

Abstract
Genetic constraints can block many mutational pathways to optimal genotypes in real fitness landscapes, yet the extent to which this can limit evolution remains to be determined. Interestingly, mutator bacteria elevate only specific types of mutations, and therefore could be very sensitive to genetic constraints. Testing this possibility is not only clinically relevant, but can also inform about the general impact of genetic constraints in adaptation. Here, we evolved 576 populations of two mutator and one wild-type Escherichia coli to doubling concentrations of the antibiotic cefotaxime. All strains carried TEM-1, a β-lactamase enzyme well known by its low availability of mutational pathways. Crucially, one of the mutators does not elevate any of the relevant first-step mutations known to improve cefatoximase activity. Despite this, both mutators displayed a similar ability to evolve more than 1000-fold resistance. Initial adaptation proceeded in parallel through general multi-drug resistance mechanisms. High-level resistance, in contrast, was achieved through divergent paths; with the a priori inferior mutator exploiting alternative mutational pathways in PBP3, the target of the antibiotic. These results have implications for mutator management in clinical infections and, more generally, illustrate that limits to natural selection in real organisms are alleviated by the existence of multiple loci contributing to fitness.
Keywords: mutator, epistasis, mutational pathways, TEM-1, PBP3
1. Introduction
A striking insight gained over the past decade of research is that epistatic interactions are common in experimental fitness landscapes [1]. Of particular interest is the observation of a special type of epistasis termed ‘sign epistasis’ [2], according to which mutations are beneficial or deleterious depending on the presence or absence of others [3–8]. Under sign epistasis, neighbouring mutation combinations display contrasting fitness values, introducing ‘ruggedness’ in the surface of the fitness landscape. This feature can have at least two important evolutionary consequences. First, it generates mutational pathways whose intermediate steps are no longer arranged in ascending order. Therefore, the number of pathways accessible by natural selection decreases [2] and successful adaptation becomes contingent upon the identity of first-step mutations [7]. A second, more drastic consequence is the emergence of multiple fitness peaks, which requires a particularly extreme form of epistasis known as reciprocal sign epistasis [9]. Overall, these observations led to the conjecture that natural selection may be limited by pervasive genetic constraints, contributing to explain the surprising degree of repeatability observed in many microbial evolution experiments [10,11].
One case presumably well suited to investigate the influence of genetic constraints on adaptation is that of bacterial mutators. Bacteria with a high mutation rate are frequently isolated in clinical settings [12,13] and in laboratory evolution experiments [14,15]. Their emergence is an eventual consequence of the genetic structure of asexuals, which allows mutator alleles to hitchhike with beneficial mutations occurring in the same genome [16,17]. Mutators pose a serious concern in clinical infections because they can readily evolve further adaptations, such as those promoting evasion of the immune system [18], increasing resistance to antibiotics [19] or alleviating the resistance fitness cost [20]. Different mutators, however, display a characteristic tendency to elevate only some types of transitions, transversions or frameshifts. This is because mutator phenotypes arise from alterations in specific mutation-avoidance mechanisms, whose malfunctioning permits the accumulation of particular mutational classes [21].
Here, we hypothesized that such mutational idiosyncrasy could render mutators particularly sensitive to genetic constraints. Each mutator genotype is expected to heighten the occurrence rate of only an arbitrary subset of beneficial first-step mutations [22]. If such a subset does not include the best possible mutations, mutator populations could be compelled to follow suboptimal paths—either leading to the same or a different fitness peak. Furthermore, in cases where the initial steps were drastically limited, mutators failing to raise the relevant first-step mutations may not be more able to adapt than their wild-type counterpart. Such stringent circumstances could happen when adaptation requires very specific multiple amino acid substitutions, as is observed in some cases of antibiotic resistance [8,23].
