

Abstract
The leucine transporter (LeuT) is a bacterial homolog of the human monoamine transporters, which are important pharmaceutical targets. There are no high-resolution structures of the human transporters available; however, LeuT has been crystallized in several different conformational states. Recently, an inward-facing conformation of LeuT was solved revealing an unexpectedly large movement of transmembrane helix 1a (TM1a). We have performed molecular dynamics simulations of the mutated and wild-type transporter, with and without the cocrystallized Fab antibody fragment, to investigate the properties of this inward-facing conformation in relation to transport by LeuT within the membrane environment. In all of the simulations, local conformational changes with respect to the crystal structure are consistently observed, especially in TM1a. Umbrella sampling revealed a soft potential for TM1a tilting. Furthermore, simulations of inward-facing LeuT with Na+ ions and substrate bound suggest that one of the Na+ ion binding sites is fully disrupted. Release of alanine and the second Na+ ion is also observed, giving insight into the final stage of the translocation process in atomistic detail.
Introduction
Transport proteins are essential for life because they allow selective exchange of ions and molecules across cell membranes (1). The monoamine transporters facilitate Na+-coupled reuptake of the neurotransmitters dopamine, serotonin, and norepinephrine from the synaptic cleft (2). These transporters are important drug targets because they are involved in diseases such as major depression, anxiety disorders, attention deficit hyperactivity disorder, and autism (3,4). However, the actual mechanism of transport has yet to be fully elucidated.
One of the main obstacles is the lack of experimentally determined atomistic models of the mammalian monoamine transporters (5). However, in 2005 the structure of a homologous bacterial leucine transporter (LeuT) from Aquifex aeolicus was published (6). Since then, numerous structures with similar topology have been solved, and these, along with several other LeuT structures, reveal a number of distinct transporter conformations (5). Most recently, the structure of the dopamine transporter from Drosophila melanogaster (dDAT) was published (7), and this is, as of this writing, the closest relative to the mammalian monoamine transporters for which a structure is known.
Computational studies of the bacterial transporters (5,8–15) along with homology models of the human transporters (16–19) have led to a better understanding of how these transporters function (20). The high similarity (21) between the binding mode of the inhibitor bound in the recent dDAT structure (7) and a computational model of a similar inhibitor bound to the serotonin transporter (22) also demonstrate that reliable results can be obtained from computational studies of this family of transporters. The transport process is thought to follow the alternating access mechanism where the transporter alternates between conformations in which the substrate binding site is accessible from either the extracellular or the intracellular side of the membrane (see Fig. 1 A). The core motif for this family of transporters consists of 10 transmembrane helices (TMs) arranged such that TM1–5 can be superimposed onto TM6–10 by an ∼180° rotation. This protein fold is known as the 5-TM inverted repeat or the LeuT fold (12). The first published LeuT structure captured the transporter in an outward-facing conformation with a few residues occluding the access to the central leucine binding site (6). This conformation is hence referred to as “outward-occluded”. Later, in 2008, a structure of LeuT in a fully outward-open conformation with tryptophan bound was published (23) and in 2012, the first inward-open LeuT structure was published (24). Comparison of the outward- and inward-facing crystal structures indicates that the conformational change connecting these structures can be largely described as a movement of a four-helix bundle (TM1, TM2, TM6, and TM7) with respect to a four-helix hash motif (TM3, TM4, TM8, and TM9) as well as a change in conformation of TM5 (Fig. 1 B).
Figure 1.
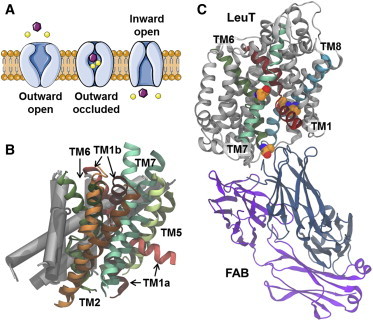
Conformational changes in the transport cycle and the crystal structure of inward-facing LeuT. (A) Schematic representation of the transport cycle divided into three conformational states with the substrate (purple) and the cotransported Na+ ions (yellow). (B) Comparison of the outward- and inward-open state of LeuT (PDB: 3TT1 and PDB: 3TT3 (24)). The five helices displaying the largest conformational change between the two states (TM1–2 and TM5–7) are shown as ribbons, with TM1 (red), TM2 (orange), TM5 (light green), TM6 (dark green), TM7 (turquoise), and TM3-4 and TM8-10 as gray cylinders. TMs representing the inward-open state (bright colors); TMs corresponding to the outward-open state (dark colors). (C) The structure of the inward-open LeuT is shown (gray) with TM1 (red), TM6 (dark green), TM7 (light green), and TM8 (cyan). The two chains in the Fab domain are shown (blue and violet). The four mutated residues (Tyr-268-Ala in TM6, Lys-288-Ala in TM7, and Thr-354-Val and Ser-355-Ala in TM8) are highlighted (as spheres) and colored by element (carbon atoms shown in orange). To see this figure in color, go online.
Whereas the previous outward-open and outward-occluded conformations have been crystallized using wild-type LeuT, the crystal structure of inward-facing LeuT contains four point mutations as well as an intimately bound antibody fragment (Fab) (see Fig. 1 C) that was added to stabilize the inward-facing conformation and enhance crystallization behavior. In general, the inward-facing conformation of LeuT is similar to structures of homologous transporters (10,25,26), and consistent with suggested transport mechanisms (10,27). However, a large difference in the arrangement of the N-terminal part of the TM1 domain (TM1a) was observed (24). The crystallization procedure, together with the large movement of TM1a, raise questions as to how well the solved structure mimics the wild-type transporter in a native environment, and whether the point mutations and the Fab fragment have perturbed the overall conformation of LeuT away from the native inward-facing conformation.
