseudocolors. Red represents high values, whereas blue represents lower values. The range of diffusion is represented in the figure inset with the mean value represented by the green square and the median by the red circle. The open rectangle is the 1st to the 3rd quartile and the bars the 0.1 to 0.9 percentile. (c) Average intensity of the ROI for the 12 stacks of images of the cell versus the corresponding isotropic diffusion rates obtained from the iMSD curves. A regression line was fitted through the data and the equation obtained is shown on the graph. The bar equals 18.56 μm. To see this figure in color, go online.
Figure 9.
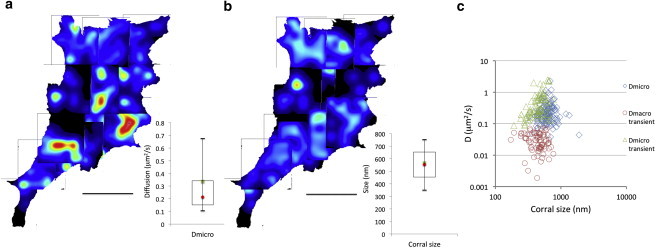
CTxB confinement in the NIH3T3 cell membrane. (a) Map of the diffusion rates in confined areas (Dmicro). The range of diffusion is represented in the figure inset with the mean value represented by the square and the median by the circle. The open rectangle is the 1st to the 3rd quartile and the bars the 0.1 to 0.9 percentile. White represents faster diffusion rates, whereas blue represents slower diffusion. Black are ROI without confined or transiently confined diffusion. (b) Linear dimension of the restricted diffusion assuming a square confinement. Red represents large confinement size, whereas blue represents smaller confinement size. Black are ROIs without confinements. The inset represents the range of corral (confinement) sizes with the mean value represented by the green square and the median by the red circle. The open rectangle is the 1st to the 3rd quartile and the bars the 0.1 to 0.9 percentile. (c) Diffusion rates versus corral (confinement) size. The diamonds are the data (Dmicro) obtained for ROI displaying confined diffusion, whereas the triangle and the circles represent the data for Dmicro and Dmacro obtained for ROI displaying transiently confined diffusion, respectively. The bar equals 18.56 μm. To see this figure in color, go online.
To verify that the measurements of CtxB diffusion were not due to an artifact, we simultaneously determined the diffusion of TR-BODIPY labeled PI(4,5)P2 and CtxB labeled with Alexa488, using two colors split with a 2-channel imager on the same charge-coupled device camera. Fig. S2 clearly shows that independent modes of diffusion and diffusion rates were obtained for the two probes in the same ROIs and are therefore not due to artifacts.
Examples of curves obtained for each type of diffusion are presented in Fig. 2. From these fits, we can extract a slow long-range diffusivity (Dmacro) for isotropic and transiently confined diffusion. For transiently confined and confined diffusion we extract a faster local diffusivity (Dmicro) and the size of the confinement or corral. Fig. 2 d illustrates the different types of diffusion occurring across the cell and shows that some areas of the cell are predominantly undergoing isotropic diffusion, whereas others have more of a mix between transiently confined and confined diffusion.
Figure 2.
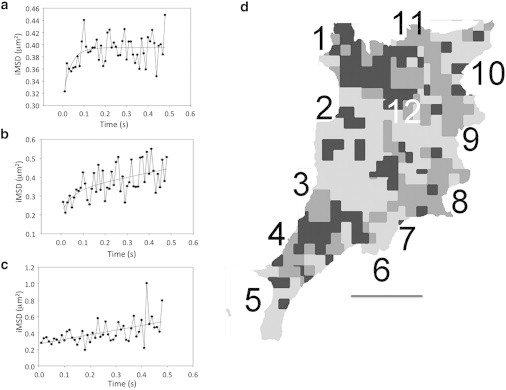
Diffusion models of CTxB on cell membrane in a single NIH3T3 cell. (a) iMSD curve obtained for ROI fitted with Eq. S7 (in the Supporting Material) for the confined diffusion model. (b) iMSD curve obtained for a different ROI fitted with Eq. S8 for a transiently confined diffusion model. (c) iMSD curve obtained for another ROI fitted with the isotropic diffusion model using Eq. S6. (d) Map of the types of diffusion observed for the entire cell. Light gray, isotropic; dark gray, transiently confined; black, confined diffusion modes. The numbers represent the sequence in which the stacks of images were obtained. Each image measures 18.56 × 18.56 μm.