To study these possibilities, we characterized the evolution of wild-type and mutator strains of Escherichia coli towards increased resistance to the third-generation β-lactam antibiotic cefotaxime (CTX). Our experimental design sought to track mutations in the gene coding for the β-lactamase enzyme TEM-1 (blaTEM-1). The original allele, well adapted to hydrolyse early β-lactams, diversified naturally through the accumulation of point mutations into hundreds of variants with greater activity against later compounds of this family [24]. This natural evolution has been extensively emulated in a variety of laboratory experiments, yielding TEM-1 as one of the best-characterized models for the study of molecular evolution [7,25–27].
In the specific case of mutations conferring CTX resistance, there are several dozens of substitutions known to increase the resistance phenotype [26]. Despite the potentially huge number of combinations among these, only a small fraction are observed repeatedly both in nature and in laboratory evolution experiments—a signature of epistatic constraints [7,24]. Interestingly, we realized that the most frequent CTX resistance substitutions observed in clinical isolates arise from G : C → A : T transitions, one of the two dominant nucleotide changes in the spectrum of mismatch repair-deficient strains, the most prevalent type of strong bacterial mutators [28]. These substitutions include the top-ranked G238S and E104K, which appear combined in many notorious naturally occurring TEM alleles, and substitutions R164H and A237T [24,26]. In addition, the other transition characteristic of these mutators (A : T → G : C) also generates clinically relevant substitutions, such as M182T, H153R and I173V [24,26]. We next examined the case of MutT-deficient mutators, the second most prevalent type of strong mutator bacteria [28]. In sharp contrast, the elevation of A : T → C : G transversions peculiar to this background does not match any of the common CTX resistance mutations found in natural isolates [26]. Substitutions of intermediate to low effect, nonetheless, have been described (S268R, E240A, E104A and F72V), yet they have exclusively been observed in laboratory settings [26].
We reasoned that CTX resistance evolution could be a suitable model to examine the extent to which limited availability of mutational pathways can hinder the evolution of bacterial mutators. Studying this possibility is doubly relevant; not only because of the clinical importance of mutators, but because it can also inform about the more general question of the impact of genetic constraints in evolution. We thus serially propagated 192 independent populations of a ΔmutT mutator, a mismatch repair-deficient ΔmutH mutator and wild-type E. coli in the presence of periodically increasing concentrations of CTX. Crucially, we expected that the performance of the ΔmutT mutator would depend on its ability to exploit rare mutational pathways compatible with its narrow mutational spectrum. We also conjectured that any spectrum-dependent differences would be levelled out by a general increase in the supply of beneficial mutations. The rationale for this is that such an increase will favour that the best mutations are available for selection, irrespective of the genetic background. To test this, in the experimental set-up, half of the populations for each strain carry a single chromosomal copy of blaTEM-1, whereas the remaining half contain approximately 18 plasmidic copies of this allele [29]. Overall, we established six experimental settings (three mutational spectra with two gene dosages), for a total of 576 experimental populations.
2. Results
(a). Dynamics of adaptation to increasing antibiotic concentrations
To test whether different mutational spectra confer disparate ability to evolve high-level, multi-step CTX resistance, we serially passaged mutator and wild-type bacteria to doubling concentrations of the drug; starting from 1/4 to 1024 times the minimal inhibitory concentration (MIC) of the ancestral strain. In general terms, the results show that an increase in mutational supply, either through a mutator phenotype or by multiple plasmidic copies of blaTEM-1, translates into a greater fraction of replicates surviving at the end of the experiment (figure 1). More quantitatively, we found a strong linear correlation between the final number of survivors and the estimated product of mutation rate and blaTEM-1 copy number (Pearson's r = 0.87, p = 0.026; electronic supplementary material, figure S1). This indicates that the ΔmutT genotype, despite not elevating mutagenesis to the typical CTX resistance blaTEM-1 mutations, is not broadly hindered to evolve high levels of resistance.
Figure 1.