Herein, we address the above questions through extensive molecular dynamics (MD) simulations of LeuT in the inward-facing conformation with the transporter embedded in a lipid membrane. Multiple repeats have been performed for each simulation for improved sampling (16,28). Despite the name of the transporter, LeuT transports a number of different hydrophobic amino acids and alanine is actually transported more efficiently than leucine (23). While it is known that the transport process involves cotransport of Na+ ions, the coupling between substrate and ion transport is not fully understood. To elucidate the sequence of these events, we present simulations that can probe release of alanine and Na+ ions from the inward-facing conformation in atomistic detail.
Materials and Methods
Preparation of simulation systems
The crystal structure of LeuT in the inward-facing conformation (Protein Data Bank (PDB) PDB: 3TT3 (24)) was prepared for MD simulations using the SCHRÖDINGER SUITE 2012 (29) and VMD (30), Ver. 1.9.1. Residues 5–10 of the N-terminal of LeuT, which were missing in the crystal structure, were added using the coordinates of an outward-occluded LeuT structure (PDB: 2A65 (6)). This was done by aligning residues 11–23 in the two structures, extracting the coordinates of residues 5–10 and including them in the model of inward-facing LeuT. The detergent molecule found close to TM9 and TM12 in the crystal structure was deleted, and for the simulations without the Fab antibody fragment, this domain was deleted as well. Atoms missing in the side chains were added using the PROTEIN PREPARATION WIZARD in the SCHRÖDINGER SUITE 2012 followed by a restrained relaxation of the entire structure. The pKa values for all titratable groups were estimated using PROPKA, Ver. 3.1 (31–34). Based on these results and the local environments with respect to hydrogen bonding, Glu-62, Glu-112, and Glu-287 were all modeled as protonated, while the remaining acidic residues were modeled as charged. All His residues were modeled as δ-tautomers. The C- and N-terminals of LeuT were capped with an N-methyl and an acetyl group, respectively. The resulting model is referred to as MUT. A wild-type version of the transporter was prepared by changing three of the mutated residues (Ala-268, Val-354, and Ala-355) back to the native residues (Tyr-268, Thr-354, and Ser-355). The Lys-288-Ala mutation was not changed, because it has been shown that this mutation enhances substrate flux in proteoliposomes (35). This model is consequently referred to as WT. End-point structures from simulations of the WT setup was further used for constructing substrate-bound models with alanine placed in the central binding site and Na+ ions in the Na1 and Na2 sites. These are referred to as ALA. Coordinates of alanine in the central binding site and the Na+ ions in the Na1 and Na2 sites were obtained from a PDB structure of outward-occluded LeuT with alanine bound in the central binding site (PDB: 3F48 (23)), by aligning the PDB structure to the WT model using the residues surrounding the substrate and ion binding sites for the alignment.
Because all crystal structures of LeuT obtained without the use of antibodies contain LeuT dimers and the homologous human serotonin transporter functions as a dimer or higher oligomer (36–38), a LeuT dimer was built for the simulations according to the symmetry of the LeuT PDB: 3F3A. From a practical point of view, the use of a dimer also reduces the computational costs because two monomer trajectories are obtained for each setup. The PSFGEN plugin in the VMD program was used to add hydrogen atoms to the dimer using CHARMM36 (39) topology files. The LeuT dimer was inserted into a POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) lipid bilayer emulating a bacterial cell membrane (40). The size of the membrane was chosen such that at least 20 Å of lipids surround the dimer. The dimer was positioned in the membrane according to the alignment of PDB: 2A65 in the Orientations of Proteins in Membranes database (41). The system was solvated by adding a 15 Å layer of water molecules on both sides of the membrane. NaCl was added to a concentration of 0.2 M. Finally, a steepest-descent minimization of 20,000 steps of the entire simulation system was performed in GROMACS 5.0 (42).
In addition to the three dimer systems (WT, MUT, and ALA), a model of monomeric, inward-facing LeuT interacting with the Fab fragment was also constructed (FAB), based on the PDB: 3TT3 crystal structure. In the light chain of Fab, disulfide bonds were added between Cys-23 and Cys-92 as well as between Cys-138 and Cys-198. His-38 and His-202 were modeled as ε-tautomers, while His-193 was modeled as the δ-tautomer. In the heavy chain, disulfide bridges were added between Cys-22 and Cys-96 as well as between Cys-146 and Cys-201. His-32, His-33, His-170, and His-205 were all modeled as ε-tautomers. The C-terminals of the light and heavy chain of the antibody were capped with N-methyl groups and the C- and N-terminals of LeuT were capped with an N-methyl and an acetyl group, respectively. The simulation system was built according to the same procedure as described above for the dimer systems. Each of the dimer systems contain ∼130,000 atoms and have a box size of ∼140 × 120 × 100 Å3, while the monomer system with the LeuT-Fab complex contains ∼160,000 atoms and has a box size of ∼130 × 110 × 150 Å3. An overview of the setups is given in Table 1.
Table 1.
Overview of simulated systems with respect to protein setup and substrates
| Description | WT | MUT | ALA | FAB |
|---|---|---|---|---|
| Mutations | K288A | Y268A, K288A, T354V, S355A | K288A | K288A |
| Substrate/ions | None | None | Ala, 2 × Na+ | None |
| Fab fragment | No | No | No | Yes |
| Number of LeuT monomers | 2 | 2 | 2 | 1 |
Simulation protocol
The MD simulations were performed with GROMACS 5.0 (42) using the CHARMM36 force field (43,44). The water molecules were described by the TIP3P water model (39). The simulations were performed in an NPT ensemble, unless otherwise stated, and a time step of 2 fs was applied. The P-LINCS algorithm (45) was applied to constrain all bond lengths for bonds involving hydrogen. The pair-list, stating for which atom pairs the nonbonded interactions should be calculated, was updated every 10th time step. The temperature was maintained at 310 K using a Nosé-Hoover extended ensemble with τT = 1.0 ps. The pressure was kept constant at 1.0 atm by the Parrinello-Rahman method with τP = 1.0 and a compressibility of 4.5 × 10−5 bar−1. Short-range nonbonded interactions were calculated with a switching function (46) starting at 10 Å and a cutoff distance of 12 Å. Long-range electrostatics were calculated utilizing the particle-mesh Ewald algorithm (47).
