

Abstract
The renal outer medullary potassium channel (ROMK, or Kir1.1, encoded by KCNJ1) critically regulates renal tubule electrolyte and water transport and hence blood volume and pressure. The discovery of loss-of-function mutations in KCNJ1 underlying renal salt and water wasting and lower blood pressure has sparked interest in developing new classes of antihypertensive diuretics targeting ROMK. The recent development of nanomolar-affinity small-molecule inhibitors of ROMK creates opportunities for exploring the chemical and physical basis of ligand-channel interactions required for selective ROMK inhibition. We previously reported that the bis-nitro-phenyl ROMK inhibitor VU591 exhibits voltage-dependent knock-off at hyperpolarizing potentials, suggesting that the binding site is located within the ion-conduction pore. In this study, comparative molecular modeling and in silico ligand docking were used to interrogate the full-length ROMK pore for energetically favorable VU591 binding sites. Cluster analysis of 2498 low-energy poses resulting from 9900 Monte Carlo docking trajectories on each of 10 conformationally distinct ROMK comparative homology models identified two putative binding sites in the transmembrane pore that were subsequently tested for a role in VU591-dependent inhibition using site-directed mutagenesis and patch-clamp electrophysiology. Introduction of mutations into the lower site had no effect on the sensitivity of the channel to VU591. In contrast, mutations of Val168 or Asn171 in the upper site, which are unique to ROMK within the Kir channel family, led to a dramatic reduction in VU591 sensitivity. This study highlights the utility of computational modeling for defining ligand-ROMK interactions and proposes a mechanism for inhibition of ROMK.
Introduction
The renal outer medullary K+ (ROMK, Kir1.1) channel is a product of the gene KCNJ1 and founding member of the inward rectifier K+ (Kir) channel family (1,2). ROMK is expressed in the thick ascending limb of Henle’s loop and mineralocorticoid-sensitive cortical collecting duct of the kidney tubule, where it critically regulates transepithelial Na+ and osmotically coupled H2O reabsorption and K+ excretion (3,4). Greger and Schlatter (1981) first demonstrated that NaCl reabsorption in the mammalian TAL is coupled to a Ba2+-inhibitable K+ conductance in the luminal membrane. Using isolated-perfused tubule segments, they found that perfusing the tubule lumen with Ba2+-containing or K+-free solutions reduced the transepithelial short-circuit current (Isc) by ∼36%. Perfusion with a K+-free, Ba2+-containing solution virtually eliminated the Isc (5). The investigators subsequently found that the luminal membrane is almost exclusively K+-conductive and concluded that this conductance is responsible for the recycling of K+ ions across the luminal membrane, which in turn provides substrate K+ ions needed for the activity of the Na+-K+-2Cl− cotransporter (6).
Patch-clamp recordings of the luminal membrane revealed at least two distinct populations of Ba2+-inhibitable K+ channels that could contribute to K+ recycling in the TAL: an intermediate-conductance K+ channel of ∼70 pS and a small-conductance channel of ∼30 pS. With the exception of their sensitivity to Ba2+, the two channels differ dramatically in their pharmacological properties. For example, the ∼70 pS channel is inhibited by quinidine, tetraethylammonium ions, and Ca2+, whereas the 30 pS channel is largely insensitive to these blockers. In contrast, the KATP channel antagonist glyburide (glibenclamide) inhibits the ∼30 pS channel, but not the ∼70 pS channel. This latter point is of interest because it was shown that glyburide dose-dependently increases urinary Na+ excretion in rats without affecting K+ excretion (7). Similar results were subsequently reported for the structurally unrelated KATP channel inhibitor, U37883A (8). Taken together, these data suggest that pharmacological inhibition of the ∼30 pS channel specifically, without inhibiting the 70 pS channel, is sufficient to induce diuresis in rodents.
Indeed, a growing body of genetic evidence supports the notion that ROMK represents a new diuretic target in humans. Autosomal-recessive loss-of-function (LOF) mutations in KCNJ1 give rise to antenatal (type II) Bartter syndrome, a life threatening renal tubulopathy presenting with excessive urination, low to normal blood pressure, metabolic alkalosis, and hypokalemia (9–11). The observation that antenatal Bartter syndrome patients exhibit urinary K+ wasting seems contradictory, given that ROMK constitutes an important pathway for distal K+ secretion. However, studies of KCNJ1−/− mice revealed an important role of flow-stimulated big-conductance calcium-activated K+ (BK) channels in mediating K+ secretion in the absence of ROMK (12,13). Interestingly, heterozygous carriers of KCNJ1 LOF mutations have lower blood pressure and are protected from stroke, but do not exhibit the clinical features of Bartter syndrome (14). These observations provided strong human genetic evidence for ROMK as a novel diuretic target for the treatment of hypertension.
The small-molecule pharmacology of ROMK remained largely undeveloped for more than 15 years after ROMK was cloned (1,2). To our knowledge, the first publically disclosed inhibitor, termed VU590 (7,13-bis[(4-nitrophenyl)methyl]-1,4,10-trioxa-7,13-diazacyclopentadecane dihydrochloride), was discovered by our group in a high-throughput screen (HTS) of ∼225,000 compounds from the Molecular Libraries Screening Center Network (MLSCN) library. VU590 inhibits ROMK with a 50% inhibitory concentration (IC50) of ∼220 nM but suffers from poor selectivity due to weak activity toward Kir7.1 (IC50 8 μM) (15). The second inhibitor from the MLSCN probe development campaign was VU591 (6-nitro-2-[(6-nitro-1H-benzimidazol-2-yl)methoxymethyl]-1H-benzimidazole hydrochloride) which, like VU590, contains two nitro (−NOO) groups flanking a nitrogen- and oxygen-rich linker (see Fig. 2 A). VU591 inhibits ROMK with a 240 nM IC50, is active on native ROMK channels in isolated-perfused collecting ducts, and is selective for ROMK over >70 potential off-targets, including Kir7.1 (16).
Figure 2.
