

Abstract
Diabetes associated depression is a largely understudied field which nonetheless carries a significant disease burden. The very low therapeutic efficacy of the existing conventional drugs with poor outcome may be, in part, due to uncertainty of the mechanism involved that clearly explains the existing comorbidity. The main purpose of this review was to address the sophisticated mechanisms of this comorbidity with a view of developing potential novel targets with higher efficacy and specificity. Data were collected from database searches including PubMed, references from relevant English language research/review articles and other official publications. Articles from 1990 to 2013 were included, and a broad search term criteria were followed for data mining so that relevant information was not missed out. Some of the search terms used included; diabetes-induced depression, diabetes and serotonin, hypothalamic-pituitary-adrenal (HPA) axis and diabetes and glucocorticoids in diabetes. Neuropathologically, depletion of brain monoaminergic activity specifically the serotonin (5-hydroxytryptamine [5-HT]) system, due to chronically persisting diabetic state may lead to the mood and behavioral complications that further add on worsening the quality life years. The 5-HT system through multifunctional tasks regulates neurogenesis and plasticity and by complex receptor mechanism controls the emotional and behavioral activity. Persisting hyperglycemia leads to impaired neurogenesis, decreased synaptic plasticity, undesired neuro-anatomical alterations, neurochemical deficits, and reduced neurotransmitter activity. The neurotrophic factors and secondary messenger functions affected at molecular and genetic levels indicate the impact of diabetes-mediated dysregulation on neuronal circuits. HPA activity, glycogen synthase kinase 3, and insulin signaling controls were also found to be hampered, interlinked to 5-HT system following diabetic progression.
KEY WORDS: Depression, diabetes, glucocorticoids, glycogen synthase, insulin receptors, serotonin
Introduction
Diabetes has emerged from a mild metabolic disorder to one of the major causes of morbidity and mortality in the developed world. According to International Diabetes Federation, 4.6 million deaths were reported in 2011 due to diabetes and associated complications. It is estimated that 552 million people will suffer from diabetes by 2030.[1] Diabetes associated depression is a serious condition that can aggravate both the symptoms of depression as well as diabetes associated complications. It has been reported that the incidence of depression is 2–3 times higher in diabetes than in the nondiabetic population and 1 in 3 diabetes patients suffer from one or the other form of depression.[2,3] More specifically, it has been found that diabetic patients with poor glycemic control was at greater risk of depression than those with well-controlled sugar levels.[4] Patients with diabetes associated depression has a significantly reduced prognosis as they have less adherence to antidiabetic/antidepressant medications and are predisposed to have related risk factors like obesity, poor hyperglycemic control, smoking, and a sedentary lifestyle.[5,6] A multidirectional approach may explain the contribution of depression to diabetes and the effect of diabetes on aggravating or triggering a depressive episode.[7]
Some of the major theories on the origins and causes of depression include the genetic vulnerability, altered hypothalamic-pituitary-adrenal (HPA) activity, monoamine deficiency, dysfunction of specific brain regions, reduced gamma amino butyric acid activity and altered neurotrophic and neurotoxic process [Figure 1].[8,9] However, one of the most widely accepted and experimentally proven theories is the altered monoamine levels in the brain. Monoamines such as 5-hydroxytryptamine (5-HT), dopamine, epinephrine, and norepinephrine have been found to play a significant role in the neurobiology of depression, and a down-regulation of any of these is a characteristic of depression.[10] Of these, 5-HT is of particular interest due to the diversity of its receptor subtypes and the strong experimental evidence that correlates altered receptor regulations and 5-HT levels in diabetes associated depression. Here, we would like to detail on the multifunctional role of 5-HT system and its possible ramifications in the development of depression associated with diabetes. Further, the clinical implications of existing antidepressant drugs in the comorbidity have been focused.
Figure 1.
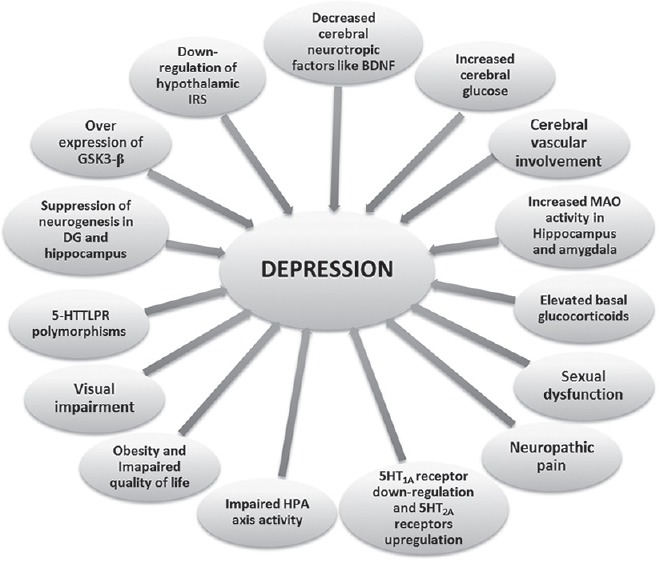
Possible diabetic factors that can contribute to depression in diabetic individuals. BDNF=Brain-derived neurotrophic factor, MAO=Monoamine oxidase, HPA=Hypothalamic-pituitary-adrenal axis, serotonin transporter gene 5’ promoter region, DG=Dentate gyrus, GSK3-β=Glycogen synthase kinase 3 β, IRS=Insulin receptor substrate
Aims and Methodology
There has been a significant development in the field of both depression and metabolic disorders like diabetes, but until recently they have been considered as separate entities with studies focusing on separate disease mechanisms and pathways. Recent research points toward the necessity to consider the comorbid nature of such disorders and study them as a single disease with intertwined mechanisms and branching pathways. The main aim of this paper was to review the recent developments in this field and understand the proposed mechanisms that interlink both diabetes and depression. Considering both are highly prevalent social diseases, the need to isolate a single efficient target that can be utilized in higher drug discovery platforms for treating both the conditions simultaneously is pressing. Medical and life science research databases such as PubMed, Medline and PubMed Central, were searched using a broad search term based criteria. Selection priority was given to preclinical studies emphasizing on mechanistic approaches rather than experimental part. Only English language articles from 1990 to 2013 that were indexed with one or more of the earlier discussed search terms were included for the study.