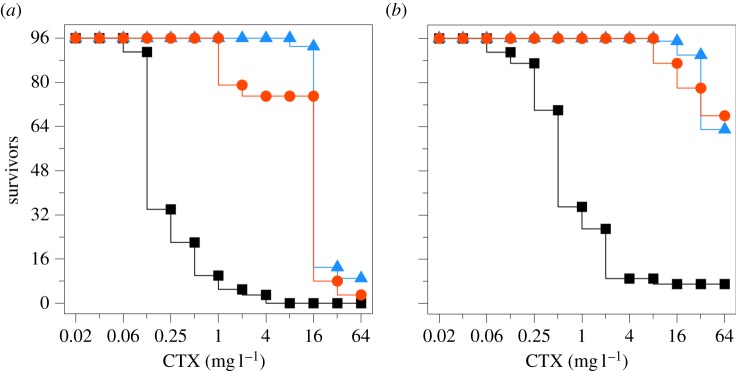
Experimental evolution of wild-type and mutator E. coli to CTX resistance. The figure shows the number of surviving lineages against antibiotic concentration, which was doubled every 2 days. The different curves correspond to wild-type (black squares), ΔmutH (red circles) and ΔmutT (blue triangles) lineages; carrying either one (a) or multiple (b) copies of the gene coding for the β-lactamase TEM-1. Overall, an increase either in mutation rate or in blaTEM-1 copy number translates into a higher number of final survivors. Note that the anticipated advantage of the ΔmutH over the ΔmutT mutator was not realized, despite being the only one elevating mutagenesis to the most-beneficial known substitutions in TEM-1 (see text for details).
In the blaTEM-1 single-copy setting, the anticipated advantage of the ΔmutH over the ΔmutT genotype was not observed (figure 1a). Indeed, the final number of survivors was greater in the ΔmutT than in the ΔmutH background (9/96 versus 3/96), although the values are not statistically different (Fisher's exact test, p = 0.1332). It is worth noting that 18 ΔmutH populations went prematurely extinct at 2 mg l−1 of CTX, a significant drop in the survival curve (log-rank test, p < 0.001) which may reflect an early evolutionary dead-end associated with its mutational spectrum. Regarding the blaTEM-1 multiple-copy setting, we found no discrepancies between the two mutator backgrounds (figure 1b), neither in terms of final number of survivors (Fisher's exact test, p = 0.5354) nor in survival curves (log-rank test, p = 0.808).
As pointed out in the Introduction, the most common blaTEM-1 CTX resistance substitutions arise mainly from transitions, which are only elevated in the ΔmutH genotype [21]. Taking this into account, it is revealing to compare the performance of the plasmid-harbouring wild-type (figure 1b) with that of both mutator populations carrying a single-copy blaTEM-1 (figure 1a). An increased copy number confers a capacity to reach high resistance similar to hypermutability (7/96 versus 9/96 and 3/96 final survivors; Fisher's exact test, p > 0.3), although the wild-type's survival curve is notably unique (log-rank test, p < 0.0001). This suggests that the evolutionary paths followed by the different strains are not simply the result of accumulating the well-known blaTEM-1 CTX resistance substitutions. If that were the case, the wild-type would be expected to perform better than the ΔmutT strain, because the latter is effectively non-mutator for these typical substitutions whereas the former should produce them at an approximately 18-fold higher rate [29]. In addition, the ΔmutH genotype should enjoy the best outcome, because it elevates approximately 100- to 300-fold the required transitions [30,31].
At least two non-mutually exclusive possibilities might explain these results. First, both ΔmutT and the wild-type strains could be substituting non-canonical blaTEM-1 CTX resistance mutations. The wild-type may thus profit from the fact that an increase in copy number elevates not only the two transitions, but also all the six possible base substitutions. Although the effect on each mutation type is moderate, the overall elevation in mutational supply becomes comparable to that of the ΔmutH background. Additionally, the greater allelic diversity generated may be fuelling evolution through homologous interplasmid recombination [32] (although the electronic supplementary material, figure S1 gives no clear support for this hypothesis). In turn, the ΔmutT strain elevates only one type of base substitution (A : T → C : G transversions), but it does so by approximately 500- to 10 000-fold [30,33]; a huge increment that may allow this genotype to accumulate rare blaTEM-1 CTX resistance substitutions.