The APO, MUT, and FAB systems were equilibrated in three stages. First, the lipid tails were melted for 0.5 ns while all other atomic positions were restrained (k = 1000 kJ/mol/nm2). This step was done in an NVT ensemble using a modified Berendsen thermostat, which includes a stochastic term. Second, a 2-ns equilibration was performed with the heavy atoms of the protein restrained at their positions through a harmonic potential with a force constant of 1000 kJ/mol/nm2. Third, a 4-ns free equilibration was performed. The ALA models were equilibrated through a 5-ns MD simulation with the heavy atoms of the substrate and Na+ ions in the binding sites restrained (k = 1000 kJ/mol/nm2). Two production runs of 200 ns each were performed for each of the four LeuT systems entailing a total of 1.6 μs of unbiased MD simulation and 2.8 μs of trajectories for analysis due to the dimeric nature of three of the systems. Coordinates for all atoms were saved every 5 ps and, unless otherwise stated, 10,000 snapshots from the trajectories (corresponding to one snapshot per 20 ps) were used for analysis. The individual trajectories are referred to by the setup name (WT, MUT, ALA, and FAB), the repeat number (1 or 2), and the monomer of the complex (A or B) such that, e.g., WT-1A corresponds to monomer A from repeat 1 of the WT system.
Umbrella sampling
Umbrella sampling was performed using snapshots from the WT-1 simulation. The distance between the center of mass of the Cα atoms in residues 11–14 in TM1 and residues 89–98, 361–370, and 495–504 in TM3, TM8, and TM12, was used as a measurement of the extent of kinking of TM1a. Simulations were performed for 31 equally spaced windows for distances between 37.0 and 43.0 Å. Three simulations with a simulation time of 5 ns were performed at each window with each simulation starting from an individual set of randomly assigned velocities. A force constant of k = 10,000 kJ/mol/nm2 was used to restrain the distance. A free energy profile was constructed with the weighted histogram analysis method (48) implemented in the G_WHAM tool (49) in GROMACS using only the last 4 ns of each simulation. Errors were estimated using bootstrapping.
Analysis
Analysis of simulation trajectories was performed using VMD (30). Before analysis, each trajectory was aligned to the initial simulation system. Only translation in the membrane plane and rotation around the membrane normal (corresponding to the z axis) were applied to ensure that the orientation of the membrane was not changed by the alignment.
The angle of the transmembrane helices in LeuT with respect to the membrane normal was determined in the following way. For each helix, a vector was assigned by minimizing the distance from the Cα atoms in the helix to the vector. Vectors were determined for TM1–10, with TM1 and TM6 each described by two vectors due to their unwound middle regions. An overview of the residues used for each helix is given in Table S1 in the Supporting Material. Snapshots from the last 100 ns of the simulations (5000 per simulation) were used for the calculation. The helix tilt angles of PDB: 3TT1 and PDB: 3TT3 were also determined after aligning each of the two structures according to their orientation in the Orientations of Proteins in Membranes database (41).
The surface representing the average position of the carbonyl oxygen atoms in the two ester linkages in the POPE lipids was determined using the protocol described by Sonntag et al. (50). In short, the z positions of the carbonyl atoms are mapped onto an x,y grid for each simulation frame and an average position is determined for each grid point. A grid spacing of 4 Å was used for the procedure. For each leaflet, the deviation in the z position in each grid point relative to the most frequently observed z position in the leaflet was plotted.
The occupancy of the two Na+ ion binding sites (Na1 and Na2) was determined by identifying Na+ ions within 4 Å of the center of mass of Asn-27, Thr-254, and Asn-286 (Na1 site) or Val-23, Ala-351, and Ser-355 (Na2 site).
Results and Discussion
The stability of the inward-facing structure of LeuT, derived by x-ray, is assessed by comparing simulations of a wild-type-like variant (referred to as WT) with simulations of the mutated variant, which was used in the x-ray experiments (referred to as MUT), as well as simulations of apo wild-type-like LeuT in complex with the antibody fragment from the x-ray experiments (i.e., FAB). Furthermore, the release of Na+ ions and substrate from the inward-facing conformation is investigated in atomistic detail. For three of the setups (WT, MUT, and ALA), the simulation system contains two LeuT monomers, which yields two protein trajectories for analysis from each simulation. An overview of the setups can be found in Table 1.
Stability of the simulation systems
To estimate the overall stability of each simulation system, the root-mean-square deviation (RMSD) and fluctuation (RMSF) was determined for the Cα atoms in LeuT (see Fig. S1 in the Supporting Material). The RMS deviation values level off at ≤2.0 Å for all simulations, indicating that all four types of simulation systems are stable on the simulated timescale. The RMS fluctuation analysis shows that the transmembrane parts are rigid with an average fluctuation of ∼1 Å, and the terminal and loop regions are flexible, as expected. Furthermore, it should be noted that the extracellular gate consisting of a salt-bridge interaction between Arg-30 and Asp-404 remains closed in all simulations.