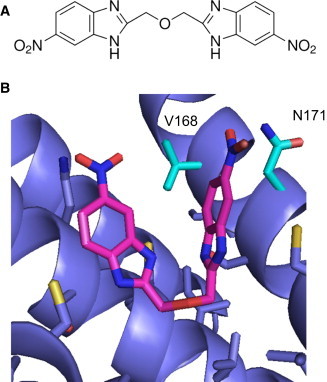
In silico docking of VU591 into a model of ROMK (A) Chemical structure of VU591. (B) Final binding pose. The model most consistent with the mutagenesis and electrophysiology data is shown in detail. The inhibitor VU591 is shown as magenta sticks, and ROMK is shown in blue cartoon, with neighboring side chains depicted as sticks. In addition, nitrogen atoms are colored blue, oxygen red, and sulfur yellow. Two specific residues discussed in the text, V168 and N171, are highlighted in cyan. To see this figure in color, go online.
The recent development of potent and highly selective small-molecule ROMK inhibitors by our group and Merck Research Laboratories (17,18) has created unprecedented opportunities not only for probing the druggability of ROMK, but also for understanding the chemical and physical interactions that underlie high-affinity ligand interactions with these important channels. Inward rectifier K+ channels are tetramers of identical (homotetramers) or similar (heterotetramers) subunits. Each subunit contains two membrane-spanning domains flanking a reentrant pore loop and a cytoplasmic domain comprised of intracellular amino- and carboxyl-termini. Rectification, or the propensity to pass inward current more readily than outward current, is due to pore block by intracellular polyvalent cations (e.g., Mg2+ and polyamines) interacting with negatively charged amino acid side chains lining the channel pore. ROMK is a homotetramer that exhibits weak rectification compared to the classical strong rectifier Kir2.1. In ROMK, several negatively charged rectification residues are replaced with uncharged amino acids that coordinate pore blockers less strongly.
A detailed understanding of how small-molecule inhibitors participate in high-affinity ligand interactions with ROMK will be valuable for rationalizing future efforts at drug design. Although the x-ray crystal structure of ROMK has not been determined, there are several closely-related channels of known structure, making it possible to develop a comparative model of ROMK. In this study, we generate such a model and use it to perform in silico docking calculations guided by concurrent electrophysiology experiments to arrive at a physical model for inhibition of ROMK by VU591.
Materials and Methods
Construction of comparative models of ROMK1
An ensemble of 10 comparative ROMK models was constructed in distinct conformations to simulate receptor flexibility for the docking studies (Fig. S1 in the Supporting Material). The experimental structures used as model templates, 3JYC (Kir2.2), 3SPG (Kir2.2), 3SPH (Kir2.2), 3SPI (Kir2.2), 3SPJ (Kir2.2), 3SYA (Kir3.2), 3SYC (Kir3.2), 3SYO (Kir3.2), 3SYP (Kir3.2), and 3SYQ (Kir3.2) (19–21), were obtained from the Protein Data Bank (PDB). The sequence of these 10 channels was aligned with the sequence of ROMK using ClustalW (22). Sequence identity ranges from 23% to 49%. From this multiple sequence alignment, all-atom comparative models were generated using Modeler v.9.10 (23). These initial models were refined using the Relax application in Rosetta v.3.2 (24) with a modified energy potential to mimic the membrane environment (25,26). Transmembrane residue definitions for input to Rosetta were derived from the Octopus webserver (27) and confirmed visually. The lowest-energy model from each template was selected out of 1000 refined models generated. The 10 resulting homology models were then used as input for the subsequent ligand-docking calculations. Further details and command lines are provided below, and as a protocol capture (http://structbio.vanderbilt.edu/∼sheehajh/denton/swale_protocol_capture.tgz) as part of the Supporting Material.
In silico docking of VU591
RosettaLigand (28) docking was used to predict the lowest-energy binding mode of VU591 in ROMK. To account for ligand flexibility, 200 low-energy conformations of VU591 were generated using the systematic conformational search tool in Molecular Operating Environment (MOE) 2013.08 (Chemical Computing Group, Montreal, Quebec, Canada). Rotation and flexibility about the central ether oxygen and proximal carbons were permitted, but the flanking nitro groups on VU591 required additional restraints in MOE to maintain coplanarity with the aromatic ring systems. The ligand ensemble was docked 9900 times into each of the 10 ROMK models. To ensure complete sampling, the ligands were manually placed in the upper, middle, or lower regions of the ROMK channel pore (Fig. 1, blue poses) before initiating the random search for low-energy binding poses. The ligand was allowed to move up to 5 Å per step and completely reorient. VU591 was docked into the ROMK models a total of 99,000 times. Fig. 1 (middle image) demonstrates that the entire channel pore was sampled (red). All details of the Rosetta command lines and options used are included in the Supporting Material.
Figure 1.
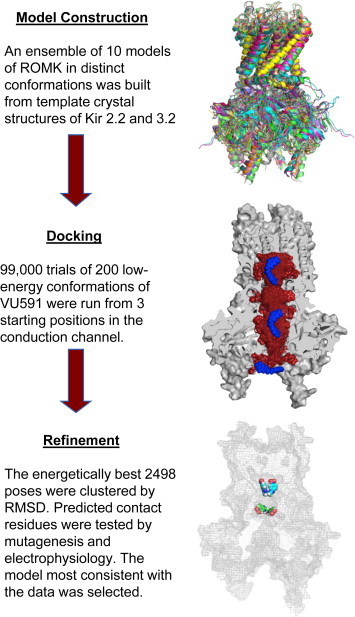
Overview of docking procedure. The flowchart shows the three parts of the process. (Top) Ten comparative models of ROMK were built using crystal structures of Kir2.2 and Kir3.2 as templates. This ensemble of models was used to represent some of the conformational diversity in ROMK, which can be seen clearly when the models are overlaid. (Middle) RosettaLigand was used to predict binding locations of the inhibitor. One model is shown in gray, using cutaway mode to expose the conduction channel. Three starting positions for VU591 (blue) were used to sample the entire interior of the channel. Red spheres are used to show the extent of sampling (only 300 out of 99,000 placements are shown, for clarity). An ensemble of 200 low-energy conformations was also used for the inhibitor, to account for flexibility in binding. (Bottom) The lowest-energy binding predictions were refined using clustering by similarity and ranking by score. Two of the best predictions are shown as colored spheres in the context of the complete protein shown in gray outline. To see this figure in color, go online.