Effect of illumination on the diffusion mode
We investigated the changes in diffusion of CTxB/GM1 following continuous illumination. A series of 9 (MDA) and 10 (NIH3T3) stacks of images were obtained at a 1 min interval of the same region of the cells. Interestingly, the MDA cell is undergoing a retraction of the area and large aggregates of CTxB/GM1 are entering the imaging frame (Fig. S2 a). This is also reflected in a decreased number of molecules in the area of high brightness. On the other hand, the NIH3T3 cell does not show any displacement and clusters of CTxB/GM1 are moving out of the imaging area (Fig. S2 b). The illumination power used did not result in bleaching with the ratios of the average intensity of the last frames of the stacks and the first frames of the stacks equal to 0.99 ± 0.01 (mean ± SD). Fig. 3 a shows the fraction of ROI displaying a confined mode of diffusion as a function of continuous illumination. For both cells, we observe a decreased proportion of areas undergoing confined diffusion to ∼10% of the ROI after 2–3 stacks of images through the disappearance of fast local diffusion (Dmicro) and hence an increase in isotropic diffusion (see Fig. S2). However, there are no significant changes in the diffusion rates Dmacro and Dmicro with illumination (data not shown).
Figure 3.

Effect of illumination on the mode of diffusion. (a) Proportion of ROI showing confined (corralled) diffusion for NIH3T3 (solid line) and MDA 231 MB (dashed line) cells as a function of image stack acquisition time. The time interval between the start of each stack is 1 min. Ten consecutive stacks of images were obtained for the NIH3T3 cells and nine for the MDA cells. (b) Changes in diffusion between the first stack and the last stack of images from the entire cell. Intensity images of the first stack and the last stack of images. The images are scaled so that the maximum intensity (red) is 1516 (arbitrary units) and the minimum (dark blue) is 1150 (arbitrary units). Map of the types of diffusion observed for image 1 in the cell. Light gray, isotropic; dark gray, transiently confined; black, confined diffusion modes. Isotropic diffusion (Dmacro) rates and confined diffusion (Dmicro) rates mapped on the cell in pseudocolors. Red represents high values, whereas blue represents lower values. Each image measures 18.56 × 18.56 μm. To see this figure in color, go online.
To assess the effect that illumination has had on the imaging of the whole cell, an additional stack of images was acquired at the first position. When the diffusion maps are compared between the first and the last stacks of images obtained at the same position (Fig. 3 b), one can notice an increase in ROI displaying isotropic diffusion. This is also associated with a decrease in confined diffusion but to a much lesser extent (from 70% of ROI displaying confinement down to 50% of ROI) than the decrease observed following continuous illumination. In addition, most of the ROI that were displaying a given mode of diffusion are still presenting the same mode of diffusion. The long-range diffusion rates (Dmacro) have increased on average from 0.045 ± 0.03 μm2/s to 0.071 ± 0.03 μm2/s (mean ± SD), although there are no changes in the average local confined diffusion rates (0.261 ± 0.161 μm2/s for the first stack compared to 0.256 ± 0.119 μm2/s for the last stack; mean ± SD).
Although these changes could result from light exposition, some changes in diffusion might also be associated with the time interval between the first and last stack of images. Indeed, when two overlapping areas of image taken at 3–4 min interval (i.e., images 3 and 6) are compared, we can already notice some variations in diffusion modes and rates of diffusion (Fig. S4) illustrating the dynamic nature of the cell membrane.