Stability of transmembrane helices
The stability of the domains of the inward-facing conformation, as found in the crystal structure, has been assessed from the MD simulations through measurements of helix tilt angles with respect to the membrane normal. In this analysis, each helix is represented by a least-squares fit vector with respect to the position of the Cα carbon atoms (see details in Materials and Methods). Due to the unwound regions in the central parts of TM1 and TM6, these two helices have each been separated into two halves in the analysis. Only the final 100 ns of each simulation were used for analysis to ensure that the systems have reached fully equilibrated conformations. The average helix tilt angles of TM1–10, which constitute the LeuT fold, are plotted in Fig. 2. Along with the simulation results, the corresponding helix tilt angles in the inward-open (PDB: 3TT3) and outward-open LeuT structures (PDB: 3TT1) are also shown for comparison. TM1a and TM6b, both of which are part of the bundle, exhibit large fluctuations in the simulations. TM1a generally moves toward smaller tilting angles than what is observed in the crystal structure of the inward-facing state, corresponding to structures that are less inward-open. On the other hand, for the WT and MUT setups, TM6b exhibits a larger angle than that in the crystal structure. In effect the two helices come closer together during the MD simulations, resulting in a more occluded conformation. However, the FAB simulation stands out as the only one with indications of a slightly smaller tilt angle of TM6b. Although the large error bars makes it impossible to draw any solid conclusions, this does suggest that the presence of the Fab domain during the crystallization procedure may have affected the position of TM6b. The large flexibility of TM1a is consistent with the weak electron density observed for TM1a in the x-ray structure compared to the remaining parts of the solved structure (24). The smallest tilt angles for TM1a are observed in the apo WT simulations. However, a shift toward smaller angles is observed for all three types of simulation systems, and it appears to be independent of the point mutations as well as the presence of the Fab antibody, although the value of the most favored tilt angle does appear to vary between the setups.
Figure 2.
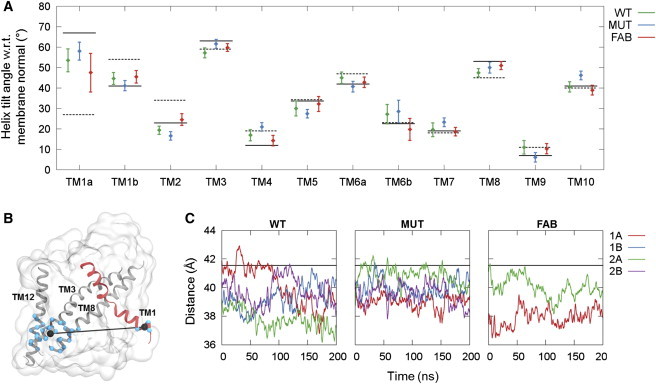
Helix tilt angles and fluctuation of TM1a. (A) Plot of the average angle between the membrane normal and TM1–10 during the last 50 ns of the simulations. (Error bars) Standard deviations. (Solid black line) Helix tilt angle for the inward-open starting structure (PDB: 3TT3 (24)); (dashed black line) corresponding angle for the outward-open conformation (PDB: 3TT1 (24)). (B) Depiction of the TM1a-scaffold distance used as a simple descriptor of TM1a tilting. The distance is measured between the center of mass of the Cα atoms in residues 11–14 in TM1 (shown as blue spheres on red ribbon) and residues 89–98, 361–370, and 495–504 in TM3, TM8, and TM12 (shown as blue spheres on gray ribbons). The two center-of-mass positions are shown (black spheres) and the measured distance is displayed (black line). (C) Plots of the time evolution of the TM1a-scaffold distance. To see this figure in color, go online.
To further investigate the flexibility and preferred position of TM1a, a simpler descriptor of the tilting of TM1a was introduced that allows us to explore the energy landscape associated with the observed tilting of TM1a. The descriptor corresponds to the center-of-mass distance between residues in the N-terminal end of TM1a and residues in the intracellular ends of TM3, TM8, and TM12, as shown in Fig. 2 B, and will be referred to as the “TM1a-scaffold distance”. As seen in Fig. 2 C, the distance varies from ∼36 to 43 Å during the simulations, corresponding to sampling of tilting angles between ∼40 and 70°. It is clear that the TM1a helix is very flexible and samples a large range of angles during each 200-ns simulation. The largest variation for the WT system is found for WT-1A, and snapshots from this simulation were used to perform umbrella sampling along the TM1a-scaffold distance. The resulting energy profile is shown in Fig. 3 A. In accordance with the results in Fig. 2 C, the energy minimum is found at a distance of 41.2 Å, which is shorter than the distance observed in the crystal structure of inward-facing LeuT, although only by ∼0.5 Å. The rather gentle slope of the energy profile on either side of the minimum suggests that TM1a is very dynamic, and does not encounter any large energy barriers. The slope toward smaller distances, and hence smaller tilting angles, is more gentle than toward larger distances, which most likely explains why distances smaller than the optimal distance dominate the plots in Fig. 2 C. In Fig. 3 B, the simulations results are compared with the inward-facing state as observed in crystal structures of LeuT and three homolog transporters Mhp1 (10), BetP (25), and vSGLT (26). It is obvious that while the position of TM1a is similar for the three latter transporters, TM1 is much more kinked in the crystal structure of LeuT. The snapshots of the smallest and largest distances observed during the WT simulations highlight that angles as small as those observed in the three homologs, as well as angles as large as observed in the LeuT crystal structure, are sampled during the simulations. However, the optimal angle, corresponding to the minimum energy distance in Fig. 3 B, is closer to the LeuT crystal structure than to any of the other three transporters. This suggests that LeuT differs from these three transporters with respect to how far upwards TM1a is able to move during the conformational cycle.
Figure 3.
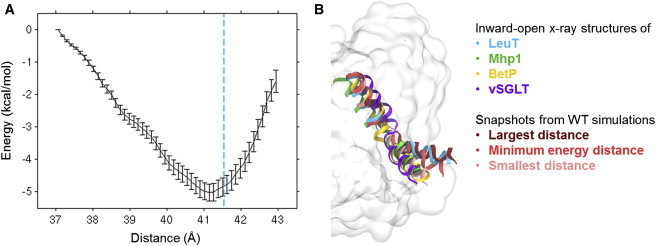
Energy profile for TM1a and comparison with other LeuT-fold transporters. (A) Energy profile obtained from umbrella sampling along the TM1a-scaffold distance. (Dashed blue line) Distance found in the crystal structure of inward-facing LeuT. (B) Comparison of TM1a positions observed in the simulations and TM1a positions found in crystal structures of the inward-facing state of four LeuT-fold transporters, namely LeuT (PDB: 3TT3 (24)), Mhp1 (PDB: 2X79 (10)), BetP (PDB: 3P03 (25)), and vSGLT (PDB: 2XQ2 (26)). The structures have been aligned using the Cα atoms in TM1b, TM2, TM6, and TM7. To see this figure in color, go online.