Abnormal Neuronal Functioning in Diabetes
There have been numerous reports validating a strong correlation between diabetes and a significant increase in neuronal damage.[11] One of the most studied and well-established alterations is the impairment to the stress reactivity and the HPA axis activity dysregulation associated with depression.[12] Other established alterations include changes in the synaptic plasticity, neuroanatomical alterations, neurochemical changes and deficits in insulin signaling.[13,14] Moreover, the hyperglycemia induced neuroanatomical changes in the hippocampus of streptozotocin (STZ) rats may trigger a series of events that may lead to irreversible neuronal damage. Inhibition of neurogenesis and neuronal apoptosis has been detected in rodent brains with uncontrolled hyperglycemia.[15,16] This correlates well with clinical findings that the probability of depression in diabetes decreases with increased control of the hyperglycemia. The hippocampal long-term potentiation is an important attribute to normal neuronal functioning, and this has been found to be impaired in STZ diabetic rats leading to loss of performance in most standard cognitive tests.[17] Furthermore, there is significant suppression of the insulin receptors (IRs) in the hippocampal areas in STZ diabetic rats, which may be the reason for the poor cognitive performance.[18]
Diabetes and Depression: The Role of the 5-Hydroxytryptamine System
One of the most studied and established neurological links between depression and diabetes is the involvement of the 5-HT system. The role of 5-HT in modulation of cerebral glucose levels may be multifacetedly ranging from changes in transporter polymorphisms to downstream signaling as a result of direct receptor activation.[19] A definite correlation has been established between 5-HT transporter (5-HTT) polymorphisms and depression in diabetic individuals, further strengthening the possible role of 5-HT in diabetic associated depression.[20] The polymorphisms in the lower expressing allele of the 5-HTT gene 5’ promoter region can contribute to a higher susceptibility to depression in response to stress full environmental stimulus. It was also established that compared to the higher expressing allele, the lower expressing ones had a greater contribution to severe depression in response to stress events like diabetes. However, the exact mechanism mediating the heightened sensitivity to depression in response to stress is still unresolved. The decreased 5-HTT gene expression in depressed suicide victims indicates a role of transporter polymorphisms in depression.[21] As discussed earlier, the polymorphisms in the 5-HT promoter region have been associated in many chronic disease conditions like coronary artery disease. The polymorphisms are associated with an increased vulnerability for developing depressive symptoms which is compounded in patients with comorbid disorders like diabetes. All these data indicate that there might be an important role for genetics in comorbid depressive symptoms.
The exact cellular mechanism that conclusively explains the development of depression in diabetes is still under investigation. However, one possible mechanism may be the enzymatic alterations in the brain including decreased levels of free fraction of L-tryptophan (FFT) and the inhibition of tryptophan-5-hydroxy-lase 2 (TPH 2) that is responsible for the conversion of FFT to 5-HT. It has been confirmed that diabetes disturbs the equilibrium of FFT with other neutral amino acids that plays an important role in determining the amount of FFT available for conversion to 5-HT, ultimately leading to a suppression of 5-HT synthesis.[22,23] Other findings suggest the alterations in 5-HT receptor levels and functions that occur as a consequence of diabetes. The 5-HT1A receptors have been found to be dysregulated in diabetes, and the effect of antidepressant drugs that act primarily through this receptor is diminished in STZ-induced diabetic animals.[24] The 5-HT2A receptors were found to be up-regulated in STZ diabetic rat models confirming clinical findings of increased 5-HT2A receptor density in the frontal cortex of depressed subjects.[20] Studies have further confirmed the increased 5-HT1A and 5-HT2 receptor density in STZ diabetic rat brains that may be attributed to a compensatory mechanism for the reduced 5-HT activity.[25] These data clearly indicate a strong and rational correlation between the increased epidemiology of affective mood disorders like depression in diabetic conditions and possible mechanistic link of 5-HT.
Molecular Interplay and Putative Mechanisms
The molecular investigations indicate the existence of a complex mechanism regulating mood and behavioral activities. These include HPA axis controlling mechanism, insulin signaling regulation, and neuro-anatomical and neurogenetic factors activity that maintain the neuronal stability and dynamics. Diabetes being a metabolic disorder affects these functions and alters the regulatory mechanisms. However, detailing the complexity is beyond the scope of this review, the putative mechanism linking 5-HT system has been described below.
Glucocorticoid Mediated Modulation of 5-Hydroxytryptamine System through the Hypothalamic-pituitary-adrenal Axis
One of the hypothesized mechanisms that correlates diabetes and depression is the activation of the HPA axis with the involvement of brain glucocorticoid (GC) system.[26] Neuronal stress induces the release of corticotrophin-releasing factor (CRF) by the neurons of the paraventricular nucleus (PVN) in the hypothalamus and the CRF stimulates the release of adrenocorticotropin via pituitary gland to stimulate the synthesis and release of GCs in adrenals (cortisone in humans and corticosterone in rodents) that can have wide ranging effects on the brain mood and behavioral functions. The released GCs control HPA axis by negative feedback mechanism and a sustained elevation in GCs as seen in diabetes can damage the hippocampal neurons through reduced dendritic branching and hippocampal atrophy.[27] The destruction of the hippocampal regions can further disturb the feedback inhibition of the GCs producing a prolonged hypercortisolemic state that can further lead to depressive symptoms.[27,28,29] Furthermore, it has been suggested that postsynaptic 5-HT1A, 5-HT2A, and 5-HT2C are regulated by brain corticosterone leading to 5-HT mediated behavioral, neuroendocrine and temperature regulatory effects, though the exact mechanism is still uncertain.[30,31,32] The presence of GC receptors on the cell bodies of the ascending 5-HT neurons further indicates a direct GC modulatory mechanism of presynaptic 5-HT neurons.[31] The GC receptors have also been identified in specific brain areas that has been found to play key role in cognitive and mood related disorders including amygdala, prefrontal cortex, and hippocampus, indicating a possible regulation of the 5-HT system by the brain GC.[33] 5-HT was reported to stimulate the HPA axis via activation of the 5-HT2C receptors in the PVN of the hypothalamus.[34] Stimulation of 5-HT1A receptors has been found to activate the HPA axis and induce the release of GC. Down-regulation of the 5-HT1A receptors can lead to inhibition of the HPA axis, thereby inhibiting the GC release that can stimulate the insulin secretion.[35] The 5-HT1A receptors are potential targets for novel antidepressant agents with efficacy in diabetes-induced depression as they can regulate the GC levels which in turn affect glycemic control.