Fluorescence intensity and isotropic diffusion (Dmacro)
The intensity distribution of the CTxB/GM1 is presented in Fig. 4 a. The distribution of CTxB is not uniform across the cell with areas of high intensity and areas of low intensity. The average isotropic diffusion value obtained is 0.045 μm2/s (inset, Fig. 4 b) but varies between 0.0003 and 0.3 μm2/s depending on the cell area. When the area undergoing isotropic diffusion was mapped onto the cell (Fig. 4 b), there is an apparent inverse relationship between the area of high fluorescence intensity and areas of high rates of isotropic diffusion. This trend is confirmed when the average intensity of the ROIs is plotted against Dmacro (Fig. 4 c). Interestingly, 62% and 61% of NIH3T3 and MDA 231 MB cells displayed a similar trend, respectively. This result in an overall increase in Dmacro with a decrease in average intensity for all the NIH cells analyzed (Fig. S5 a). However, there was no decrease of intensity with increased diffusion rates when all the MDA 231 MB data were pooled together (Fig. S5 b). This difference simply reflects the variations in the number of data points obtained in each cell analyzed. The reduced rate of diffusion with increased average intensity observed in 60% of the cells could result partly from the formation of clusters of CTxB/GM1 in the cell membrane. To investigate the formation of clusters, we further analyzed the images using brightness analysis.
Brightness analysis
The brightness map obtained is presented in Fig. 5 a. Assuming that the majority of CTxB are not aggregating, for monomers, we then obtained a median brightness of 0.022 counts/dwell time/molecule where the dwell time was 0.01 s. We observed an increased brightness for 3.5% of the total pixels in the image of the cell with 3.3% of the pixels having a median brightness of 0.63 counts/dwell time/molecule corresponding to ∼30 mers, 0.2% of the pixels having a median brightness of 1.08 counts/dwell time/molecule corresponding to ∼50 mer aggregates, and 0.02% of the pixels having a median brightness of 1.52 counts/dwell time/molecule corresponding to ∼70 mers.
Figure 5.

Clustering and dynamic partitioning of CTxB on the NIH3T3 cell membrane. (a) Brightness map of the cell. The green pixels are the brightness value attributed to monomers (or small aggregates), the red (30 mers), and blue (50 mers) pixels are pixels displaying higher level aggregates. (b) Map of corrected offset values. Yellows are larger values and blue smaller values. The inset shows the mean value represented by the green square and the median by the red circle. The open rectangle is the 1st to the 3rd quartile and the bars the 0.1 to 0.9 percentile. The bar equals 18.56 μm. (c) Effect of particle size on the diffusion rates. The linear regression represented by the dashed line is the trend observed for all the points (diamonds and triangle) regardless of the aggregation state of the molecules. The triangles highlight the points obtained for ROI containing high levels of aggregates and the solid line represents the linear regression obtained for those points. (d) Subset of the data presented in (c) but for ROI void of CTxB aggregates. Because in this case, there is no influence of the particle size on the offset, the data can be plotted as a function of offset converted in domain size (i.e., SQRT(offset)). The solid line is the linear regression. To see this figure in color, go online.
Fig. 5 a also shows that there are ROI in the cell displaying higher densities of clusters (up to 27% of pixels) than others (some ROI have no clusters) and that those areas with higher cluster density correspond to areas of higher fluorescence intensities (Fig. 4 a).
Offset of the iMSD plots
Di Rienzo et al. (10), reported that the offset of the iMSD analysis (i.e., the iMSD value at delay time 0), is due to the size of the PSF but could also be due to the size of the particles under observation and the trapping component of dynamic partitioning of the diffusing molecules into fixed nanodomains smaller than the observation volume. The authors also mentioned that the exposure time for a fast diffusing particle affects the iMSD plot due to a broadening effect resulting from particle diffusion during the exposure time, which thus changes the offset value. Therefore, for a given exposure time, the offset value will also change as a function of the rate of diffusion of the particles. Fig. S6 shows the offset values obtained from iMSD simulations using the parameters of the cell experiments with a PSF radial waist of 280 nm for different rates of particle diffusion. The simulated data points were fitted and the equation obtained was then used to subtract the offset value due to the size of the PSF from the iMSD data obtained in the cells. Having subtracted the PSF contribution to the offset, the remaining offset value is now due to either the size of the particles under observation or the trapping component of the dynamic partitioning. Fig. 5 b shows a map of the offset due to the last two factors. The offset ranges from 0.0001 to 0.298 μm2 with an average value of 0.07 ± 0.06 μm2 (mean ± SD) (inset, Fig. 5 b). Again, one can see that the larger offset values are obtained in areas that have a higher density of high-brightness particles (Fig. 5 a).