A second possibility is that other chromosomal loci played a substantial role in the acquisition of high-resistance phenotypes. Expanding the mutational target size should, in principle, mitigate spectrum-dependent differences, which could explain the lack of impairment observed in the chromosomally blaTEM-1-encoding ΔmutT genotype. Candidate loci include genes controlling multi-drug resistance determinants, such as efflux pumps and porin channels; and those coding for the penicillin-binding proteins, the target of β-lactam antibiotics [34].
(b). Phenotypic characterization of evolved lineages
To gain insights into putative non-β-lactamase-mediated resistance mechanisms, we assembled a collection of 160 clonal isolates and tested them against a panel of chemicals, antibiotics and bacteriophage viruses. The largest part of the collection consisted in endpoint isolates from: (i) lineages that survived at the end of the experiment (n = 50), and (ii) the highest resistant lineages within each setting that went extinct before the end of the experiment (n = 57). To compensate that most mutator populations went extinct very late, the collection was completed with 43 clones from random lineages isolated at low CTX concentrations (less than or equal to 0.5 mg l−1). The battery of assays (see Material and methods) was designed to examine the status of AcrAB, the main efflux pump involved in β-lactam export [35]; and the outer membrane porins OmpF and OmpC, the primary routes of entry of these drugs [36]. To assess the validity of the tests, the loci acrA, acrB, acrR, envZ, ompR, ompF and ompC were sequenced in two independent strains positive for efflux and porin alterations. Both strains confirmed non-synonymous point mutations in several of these genes: acrB and acrR in both cases, together with envZ and ompC in one case and ompR in the other (see the electronic supplementary material, table S1).
The collection exhibited a high frequency of altered porin phenotypes (79%) and, to a lesser extent, increased efflux phenotypes (43%; figure 2)—a combination usually observed in multi-drug-resistant isolates [37]. Intriguingly, we found that 20% of the strains exhibit hypersusceptibility to erythromycin, a well-known substrate of AcrAB [38]. This phenotype could be a by-product of mutations improving the specificity of AcrAB for CTX, at the cost of reducing erythromycin extrusion [39]. Alternatively, it could emerge from mutations in the peptidoglycan synthesizing apparatus (including but not necessarily limited to PBP3, the molecular target of CTX), with pleiotropic effects in the organization of the outer membrane that result in enhanced passive diffusion of hydrophobic agents such as erythromycin [40]. Whatever the mechanism, we reported this phenotype owing to its prevalence, which might well be an underestimation given the high frequency of increased efflux phenotypes observed. Only 11% of the isolates showed no apparent alteration in any of these phenotypes.
Figure 2.
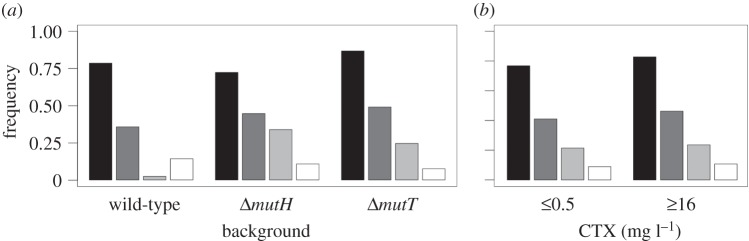
Phenotypic profiles of representative evolved clones. Bars represent the frequency of isolates displaying alterations in permeability (black), efflux (dark grey) and erythromycin hypersusceptibility (light grey). White bars correspond to isolates without alterations. (a) Phenotypic profiles of wild-type and mutator strains. Only differences in erythromycin hypersusceptibility were found to be statistically significant (see main text). (b) Phenotypic profiles according to the antibiotic concentration from which the clones were isolated. No statistically significant differences between early and late clones were observed. These results suggest that general multi-drug resistance evolved early and in parallel across the different backgrounds, and therefore is not sufficient to explain the final survival patterns.