Neurotrophic Factors and Regulation of 5-Hydroxytryptamine System
Another major link between diabetes and depression is the alteration in the neurotrophic mechanisms including suppression of the brain-derived neurotrophic factor (BDNF) possibly by pathways that involve GC excess. The regulation of BDNF and other similar neuropeptides by GCs has been well-established and can take place through a variety of cellular pathways.[36] Furthermore, BDNF and other neuropeptides have been found to be involved in the regulation of 5-HT system.[37] One of the most direct involvements of the GC in BDNF regulation is the modulation of BDNF transcription by binding of GC on the glucocorticoid response element in the promoter regions. Another mechanism is the interference in the proteolytic conversion of pro-BDNF to mature-BDNF thereby altering the levels of matured BDNF in the brain [Figure 2]. Further, the binding of BDNF to its tyrosine kinases B leads to activation of phospholipase C-γ (PLC-γ), phosphatidylinositol-3-kinase (PI3K), and mitogen-activated protein kinase pathways leading to downstream activation resulting in many physiological process including synaptic plasticity, neuronal excitability, and survival [Figure 3]. The GCs have been found to regulate these pathways at multiple points resulting in the overall depression of BDNF activity.[38]
Figure 2.
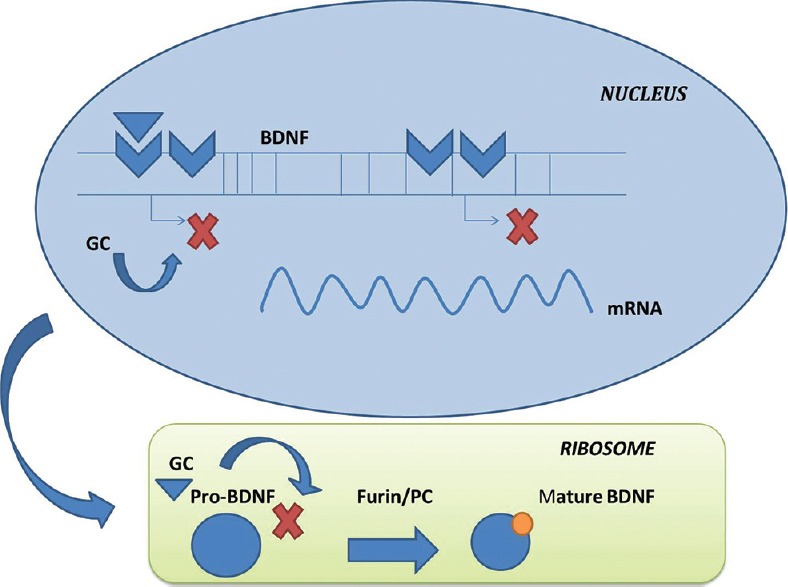
Glucocorticoids (GCs) mediated regulation of brain-derived neurotrophic factors (BDNF) transcription. GCs alter the transcription in nucleus and ribosomal translation process of BDNF and hence affect its synthesis. mRNA= micro RNA[67]
Figure 3.
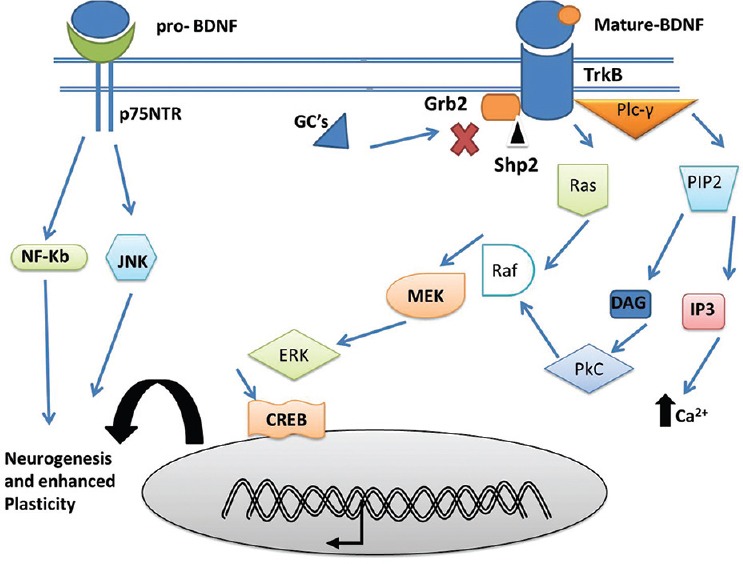
Schematic representation of the modulation of downstream signaling pathways of the brain derived neurotrophic factors (BDNF) by glucocorticoids (GCs). BDNF bind to their cognate tyrosine kinases receptor to initiate downstream signaling events which is inhibited by GCs. BDNF through Phospholipase C-γ/protein kinase C pathway through a cascade involving phosphatidylinositol 4,5-bisphosphate, inositol triphosphate, leads to release of intracellular calcium (Ca2+) and regulates synaptic plasticity. BDNF activated Ras/extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase/ERK kinases)/cyclic AMP response element-binding protein signaling is involved in cell proliferation, differentiation and protection of neurons. BDNF signaling causes deactivation of C-jun N-terminal kinase, thereby inhibiting destabilization of microtubules and maintaining neuronal dynamic stability. DAG=Diacylglycerol, Nf-Kb=Nuclear factor kappalight- chain-enhancer of activated B cells, Src homology region two domain-containing phosphatase-2, p75 neurotrophin receptor, Grb- 2=Growth factor receptor-bound protein 2[68]
There is conclusive evidence for the strong regulation of 5-HT system by BDNF, including morphological differentiation of 5-HT neurons, increased expression of TPH 2 and enhanced 5-HT uptake that may all point to an activation of 5-HT system by BDNF.[39] The substantially increased GC levels in diabetes patients may point towards the possible alterations of neurotrophic factors that ultimately results in depression.