Fig. 5 c shows a plot of the isotropic diffusion rate (Dmacro) as a function of offset values. There is an exponential decrease of the diffusion rate as the value of the offset increases. To evaluate whether this decrease in diffusion rate could be due to the increase in the size of the cluster, the points corresponding to the region of higher cluster density (image 2, Fig. 5 a) are highlighted and represented by the triangles in Fig. 5 c. Most of the data points corresponding to these areas of large clusters have a substantially lower diffusion coefficient than what is observed for the whole cell.
When ROIs that do not have any clusters are selected, the offset value can then be solely attributed to the trapping component of the dynamic partitioning into nanodomains and the size of the domains can be estimated from the offset using . For these ROIs, we calculated domain size ranging from 59 to 368 nm with an average value of 266 nm. Interestingly, when the diffusion coefficient is plotted as a function of the domain size, we still observe a decrease in diffusion associated with the increase in domain size (Fig. 5 d).
Apparent negative isotropic (Dmacro) diffusion
Two different types of iMSD curves were obtained for several areas of the cell. These are illustrated in Fig. 6, a and b. In the first one, we observe a linear decrease in iMSD with time, whereas in the other we observe an initial increase followed by a decrease in iMSD as a function of time. This type of plot results in an apparent negative long-range diffusion of CTxB/GM1 and Fig. 6 c shows the areas of the cell undergoing this process. In the iMSD simulations below, we investigate the effect of diffusion rates and binding to immobile structures.
Figure 6.
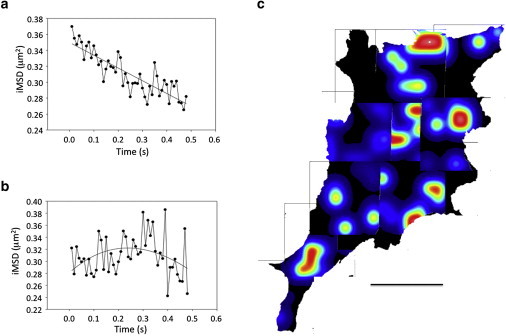
CTxB showing negative iMSD curves. (a and b) The solid lines are the fit obtained with (a) Eq. S6 in the Supporting Material (isotropic diffusion) and (b) Eq. S8 (transiently confined diffusion), respectively. In both cases, the Dmacro obtained is negative. (c) Map of the negative diffusion rates. White represents larger negative slopes of the iMSD curves, whereas blue represents lower slopes. The black area represents ROI not displaying binding or very slow diffusion. The bar equals 18.56 μm. To see this figure in color, go online.
Effect of diffusion rates
Fig. 7 shows the recovered diffusion rates after simulations and the fractions of ROI displaying a negative iMSD as a function of diffusion rates. The average diffusion rates recovered from the iMSD are in excellent agreement with the diffusion rates selected for the simulations (Fig. 7 a). Furthermore, it is evident that as the diffusion rates are decreasing to very low values, there is progressively more chances to obtain ROIs with negative iMSD, and this is reflected by the increased fraction of negative iMSD as we approach zero (Fig. 7 b). In a regime of very low diffusion, the changes in the variance as a function of delay time (Eq. S4 in the Supporting Material) become so small that the signal is dominated by the noise and can give rise to negative iMSD.
Figure 7.
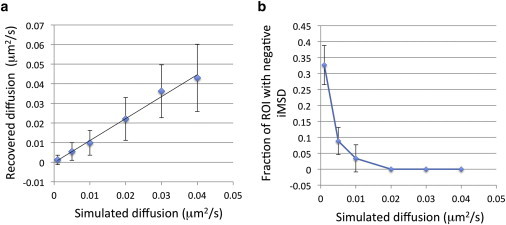
iMSD analysis of simulation of particles diffusion. (a) Calculated diffusion values from the iMSD curves versus the diffusion value used for the simulation. (b) Fractions of ROI displaying negative iMSD as a function of diffusion rates. The error bars are the SD obtained across all ROIs (n = 147) for the given diffusion rate. To see this figure in color, go online.