It has been suggested from both single-molecule fluorescence resonance energy transfer experiments (51) and from structures of other LeuT fold transporters that the movement of TM1a is involved in the transport cycle, although the extent of the movement is not fully known. Based on our simulations, we suggest that in the inward-facing conformation, which LeuT goes through during the transport cycle, TM1a adopts a less kinked conformation than that of the crystal structure, most likely with a tilt angle fluctuating at ∼60° rather than at ∼68°. This is also in accordance with a recent MD study of LeuT in which TM1a tilting angles slightly smaller than what is found in the crystal structure were observed for the inward-facing state of LeuT (15). Furthermore, the shift of TM6b toward larger angles indicates that the position of TM6b in the crystal structure may be slightly perturbed with respect to the inward-facing conformation that is part of the transport cycle. Overall, our results suggest that in future studies based on the inward-facing crystal structure of LeuT such as homology modeling of human homologs, one should be extremely careful in drawing any solid conclusions based on the conformation of TM1a, and that relaxation of the LeuT crystal structure by MD simulations before homology modeling may be advantageous.
Membrane adaption around the inward-facing conformation of LeuT
Large conformational changes in membrane proteins may also lead to changes in the lipid packing around the protein (50). To quantify how the lipids adapt to the inward-facing conformation of LeuT, the average displacement of the ester carbonyl oxygen atoms in the POPE lipids with respect to the overall average position for all the ester carbonyl oxygen was calculated on a grid around the protein dimer for each of the two WT trajectories. The large kink in TM1 could be expected to induce a drastic change in the local membrane environment. However, as seen in Fig. 4, only small membrane adaptations in the inner leaflet are observed around TM1a during the simulations. Thus, positioning of TM1 in the kinked conformation found in the inward-facing state does not seem to require significant adaptions of the surrounding lipid environment. The most N-terminal residue of TM1a, Arg-11, consistently interacts with the lipid headgroups during the simulations, revealing that TM1a is not placed centrally in the hydrophobic part of the membrane. It should be noted that the energy barrier associated with deformation of the POPE bilayer might be too high for the process to be observed on a 200-ns timescale. Thus, it is possible that additional membrane adaption around TM1a would be observed for longer simulation times. On the other hand, it has previously been shown for a number of different bilayers that lipid adaption around proteins can be observed in 50–100-ns MD simulations (50,52).
Figure 4.
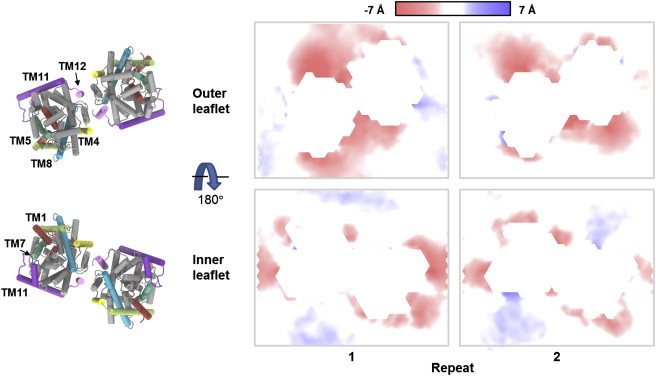
Local deviations from the average lipid membrane thickness. The average position of the ester oxygen atoms in the POPE lipids has been calculated on a grid with a 4 Å spacing and is shown as a surface colored according to deviations along the membrane normal. (Red and blue areas) Membrane thinning and thickening, respectively. The LeuT dimer at the end point of the WT-2 simulation is also shown with TM1 and the N-terminal (red), TM4 (yellow), TM5 (lime green), TM7 (green), TM8 (blue), TM11 and intracellular loop 5 (purple), TM12 (pink), and the remaining protein (gray). To see this figure in color, go online.
Interestingly, a recently published structure of the dopamine transporter (7) in an outward-open conformation included a cocrystallized cholesterol molecule on top of TM1a, and it was suggested that cholesterol has an inhibitory effect on the upward movement of TM1a. Thus, even though our data suggests that no significant rearrangement of the lipids around TM1a occurs during the conformational change to the inward-facing conformation, other components of the membrane may affect the transport process. The most distinct distortion observed in the simulations is a thinning of the outer leaflet around the dimer interface and TM11. An analysis of the protein-lipid interactions revealed that this is caused by a hydrophobic mismatch between the POPE lipids and a number of charged and polar residues in the extracellular ends of TM4, TM11, and TM12. Specifically, the hydrophilic residues Ser-165, Arg-469, Glu-470, and Trp-484, which were all positioned in the hydrophobic part of the bilayer in the beginning of the simulation, interact with the lipid headgroups causing a downward movement of the lipids around these residues.
In a recent computational study by Mondal et al. (52) aiming at investigating the effect of a LeuT monomer on a POPC bilayer, local deformations of the bilayer around the extracellular end of TM11 were also detected. Thus, the observed membrane thinning around TM11 is seemingly independent of whether the bilayer consists of POPC or POPE lipids and whether a monomer or a dimer of LeuT is used in the simulations. Membrane thinning is also seen around the extracellular ends of TM5 and TM8, where it is mainly caused by interactions between the lipid headgroups and Arg-212 and Thr-338. Additionally, residues at the intracellular end of TM11 and in the loop connecting TM10 and TM11 (Asp-430, Arg-446, Tyr-450, Arg-453, and Tyr-454) are also observed to interact with the lipid headgroups, which results in a local thinning of the inner leaflet around the intracellular end of TM11. In the study by Mondal et al. (52), local deformations in the inner membrane leaflet around TM1 and TM7 occurred as a consequence of the charged Lys-288 residue positioned centrally in TM7. Because all of our simulations are performed with the K288A mutant, also used for crystallization, such deformations are not expected to occur. Indeed, as is seen from Fig. 4, thinning of the bilayer around intracellular end of TM7 is not observed.