Clustering of low-energy docking poses of VU591
The 1000 lowest-energy docking poses from each template were pooled, for a total of 10,000 poses for further analysis. The lowest predicted binding energy (ΔΔG) observed was −16 Rosetta energy units (REUs). Because we docked into comparative models, we expect that some inaccuracies in the models will result in inaccurate energy readout. Therefore, we considered not just the lowest-scoring binding pose, but all 2498 poses that displayed a ΔΔG >−10 REUs. To analyze the conformational space, we performed hierarchical clustering analysis (29). The clustering protocol used a similarity cutoff of root mean-square deviation 4 Å between two ligand poses. Further, it was adapted to take advantage of the fourfold symmetry of the channel by combining symmetry-related clusters. This increased the cluster sizes substantially, thereby reducing the number of clusters that need to be considered, and increased the size differential between the largest and all other clusters (Fig. S2 and Table S1). The best-scoring ligand poses from the largest four clusters were visually inspected. Clustering was performed using the Biological and Chemical Library (BCL) (30) software suite from the Meiler Lab. Molecular structure images were produced using PyMOL [The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.] and UCSF Chimera (31).
Heterologous ROMK1 expression in HEK-293T cells
The rat ROMK1 open reading frame (ORF) ligated into pcDNA3.1 was provided by Dr. Chun Jiang, (Georgia State University, Atlanta, GA). Site-directed mutagenesis was performed using a QuickChange II kit (Stratagene, La Jolla, CA) and fully sequenced to ensure that only the intended codon change was incorporated into the ROMK1 ORF. Alanine-scanning mutagenesis was used for most mutations. Given the close proximity of C175 to N171 (which is required for inhibition by VU591), C175 was mutated to alanine, threonine, and isoleucine to more fully explore its potential role in VU591 sensitivity. Isoleucine 182 was mutated to methionine (which is found in Kir2.1 at the equivalent position) to determine if a methionine accounts for the lack of VU591 sensitivity in Kir2.1. HEK-293T cells (American Type Culture Collection; Manassas, VA) were transiently cotransfected with pcDNA3.1-ROMK1 and pcDNA3.1-EGFP (transfection marker) using Lipofectamine LTX plus (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Patch-clamp electrophysiology
Patch-clamp experiments were performed 24 h posttransfection, essentially as described previously (15,16). Briefly, the cells were rinsed with divalent-free Hank’s balanced salt solution (145 mM NaCl, 5 mM KCl, 4 mM NaHCO3, 0.3 mM Na2HPO4, 0.3 mM KH2PO4, and 5.6 mM D-glucose, pH 7.4, 290 mOsmol/kg of water), dissociated by brief (∼60 s) exposure to 0.25% trypsin containing 1 mM EDTA, plated on poly-L-lysine-coated round glass coverslips, and allowed to recover at 37°C in a 5% CO2 incubator for at least 1 h before experiments. The coverslips were transferred to a small-volume perfusion chamber (Warner Instruments, Hamden, CT) and mounted on the stage of a Nikon Eclipse TE2000-U inverted microscope (Nikon, Tokyo, Japan). Patch electrode resistances ranged from 3 to 5 MΩ when filled with an intracellular solution of (in mM) 135 KCl, 2 MgCl2, 1 EGTA, 10 HEPES-free acid, and 2 Na2ATP (Roche Diagnostics, Indianapolis, IN), pH 7.2, 275 mOsmol/kg water. The standard bath solution contained (in mM) 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES-free acid, pH 7.4.
Whole-cell currents were recorded under voltage-clamp conditions using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Cells were maintained at a holding potential of −75 mV and stepped every 5 s to −120 mV for 200 ms before ramping to 120 mV at a rate of 1.2 mV/ms. Data were collected at 5 kHz and filtered at 1 kHz. Data acquisition and analysis were performed using pClamp 9.2 software (Molecular Devices).
VU591 preparation
VU591 powder was purchased from Sigma-Aldrich (St. Louis, MO), dissolved in anhydrous dimethyl sulfoxide up to 100 mM, and diluted in bath solution each day before use. For Kir2.1 studies, inhibitor solutions (50 μM and 100 μM) were made fresh before each recording to reduce the potential for precipitation of VU591 out of the bath solution. Final DMSO concentration used in experiments was <0.1% (v/v). Both recordings were terminated after steady state was reached by applying 2 mM barium (Ba2+) to allow measurement of residual leak current. Cells exhibiting <90% block by Ba2+ were excluded from analysis.
Statistical analyses
Wild-type and mutagenized ROMK1 channels were initially assayed for sensitivity to 300 nM VU591. The mean current amplitude recorded over five successive steps to −120 mV in control or experimental solutions were compared using a two-tailed, unpaired t-test, with statistical significance defined at P < 0.05 (GraphPad InStat, GraphPad Software, San Diego, CA). IC50 values were determined by fitting the Hill equation to concentration-response curves (CRCs) using variable-slope, unconstrained, nonlinear regression analyses performed with GraphPad Prism.
Results
Comparative molecular modeling and docking identify putative VU591 binding pockets
VU591 exhibits voltage-dependent knock-off at hyperpolarizing voltages, leading us to postulate that the binding site is located within the ion conduction pathway (16). In an effort to test this hypothesis and elucidate the mechanism of action of VU591, we used a coupled strategy of molecular modeling and docking for hypothesis generation, and site-directed mutagenesis and electrophysiology to test, refine, and validate the models (Fig. 1). A challenge in the application of protein-ligand docking is the computational expense of incorporating flexibility of both the ligand and the protein. This challenge was addressed by using an ensemble-docking approach to simulate the flexibility of each partner. An ensemble of 10 conformations of ROMK was constructed by selecting 10 template structures reflecting Kir3.2 and Kir2.2 in the presence or absence of different ligands, ions, and functionally important mutations (19–21) (Fig. S1). The flexibility of VU591 was simulated in a similar way, using 200 low-energy conformers as input to the docking calculation. To ensure complete sampling of the entire length of the ROMK1 channel (Fig. 1), which extends >80 Å from the extracellular to the intracellular extremity, VU591 was manually placed at three different starting positions at the outset of the docking simulations (Fig. 1).