Effect of binding to immobile structures
We further investigated the effect of binding of the particles to immobile structures. For these simulations we used a constant diffusion rates of 0.01 μm2/s, we created an additional immobile fraction of 1000 particles and simulated different rates of binding to these immobile particles, from no binding (no reaction) to binding with reaction rates of 0.03/s, 0.33/s, and 3.33/s. Again three stacks of 1000 frames of 128 × 128 pixels were acquired for each condition and each stack analyzed using 49 ROIs. As expected, because the iMSD analysis removes the immobile component of the images (10), the presence of 1000 immobile particles did not affect the iMSD analysis and the diffusion coefficient recovered in the absence of binding matches the diffusion rates of the simulated particles (Fig. 8 a). With increasing reaction rates, there is a small tendency to recover a slightly lower diffusion coefficient but the differences are not significant. However, when the rate of binding is high (i.e., 3.33/s), there is a large increase in the variations between the diffusion coefficients recovered in each ROI, with many ROI showing negative iMSD. This trend can be better illustrated when looking at the fractions of negative ROI as a function of reaction rate presented in Fig. 8 b. We find <1% negative iMSD in the absence of reaction with an increase to nearly 40% for the faster binding rate simulated. Similar to the case of the very slow diffusion, as the binding rates increase, molecules cannot diffuse very far before they become immobile. Therefore, as the binding rates increase, the changes in the variance as a function of delay time of the STIC function becomes smaller until the signal becomes dominated by the noise resulting in increased probability of negative iMSD.
Figure 8.

iMSD analysis of simulation of particles diffusion upon binding to immobile structures. The simulated diffusion rate was 0.01 μm2/s. (a) Diffusion rates recovered from the iMSD analysis for increasing forward reaction rates (binding rates). (b) Fractions of ROI displaying negative iMSD as a function of forward reaction rates (binding rates). The error bars are the SD obtained across all ROIs (n = 147) for the given reaction rate. To see this figure in color, go online.
Therefore, both very slow diffusion and fast binding (and the combination of both) will result in a higher probability of obtaining negative iMSD. Because these are due to noise, the actual diffusion values recovered from the slope of the iMSD as a function of time are meaningless. However, the presence of ROI with negative iMSD indicates areas of very slow diffusion and/or fast binding of the fluorophores to immobile structures in the cell membrane.
Corral size and corralled diffusion (Dmicro)
Fig. 9 a maps the cell diffusion values obtained when diffusion of CTxB/GM1 is confined into corrals or is undergoing transiently confined diffusion. In these cases, the diffusion rates range between 0.04 and 2.44 μm2/s with an average of 0.34 μm2/s and a median value of 0.21 μm2/s (inset, Fig. 9 a). These molecules are diffusing in corrals or confined areas represented on the map in Fig. 9 b of sizes between 170 and 1896 nm with average and median values of 566 and 553 nm, respectively (inset, Fig. 9 b). When CTxB/GM1 is transiently confined there is no apparent correlation between diffusion rates (Dmacro and Dmicro) and corral sizes (Fig. 9 c). Although there are still large variations of corral sizes for the same diffusion and conversely a large range of Dmicro diffusion rates for the same corral size, there is nonetheless a trend toward a higher diffusion rate for larger corral sizes when all cells are plotted (Fig. S7).
Discussion
iMSD analysis gives us a unique insight into the diffusion modes of CTxB/GM1 in the cell membrane by investigating single-particle behavior in a sea of many across the whole cell membrane. In particular, we demonstrate in this work that the diffusion modes vary widely between different regions of the cell and that depending on the question asked, it may make little sense to analyze the diffusion modes in isolated cell regions without an overview of these diffusion modes across the whole cell membrane. Interestingly, although the STICS analysis was developed to investigate directed diffusion and slow protein fluxes in cells (14), in this study, we do not find evidence of this type of particle diffusion for CTxB/GM1 complexes. It is worth noting that the experimental iMSD correlation function has a symmetric Gaussian shape if the diffusion is isotropic in the region analyzed. If the diffusion is not isotropic, for example faster along one direction, i.e., due to the presence of obstacles or preferential diffusion paths, the Gaussian function will reflect this situation by elongating along one particular direction. Curved surfaces will produce additional deformation of the Gaussian shape. Although we noted in a few areas what appears to be anisotropic diffusion (such as the edges of the cells), in this work we averaged the Gaussian variances along the different directions.