Na+ and substrate transport
The transport of substrate through LeuT and other related transporters is coupled to the cotransport of Na+ ions (53). However, the details of how the transport of the substrate is linked to the transport of Na+ are yet to be fully elucidated, as is the sequence of events. The crystal structure of LeuT in the inward-facing conformation contains neither substrate nor ions (24). While the Na2 sodium ion binding site is dramatically different between the outward- and inward-facing conformation of LeuT due to the large movement of TM1a, the Na1 sodium ion binding site and, in particular, the central substrate binding site are quite similar in the two states (see Fig. 5 A). Thus, it is not clear from the structures alone which of the events lead to the release of substrate and ions to the intracellular side. To investigate this in atomic detail, we performed MD simulations of the inward-facing conformation of wild-type like LeuT with Na+ ions added to the two Na+ ion binding sites (Na1 and Na2) and alanine bound in the central binding site (ALA setup). The substrate and ion were added to the end-point structure of the WT simulations to ensure full equilibration of LeuT before the insertion.
Figure 5.
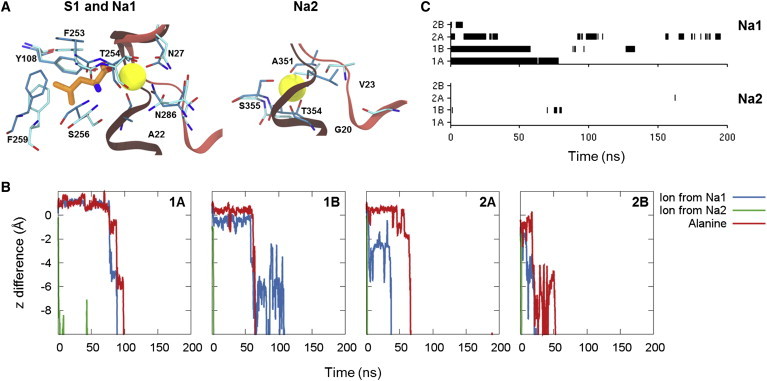
Substrate and ion binding sites and release. (A) Substrate and ion binding sites for the outward- and inward-facing state of LeuT. The structure of the outward-occluded conformation of LeuT (PDB: 2A65 (6)) is shown in darker colors and the structure of the inward-open conformation (PDB: 3TT3 (24)) is shown in lighter colors. TM1 (red) and the residues surrounding the substrate and ion binding sites are shown (sticks colored according to element with the carbon atoms in cyan). The substrate bound in the outward-occluded conformation is shown (sticks colored according to element with the carbon atoms in orange and the two Na+ ions are shown as yellow spheres). (B) Plot of position relative to the starting position for the two Na+ ions initially bound in the Na1 and Na2 site and the center of mass of alanine. (C) Plot of occupancy of each of the two Na+ ion binding sites during the ALA simulations. To see this figure in color, go online.
From these simulations, we are able to observe the stability of substrate and Na+ ion binding, as well as possible release pathways. Fig. 5 B shows the movement of the center of mass of the substrate and each of the two initially bound Na+ ions along the direction of the membrane-normal relative to the initial position. As seen from the figure, the sodium ion in the Na2 site is rapidly released in all of the simulations. In the LeuT construct used to obtain the inward-facing structure of LeuT, the Na2 site is destructed through mutations of T354 and S355. The residues in the Na2 site have been reconstructed in the ALA simulations, but it is clear from the simulations that Na+ ion binding in the Na2 site is not very favorable when the transporter is in the inward-facing conformation. Two of the residues coordinating Na+ in the Na2 site in the outward-facing state, Gly-20 and Val-23, belong to TM1a and are displaced from the site in the inward-facing conformation due to the large kink in TM1 (see Fig. 5 A). Although TM1 is seen to become less kinked during our simulations, the Na2 site is not completely reformed in any of the ALA simulations (Fig. S2), supporting that Na+ binding in the Na2 site is unfavorable for the inward-facing state.
In all of the four trajectories, release of a Na+ ion from the Na1 site is also observed. In all instances, the release occurs after the release of the Na+ ion from Na2. Generally the release of Na+ from the Na1 site is observed to be much slower than release from the Na2 site, suggesting that the Na1 site is less disrupted in the inward-facing state. This is also supported by the observation that binding of ions from the solvent is mainly observed for the Na1 site rather than the Na2 site for all of the simulation setups (Figs. 5 C and S3).
In all of the four substrate-bound LeuT trajectories, we observed the release of not only the Na+ ions, but also the alanine substrate (Fig. 5 B). The release of substrate occurs after the release of Na+ from the Na2 site and either after or along with the release of Na+ from the Na1 site. In the central binding pocket, the substrate interacts with the Na+ ion in the Na1 site through its charged carboxyl group. Based on our results, it seems likely that the loss of this interaction, which occurs when Na+ diffuses away from the Na1 site, enables the substrate to be released from the binding pocket. Due to the limitations of MD in terms of the timescales that can be sampled, it is possible that the ALA simulations have not been initiated from fully equilibrated inward-facing structures, which may affect the obtained results. As seen from Fig. 2 C, the end points of the WT simulations, which have been used as starting point for the ALA simulations, correspond to four different positions of TM1a. Yet, consistent results are observed from the ALA simulations, suggesting that the observed trends are not highly dependent on the exact position of TM1a. Other studies have previously focused on the release of Na+ ions from vSGLT, showing that the crystal structure of inward-facing conformation of vSGLT corresponds to an ion-releasing state (54) and that release of Na+ from the Na2 site in vSGLT triggers the release of substrate to the intracellular site (26). Accelerated MD simulations of LeuT has also shown that the removal of Na+ from the Na2 site leads to transitions toward the inward-facing state (55), suggesting that release of Na+ from Na2 is necessary for the formation of the inward-facing state. Furthermore, based on steered MD simulations of LeuT, Shi et al. (13) observed that pulling the substrate out of the binding pocket and toward the cytoplasm is easier in the absence of Na+ in the Na2 site, which also suggests that release of Na+ from the Na2 site occurs before substrate release. These studies on the vSGLT and LeuT are consistent with our results, as we also observe rapid release of Na+ from the Na2 site and release of substrate subsequent to release of Na+ from Na2, and points to a common mechanism for the two homologous transporters. The release pathway for the substrate is mainly along the intracellular parts of TM1, TM6, and TM8, which is in agreement with the release pathway for the human serotonin transporter indicated by accessibility measurements (27) and consistent with observations from MD simulations of the human serotonin transporter (16). Cotransport of the substrate and Na+ ion from the Na1 site has previously been observed in random acceleration MD simulations starting from leucine-bound outward-facing LeuT (56). In MD simulations starting from an inward-facing state of alanine-bound LeuT obtained from a targeted MD simulation, Cheng and Bahar (15) also observed release of Na+ from the Na2 site followed by release of Na+ from the Na1 site and alanine from the central substrate binding site. However, in the same study, alanine release was also seen in other simulations to occur without prior release of any of the Na+ ions. Thus, it is possible that inward release of substrate and ions from LeuT can occur by several distinct mechanisms, which differ in the sequence of events. Because the release of substrate is generally a slow process in comparison with the timescales obtainable by MD simulations, most simulation studies only explore one or a few release events, making it difficult to obtain a complete picture of the process. Therefore, very long simulation times, such as the recent microsecond simulations of ion and substrate binding events published by Zomot et al. (14), may help to give further insight into this question in the future.