No significant profile differences were detected among the three genotypes (Fisher's exact test, p > 0.15; figure 2a). The only exception is erythromycin hypersusceptibility, at least 10 times more frequent in either mutator than in the wild-type background (Fisher's exact test, p = 0.00014); suggesting that this phenotype may be arising only from a few, very specific point mutations. We also found no significant differences among clones isolated from early and late CTX concentrations (less than or equal to 0.5 versus more than or equal to 16 mg l−1, Fisher's exact test, p > 0.3; figure 2b). Taken together, the results indicate that efflux and permeability adaptations were acquired early and in parallel across the different genotypes, and therefore are not sufficient to explain the final survival patterns.
(c). Genotypic characterization of evolved lineages
Given the evidence provided above, we reasoned that high-level resistance may presumably be the result of either non-canonical blaTEM-1 substitutions or the combination of blaTEM-1 substitutions with alterations in ftsI, the gene coding for the main target of CTX (the transpeptidase PBP3). To explore these possibilities, we sequenced both loci from a subset of 70 strains from the collection of isolates used for the phenotypic assays (see the electronic supplementary material, table S2).
We found 21 out of 70 strains to be positive for non-synonymous blaTEM-1 substitutions, all of which belonged to the multiple-copy setting (n = 44; see the electronic supplementary material, table S2). Loss or insufficiency of expression of the chromosomal bla gene in the single-copy setting (n = 26) was discarded because all the analysed isolates retained inhibitor-susceptible resistance to ampicillin (data not shown). In total, we identified 27 point mutations corresponding to just five amino acid changes (E104K, H153R, R164S, M182T and G238S; figure 3a). These substitutions gave rise to five different alleles, including the double-mutant TEM15 and TEM112 alleles, and the triple-mutant TEM52 allele (see the electronic supplementary material, tables S2 and S3). All these substitutions and alleles have already been described both in clinical and experimental studies, and details about them can be read elsewhere [24]. The most frequent amino acid change in our dataset was G238S (20/27), in agreement with its prevalence in previous reports [7,41]. As anticipated, most of the mutations arose from either G : C → A : T or A : T → G : C transitions. The only exception was R164S, arising from a C : G → A : T transversion, detected just in one wild-type isolate. In consequence, it is not surprising that ΔmutH was the background where the greatest portion of substitutions were observed (16/27; figure 3a), and also that exhibited the highest average number of substitutions per sequence (1.25; electronic supplementary material, figure S2).
Figure 3.
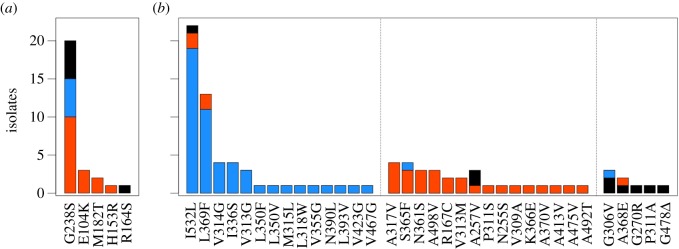
Substitutions in TEM-1 and PBP3 generated by wild-type and mutator strains. The histogram shows abundance of different amino acid substitutions among 70 isolates of wild-type (black), ΔmutH (red) and ΔmutT (blue) populations. (a) Substitutions in TEM-1. (b) Substitutions in PBP3, grouped by matching of the underlying nucleotide change with the mutational spectrum of ΔmutT (A : T → C : G, left), ΔmutH (G : C → A : T and A : T → G : C, centre) and wild-type (others, right) backgrounds. Note the contrasting levels of molecular idiosyncrasy observed in both proteins, indicating that mutational pathways are more constrained in TEM-1 than in PBP3.