Isotropic, confined, and transiently confined diffusion modes were identified across the cell membrane. Some areas display large proportions of confined and transiently confined diffusion such as images 1 and 4 in Fig. 2 d, whereas others have large areas of isotropic diffusion (e.g., images 2 and 3), and again others display a mix of the three types of diffusion (e.g., images 8, 10, and 11).
Nevertheless, we observed large variations of these diffusion modes across the cell. Therefore, we investigated the changes in diffusion of a single area as a function of time. Although there is little change in diffusion rates and modes within the first 2–3 min, after the acquisition of 2–3 stacks of images of the same cell area, we observed changes in diffusion that are not related to either the formation of clusters or the movement of the cells and could be due to either the dynamic nature of the cell membrane or an effect of the illumination of the cell with laser light, or both. Indeed, in both NIH3T3 and MDA 231 MB cells, continuous illumination induces the disappearance of corrals as illustrated by the decrease in areas undergoing Dmicro (Fig. 3) and as a result changes from confined diffusion to purely isotropic diffusion. Because both cells show a similar decrease, although not from the same starting point, it is likely that the changes of diffusion mode observed are due to the continuous illumination of the same area of the cell.
This finding may have implications when membrane diffusion processes are studied using high-powered illumination such as FRAP.
Indeed, if continuous illumination of the cell without significant photobleaching, as shown in this work, results in a change in diffusion mode then, photobleaching of an area of the cell, such as the one produced during the FRAP experiment, might perturb and modify the process under investigation and could potentially reduce the complexity of the diffusion processes.
Although we have tried to minimize the cell exposure to light when imaging the entire cell membrane, we do observe a change in diffusion mode toward isotropic diffusion when the iMSD data are compared between the beginning and the end of the cell imaging or when the same areas of the cells are imaged again after a few minutes. However, several of the ROI that were displaying confined diffusion remains. In addition, the average rate of confined diffusion (Dmicro) has not changed significantly during the time of the imaging. This suggests that in our conditions of acquisition, there is only a minimum perturbation of the diffusion modes due to illumination of the membrane. However, there is a small shift of the diffusion modes toward isotropic diffusion between image 1 and 12. In addition, we also observe an increase in the long-range isotropic diffusion rate. Because no changes of diffusion rates were observed for the continuous illumination, it is not clear what causes this increase but could simply be due to normal fluctuations in the rates of diffusion and further experiments are needed to address this question.
Despite this shift in diffusion modes, the long-range isotropic diffusion obtained by iMSD is in excellent agreement with previously reported values using FRAP microscopy (5,7).
It is interesting that the areas displaying high densities of high brightness particles (Fig. 5 a) predominantly show isotropic diffusion suggesting that aggregation of CTxB/GM1 favors this type of diffusion. However, aggregation of complexes is not the sole factor responsible for isotropic diffusion because other areas with low brightness also display this type of diffusion.
The slow diffusion of CTxB does not seem to be related to one factor in particular but is probably the result of a combination of multiple factors. We showed that CTxB/GM1 form clusters in the membrane and that these clusters represent a small fraction (3.5%) of the total number of pixels in the entire cell image. These results are in agreement with FRET data between CTxB in the membrane showing the existence of clusters (15) and also with the correlation of the FRET with the surface density of the CTxB (16) reflecting the presence of a large fraction of CTxB not in clusters. We also demonstrate that most of the CTxB/GM1 clusters are formed on average by the aggregation of 30 CTxB/GM1 complexes. Given that the diameter of CTxB is ∼6 nm and if we assume that the clusters are organized as a circular assembly this would produce nanodomains of ∼400 nm in diameter, whereas the larger clusters observed would result in nanodomains of the order of 500–600 nm. The presence of large domains is also confirmed by the increase in the iMSD offset for areas of high density of clusters determined by number and brightness analysis. The formation of such domains will result in a decrease in the diffusion rate of the CTxB/GM1 and we indeed observed substantially lower diffusion rates of the CtxB/GM1 complex. However, this effect does not solely explain the slow diffusion rate of CTxB because a slow diffusion is also observed in areas with low densities of clusters or in areas where there are no clusters of CTxB. Surprisingly, there was also an effect of the size of the dynamic partitioning into fixed nanodomains smaller than the PSF with slower diffusion rates for areas displaying larger domains (Fig. 5 d). One possible explanation is that as the lipid domain size increases, there is a higher probability for the particles diffusing outside of the domains to be incorporated into them. This would reduce the slope of the iMSD therefore leading to an apparent decrease in diffusion rates. The extrapolation of the trend line in Fig. 5, c and d, to the absence of domains (i.e., domain size = 0) results in a corrected long-range diffusion coefficient of ∼0.1 μm2/s. Therefore, both the formation of clusters and the dynamic partitioning can explain the spread of diffusion observed for the long-range (Dmacro) processes. It follows that the size of the confinement (corrals) is not affecting the long-range diffusion rates because we observe the same diffusion rates regardless of corral sizes (Fig. 7 c). However, if the confinement hinders the diffusion of CTxB/GM1 complexes and act as a barrier for diffusion, the number of barriers that the complexes will have to cross will then affect the long-range diffusion.
