

Abstract
Recent analyses have found that a substantial amount of the Neandertal genome persists in the genomes of contemporary non-African individuals. East Asians have, on average, higher levels of Neandertal ancestry than do Europeans, which might be due to differences in the efficiency of purifying selection, an additional pulse of introgression into East Asians, or other unexplored scenarios. To better define the scope of plausible models of archaic admixture between Neandertals and anatomically modern humans, we analyzed patterns of introgressed sequence in whole-genome data of 379 Europeans and 286 East Asians. We found that inferences of demographic history restricted to neutrally evolving genomic regions allowed a simple one-pulse model to be robustly rejected, suggesting that differences in selection cannot explain the differences in Neandertal ancestry. We show that two additional demographic models, involving either a second pulse of Neandertal gene flow into the ancestors of East Asians or a dilution of Neandertal lineages in Europeans by admixture with an unknown ancestral population, are consistent with the data. Thus, the history of admixture between modern humans and Neandertals is most likely more complex than previously thought.
Main Text
As modern humans migrated out of Africa and dispersed throughout the world, they encountered and hybridized with Neandertals.1,2 The similarly low levels of Neandertal ancestry found in all modern non-African populations studied to date have been parsimoniously interpreted to be the result of a single pulse of admixture into the population ancestral to all non-Africans. However, recent reports show that East Asians have, on average, inherited ∼20% more Neandertal ancestry than Europeans have.3–6 Two explanations have been proposed to account for this observation. Sankararaman et al.5 suggested that because Neandertal lineages appear to be subject to widespread purifying selection in modern humans, differences in the efficiency of purifying selection could account for higher levels of Neandertal ancestry in East Asians. This hypothesis is supported by previous studies that have shown that East Asians have a smaller effective population size than do Europeans.7 In contrast, through extensive simulations, Vernot and Akey4 found that the excess of Neandertal ancestry in East Asians could not be explained by a single ancestral introgression event. Rather, the data were better explained by a two-pulse model, where introgression occurred in the common ancestor of East Asians and Europeans and was followed by additional gene flow into East Asians. However, these simulations did not account for the potential confounding effects of natural selection, and thus ambiguity remains about whether a simple single pulse of admixture between modern humans and Neandertals can explain the data.
To investigate how patterns of introgressed Neandertal sequences in East Asians and Europeans are influenced by potential differences in the efficiency of purifying selection between populations, we first partitioned the genome by using B-values,8 which measure the degree to which neutral variation has been reduced as a result of linked selected sites. Specifically, we selected 50-kb windows from Vernot and Akey4 and binned each window according to the minimum B-value at any base in that window. B-values range from 0 (all neutral diversity eliminated by background selection) to 1 (no reduction of neutral diversity by background selection). Next, we calculated values of two summary statistics of Neandertal introgression,4 Rind and Rpop (Figure 1), as a function of B-values. Rind is the ratio of the amount of introgressed Neandertal sequence per individual in East Asians to that in Europeans (Figure 1). Rpop is the ratio of the number of genomic bases covered by introgressed Neandertal sequence in any East Asian individual to that in any European individual (we corrected for differences in sample size by subsampling sets of 20 individuals from each population; Figure 1). Our previously measured genome-wide values of Rind and Rpop were 1.21 and 1.05, respectively.4 Moreover, Rind has consistently been reported to be greater than 1, including values of 1.19,3 1.20,5 and 1.4.6 These estimates indicate that although approximately the same amount of the Neandertal genome survives in Europeans and East Asians, a given Neandertal haplotype in East Asians is on average at higher frequency (Figure 1).
Figure 1.