RosettaLigand uses a statistically-derived potential to score each docking pose according to its predicted energy (28). In theory, the lowest-energy result should identify the native binding mode. However, because the energy function is an imperfect approximation based on physics- and knowledge-based terms, because we dock into comparative models with an unknown amount of intrinsic inaccuracy, and because the sampling density is finite, the predicted energy score is insufficient to discriminate among binding poses by itself. This limitation can be addressed by clustering the resulting docking poses according to a similarity metric, in this case the root mean-square deviation between poses. Previous work has shown that the depth of the native energy well correlates with its width, and as a result, relatively more low-energy models tend to accumulate near the native conformation (32). Our clustering strategy took into account the fourfold symmetry of the channel, and achieved significant discriminatory power (Fig. S2 and Table S1). The selection of top binding poses was thus a combination of cluster size and low energy. The two largest clusters stood out by sizes and predicted binding energy.
Binding pose
The largest clusters of low-energy binding poses were found 16 Å below the selectivity filter (middle; 351 poses) and 6.5 Å below the selectivity filter (upper; 109 poses), with additional smaller clusters as far as 42 Å away from the selectivity filter (lower). Representatives of the upper and middle binding poses are shown in Fig. 1. The upper binding pocket is located in the ion conduction channel, between the selectivity filter and the inner helix gate. The ligand VU591 adopts a V-shaped, or partially folded conformation within the cavity and displays remarkable shape complementarity with the cup-shaped pocket. As shown in Fig. 2 B, VU591 is predicted to contact backbone and side-chain atoms of inner helix residues 167–184. This is a surface of mixed hydrophobic and hydrophilic character. Without additional experimental distance restraints, the prediction of specific atom-atom contacts based on only the lowest-energy result would be overly ambitious. Therefore, an extensive likelihood analysis was performed by combining contact statistics from all members of each cluster (see Supporting Material for details). The reasoning is that we have more confidence in ligand-residue contacts that are found in more of the predicted binding conformations. This analysis identified pore-lining residues in ROMK including N171 and V168, in proximity to the nitro group of VU591. The compound binds to the surfaces of two adjacent subunits of the ROMK tetramer simultaneously. The second binding pocket is located 10 Å below the upper site but still above the intracellular vestibule, and it is slightly larger in diameter, allowing the ligand to adopt a more extended conformation. The list of contact residues included some in the gate and flexible loop regions, but none of these correlated well with the mutagenesis data (see below) and were not further pursued.
The best 2498 docking results were found to be distributed among the H,M,L cavities differently, according to the conformation of the template from which the model was built (Table S2). This is consistent with the idea that the state of the protein affects both the binding affinity and location of the probe. It is noteworthy that the diameter of the selectivity filter is too narrow to allow passage of VU591 from the extracellular side. In models that were built on closed templates, the conduction channel from the cytoplasmic side of the pore to the binding site is also too narrow. However, in the models built in the open conformation (templated on, e.g., PDB 3SYQ, which is Kir3.2 with PIP2 bound, and the activating mutation R201A), it is wide enough for the ligand to reach the binding site.
To experimentally validate the docking predictions, residues were prioritized for site-directed mutagenesis using the likelihood analysis mentioned above. Briefly, residues in contact with the probe (within 4 Å of a ligand atom) were identified in each of the 10 lowest-energy conformations of each cluster, and the relative frequency of the contacts summed over all conformations was used to rank the contact residues in the putative binding pocket for experimental testing (for greater detail, see the link to protocol capture in Materials and Methods).
Functional analysis of the putative VU591 binding pockets in ROMK1
Patch-clamp electrophysiology and site-directed mutagenesis were used to test whether residues populating the middle and upper binding sites identified in docking simulations contribute to ROMK sensitivity to VU591. As shown in Fig. 3, B and C, superfusion of cells with 10 μM VU591completely inhibited ROMK1 current, and the inhibited current was indistinguishable from the current trace after the application of the nonspecific Kir channel inhibitor, barium. The residual outward current observed in the presence of VU591 is carried by endogenous Kv currents expressed in HEK293 cells, as VU591 has no effect on this current (16). Some ROMK1 mutants (specifically, T304A and N171D/E/Q) failed to express channel activity that could be distinguished from the endogenous inward rectifier current expressed in HEK293 cells. To circumvent this problem, the loss-of-function mutations were introduced into ROMK1 carrying a mutation of lysine 80 to methionine (K80M). We and others have shown that the K80M mutation can rescue the function of some ROMK1 mutants, presumably by stabilizing the helix-bundle crossing gate in the open state (33,34). It is important to note that the IC50 values for VU591 were indistinguishable between ROMK-wild-type (IC50 297 nM) and ROMK-K80M (IC50 307 nM) (Fig. 3 G).
Figure 3.
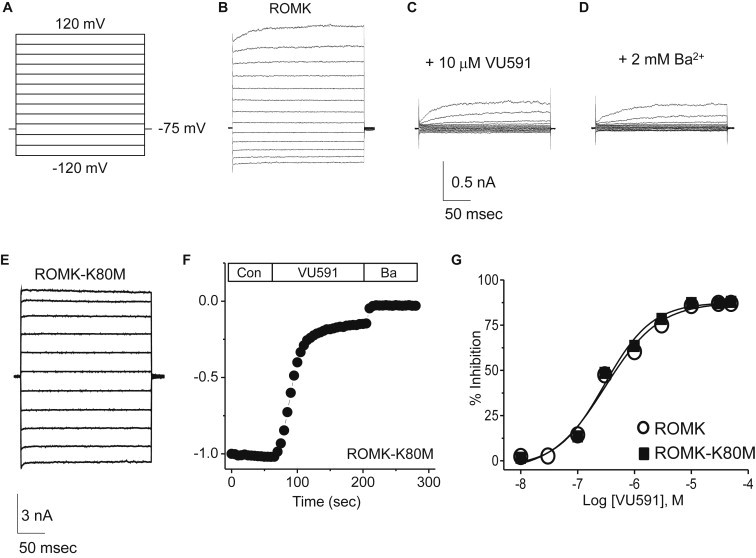
Inhibition of ROMK-WT and ROMK-K80M by VU591. (A–E) Voltage-clamp protocol (A) used to evoke ROMK-WT and ROMK-K80M currents (B and E, respectively). ROMK-WT activity was recorded in control buffer (B), in the presence of 10 μM VU591 (C), and in the presence of 2 mM Ba2+ (D). (F) Representative time course of ROMK-K80M inhibition by 10 μM VU591 and by 2 mM Ba2+. (G) CRC for VU591-dependent inhibition of ROMK-WT and ROMK-K80M. Data represent the mean ± SE steady-state inhibition by the indicated concentration of VU591 (n = 5–15). The IC50 values for ROMK-WT and ROMK-K80M derived from Hill fits to the data are 297 nM and 307 nM, respectively.