Central Insulin Receptors and the 5-Hydroxytryptamine System
Down-regulation of the hypothalamic IRs causes obesity, insulin resistance and diabetes. New evidence suggests that the adipocyte-derived hormone leptin plays a role in linking obesity, diabetes, and depression. The down-regulation of the hypothalamic IRs leads to increased plasma leptin levels, producing to hyperleptinemia.[40] There are currently two hypotheses regarding the mechanistic role of leptin in depressive behavior in diabetes. The first one points at the hyperleptinemia caused due to the down-regulation of the hypothalamic IRs that impairs the leptin transport across the blood-brain barrier leading to a suppressed central nervous system leptin availability in spite of high plasma leptin levels.[41] The second hypothesis identifies the regulatory role of leptin on the 5-HT system. An increased brain leptin levels have found to decrease the expression of the TPH 2 enzyme involved in the synthesis of 5-HT [Figure 4]. Furthermore, it has been demonstrated that TPH 2 levels and 5-HT concentration in the brain stem was found to be high in ob/ob mice where the leptin gene has been knocked out.[42] All these data indicate a significant role of leptin in depressive behavior in diabetic subjects involving a dysegulated 5-HT system.
Figure 4.
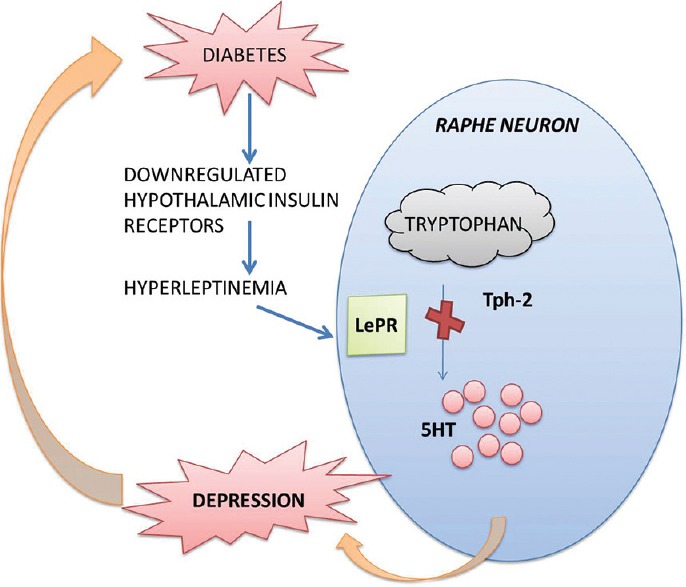
Regulation of serotonin synthesis by brain leptin levels. Excessive brain leptin levels directly inhibit tryptophan hydroxylase-2 thereby decreasing the circulating neuronal levels of serotonin (5-hydroxytryptamine)[41]
Glycogen Synthase Kinase 3 Regulation and 5-Hydroxytryptamine System
Another major cellular mechanism that can suggest the arousal of depressive symptoms in diabetic individuals is the glycogen synthase kinase 3 (GSK3) pathway and the regulatory influence of 5-HT system on GSK3. GSK3 has been known to exert significant influences on the neuronal plasticity, cellular architecture as well as regulation of apoptosis and cell survival.[43] GSK3 is basically a cytosolic serine/threonine protein kinase with two isoforms α/β which arrest glycogen synthesis as one of its regulatory processes by inhibiting the rate-limiting enzyme glycogen synthase. Several metabolic disorders like diabetes are associated with an increased GSK3 expression, and GSK3 mediated multifunctional process may add on the neuropsychiatric complications such as depression to be associated with diabetes. The phosphorylation and dephosphorylation mechanisms are the most studied regulatory process of GSK3 [Figure 5]. The α and β isoforms of GSK are inhibited by phosphorylation of an N-terminal serine (serine-9 in GSK3 β and serine-21 in GSK3α) that can be mediated by a number of different kinases including protein kinase A (PKA), protein kinase Cand Akt.[44] This phosphorylation mediated inhibition of GSK is found to be impaired in diabetes which can have a profound impact on the neuronal plasticity and neurogenesis, culminating in neuropsychiatric disorders like depression. Another interesting finding is the association of the 5-HT activity with inhibitory control of GSK3 expression. It has been identified that stimulation of 5-HT1A receptors and antagonism of 5-HT2A receptors can increase the serine-9 mediated phosphorylation of GSK3 β, indicating a dynamic regulatory balance between the two 5-HT receptors in the brain.[45] The 5-HT1A receptors coupled to Gi can activate the PI3K and Akt, which in turn can trigger the phosphorylation of GSK3 β. Interestingly the brain samples from depressed suicide victims indicated a suppressed activity of PI3K and Akt that may account for the higher GSK3 β activity that might have led to depression.[46] Though there is no conclusive report, 5-HT2A receptors are classically coupled with Gq and may involve PLC/PKC signaling mechanism mediated regulation of GSK3.[47] An increased uncontrolled expression of GSK3 in diabetes may reflect the impaired regulatory effect of 5-HT system leading to depression-like condition in diabetes patients.
Figure 5.