Examination of the ftsI locus revealed 54 out of 70 strains displaying non-synonymous substitutions, with a remarkably high degree of polymorphism. We identified 92 point mutations (34 unique), affecting 31 positions, accounting for a total of 39 different alleles (figure 3b and electronic supplementary material, tables S2 and S3). Some of these variants have been already associated with resistance to β-lactam antibiotics, either in laboratory strains of E. coli (A257V and N361S) [42,43] or in related species such as Salmonella enterica (P311S) [44] and Haemophilus influenzae (L369F) [45]. All the substitutions but one mapped within the transpeptidase module (D237–V577) of the PBP3 protein, clustered in the surroundings of the three catalytic motifs (STVK310, SSN361 and KTG496) [46]. The sole mutation observed in the non-catalytic module (G57-D237), R167C, affected the first position of the conserved motif RYYPSG172. This module is thought to function as an intramolecular chaperone, and thus mutations in it could contribute to resistance by influencing the folding or stability of the whole protein [46]. Mutations in this module, indeed, have been reported in E. coli to confer resistance to ceftazidime, a β-lactam antibiotic structurally very close to CTX [47].
In contrast to what was observed in the bla locus, both mutators accumulated highly idiosyncratic substitutions in ftsI, including double- and triple-mutant alleles where all mutations correspond to each specific mutational spectrum (figure 3 and electronic supplementary material, tables S2 and S3). Regarding to this, only 7 out of 83 substitutions found in both mutators did not arise from spectrum-specific base changes, suggesting that these are mutations with strong beneficial effects. Interestingly, the ΔmutT background was the one where the greatest portion of substitutions were observed (52/92), exhibiting the highest average number of substitutions per isolate (2.08; electronic supplementary material, figure S2) and the highest total number of triple mutant alleles (eight in total, six unique; see the electronic supplementary material, tables S2 and S3).
Two lines of evidence indicate that these observed polymorphisms in the loci bla and ftsI are likely to explain high-level resistance patterns. On the one hand, all sequenced strains contained substitutions in at least one of these two genes, with the unsurprising exception of five clones from the wild-type background isolated at low concentrations (less than or equal to 0.5 mg l−1; electronic supplementary material, table S2). On the other hand, although some isolates exhibited alterations in both TEM and PBP3 (12/70), there was a statistically significant tendency to lack changes in PBP3 when TEM substitutions are already present (Fisher's exact test, p < 0.0001). Actually, whenever a triple-mutant allele was observed at either loci, alterations at the other locus were completely absent (electronic supplementary material, table S2).
3. Discussion
This work was motivated by the question of whether limits to natural selection posed by genetic constraints could become particularly apparent in mutator bacteria. To gain insights into this issue, we monitored the evolution of two distinct mutators in an experimental system where only one of them elevates known beneficial mutations. Under such conditions, the performance of the a priori inferior mutator would depend on its ability to exploit secondary mutational pathways, if available. In the extreme case, we hypothesized that its performance could be as limited as that of the wild-type.
We found that both mutators were indistinguishable in terms of their ability to evolve high-level CTX resistance. In view of the above considerations, a reasonable explanation for these results relies on the seemingly large size of the global mutational target for CTX resistance evolution. Such large size provides multiple alternative evolutionary routes and, consequently, increases the likelihood that a particular mutational spectrum raises fitting beneficial mutations. The causes for this large mutational target size are at least twofold. First, adaptation can proceed via changes at loci other than bla, most notably at ftsI. Second, the mutational target within the ftsI locus is apparently very broad in itself. In this respect, it is worth mentioning the high proportion of PBP3 substitutions observed only once (22/34; electronic supplementary material, table S2), which points at the existence of many other possible mutations that just went unsampled [48]. This is consistent with the fact that more than 30 PBP3 substitutions not detected here have been described elsewhere to confer resistance to a variety of β-lactams, including CTX [42–45,49].