The binding-site mutants were screened at a concentration of 300 nM VU591, which is close to the IC50 for the inhibitor. As shown in Fig. 4 E, mutating the top-scoring residues within the middle site, either alone (S183A, T304A/K80M, and S305A) or in combination (S183A/S305A), had no effect on VU591 sensitivity compared to wild-type ROMK1, which was inhibited by 49 ± 2.0% (n = 25). We therefore assessed residues in the predicted upper binding site, as well as those populating the intervening region of the pore. S172A, C175A, C175I, C175T, and I182M were without effect, but the I178A mutation caused a small (∼11%), but significant (P < 0.05), increase in VU591 sensitivity. L179A and A180L failed to generate measureable currents in wild-type ROMK1 or the K80M mutant (data not shown).
Figure 4.
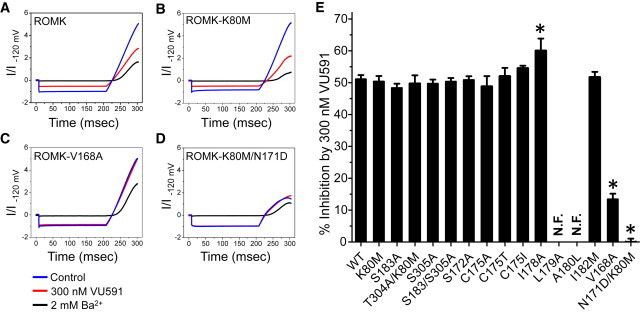
Mutagenesis analysis of the putative middle and upper VU591 binding sites in ROMK-WT and ROMK-K80M. Currents were evoked by a step-ramp protocol (see Materials and Methods) before (control – blue) and after bath perfusion with 300 nM VU591 (red) or 2 mM Ba2+ (black). (A–D) ROMK-WT (A), ROMK-K80M (B), ROMK-V168A (C), and ROMK-K80M/N171D (D). (E) Mean ± SE percent (%) inhibition of the indicated ROMK variant by 300 nM VU591. The asterisks indicate a significant difference (P < 0.05) from control (ROMK-WT or ROMK-K80M) recordings performed on the same day. N.F., nonfunctional mutant. To see this figure in color, go online.
Mutation of two residues in the upper site led to striking changes in the VU591 sensitivity of ROMK1. In single-dose experiments, 300 nM VU591 inhibited ROMK1-V168A by only 13 ± 1.8% (n = 10). Representative current traces of ROMK1, K80M, V168A, and N171D-K80M mutants exposed to 300 nM are shown in Fig. 4, A–D. CRCs revealed an approximate fivefold rightward shift in the IC50 of V168A (IC50 1612 nM) compared to the wild-type channel (IC50 297 nM). Mutation of V168 to C or L led to IC50 values of 1178 nM and >50 μM, respectively (Fig. 5 A).
Figure 5.
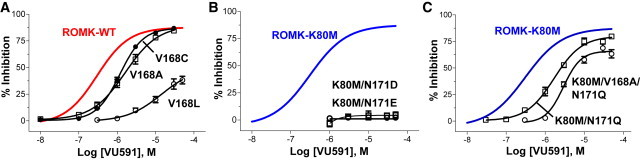
Effect of mutations of V168 and N171 on ROMK sensitivity to VU591. (A) Mean ± SE percent inhibition by VU591 of ROMK-V168C (solid circle), V168A (open square), and V168L (open circle). (B) Mean ± SE percent inhibition by VU591 of ROMK-K80M/N171D (open squares) or K80M/N171E (open circle). (C) Mean ± SE percent inhibition by VU591 of ROMK-K80M/N171Q (open squares) or ROMK-K80M/V168A/N171Q (open circle). Solid lines in A–C are Hill fits taken from Fig. 2G for comparison of ROMK-WT (red) and ROMK-K80M (blue) channels (n = 5–15). To see this figure in color, go online.
Asparagine 171 (N171) of ROMK1 comprises the so-called rectification controller and corresponds to aspartate 172 (D172) in the classical inward rectifier Kir2.1. Because Kir2.1 is insensitive to VU591 (16), and given the proximity and importance of V168 in VU591 sensitivity, we assessed whether mutation of N171 to D (D172) affects ROMK sensitivity to VU591. However, ROMK1-N171D carried no measurable current, prompting us to examine the mutation in the K80M background. Whereas 300 nM VU591 inhibited K80M by 48 ± 1.6% (n = 10), which was not statistically different (P > 0.05) from wild-type ROMK1 (Fig. 4 E), the N171D mutation abolished channel sensitivity to this concentration of VU591 (Fig. 4, D and E). IC50 values for N171D (or N171E) could not be reliably determined, because <5% inhibition was observed at a concentration of 50 μM VU591 (Fig. 5 B). Introduction of the polar uncharged residue glutamine at position 171 (N171Q/K80M) resulted in a 5.5-fold loss in channel sensitivity to VU591 (IC50 1692 nM) compared to ROMK1-K80M controls (307 nM). Interestingly, the double mutation V168A/N171Q right-shifted the CRC by 9.5-fold (IC50 2926 nM, 95% CI, 2264–3782; Hill slope, 1.9; r2 = 0.92) compared to K80M (Fig. 5 C), suggesting that both residues contribute to VU591-dependent block independently. To increase confidence in the binding site prediction, N171 was mutated to D in the ROMK model. This in silico mutagenesis resulted in a significant (P = 1.8−9) decrease in the predicted VU591 binding energy from −14.1 REUs to −11.0 REUs, in agreement with results from patch-clamp experiments.