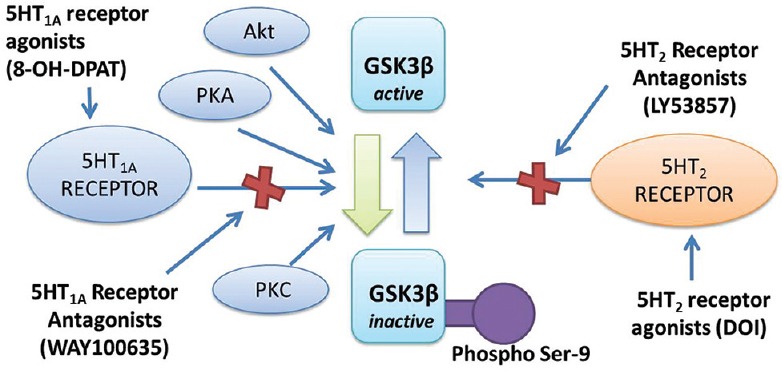
Phosphorylation-dephosphorylation mediated regulation of glycogen synthase kinase 3 β by serotonin and other cellular kinases. Serotonin type-1A (5-hydroxytryptamine [5-HT1A]) receptors activation by agonists as 8-hydroxy-N, N-dipropyl-2-aminotetralin inactivate the enzyme by phosphorylation at serine-9 residues through a series of cascade process involving protein kinase C (PKC), PKA, and Akt pathway, while antagonist (WAY100635) inhibit it. Serotonin type-2 (5-HT2) receptor agonists as 2, 5-dimethoxy-4-iodo-phenylisopropylamine by dephosphorylation activates the enzyme, while antagonists (LY53857) inhibit it[45]
5-Hydroxytryptamine Regulation of Neurogenesis
Diabetes can induce significant alterations in the structural as well as functional integrity of the brain, like changes in hippocampal synaptic plasticity, neurotoxicity.[48] As previously discussed, the alterations in dendritic branching and hippocampal plasticity are associated with higher circulating levels of GCs as occur in diabetes.[49] One of the central regions most affected by diabetes in the brain is the dentate gyrus (DG) exhibiting a reduced dentate gyral volume and a significant decrease in the granule cell density following diabetes.[50] The 5-HT system has been found to play an important role in regulating the proliferation of dendritic neurons. A depletion of 5-HT in the adult rat brain leads to a decrease in the newly synthesized neurons, indicating a possible role of 5-HT in the dendritic neurogenesis. Furthermore, injecting a 5-HT neurotoxin, 5, 7-dihydroxytryptamine into the raphae nuclei of rat brain led to a dramatic reduction in the newly generated neurons. The injection of 5-HT synthesis inhibitors that suppress 5-HT production, without destruction of the 5-HT neurons also reduced neurogenesis indicating a direct role of 5-HT in neurogenesis.[51]
The 5-HT1A receptors have been implicated in the regulation of neurogenesis in the dentate and hippocampal areas. The 5-HT1A receptors located on the astrocytes have been found to regulate the expression of neurotrophic factors like insulin-like growth factor-1 that can further assist in cell proliferation in the central regions.[52] The 5-HT1B autoreceptors are implicated in subventricular zone and DG while the hetero receptors are involved in the DG alone.[53] All these data indicated a clear regulatory role of 5-HT and its various receptors in the neurogenesis in various brain regions that have been implicated in several mood disorders like depression. The overall suppression of the 5-HT system that has been associated with diabetic states is one of the major cellular mechanisms for depression and other mood-related disorders associated with diabetes.
Clinical Implications
The complicated association between diabetes and depression has wide-ranging clinical implications in terms of pharmacological therapy as well as comorbid disease management. It has been identified that selective serotonin 5-HT reuptake inhibitors (SSRIs) such as fluoxetine and sertraline inhibited the insulin-induced tyrosine phosphorylation of the IR substrate-2 (IRS-2) protein leading to deactivation of its targets like Akt. SSRIs were also associated with increased phosphorylation and activation of GSK3 β which is functionally an IRS kinase. The activated GSK3 β promote phosphorylation of the inhibitory serine sites that result in inhibition of insulin action. The SSRIs inhibited the glucose-stimulated insulin release in a dose dependent manner and prolonged treatment lead to endoplasmic reticulum stress responses, induction of nitric oxide synthase, apoptotic process with increased caspase3/7 activity finally leading to β cell death, culminating in a diabetic state.[54] Furthermore, epidemiological studies have shown that SSRIs like sertraline, fluoxetine, and fluvoxamine use is related with increased risk of obesity and hypercholesterolemia.[55] All these point to an increased risk of hyperglycemia with SSRIs that can be especially fatal in comorbid conditions like diabetes.
As previously discussed, the GSK3 has diverse functional dynamics in association with diabetes and depression and is an effective target for mood-related disorders including depression. GSK inhibition activates the glycogen synthase, and it is one of the targets of the various downstream signaling molecules of the neurotrophic factors like BDNF. Akt and PI3K phosphorylates GSK leading to its inhibition.[56] Apart from the various metabolic regulatory pathways that it is involved in; the cognitive implications of this molecule make it a suitable target for comorbid disorders like diabetes associated depression. However, drugs like lithium that directly target GSK have been found to reduce the fasting plasma glucose level significantly and may prove to be effective in depression but its weight gain potential need to be evaluated in clinical settings, especially with regard to comorbid associations with metabolic dysregulation like diabetes.[57] Drug-induced weight gain in high-risk population like type-2 diabetes can worsen the condition of the patients. Furthermore, other clinically used antidepressants like monoamine oxidase inhibitors (MAOIs) have a significant effect on glucose metabolism.[58] Reversible inhibitors of MAO like moclobemide do not exert any adverse effect on glucose metabolism and are sometimes an advantage over the conventional MAOIs.[59] Bupropion used for its weight loss effects does not have any glucose regulatory effects and is a safe alternative for MAOIs.[60] Tricyclic antidepressants (TCA) can cause hyperglycemia and can be potentially dangerous in diabetic patients. Thus, all possible complications must be evaluated before starting therapy. A randomized control study of the effect of TCA on glycemic control and depression indicated an efficient antidepressant effect but a lower degree of glycemic control.[61]
A few antidiabetic medications like the peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists like rosiglitazone and pioglitazone has been found to be effective in depressive symptoms. Animal models of depression like mouse tail suspension tests and rat forced swimming tests gave positive results for the antidepressant effects of these agents.[62] Furthermore in humans, pilot studies have confirmed a significant decline in the depressive symptoms in diabetic individuals with a 12 weeks dosing regimen of PPAR-γ agonists like pioglitazone.[62,63] Some of the possible mechanisms for the antidepressant activity of this class of antidiabetic agents might be decreased in visceral adiposity, improved insulin sensitivity or central effects on the neurogenesis and regulation of the calcium-dependent pathways.[63] Thus, inclusion of PPAR-γ agonists as monotherapy or in combination with other antidepressants in treatment regimen of diabetes-induced depression can have an improved prognosis of comorbid conditions. Furthermore, the effectiveness of combination therapy with metformin and milnacipran (a serotonin and norepinephrine reuptake inhibitor) in depressed diabetic individuals indicates a strong clinical significance of this approach. The study reported a significant reduction of glycosylated haemoglobin from 46% to 7% in patients who responded to milnacipran while there was no improvement in glucose levels in patients who did not respond to their depressive symptoms.[64]
The pharmacological management of depression in diabetic patients need critical evaluation of the comorbid conditions; adverse drug reactions, drug interactions, and some rare side effects like drug induced hypoglycemia as seen with SSRIs. Impaired sexual functions are very common in both diabetes and depression and may be improved using bupropion. Diabetic nephropathy and other pain related complications can be managed adequately by duloxetine and venlafaxine.[63]
The implication of depression associated with diabetes (gestational diabetes) has not been well-established in pregnancy, and necessary precautions need to be taken before commencing therapy.[65] In short, the treatment strategy should be a multifaceted one by considering all possible factors that may contribute to the aggravation of either of the comorbidities. The stress of long-term therapy associated diabetic complications such as nephropathy, neuropathy, retinopathy, forced lifestyle modifications, and prolonged hospitalization periods may all contribute to depression and measures need to be taken to address these issues. Pharmacologic treatments should be individualized based on the extent of glycemic control and depression and patients with severe depression should not be prescribed drugs that have a chance of further deteriorating the mental status due to the high propensity for suicidal behavior. Also continuous monitoring of depressed patients for alcohol or substance abuse, overeating, smoking, cardiovascular complications, and other related behavioral effects observed in depression is required to maintain a good prognosis for diabetes and related complications including mood disorders like depression.[66]
Footnotes
Source of Support: Nil.