The diffusion rates observed within the confinement or corrals are an order of magnitude larger than the ones observed for the long-range diffusion. Taking into account the clustering of 5 GM1 per CTxB subunit, these values are comparable to the values obtained by svFCS for FL-labeled GM1 (8).
Our data on diffusion rates within corrals (Dmicro) show that the diffusion coefficient is only marginally affected by the size of the corrals because there is little correlation between Dmicro and the corral size (Fig. 7 c). In addition, for Dmicro there are no correlations between the offset and the diffusion rates (data not shown). On the other hand, Dmicro varies 10-fold for the same confinement size. Because the amount of CTxB on the cell surface can be modulated by the accessibility of GM1 (17) and that a single native binding site is sufficient for CTxB to bind to the cell membrane (6), it is highly probable that not all CTxB have 5 GM1 bound and that depending on the availability of GM1, a range of complexes can be formed. This would result in a range of diffusion rates as observed for the rates of diffusion within the corrals (Dmicro).
Finally, our simulations (see Supporting Material) show that apparent negative iMSD curves are the result of either very slow diffusion or CTxB/GM1 binding to immobile structure in the membrane. Because FRAP microscopy analysis showed that cell caveolae are immobile in the plasma membrane and held in place by the cortical actin cytoskeleton (18,19), negative iMSD, as we observed, could result in the interaction of CTxB/GM1 complexes with these structures. In addition, partitioning into very slow diffusing lipid rafts or cholesterol-rich domains could also result in negative iMSD.
We also find that these negative iMSD occur both in areas of clusters or in areas without or with a very low cluster density. Therefore, the formation of clusters of CTxB/GM1 is not a prerequisite to these interactions.
If the formation of the CTxB/GM1 complex initiate a signaling event that transiently connects the complex to the cytoskeleton as suggested by Day and Kenworthy (7), the number of GM1 binding per CTxB and/or the aggregation between CTxB/GM1 complexes might modulate this signal. Once the signal is activated, the CTxB/GM1 complex could escape from the confinement area and bind to the immobile structure of the membrane, such as caveolae or other nearly immobile membrane fraction (lipid rafts), thus providing a mechanism to facilitate the uptake of the toxin by the cell (20,21).
Acknowledgments
The authors thank Mrs. Milka Stakic for the cell cultures used in this work.
P.M. was supported by a grant from the School of Science & Technology at the University of New England. M.A.D. and E.G. were supported in part by National Institutes of Health (NIH) grants P41 GM103540 and P50-GM076516.
Supporting Material
Supporting Citations
Reference (22) appears in the Supporting Material.