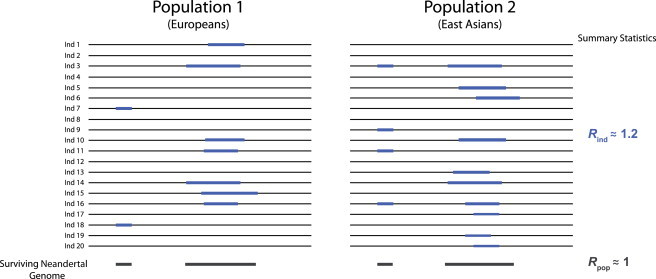
Schematic Representation of Rind and Rpop
Two hypothetical populations, each with 20 sampled individuals, are shown. Each individual’s Neandertal ancestry is shown in blue (top), and the total amount of the Neandertal genome present in this region in each population is shown in dark gray (bottom). Estimates of summary statistics for these hypothetical regions are given (left). Note that the amount of Neandertal sequence per individual is quite different between populations—population 2 contains more Neandertal sequence per individual, as reflected by an Rind value of ∼1.2. However, in each population the same amount of the Neandertal genome survives, as reflected by an Rpop value of ∼1.
If elevated levels of Neandertal ancestry in East Asians are due to differences in the efficiency of purifying selection between East Asians and Europeans, then Rind should vary significantly by B-value. Genomic regions under strong purifying selection would be more strongly depleted in Neandertal ancestry in the historically larger European population, leading to high Rind at low B-values and Rind closer to 1 at high B-values. In contrast, we observed that Rind was fairly stable with increasing B-value cutoffs (Figure 2). For example, the estimate of Rind in regions with a minimum B-value of 0.975 (spanning ∼106 Mb of the genome), which indicates that neutral variation was reduced by <2.5% as a result of background selection, was 1.175. Thus, in this more neutral subset of the genome, East Asian individuals have on average 17.5% more introgressed sequence than Europeans. This percentage closely parallels genome-wide estimates.
Figure 2.
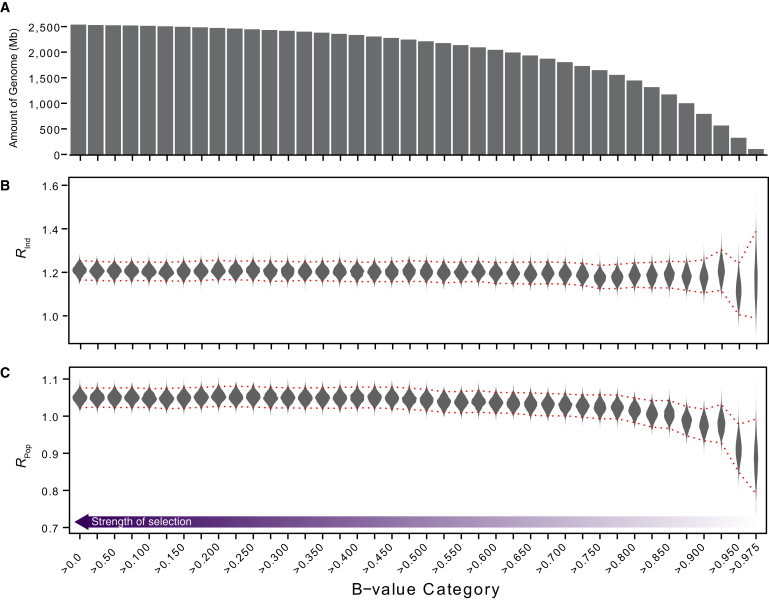
Estimates of Rind and Rpop as a Function of B-Value Cutoffs
(A) Amount of the genome in each B-value category. Note that each category is a subset of the category to its left (i.e., the category B ≥ 0 contains the entire genome).
(B and C) Rind (B) and Rpop (C) calculated over genomic regions with progressively higher B-value thresholds (i.e., less functional constraint; see color bar). Violin plots show bootstrap resamples, and red dotted lines denote the 95% confidence intervals (CIs).