A recent study with Pseudomonas aeruginosa reported minor differences between hypermutability and stress-induced mutagenesis in promoting adaptation, a result akin to ours in that both experiments involved different mutational spectra [50]. However, the negligible effect on evolvability observed here is particularly revealing, because our experimental design purposely involved both a narrow-spectrum mutator and a well-characterized model of constrained molecular evolution. A comparison with the literature lead us to believe that this result is probably not peculiar to our specific conditions, but rather it could be fairly general. While our system, in fact, exhibited substantial flexibility in evolutionary trajectories, this flexibility is similar to what is typically observed in many bacterial evolution experiments [51]: hard adaptive constraints are rarely observed at the nucleotide level [52], and even in the examples that reported the highest degree of molecular convergence, adaptation involved more than one loci and featured a considerable amount of intralocus diversity [53–55]. Therefore, given that mutational target sizes seem not strictly limited, we expect spectrum effects on evolvability to be modest at best in most cases.
In a broader evolutionary context, this work contributes to the question to what extent the ruggedness of fitness landscapes can hinder natural selection. Several reports have documented both inter- [4,5,56] and intralocus [27,57,58] sign epistasis in different model systems. These observations promoted the idea that genetic constraints could be prevalent and hence adaptation could proceed through very few mutational paths to optimal genotypes [8,11]. This possibility implies that evolution may be largely repeatable and, perhaps, even predictable [11]. It has been argued, however, that adaptive limitations can be particular to low-dimensional fitness landscapes, which to date have been the only ones amenable to empirical examination [11]. The rationale for this is that, because increasing dimensionality implies that genotypes have more neighbours, what might look like an isolated fitness peak at some scale can actually be connected at higher dimensions [59]. An example along these lines can be found in the case of substitution G238S in TEM-1 β-lactamase. In vitro experiments showed that this substitution is the first-step mutation that confers the highest resistance to CTX [26]. Most interestingly, they revealed that once it becomes fixed, epistatic interactions strongly restrict which other mutations can be substituted next [7]. In contrast to these limitations, our results uncovered the existence of multiple alternative pathways emerging from the combination of G238S with various substitutions in PBP3 (electronic supplementary material, table S2); illustrating how high dimensionality facilitates the bypass of local adaptive constraints in real fitness landscapes.
Apart from evolvability, there are other possible long-term consequences to mutational spectrum differences. Mutational biases forcibly impose a high level of divergence at the molecular level. This idiosyncratic variability, although perhaps equivalent in fitness under the selective conditions, might confer distinct properties in other environments. In our experiment, each mutator exhibited a marked preference for accumulating amino acid changes either in TEM or in PBP3 (figure 4). It is easy to imagine different long-term advantages and disadvantages for both resistance mechanisms. Some of the observed TEM alleles, for instance, are able to hydrolyse β-lactams not primarily targeting PBP3, thus conferring resistance to a broader range of compounds [60]. In addition, given that these enzymes are commonly plasmid-encoded in natural isolates, they could recombine at a higher rate and also be more readily transmissible through horizontal-gene transfer [61]. In turn, being an essential gene, PBP3 alleles are likely to entail fitness costs in other conditions. Such costs have already been reported in some cases, including reduced growth at high temperatures [42] and impaired biofilm formation [62]. Alterations in the cell wall structure, however, could also have an adaptive value: a recent report showed that PBP3 substitutions can increase the ability of H. influenza to invade epithelial cells [63].
Figure 4.