References
- 1.Owen D.M., Williamson D., Gaus K. Quantitative microscopy: protein dynamics and membrane organization. Traffic. 2009;10:962–971. doi: 10.1111/j.1600-0854.2009.00908.x. [DOI] [PubMed] [Google Scholar]
- 2.Simons K., Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 3.Hammond A.T., Heberle F.A., Feigenson G.W. Cross-linking a lipid raft component triggers liquid ordered-liquid disordered phase separation in model plasma membranes. Proc. Natl. Acad. Sci. USA. 2005;102:6320–6325. doi: 10.1073/pnas.0405654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson S.A., Stinson B.M., Baumgart T. Temperature-dependent phase behavior and protein partitioning in giant plasma membrane vesicles. Biochim. Biophys. Acta. 2010;1798:1427–1435. doi: 10.1016/j.bbamem.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolf A.A., Jobling M.G., Lencer W.I. Attenuated endocytosis and toxicity of a mutant cholera toxin with decreased ability to cluster ganglioside GM1 molecules. Infect. Immun. 2008;76:1476–1484. doi: 10.1128/IAI.01286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jobling M.G., Yang Z., Holmes R.K. A single native ganglioside GM1-binding site is sufficient for cholera toxin to bind to cells and complete the intoxication pathway. MBio. 2012;3:e00401–e00412. doi: 10.1128/mBio.00401-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Day C.A., Kenworthy A.K. Mechanisms underlying the confined diffusion of cholera toxin B-subunit in intact cell membranes. PLoS ONE. 2012;7:e34923. doi: 10.1371/journal.pone.0034923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lenne P.F., Wawrezinieck L., Marguet D. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 2006;25:3245–3256. doi: 10.1038/sj.emboj.7601214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller V., Ringemann C., Eggeling C. STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys. J. 2011;101:1651–1660. doi: 10.1016/j.bpj.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Rienzo C., Gratton E., Cardarelli F. Fast spatiotemporal correlation spectroscopy to determine protein lateral diffusion laws in live cell membranes. Proc. Natl. Acad. Sci. USA. 2013;110:12307–12312. doi: 10.1073/pnas.1222097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown C.M., Dalal R.B., Gratton E. Raster image correlation spectroscopy (RICS) for measuring fast protein dynamics and concentrations with a commercial laser scanning confocal microscope. J. Microsc. 2008;229:78–91. doi: 10.1111/j.1365-2818.2007.01871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedde P.N., Dörlich R.M., Nienhaus G.U. Stimulated emission depletion-based raster image correlation spectroscopy reveals biomolecular dynamics in live cells. Nat. Commun. 2013;4:2093. doi: 10.1038/ncomms3093. [DOI] [PubMed] [Google Scholar]
- 13.Unruh J.R., Gratton E. Analysis of molecular concentration and brightness from fluorescence fluctuation data with an electron multiplied CCD camera. Biophys. J. 2008;95:5385–5398. doi: 10.1529/biophysj.108.130310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hebert B., Costantino S., Wiseman P.W. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 2005;88:3601–3614. doi: 10.1529/biophysj.104.054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nichols B.J. GM1-containing lipid rafts are depleted within clathrin-coated pits. Curr. Biol. 2003;13:686–690. doi: 10.1016/s0960-9822(03)00209-4. [DOI] [PubMed] [Google Scholar]
- 16.Kenworthy A.K., Petranova N., Edidin M. High-resolution FRET microscopy of cholera toxin B-subunit and GPI-anchored proteins in cell plasma membranes. Mol. Biol. Cell. 2000;11:1645–1655. doi: 10.1091/mbc.11.5.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lingwood D., Binnington B., Simons K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011;7:260–262. doi: 10.1038/nchembio.551. [DOI] [PubMed] [Google Scholar]
- 18.Pelkmans L., Kartenbeck J., Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001;3:473–483. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 19.Thomsen P., Roepstorff K., van Deurs B. Caveolae are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking. Mol. Biol. Cell. 2002;13:238–250. doi: 10.1091/mbc.01-06-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujinaga Y., Wolf A.A., Lencer W.I. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to endoplasmic reticulum. Mol. Biol. Cell. 2003;14:4783–4793. doi: 10.1091/mbc.E03-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nichols B.J., Lippincott-Schwartz J. Endocytosis without clathrin coats. Trends Cell Biol. 2001;11:406–412. doi: 10.1016/s0962-8924(01)02107-9. [DOI] [PubMed] [Google Scholar]
- 22.Qian H., Sheetz M.P., Elson E.L. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 1991;60:910–921. doi: 10.1016/S0006-3495(91)82125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.