Reverse-engineering a VU591 binding site into Kir2.1
To further assess the role of V168 and N171 in the mechanism of ROMK inhibition, we attempted to reverse-engineer VU591 sensitivity by mutating corresponding amino acids, C169V and D172N, respectively, in Kir2.1. Representative I-V plots for Kir2.1-WT and Kir2.1-C169V/D172N are shown in Fig. 6, A and B. As reported previously (16), the wild-type channel is largely insensitive to VU591 (2 ± 1.5% and 9.6 ± 1.4% by 50 μM and 100 μM VU591, respectively; n = 5 each). However, the D172N mutation increased Kir2.1 sensitivity to VU591 by approximately threefold (i.e., 6.8 ± 1.1% and 22.6 ± 3.2% inhibition at 50 μM and 100 μM VU591, respectively), which was statistically (P < 0.05) different from that observed for wild-type Kir2.1 (Fig. 6 C). Although mutation of Kir2.1-C169 to valine (i.e., Kir2.1-C169V), which corresponds to ROMK1-V168, did not significantly (P > 0.05) increase the sensitivity of the channel to VU591 as compared to Kir2.1-D172N, the double mutant was inhibited by 9 ± 1.3% and 28 ± 2.8% by 50 μM and 100 μM VU591, respectively (n = 5 each). Taken together, these data support the conclusion that the VU591 binding site in ROMK1 includes V168 and N171.
Figure 6.
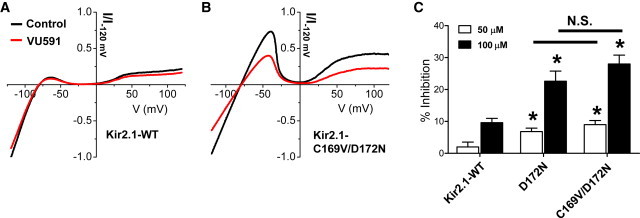
Reverse engineering a VU591-sensitive Kir2.1 mutant. (A and B) I-V plot for Kir2.1-WT (A) or Kir2.1-C169V/D172N mutant (B) recorded in the absence (black trace) or presence (red trace) of 100 μM VU591. (C) Summary of the mean ± SE percent inhibition of Kir2.1-WT, Kir2.1-D172N, and Kir2.1C169/D172N by 50 μM (open bars) or 100 μM VU591 (solid bars). Statistical significance (P < 0.05) from Kir2.1-WT at the same dose of VU591 is indicated with an asterisk. N.S., no significant difference between D172N and C169V/D172N. To see this figure in color, go online.
Discussion
The molecular pharmacology of the inward rectifier family has remained underdeveloped since the founding member, ROMK, was cloned more than 20 years ago (1,2). To date, nanomolar-affinity antagonists have been developed for ROMK, Kir2.1, and KATP (Kir6.x/SURx). Potent and selective KATP channel inhibitors (glibenclamide, tolbutamide) as well as openers (diazoxide, pinacidil) have been used clinically for decades to treat disorders of insulin and glucose homeostasis (35). These compounds interact with high-affinity binding sites located on regulatory SUR subunits to induce conformational changes that open or close the Kir6.x channel pore (36,37). Although there is some evidence that SUR2B interacts with ROMK and confers weak sensitivity to glibenclamide (38,39), others have proposed that ROMK is intrinsically sensitive to the inhibitor (40).
The small-molecule pharmacology of the Kir family is dominated by drugs mostly directed toward cardiac and neurological targets but that also act directly on Kir channels at higher doses (for review, see Bhave et al. (41) and Swale et al. (42). For example, the tricyclic antidepressant nortriptyline and selective serotonin reuptake inhibitor fluoxetine inhibit Kir4.1 with IC50 values of 28 μM and 15 μM, respectively. Furutani et al. (43) found that mutation of E158, which corresponds to N171 in ROMK, weakened inhibition of Kir4.1 by these drugs. Mutation of ROMK-N171 to glutamine (N171Q) had no effect on channel pharmacology, whereas ROMK carrying N171E or N171D mutations exhibits enhanced sensitivity to both blockers.
Similarly, the Kir2.x-selective inhibitor ML133 also requires a negatively charged residue at this position (44), which is occupied by an aspartate residue (i.e., D172) in Kir2.1 (44). Mutation of D172 to asparagine (D172N) in Kir2.1 leads to a nearly eightfold loss of ML133 potency. In contrast, introduction of a single N171D mutation in ROMK is sufficient to confer ML133 sensitivity to the channel.
In this study, we found that introduction of a negatively charged residue (E or D) at position N171 in ROMK, which corresponds to Kir4.1-E158 and Kir2.1-D172, obliterates the sensitivity of the channel to VU591. Robertson and colleagues proposed that the introduction or neutralization of charged residues in the Kir channel pore can influence the electrostatic potential along the pore, and thus interactions with K+ ions as well as charged pore blockers, such as Mg2+, as far as 40 Å away (45). It is therefore conceivable that the N171D or N171E mutations disrupt interactions of VU591 with a binding site some distance away from the mutation site. We propose that the VU591 binding site is localized near N171 for the following reasons. First, in silico docking simulations identified an energetically favorable interaction between VU591 and N171. Second, extending the side-chain length by two methyl groups at position 171 without introducing a charge at this site, led to a ∼5.5-fold loss of VU591 sensitivity in the ROMK-N171Q mutant. Third, introduction of an asparagine residue into Kir2.1 at position confers VU591 sensitivity to this channel. Fourth, neutral mutations of the adjacent pore-lining residue, V168, also affect channel block. Substitution of the smaller hydrophobic residues alanine (V168A) or cysteine (V168C) produced ∼5.4- and ∼4-fold losses in potency, respectively. Interestingly, lengthening the side chain by only one methyl group with a V168L mutation virtually abolished sensitivity to VU591. Finally, the double mutation V168A/N171Q produced a greater loss in VU591 sensitivity than did the N171Q mutation alone. These results are consistent with our computational modeling results suggesting that VU591 is capable of interacting with both residues simultaneously.