Conflict of Interest: No.
References
- 1.Egede LE, Ellis C. Diabetes and depression: Global perspectives. Diabetes Res Clin Pract. 2010;87:302–12. doi: 10.1016/j.diabres.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 2.Anderson RJ, Freedland KE, Clouse RE, Lustman PJ. The prevalence of comorbid depression in adults with diabetes: A meta-analysis. Diabetes Care. 2001;24:1069–78. doi: 10.2337/diacare.24.6.1069. [DOI] [PubMed] [Google Scholar]
- 3.Judd LL, Rapaport MH, Paulus MP, Brown JL. Subsyndromal symptomatic depression: A new mood disorder? J Clin Psychiatry. 1994;55(Suppl):18–28. [PubMed] [Google Scholar]
- 4.Lustman PJ, Freedland KE, Carney RM, Hong BA, Clouse RE. Similarity of depression in diabetic and psychiatric patients. Psychosom Med. 1992;54:602–11. doi: 10.1097/00006842-199209000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Hirai FE, Tielsch JM, Klein BE, Klein R. Relationship between retinopathy severity, visual impairment and depression in persons with long-term type 1 diabetes. Ophthalmic Epidemiol. 2012;19:196–203. doi: 10.3109/09286586.2012.692006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldney RD, Phillips PJ, Fisher LJ, Wilson DH. Diabetes, depression, and quality of life: A population study. Diabetes Care. 2004;27:1066–70. doi: 10.2337/diacare.27.5.1066. [DOI] [PubMed] [Google Scholar]
- 7.Lin EH, Katon W, Von Korff M, Rutter C, Simon GE, Oliver M, et al. Relationship of depression and diabetes self-care, medication adherence, and preventive care. Diabetes Care. 2004;27:2154–60. doi: 10.2337/diacare.27.9.2154. [DOI] [PubMed] [Google Scholar]
- 8.Hasler G. Pathophysiology of depression: Do we have any solid evidence of interest to clinicians? World Psychiatry. 2010;9:155–61. doi: 10.1002/j.2051-5545.2010.tb00298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maletic V, Robinson M, Oakes T, Iyengar S, Ball SG, Russell J. Neurobiology of depression: An integrated view of key findings. Int J Clin Pract. 2007;61:2030–40. doi: 10.1111/j.1742-1241.2007.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirschfeld RM. History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry. 2000;61(Suppl 6):4–6. [PubMed] [Google Scholar]
- 11.Kamal A, Biessels GJ, Duis SE, Gispen WH. Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: Interaction of diabetes and ageing. Diabetologia. 2000;43:500–6. doi: 10.1007/s001250051335. [DOI] [PubMed] [Google Scholar]
- 12.Magariños AM, McEwen BS. Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc Natl Acad Sci U S A. 2000;97:11056–61. doi: 10.1073/pnas.97.20.11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiehuis AM, van der Graaf Y, Visseren FL, Vincken KL, Biessels GJ, Appelman AP, et al. Diabetes increases atrophy and vascular lesions on brain MRI in patients with symptomatic arterial disease. Stroke. 2008;39:1600–3. doi: 10.1161/STROKEAHA.107.506089. [DOI] [PubMed] [Google Scholar]
- 14.Kopf SR, Baratti CM. Memory-improving actions of glucose: Involvement of a central cholinergic muscarinic mechanism. Behav Neural Biol. 1994;62:237–43. doi: 10.1016/s0163-1047(05)80022-6. [DOI] [PubMed] [Google Scholar]
- 15.Vileikyte L, Leventhal H, Gonzalez JS, Peyrot M, Rubin RR, Ulbrecht JS, et al. Diabetic peripheral neuropathy and depressive symptoms: The association revisited. Diabetes Care. 2005;28:2378–83. doi: 10.2337/diacare.28.10.2378. [DOI] [PubMed] [Google Scholar]
- 16.Beauquis J, Roig P, Homo-Delarche F, De Nicola A, Saravia F. Reduced hippocampal neurogenesis and number of hilar neurones in streptozotocin-induced diabetic mice: Reversion by antidepressant treatment. Eur J Neurosci. 2006;23:1539–46. doi: 10.1111/j.1460-9568.2006.04691.x. [DOI] [PubMed] [Google Scholar]
- 17.Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: Effects of insulin treatment. Brain Res. 1998;800:125–35. doi: 10.1016/s0006-8993(98)00510-1. [DOI] [PubMed] [Google Scholar]
- 18.Dou JT, Chen M, Dufour F, Alkon DL, Zhao WQ. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn Mem. 2005;12:646–55. doi: 10.1101/lm.88005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith GS, Kramer E, Hermann C, Ma Y, Dhawan V, Chaly T, et al. Serotonin modulation of cerebral glucose metabolism in depressed older adults. Biol Psychiatry. 2009;66:259–66. doi: 10.1016/j.biopsych.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilhelm K, Gillis I, Reddy J, Mitchell PB, Campbell L, Dobson-Stone C, et al. Association between serotonin transporter promoter polymorphisms and psychological distress in a diabetic population. Psychiatry Res. 2012;200:343–8. doi: 10.1016/j.psychres.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 21.Zalsman G, Huang YY, Oquendo MA, Burke AK, Hu XZ, Brent DA, et al. Association of a triallelic serotonin transporter gene promoter region (5-HTTLPR) polymorphism with stressful life events and severity of depression. Am J Psychiatry. 2006;163:1588–93. doi: 10.1176/ajp.2006.163.9.1588. [DOI] [PubMed] [Google Scholar]
- 22.Manjarrez-Gutiérrez G, Herrera-Márquez JR, Molina-Hernández A, Bueno-Santoyo S, González-Ramírez M, Hernández J. Changes in cerebra serotonin synthesis induced by insulin-dependent diabetes mellitus. Rev Invest Clin. 1999;51:293–302. [PubMed] [Google Scholar]