Unlike Rind, which remained stable with increasing B-values, Rpop showed a marked decline as the B-value cutoff exceeded 0.850, indicating that in more neutrally evolving genomic regions, less of the Neandertal genome survives at the population level in East Asians than in Europeans. This observation is consistent with previous studies, which have found that a smaller ancestral effective population size in East Asians than in Europeans results in more intense genetic drift.7 Higher genetic drift would result in the loss of low-frequency Neandertal haplotypes; this effect would be strongest in regions where the competing force of purifying selection is weakest.9 Qualitatively, patterns of Rind and Rpop as a function of B-value suggest that the excess of Neandertal ancestry in East Asians cannot be explained by differences in selective forces alone and that a model of a single ancestral pulse of Neandertal introgression is unlikely.
To more formally evaluate demographic models compatible with patterns of Neandertal ancestry in East Asians and Europeans, we performed approximate Bayesian computation (ABC)10,11 on putatively neutral sequence by calculating Rind and Rpop in genomic regions with a minimum B-value ≥ 0.975. ABC analysis involves simulating data under different demographic models, calculating summary statistics from these simulations, and selecting simulations in a principled manner that best matches the observed summary statistics. We first simulated neutral sequence data under (1) a one-pulse model where all Neandertal sequence introgressed in a single pulse into the common ancestor of East Asians and Europeans (m1; Figure 3A) and (2) a two-pulse model with varying amounts of additional introgression into either Europeans or East Asians (m2; Figure 3A) after population splitting. Specifically, we performed simulations in which m2 / m1 ranged from −2% to 33% (negative values indicate simulations with additional introgression into Europeans; m2 = 0 is a one-pulse model). For summary statistics, we used both Rind and Rpop. We have previously shown that these statistics can distinguish between archaic admixture demographic models.4 We performed >30,000 simulations and estimated demographic parameters by using ABC with the R package “abc.”13 Specifically, we used the “abc” function with a non-linear neural-network regression method of correcting accepted parameter values.10 Results were similar when we corrected values by local linear regression. A complete description of the simulated demographic models can be found in Appendix A.
Figure 3.
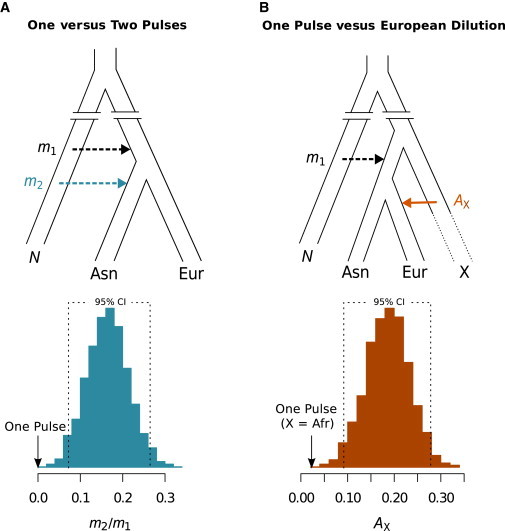
Inference of Admixture Models from Genomic Regions with Little or No Selective Constraint
(A) Schematic illustration of the one- and two-pulse models of Neandertal introgression. The majority of introgression occurred in the common ancestor of all non-Africans (m1), and a smaller additional amount of introgression occurred in East Asians (m2).
(B) Schematic illustration of the one-pulse and European-dilution models. All Neandertal introgression occurred in the common ancestor of Europeans and East Asians (m1), and a substantial portion of modern-day European ancestry (AX) derived from a second population (X) with no Neandertal ancestry.
Below each demographic model are histograms of m2/ m1 (left; the proportion of additional introgression into East Asians) and AX (right; the proportion of European ancestry derived from population X) as estimated by 30,000 simulations and ABC analysis (adjusted values of 1,500 accepted simulations are shown). Dashed lines demark 95% confidence intervals (CIs). Black arrows show the expected values of m2/ m1 (0) and AX (0.023)12 under the one-pulse model.