It is also conceivable that mutations of V168 or N171 alter the secondary and tertiary structures of ROMK and consequently the binding site for VU591 elsewhere in the channel pore. However, even conservative substitutions such as V168L and N171Q led to striking changes in channel pharmacology. Furthermore, no other mutations in the pore reduced sensitivity to VU591, indicating that the effect is specific to V168 and N171. Rodriguez-Menchaca found that mutation of residues throughout the membrane-spanning pore, including D172 (equivalent to ROMK-N171) and C169 (equivalent to ROMK-C168), had no effect on the chloroquine binding site located in the cytoplasmic domain of the channel (46). Finally, among six members of the inward rectifier family tested for sensitivity to the VU591, the only two channels inhibited were ROMK (IC50 ∼290 nM) and Kir6.2/SUR1 (IC50 >50 μM) (16). An alignment of the primary sequences of the subunits reveals that Kir6.2 is the only subunit that contains an asparagine residue at the position equivalent to that of ROMK-N171. Kir6.2 contains a methionine residue in the position corresponding to ROMK-V168. Taken together, these data support the notion that residues V168 and N171 of ROMK contribute directly to the binding site for VU591.
Results from our molecular docking studies may provide an explanation for why VU591 is ∼30-fold more potent than its precursor inhibitor 1,3-bis(5-nitro-1H-benzo[d]imidazol-2-yl)propane BNBI (16). The only difference between the VU591 and BNBI chemical structures is the presence of an ether oxygen in the linker region of VU591 (Fig. 7) (16). In a previous study, we postulated that the oxygen addition improves the potency of VU591 by increasing the electron donor potential (and hydrogen-bond acceptor potential) of the ether linker and thereby stabilizes ligand interactions with a polar portion of the ROMK channel pore. Although this may also play a role in determining the potency of VU591, the modeling results offer another potential explanation. The ROMK channel pore near N171 and V168 is too narrow to accommodate the fully extended small molecule, requiring that VU591 assume a horseshoe conformation to reach its binding site. Substitution of a methyl group in BNBI with an oxygen atom in VU591 may support greater conformational flexibility and enable VU591 to interact more intimately with its binding site. Interestingly, Merck Research Laboratories recently disclosed an ROMK channel inhibitor that bears some structural similarities to VU591. Notably, the linker region of this Merck compound contains a flexible piperazine group, suggesting that it, too, may assume a horseshoe conformation in the pore to reach its binding site near N171 in the ROMK channel pore. Garcia and colleagues reported that N171 is also essential for block of ROMK by the Merck inhibitor, (5-(2-(4-(2-(4-(1H-tetrazol-1-yl)phenyl)acetyl)piperazin-1-yl)ethyl)isobenzofuran-1(3H)-one), termed compound A (47).
Figure 7.
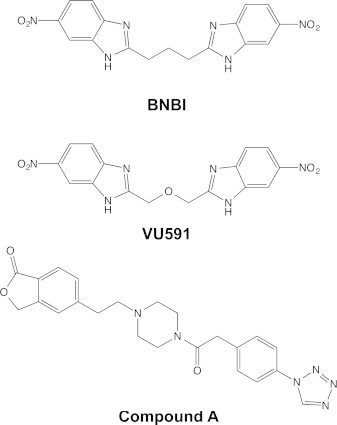
Chemical structures of BNBI, VU591, and Compound A.
Development of additional specific high-affinity inhibitors targeting Kir channels would have value for both further probe development and potential therapeutic applications. In addition to HTS approaches being used to identify new compounds, an attractive method for facilitating the discovery of additional molecules is structure-based in silico screening. Given a high-resolution crystal structure of the target protein, in-silico predictions can accelerate the screening procedure and aid in design and lead development. This study demonstrates that the challenges of using a comparative model in place of a crystal structure can be overcome by using computational and experimental techniques in concert to leverage the limited structural information available for ROMK. This validated model is expected to improve in quality as additional experimental data are incorporated.
Author Contributions
D.R.S., J.H.S., J.M., and J.S.D. designed the research; D.R.S., J.H.S., S.B., A.S.H., and T.T.N. performed the research; S.B. and J.M. contributed analytic tools; D.R.S., J.H.S., J.M., and J.S.D. analyzed data; and D.R.S., J.H.S., J.M., and J.S.D. wrote the manuscript.
Acknowledgments
Calculations were performed on Vanderbilt’s VAMPIRE cluster at the Advanced Center for Computational Research and Excellence (ACCRE) and the Piranha cluster at the Vanderbilt Center for Structural Biology (CSB).
This work was supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK082884 to J.S.D.).
Supporting Material
References
- 1.Ho K., Nichols C.G., Hebert S.C. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362:31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 2.Zhou H., Tate S.S., Palmer L.G. Primary structure and functional properties of an epithelial K channel. Am. J. Physiol. 1994;266:C809–C824. doi: 10.1152/ajpcell.1994.266.3.C809. [DOI] [PubMed] [Google Scholar]
- 3.Hebert S.C., Desir G., Wang W. Molecular diversity and regulation of renal potassium channels. Physiol. Rev. 2005;85:319–371. doi: 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welling P.A., Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am. J. Physiol. Renal Physiol. 2009;297:F849–F863. doi: 10.1152/ajprenal.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greger R., Schlatter E. Presence of luminal K+, a prerequisite for active NaCl transport in the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch. 1981;392:92–94. doi: 10.1007/BF00584588. [DOI] [PubMed] [Google Scholar]
- 6.Greger R., Schlatter E. Properties of the lumen membrane of the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch. 1983;396:315–324. doi: 10.1007/BF01063937. [DOI] [PubMed] [Google Scholar]
- 7.Clark M.A., Humphrey S.J., Ludens J.H. Unique natriuretic properties of the ATP-sensitive K+-channel blocker glyburide in conscious rats. J. Pharmacol. Exp. Ther. 1993;265:933–937. [PubMed] [Google Scholar]
- 8.Wang T., Wang W.H., Giebisch G. Effects of a novel KATP channel blocker on renal tubule function and K channel activity. J. Pharmacol. Exp. Ther. 1995;273:1382–1389. [PubMed] [Google Scholar]
- 9.Bartter F.C., Pronove P., MacCardle R.C. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am. J. Med. 1962;33:811–828. doi: 10.1016/0002-9343(62)90214-0. [DOI] [PubMed] [Google Scholar]
- 10.Peters M., Jeck N., Seyberth H.W. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am. J. Med. 2002;112:183–190. doi: 10.1016/s0002-9343(01)01086-5. [DOI] [PubMed] [Google Scholar]