- 23.Herrera R, Manjarrez G, Hernandez J. Inhibition and kinetic changes of brain tryptophan-5-hydroxylase during insulin-dependent diabetes mellitus in the rat. Nutr Neurosci. 2005;8:57–62. doi: 10.1080/10284150400027115. [DOI] [PubMed] [Google Scholar]
- 24.Miyata S, Hirano S, Kamei J. Diabetes attenuates the antidepressant-like effect mediated by the activation of 5-HT1A receptor in the mouse tail suspension test. Neuropsychopharmacology. 2004;29:461–9. doi: 10.1038/sj.npp.1300354. [DOI] [PubMed] [Google Scholar]
- 25.Sandrini M, Vitale G, Vergoni AV, Ottani A, Bertolini A. Streptozotocin-induced diabetes provokes changes in serotonin concentration and on 5-HT1A and 5-HT2 receptors in the rat brain. Life Sci. 1997;60:1393–7. doi: 10.1016/s0024-3205(97)00084-2. [DOI] [PubMed] [Google Scholar]
- 26.Mocking RJ, Ruhé HG, Assies J, Lok A, Koeter MW, Visser I, et al. Relationship between the hypothalamic-pituitary-adrenal-axis and fatty acid metabolism in recurrent depression. Psychoneuroendocrinology. 2013;38:1607–17. doi: 10.1016/j.psyneuen.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry. 2000;57:925–35. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- 28.Bruehl H, Rueger M, Dziobek I, Sweat V, Tirsi A, Javier E, et al. Hypothalamic-pituitary-adrenal axis dysregulation and memory impairments in type 2 diabetes. J Clin Endocrinol Metab. 2007;92:2439–45. doi: 10.1210/jc.2006-2540. [DOI] [PubMed] [Google Scholar]
- 29.McEwen BS. Allostasis and allostatic load: Implications for neuropsychopharmacology. Neuropsychopharmacology. 2000;22:108–24. doi: 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- 30.Celada P, Puig M, Amargós-Bosch M, Adell A, Artigas F. The therapeutic role of 5-HT1A and 5-HT2A receptors in depression. J Psychiatry Neurosci. 2004;29:252–65. [PMC free article] [PubMed] [Google Scholar]
- 31.Hesen W, Joëls M. Modulation of 5HT1A responsiveness in CA1 pyramidal neurons by in vivo activation of corticosteroid receptors. J Neuroendocrinol. 1996;8:433–8. doi: 10.1046/j.1365-2826.1996.04724.x. [DOI] [PubMed] [Google Scholar]
- 32.Espallergues J, Teegarden SL, Veerakumar A, Boulden J, Challis C, Jochems J, et al. HDAC6 regulates glucocorticoid receptor signaling in serotonin pathways with critical impact on stress resilience. J Neurosci. 2012;32:4400–16. doi: 10.1523/JNEUROSCI.5634-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han F, Ozawa H, Matsuda K, Nishi M, Kawata M. Colocalization of mineralocorticoid receptor and glucocorticoid receptor in the hippocampus and hypothalamus. Neurosci Res. 2005;51:371–81. doi: 10.1016/j.neures.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 34.Heisler LK, Pronchuk N, Nonogaki K, Zhou L, Raber J, Tung L, et al. Serotonin activates the hypothalamic-pituitary-adrenal axis via serotonin 2C receptor stimulation. J Neurosci. 2007;27:6956–64. doi: 10.1523/JNEUROSCI.2584-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minton JE. Function of the hypothalamic-pituitary-adrenal axis and the sympathetic nervous system in models of acute stress in domestic farm animals. J Anim Sci. 1994;72:1891–8. doi: 10.2527/1994.7271891x. [DOI] [PubMed] [Google Scholar]
- 36.Krabbe KS, Nielsen AR, Krogh-Madsen R, Plomgaard P, Rasmussen P, Erikstrup C, et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia. 2007;50:431–8. doi: 10.1007/s00125-006-0537-4. [DOI] [PubMed] [Google Scholar]
- 37.Martinowich K, Lu B. Interaction between BDNF and serotonin: Role in mood disorders. Neuropsychopharmacology. 2008;33:73–83. doi: 10.1038/sj.npp.1301571. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto K. Understanding depression: Linking brain-derived neurotrophic factor, transglutaminase 2 and serotonin. Expert Rev Neurother. 2013;13:5–7. doi: 10.1586/ern.12.140. [DOI] [PubMed] [Google Scholar]
- 39.Grillo CA, Piroli GG, Kaigler KF, Wilson SP, Wilson MA, Reagan LP. Downregulation of hypothalamic insulin receptor expression elicits depressive-like behaviors in rats. Behav Brain Res. 2011;222:230–5. doi: 10.1016/j.bbr.2011.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burguera B, Couce ME, Curran GL, Jensen MD, Lloyd RV, Cleary MP, et al. Obesity is associated with a decreased leptin transport across the blood-brain barrier in rats. Diabetes. 2000;49:1219–23. doi: 10.2337/diabetes.49.7.1219. [DOI] [PubMed] [Google Scholar]
- 41.Oury F, Karsenty G. Towards a serotonin-dependent leptin roadmap in the brain. Trends Endocrinol Metab. 2011;22:382–7. doi: 10.1016/j.tem.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phukan S, Babu VS, Kannoji A, Hariharan R, Balaji VN. GSK3beta: Role in therapeutic landscape and development of modulators. Br J Pharmacol. 2010;160:1–19. doi: 10.1111/j.1476-5381.2010.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology. 2004;29:1426–31. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7:1421–34. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carr DB, Cooper DC, Ulrich SL, Spruston N, Surmeier DJ. Serotonin receptor activation inhibits sodium current and dendritic excitability in prefrontal cortex via a protein kinase C-dependent mechanism. J Neurosci. 2002;22:6846–55. doi: 10.1523/JNEUROSCI.22-16-06846.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gispen WH, Biessels GJ. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000;23:542–9. doi: 10.1016/s0166-2236(00)01656-8. [DOI] [PubMed] [Google Scholar]