Only simulations with additional introgression into East Asians (two-pulse models) were accepted as plausible in the ABC analysis (Figure 3A). We estimated that a second pulse of 15% more introgression into East Asians could explain the observed excess of Neandertal introgression (95% confidence interval [CI] of m2 / m1 = 6.8%–26.6%). Under the null hypothesis of a single pulse of admixture, the ratio m2 / m1 = 0%, which is well outside the preferred range. Given these results, the two-pulse model is significantly favored, and we can strongly reject the null hypothesis of a one-pulse model (p < 6.7 × 10−4). For completeness, we repeated our ABC analysis by using summary statistics from regions with a minimum B-value ≥ 0.950 (spanning 326 Mb of the genome), which again significantly favored the two-pulse model (Figure S1). Therefore, a one-pulse model is rejected both for genome-wide calculations of Neandertal ancestry4 and when regions that might have been subjected to selective constraint are excluded.
It is important to stress that although a two-pulse model of admixture explains the empirical data significantly better than the simple one-pulse model considered here, it does not necessarily mean that the two-pulse model is correct. To investigate additional plausible demographic models, we also considered a model in which a single ancestral pulse of introgression was followed by admixture between the European population and a third modern human population that had not interbred with Neandertals. In this scenario, the amount of Neandertal ancestry in Europeans is effectively diluted by admixture with a population not carrying Neandertal lineages. Such a population could be from Africa, where there is expected to have been no, or little, Neandertal ancestry.14 Alternatively, it could be an unknown “ghost” Eurasian population that was entirely absorbed into Europeans.
To determine whether a European-dilution model could explain the data, we again performed ABC by using the summary statistics Rind and Rpop, calculated from neutral subsets of the genome (B-value ≥ 0.975) as described above. Specifically, we simulated sequence data under a one-pulse model where all Neandertal sequence introgressed in a single pulse into the common ancestor of East Asians and Europeans (m1; Figure 3B) and the ancestral European population then admixed with a third population denoted as X (Figure 3B). We varied the proportion of modern-day European ancestry derived from population X (AX) from 0% to 35%. We estimated that the observed patterns of Neandertal introgression are compatible with a European-dilution model if an average of 18.2% of modern European ancestry (AX) was contributed by this third population (95% CI of AX = 9.2%–27.6%). This is significantly larger than current estimates of African ancestry in Europeans (1%–3%12 and 2.3%;15 p = 5.7 × 10−4), suggesting that migration from Africa to Europe cannot explain the larger amount of Neandertal ancestry in East Asians than in Europeans. Tantalizingly, recent work suggests that modern Europeans might comprise admixture of three ancestral groups.16 However, each of these groups is estimated to contain ∼2% Neandertal ancestry16 and thus could not have diluted the amount of Neandertal ancestry in modern Europeans enough to account for the differences with East Asians. Thus, on the basis of current evidence, differential migration seems less likely to explain the data, increasing the likelihood of multiple-pulse models.
In addition to evaluating models of the interactions between modern humans and Neandertals, we used these analyses of Neandertal ancestry to elucidate other aspects of human demographic history. For example, in addition to estimating the parameters m2 / m1 and AX, we found that the ratio of ancestral effective population sizes between Europeans and East Asians, NeEUR / NeASN, had a significant effect on the fit of our models. Using the same ABC analyses, we estimated NeEUR / NeASN to be 1.93 (95% CI = 1.57–2.73) under a two-pulse model and estimated NeEUR / NeASN to be 1.59 (95% CI = 1.35–1.89) under the European-dilution model (Figure S1). Both of these estimates are consistent with previously accepted values.7,15
In summary, by focusing on putatively neutral regions of the genome, we have shown that the observed patterns of Neandertal ancestry in Europeans and East Asians are not consistent with a simple one-pulse model of admixture. Thus, differences in the efficiency of purifying selection among populations are unlikely to account for higher levels of Neandertal ancestry in East Asians than in Europeans. We have shown that more complex and nuanced models are necessary to explain the data and have furthermore suggested two such models that are consistent with observed patterns of Neandertal introgression in Europeans and East Asians. Additionally, we have shown that studies of Neandertal ancestry can be informative about other aspects of human history. Combined with the analysis of ancient DNA of archaic and modern humans, additional studies in geographically diverse populations will help narrow the space of plausible demographic models. Such models will provide critical insights into hominin evolutionary history and the key parameters governing admixture dynamics between modern humans and Neandertals.