- 11.Simon D.B., Karet F.E., Lifton R.P. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 12.Bailey M.A., Cantone A., Malnic G. Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of Type II Bartter’s syndrome and in adaptation to a high-K diet. Kidney Int. 2006;70:51–59. doi: 10.1038/sj.ki.5000388. [DOI] [PubMed] [Google Scholar]
- 13.Grimm P.R., Sansom S.C. BK channels in the kidney. Curr. Opin. Nephrol. Hypertens. 2007;16:430–436. doi: 10.1097/MNH.0b013e32826fbc7d. [DOI] [PubMed] [Google Scholar]
- 14.Ji W., Foo J.N., Lifton R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis L.M., Bhave G., Denton J.S. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol. Pharmacol. 2009;76:1094–1103. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhave G., Chauder B.A., Denton J.S. Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol. Pharmacol. 2011;79:42–50. doi: 10.1124/mol.110.066928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang H., de Jesus R.K., Pasternak A. Discovery of a novel sub-class of ROMK channel inhibitors typified by 5-(2-(4-(2-(4-(1H-Tetrazol-1-yl)phenyl)acetyl)piperazin-1-yl)ethyl)isobenzofuran-1(3H)-one. Bioorg. Med. Chem. Lett. 2013;23:5829–5832. doi: 10.1016/j.bmcl.2013.08.104. [DOI] [PubMed] [Google Scholar]
- 18.Tang H., Walsh S.P., Pasternak A. Discovery of selective small molecule ROMK inhibitors as potential new mechanism diuretics. ACS Med. Chem. Lett. 2012;3:367–372. doi: 10.1021/ml3000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tao X., Avalos J.L., MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 Å resolution. Science. 2009;326:1668–1674. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whorton M.R., MacKinnon R. X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature. 2013;498:190–197. doi: 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen S.B., Tao X., MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 2011;477:495–498. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 24.Leaver-Fay A., Tyka M., Bradley P. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yarov-Yarovoy V., Schonbrun J., Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins. 2006;62:1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barth P., Wallner B., Baker D. Prediction of membrane protein structures with complex topologies using limited constraints. Proc. Natl. Acad. Sci. USA. 2009;106:1409–1414. doi: 10.1073/pnas.0808323106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viklund H., Elofsson A. OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics. 2008;24:1662–1668. doi: 10.1093/bioinformatics/btn221. [DOI] [PubMed] [Google Scholar]
- 28.Meiler J., Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 29.Hastie T., Tibshirani R., Friedman J. Springer; New York: 2001. The Elements of Statistical Learning. [Google Scholar]
- 30.Alexander, N., N. Woetzel, and J. Meiler. 2011. Bcl::cluster: a method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System. IEEE Intl. Conf. Comput. Adv. Bio Med. Sci. (ICCABS), 1st, Orlando, FL. 13–18. [DOI] [PMC free article] [PubMed]
- 31.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Shortle D., Simons K.T., Baker D. Clustering of low-energy conformations near the native structures of small proteins. Proc. Natl. Acad. Sci. USA. 1998;95:11158–11162. doi: 10.1073/pnas.95.19.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fallen K., Banerjee S., Denton J.S. The Kir channel immunoglobulin domain is essential for Kir1.1 (ROMK) thermodynamic stability, trafficking and gating. Channels (Austin) 2009;3:57–68. doi: 10.4161/chan.3.1.7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schulte U., Hahn H., Ludwig J. pH gating of ROMK (K(ir)1.1) channels: control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proc. Natl. Acad. Sci. USA. 1999;96:15298–15303. doi: 10.1073/pnas.96.26.15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gribble F.M., Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46:875–891. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- 36.Aguilar-Bryan L., Nichols C.G., Nelson D.A. Cloning of the β cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- 37.Tucker S.J., Gribble F.M., Ashcroft F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 38.Tanemoto M., Vanoye C.G., Xu J.Z. Rat homolog of sulfonylurea receptor 2B determines glibenclamide sensitivity of ROMK2 in Xenopus laevis oocyte. Am. J. Physiol. Renal Physiol. 2000;278:F659–F666. doi: 10.1152/ajprenal.2000.278.4.F659. [DOI] [PubMed] [Google Scholar]
- 39.Dong K., Xu J., Hebert S.C. An amino acid triplet in the NH2 terminus of rat ROMK1 determines interaction with SUR2B. J. Biol. Chem. 2001;276:44347–44353. doi: 10.1074/jbc.M108072200. [DOI] [PubMed] [Google Scholar]
- 40.Konstas A.A., Dabrowski M., Tucker S.J. Intrinsic sensitivity of Kir1.1 (ROMK) to glibenclamide in the absence of SUR2B. Implications for the identity of the renal ATP-regulated secretory K+ channel. J. Biol. Chem. 2002;277:21346–21351. doi: 10.1074/jbc.M202005200. [DOI] [PubMed] [Google Scholar]
- 41.Bhave G., Lonergan D., Denton J.S. Small-molecule modulators of inward rectifier K+ channels: recent advances and future possibilities. Future Med. Chem. 2010;2:757–774. doi: 10.4155/fmc.10.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swale D.R., Kharade S.V., Denton J.S. Cardiac and renal inward rectifier potassium channel pharmacology: emerging tools for integrative physiology and therapeutics. Curr. Opin. Pharmacol. 2014;15:7–15. doi: 10.1016/j.coph.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furutani K., Ohno Y., Inanobe A., Hibino H., Kurachi Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol. 2009;75:1287–1295. doi: 10.1124/mol.108.052936. [DOI] [PubMed] [Google Scholar]
- 44.Wang H.R., Wu M., McManus O.B. Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem. Biol. 2011;6:845–856. doi: 10.1021/cb200146a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robertson J.L., Palmer L.G., Roux B. Long-pore electrostatics in inward-rectifier potassium channels. J. Gen. Physiol. 2008;132:613–632. doi: 10.1085/jgp.200810068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodríguez-Menchaca A.A., Navarro-Polanco R.A., Sánchez-Chapula J.A. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc. Natl. Acad. Sci. USA. 2008;105:1364–1368. doi: 10.1073/pnas.0708153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia M.L., Priest B.T., Pasternak A. Pharmacologic inhibition of the renal outer medullary potassium channel causes diuresis and natriuresis in the absence of kaliuresis. J. Pharmacol. Exp. Ther. 2014;348:153–164. doi: 10.1124/jpet.113.208603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.