- 49.Perera TD, Park S, Nemirovskaya Y. Cognitive role of neurogenesis in depression and antidepressant treatment. Neuroscientist. 2008;14:326–38. doi: 10.1177/1073858408317242. [DOI] [PubMed] [Google Scholar]
- 50.Hwang IK, Yi SS, Kim YN, Kim IY, Lee IS, Yoon YS, et al. Reduced hippocampal cell differentiation in the subgranular zone of the dentate gyrus in a rat model of type II diabetes. Neurochem Res. 2008;33:394–400. doi: 10.1007/s11064-007-9440-8. [DOI] [PubMed] [Google Scholar]
- 51.Brezun JM, Daszuta A. Depletion in serotonin decreases neurogenesis in the dentate gyrus and the subventricular zone of adult rats. Neuroscience. 1999;89:999–1002. doi: 10.1016/s0306-4522(98)00693-9. [DOI] [PubMed] [Google Scholar]
- 52.Aberg MA, Aberg ND, Hedbäcker H, Oscarsson J, Eriksson PS. Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci. 2000;20:2896–903. doi: 10.1523/JNEUROSCI.20-08-02896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maura G, Marcoli M, Tortarolo M, Andrioli GC, Raiteri M. Glutamate release in human cerebral cortex and its modulation by 5-hydroxytryptamine acting at h 5-HT1D receptors. Br J Pharmacol. 1998;123:45–50. doi: 10.1038/sj.bjp.0701581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Isaac R, Boura-Halfon S, Gurevitch D, Shainskaya A, Levkovitz Y, Zick Y. Selective serotonin reuptake inhibitors (SSRIs) inhibit insulin secretion and action in pancreatic ß cells. J Biol Chem. 2013;288:5682–93. doi: 10.1074/jbc.M112.408641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raeder MB, Bjelland I, Emil Vollset S, Steen VM. Obesity, dyslipidemia, and diabetes with selective serotonin reuptake inhibitors: The hordaland health study. J Clin Psychiatry. 2006;67:1974–82. doi: 10.4088/jcp.v67n1219. [DOI] [PubMed] [Google Scholar]
- 56.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 57.Gould TD, Manji HK. Glycogen synthase kinase-3: A putative molecular target for lithium mimetic drugs. Neuropsychopharmacology. 2005;30:1223–37. doi: 10.1038/sj.npp.1300731. [DOI] [PubMed] [Google Scholar]
- 58.Cooper AJ, Ashcroft G. Potentiation of insulin hypoglycaemia by M.A.O.I. antidepressant drugs. Lancet. 1966;1:407–9. doi: 10.1016/s0140-6736(66)91399-7. [DOI] [PubMed] [Google Scholar]
- 59.Nair NP, Ahmed SK, Kin NM. Biochemistry and pharmacology of reversible inhibitors of MAO-A agents: Focus on moclobemide. J Psychiatry Neurosci. 1993;18:214–25. [PMC free article] [PubMed] [Google Scholar]
- 60.Jain AK, Kaplan RA, Gadde KM, Wadden TA, Allison DB, Brewer ER, et al. Bupropion SR vs. placebo for weight loss in obese patients with depressive symptoms. Obes Res. 2002;10:1049–56. doi: 10.1038/oby.2002.142. [DOI] [PubMed] [Google Scholar]
- 61.Lustman PJ, Griffith LS, Clouse RE, Freedland KE, Eisen SA, Rubin EH, et al. Effects of nortriptyline on depression and glycemic control in diabetes: Results of a double-blind, placebo-controlled trial. Psychosom Med. 1997;59:241–50. doi: 10.1097/00006842-199705000-00007. [DOI] [PubMed] [Google Scholar]
- 62.Sadaghiani MS, Javadi-Paydar M, Gharedaghi MH, Fard YY, Dehpour AR. Antidepressant-like effect of pioglitazone in the forced swimming test in mice: The role of PPAR-gamma receptor and nitric oxide pathway. Behav Brain Res. 2011;224:336–43. doi: 10.1016/j.bbr.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 63.Wasan AD, Ossanna MJ, Raskin J, Wernicke JF, Robinson MJ, Hall JA, et al. Safety and efficacy of duloxetine in the treatment of diabetic peripheral neuropathic pain in older patients. Curr Drug Saf. 2009;4:22–9. doi: 10.2174/157488609787354404. [DOI] [PubMed] [Google Scholar]
- 64.Hofmann P. Treatment of patients with comorbid depression and diabetes with metformin and milnacipran. Neuropsychiatr Dis Treat. 2010;6:9–15. [Google Scholar]
- 65.Canadian Diabetes Association, Dietitians of Canada, Diabète Québec, Ordre professionnel des diététistes du Québec. Recommendations for nutrition best practice in the management of gestational diabetes mellitus. Executive summary (1) Can J Diet Pract Res. 2006;67:206–8. doi: 10.3148/67.4.2006.206. [DOI] [PubMed] [Google Scholar]
- 66.Markowitz SM, Gonzalez JS, Wilkinson JL, Safren SA. A review of treating depression in diabetes: Emerging findings. Psychosomatics. 2011;52:1–18. doi: 10.1016/j.psym.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suri D, Vaidya VA. Glucocorticoid regulation of brain-derived neurotrophic factor: Relevance to hippocampal structural and functional plasticity. Neuroscience. 2013;3(239):196–213. doi: 10.1016/j.neuroscience.2012.08.065. [DOI] [PubMed] [Google Scholar]
- 68.Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci. 2010;3:1. doi: 10.3389/neuro.02.001.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]