Acknowledgments
We thank Kirk E. Lohmueller and members of the J.M.A. laboratory for helpful discussions related to this manuscript. This work was supported by NIH grant 1R01GM110068 to J.M.A.
Contributor Information
Benjamin Vernot, Email: bvernot@gmail.com.
Joshua M. Akey, Email: akeyj@uw.edu.
Appendix A: Demographic Models and Simulations
We performed coalescent simulations on the basis of previously inferred demographic models for European, East Asian, and African populations.15,17 Simulations were performed with ms,18 and coalescent trees were extracted from the output and used for calculating the summary statistics Rind and Rpop. It is important to note that sequence variation does not affect these summary statistics, and thus many aspects of these demographic models are essentially “nuisance parameters,” i.e., they do not have an effect on the final results. The base demographic model is as follows:
-
a.
Splitting between modern humans and Neandertals 700,000 years ago.
-
b.
Neandertal Ne of 1,500.
-
c.
Splitting between Africans and non-Africans 70,000 years ago.
-
d.
African Ne of 14,474 until 5,115 years ago.
-
e.
Gradual growth (23,000 until 5,115 years ago) of non-African populations to an Ne of 8,879 in East Asians and an Ne of 9,475 in Europeans, except in simulations where no growth is required to reach an Ne greater than or equal to 8,879 and 9,475, respectively.
-
f.
Rapid growth (starting at 5,115 years ago) of all populations to a present-day Ne of 424,000 in Africans, 512,000 in Europeans, and 1,370,990 in East Asians.
-
g.
A single 500-year introgression event from Neandertals to the common ancestor of Europeans and East Asians at a rate of 0.00075 (0.075% of each generation was sampled from Neandertal individuals).
Note, although a split time, T(S), of 700,000 years ago between Neandertals and modern humans is older than the upper bound of 500,000 years ago estimated from the draft Neandertal genome,1 it is within the range estimated from the high-coverage Altai Neandertal genome.2 Furthermore, we chose this time to ensure that any introgressed lineages coalesced before non-introgressed lineages, making it easier to identify introgressed lineages by examining the coalescent trees. It is important to note that this older T(S) has no effect on the amount of introgressed sequence, given that we are considering only the presence and extent of introgressed haplotypes and not variation that has arisen on those haplotypes.
To generate demographic models from this base model, we then sampled the following parameters from uniform distributions (unless otherwise noted):
-
a.
European and East Asian T(S) between 36,000 and 55,000 years ago.
-
b.
Ancestral Eurasian Ne prior to T(S) between 5,000 and 15,000.
-
c.
European Ne / East Asian Ne between 1 and 2.5 (ne_ratio).
-
d.
European + East Asian Ne after T(S) between 8,000 and 25,000 (ne_sum).
-
e.
Ne for Europeans = ne_ratio × ne_sum.
-
f.
Ne for East Asians = ne_sum / (ne_ratio + 1).
-
g.
The time of the first introgression pulse between T(S) and 65,000 years ago.
-
h.
Migration rates were set as follows: 1.5 × 10−4 between Africans and the ancestors of Europeans and East Asians, 2.5 × 10−5 × 23,000 / T(S) between Africans and Europeans, 7.8 × 10−6 × 23,000 / T(S) between Africans and East Asians, and 3.11 × 10−5 × 23,000 / T(S) between Europeans and East Asians. The above parameters were adapted from Gravel et al.15 and Tennessen et al.17 but were adjusted for varying divergence times between Europeans and East Asians and are the rate of migration per generation per chromosome (i.e., the proportion of population A originating in population B per generation).
-
i.