Conclusions
In this study, we have performed MD simulations of the inward-facing conformation of LeuT, a bacterial homolog of the human monoamine transporters. The study investigates the stability of the conformation found in the crystal structure of inward-facing LeuT, the interactions between LeuT and the surrounding membrane, and the release of substrate and ions from the transporter. Multiple repeats of equilibrium MD simulations show that overall the inward-facing conformation from the crystal structure is stable. However, a consistent downward movement of TM1a occurs, being independent of the presence of point mutations and the Fab domain. It is thus likely that the crystal structure represents an extreme conformation of TM1 and that the helix does not bend to such a high degree during the transport cycle. Hence, additional care should be taken before making conclusions based on the positioning of TM1, e.g., in homology models based on LeuT.
The simulations with substrate and ions bound in the central cavity show that the Na2 site is unstable in the inward-facing conformation, and release of Na+ occurs in all the simulations. Subsequent release of alanine and Na+ from the Na1 site is also observed in all of the simulations, with both ions and substrate following the experimentally proposed intracellular pathway for substrate release. Thus, our simulations suggest a possible sequence of release, which is initiated by unbinding of Na+ from the Na2 site followed by the release of substrate and the Na+ ion from the Na1 site into the intracellular milieu in line with the proposed mechanism for the vSGLT.
Acknowledgments
Financial support for this study was received from The Danish Council for Independent Research/Natural Sciences, The Danish Council for Independent Research/Technology and Production Sciences, and the Danish National Research Foundation (grant No. DNRF59). Computations were made possible through grants from the Danish Center for Scientific Computing.
Footnotes
Heidi Koldsø’s current address is Department of Biochemistry, University of Oxford, Oxford, United Kingdom.
Supporting Material
References
- 1.Nugent T., Jones D.T. Membrane protein structural bioinformatics. J. Struct. Biol. 2012;179:327–337. doi: 10.1016/j.jsb.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Kristensen A.S., Andersen J., Gether U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- 3.Hahn M.K., Blakely R.D. The functional impact of SLC6 transporter genetic variation. Annu. Rev. Pharmacol. Toxicol. 2007;47:401–441. doi: 10.1146/annurev.pharmtox.47.120505.105242. [DOI] [PubMed] [Google Scholar]
- 4.Immadisetty K., Geffert L.M., Madura J.D. New design strategies for antidepressant drugs. Expert Opin. Drug Discov. 2013;8:1399–1414. doi: 10.1517/17460441.2013.830102. [DOI] [PubMed] [Google Scholar]
- 5.Zdravkovic I., Zhao C., Noskov S.Y. Atomistic models of ion and solute transport by the sodium-dependent secondary active transporters. Biochim. Biophys. Acta. 2012;1818:337–347. doi: 10.1016/j.bbamem.2011.10.031. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita A., Singh S.K., Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 7.Penmatsa A., Wang K.H., Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng M.H., Bahar I. Coupled global and local changes direct substrate translocation by neurotransmitter-sodium symporter ortholog LeuT. Biophys. J. 2013;105:630–639. doi: 10.1016/j.bpj.2013.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J., Tajkhorshid E. A gate-free pathway for substrate release from the inward-facing state of the Na+-galactose transporter. Biochim. Biophys. Acta. 2012;1818:263–271. doi: 10.1016/j.bbamem.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimamura T., Weyand S., Cameron A.D. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science. 2010;328:470–473. doi: 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez C., Khafizov K., Ziegler C. The role of trimerization in the osmoregulated betaine transporter BetP. EMBO Rep. 2011;12:804–810. doi: 10.1038/embor.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forrest L.R., Rudnick G. The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology (Bethesda) 2009;24:377–386. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi L., Quick M., Javitch J.A. The mechanism of a neurotransmitter:sodium symporter—inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zomot E., Gur M., Bahar I. Microseconds simulations reveal a new sodium-binding site and the mechanism of sodium-coupled substrate uptake by LeuT. J. Biol. Chem. 2014;290:544–555. doi: 10.1074/jbc.M114.617555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng M.H., Bahar I. Complete mapping of substrate translocation highlights the role of LeuT N-terminal segment in regulating transport cycle. PLOS Comput. Biol. 2014;10:e1003879. doi: 10.1371/journal.pcbi.1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koldsø H., Noer P., Schiøtt B. Unbiased simulations reveal the inward-facing conformation of the human serotonin transporter and Na+ ion release. PLOS Comput. Biol. 2011;7:e1002246. doi: 10.1371/journal.pcbi.1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koldsø H., Severinsen K., Sinning S. The two enantiomers of citalopram bind to the human serotonin transporter in reversed orientations. J. Am. Chem. Soc. 2010;132:1311–1322. doi: 10.1021/ja906923j. [DOI] [PubMed] [Google Scholar]