In two-pulse models, a second 500-year introgression event from Neandertals to East Asians started 500 years after T(S) at a rate between 0 and 0.00025. Some simulations instead included additional introgression into Europeans at a rate between 0 and 0.000015. In our ABC analysis, we framed this as “negative” introgression into East Asians, effectively varying the amount of introgression in this second pulse from −0.000015 to 0.00025. This is equivalent to −2% to 33% more introgression into East Asians.
-
j.
In European-dilution models, Africans were treated as population X, and the rate of migration from population X (m3) was varied from 1.25 × 10−5 to 1.75 × 10−4, i.e., this rate was varied from 0.5 to 7 times the rate estimated in Gravel et al.15 By multiplying the migration rate by the duration of the migration (T(S)), we could approximate the amount of European ancestry derived from population X (AX = m3 × T(S)).
Two example ms commands are given in the Supplemental Data.
Supplemental Data
References
- 1.Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H.Y. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prüfer K., Racimo F., Patterson N., Jay F., Sankararaman S., Sawyer S., Heinze A., Renaud G., Sudmant P.H., de Filippo C. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43–49. doi: 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer M., Kircher M., Gansauge M.-T., Li H., Racimo F., Mallick S., Schraiber J.G., Jay F., Prüfer K., de Filippo C. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338:222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vernot B., Akey J.M. Resurrecting surviving Neandertal lineages from modern human genomes. Science. 2014;343:1017–1021. doi: 10.1126/science.1245938. [DOI] [PubMed] [Google Scholar]
- 5.Sankararaman S., Mallick S., Dannemann M., Prüfer K., Kelso J., Pääbo S., Patterson N., Reich D. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;507:354–357. doi: 10.1038/nature12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wall J.D., Yang M.A., Jay F., Kim S.K., Durand E.Y., Stevison L.S., Gignoux C., Woerner A., Hammer M.F., Slatkin M. Higher levels of neanderthal ancestry in East Asians than in Europeans. Genetics. 2013;194:199–209. doi: 10.1534/genetics.112.148213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keinan A., Mullikin J.C., Patterson N., Reich D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat. Genet. 2007;39:1251–1255. doi: 10.1038/ng2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McVicker G., Gordon D., Davis C., Green P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 2009;5:e1000471. doi: 10.1371/journal.pgen.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim B., Lohmueller K. Selection and Reduced Population Size Cannot Explain Higher Amounts of Neandertal Ancestry in East Asian than in European Human Populations. Am. J. Hum. Genet. 2015;96:454–461. doi: 10.1016/j.ajhg.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blum M.G.B., François O. Non-linear regression models for Approximate Bayesian Computation. Stat. Comput. 2010;20:63–73. [Google Scholar]
- 11.Csilléry K., Blum M.G.B., Gaggiotti O.E., François O. Approximate Bayesian Computation (ABC) in practice. Trends Ecol. Evol. 2010;25:410–418. doi: 10.1016/j.tree.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Moorjani P., Patterson N., Hirschhorn J.N., Keinan A., Hao L., Atzmon G., Burns E., Ostrer H., Price A.L., Reich D. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet. 2011;7:e1001373. doi: 10.1371/journal.pgen.1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Csilléry K., François O., Blum M.G.B. abc: an R package for approximate Bayesian computation (ABC) Methods in Ecology and Evolution. 2012;3:475–479. doi: 10.1016/j.tree.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Wang S., Lachance J., Tishkoff S.A., Hey J., Xing J. Apparent variation in Neanderthal admixture among African populations is consistent with gene flow from Non-African populations. Genome Biol. Evol. 2013;5:2075–2081. doi: 10.1093/gbe/evt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gravel S., Henn B.M., Gutenkunst R.N., Indap A.R., Marth G.T., Clark A.G., Yu F., Gibbs R.A., Bustamante C.D., 1000 Genomes Project Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. USA. 2011;108:11983–11988. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazaridis I., Patterson N., Mittnik A., Renaud G., Mallick S., Kirsanow K., Sudmant P.H., Schraiber J.G., Castellano S., Lipson M. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513:409–413. doi: 10.1038/nature13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hudson R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.