- 18.Shan J., Javitch J.A., Weinstein H. The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PLoS ONE. 2011;6:e16350. doi: 10.1371/journal.pone.0016350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nolan T.L., Geffert L.M., Surratt C.K. Discovery of novel-scaffold monoamine transporter ligands via in silico screening with the S1 pocket of the serotonin transporter. ACS Chem. Neurosci. 2014;5:784–792. doi: 10.1021/cn500133b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manepalli S., Surratt C.K., Nolan T.L. Monoamine transporter structure, function, dynamics, and drug discovery: a computational perspective. AAPS J. 2012;14:820–831. doi: 10.1208/s12248-012-9391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersen J., Stuhr-Hansen N., Kristensen A.S. Molecular basis for selective serotonin reuptake inhibition by the antidepressant agent fluoxetine (Prozac) Mol. Pharmacol. 2014;85:703–714. doi: 10.1124/mol.113.091249. [DOI] [PubMed] [Google Scholar]
- 22.Sinning S., Musgaard M., Wiborg O. Binding and orientation of tricyclic antidepressants within the central substrate site of the human serotonin transporter. J. Biol. Chem. 2010;285:8363–8374. doi: 10.1074/jbc.M109.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh S.K., Piscitelli C.L., Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnamurthy H., Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez C., Koshy C., Ziegler C. Substrate specificity and ion coupling in the Na+/betaine symporter BetP. EMBO J. 2011;30:1221–1229. doi: 10.1038/emboj.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe A., Choe S., Abramson J. The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature. 2010;468:988–991. doi: 10.1038/nature09580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forrest L.R., Zhang Y.W., Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. USA. 2008;105:10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aittoniemi J., de Wet H., Sansom M.S.P. Asymmetric switching in a homodimeric ABC transporter: a simulation study. PLOS Comput. Biol. 2010;6:e1000762. doi: 10.1371/journal.pcbi.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrödinger, L. 2012. SCHRÖDINGER SUITE 2012. PROTEIN PREPARATION WIZARD; EPIK, Ver. 2.3; IMPACT, Ver. 5.8; and PRIME, Ver. 3.1. Schrödinger, New York.
- 30.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 31.Li H., Robertson A.D., Jensen J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 32.Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 33.Olsson M.H.M., Søndergaard C.R., Jensen J.H. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 34.Søndergaard C.R., Olsson M.H.M., Jensen J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 35.Piscitelli C.L., Krishnamurthy H., Gouaux E. Neurotransmitter/sodium symporter orthologue LeuT has a single high-affinity substrate site. Nature. 2010;468:1129–1132. doi: 10.1038/nature09581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fjorback A.W., Pla P., Nyengaard J.R. Serotonin transporter oligomerization documented in RN46A cells and neurons by sensitized acceptor emission FRET and fluorescence lifetime imaging microscopy. Biochem. Biophys. Res. Commun. 2009;380:724–728. doi: 10.1016/j.bbrc.2009.01.128. [DOI] [PubMed] [Google Scholar]
- 37.Kilic F., Rudnick G. Oligomerization of serotonin transporter and its functional consequences. Proc. Natl. Acad. Sci. USA. 2000;97:3106–3111. doi: 10.1073/pnas.060408997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmid J.A., Scholze P., Sitte H.H. Oligomerization of the human serotonin transporter and of the rat GABA transporter 1 visualized by fluorescence resonance energy transfer microscopy in living cells. J. Biol. Chem. 2001;276:3805–3810. doi: 10.1074/jbc.M007357200. [DOI] [PubMed] [Google Scholar]
- 39.MacKerell A.D., Jr., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 40.Dowhan W. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 1997;66:199–232. doi: 10.1146/annurev.biochem.66.1.199. [DOI] [PubMed] [Google Scholar]
- 41.Lomize M.A., Lomize A.L., Mosberg H.I. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 42.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 43.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Best R.B., Zhu X., MacKerell A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 46.Steinbach P.J., Brooks B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994;15:667–683. [Google Scholar]
- 47.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 48.Kumar S., Rosenberg J.M., Kollman P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 49.Hub J.S., de Groot B.L., van der Spoel D. G_WHAM—a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput. 2010;6:3713–3720. [Google Scholar]
- 50.Sonntag Y., Musgaard M., Thøgersen L. Mutual adaptation of a membrane protein and its lipid bilayer during conformational changes. Nat. Commun. 2011;2:304. doi: 10.1038/ncomms1307. [DOI] [PubMed] [Google Scholar]
- 51.Zhao Y., Terry D., Javitch J.A. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature. 2010;465:188–193. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mondal S., Khelashvili G., Weinstein H. The cost of living in the membrane: a case study of hydrophobic mismatch for the multi-segment protein LeuT. Chem. Phys. Lipids. 2013;169:27–38. doi: 10.1016/j.chemphyslip.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forrest L.R., Krämer R., Ziegler C. The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta. 2011;1807:167–188. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 54.Li J., Tajkhorshid E. Ion-releasing state of a secondary membrane transporter. Biophys. J. 2009;97:L29–L31. doi: 10.1016/j.bpj.2009.09.005. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas J.R., Gedeon P.C., Madura J.D. LeuT conformational sampling utilizing accelerated molecular dynamics and principal component analysis. Biophys. J. 2012;103:L1–L3. doi: 10.1016/j.bpj.2012.05.002. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Merchant B.A., Madura J.D. Insights from molecular dynamics: the binding site of cocaine in the dopamine transporter and permeation pathways of substrates in the leucine and dopamine transporters. J. Mol. Graph. Model. 2012;38:1–12. doi: 10.1016/j.jmgm.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.