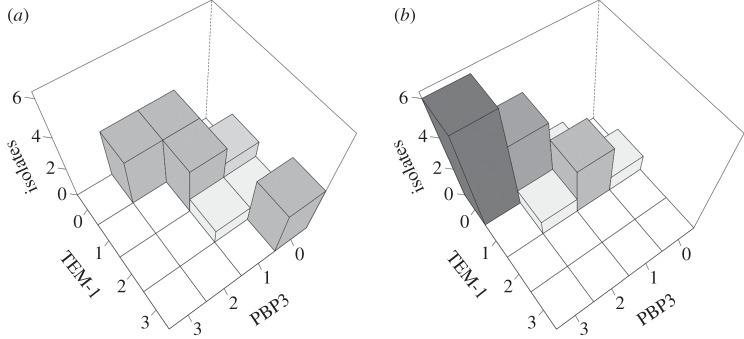
Mutational spectrum effects on divergence of mutational pathways. Bars represent number of isolates with different combinations of substitutions in TEM-1 and PBP3. Data from plasmid-carrying lineages surviving at the end of the experiment (MIC more than or equal to 64 mg l−1). (a) ΔmutH background. (b) ΔmutT background. To aid visualization, shades of grey are proportional to the number of isolates. Note how the mutational spectrum directs populations through divergent adaptive paths; each mutator preferentially accumulating substitutions either in TEM-1 or PBP3.
While these divergent prospects are specific to β-lactam resistance evolution, it seems reasonable to expect similar effects in other systems provided that adaptation can proceed through a sufficiently large mutational target. From an applied perspective, this adds an extra layer of complexity to the management of mutators in clinical infections, already recognized as a risk factor for antibiotic resistance development [64]. Future studies should consider the long-term consequences of molecular divergence when evaluating the risks of antibiotic therapy in the presence of mutators, including aspects such as cross-resistance, virulence, transmissibility or fitness in other environments.
4. Material and methods
(a). Strains and media
All strains are derivative of E. coli AB1157, obtained from the laboratory collection of Dr Bruce R. Levin. This strain naturally carries the gene coding for the β-lactamase AmpC, which was deleted to prevent interference with the experimental system. Knockouts of the genes ampC, mutH and mutT were done by P1 transduction and subsequent pCP20-mediated removal of the kanamycin resistance cassette from the appropriate Keio clone [65,66]. The single-copy blaTEM-1 strains were generated by transducing the galK::cfp/yfp, blaTEM-1 cassette from an E. coli MC4100 kindly provided by Dr Roy Kishony [67]. The multiple-copy blaTEM-1 strains were obtained by transformation with plasmid pBRACI; a pBR322 derivative which, in an attempt to reduce the fitness burden of carrying the plasmid [68], was digested with enzymes AvaI and PstII to excise the tetracycline resistance cassette. Note that these strains do not carry the blaTEM-1chromosomal copy. All genotypes were confirmed by PCR and gel electrophoresis. Bacteria were cultured in a modified version of M9 minimal medium, optimized to support maximal growth in the experimental conditions [66]. The modification consists of supplementing M9 salts with 1% glucose (G8270, Sigma-Aldrich) and 1% acid hydrolysate of casein (casamino acids, Becton Dickinson).
(b). Evolution experiment
For each of the six genotypes used here, a total of 96 parallel lineages were serially propagated in the presence of increasing concentrations of CTX (Claforan, Sanofi-Aventis). The populations were grown without agitation at 37°C in 96-well microtiter plates (1.1 ml Deep Well, Axygen), covered with loose-fitting plastic lids to allow aeration. To minimize cross-contamination, populations were arranged in a chessboard pattern across each plate, such that half of the wells contained sterile medium [67]. The fraction of these control wells that got contaminated on a daily basis was below 0.5%. Every 24 h, 16 μl of each culture was transferred into fresh 800 μl of medium, allowing a minimum of approximately 5.7 generations per day (note that this estimate represents a lower limit, owing to drops in final population density along the course of the experiment). Serial passage was conducted during 28 days. The minimal inhibitory concentration of cefotaxime (MIC) in the experimental medium for all parental strains was determined to be 0.064 mg l−1 by the microdilution method (CLSI, 2006), irrespective of plasmid carriage. The first two passages were done in the absence of antibiotic. At day three, the populations were exposed to 0.016 mg l−1 of CTX (1/4 × MIC). The antibiotic concentration was subsequently doubled every 48 h, until a final concentration of 64 mg l−1 CTX (1024 × MIC) was reached. Throughout the course of the experiment, the number of surviving populations was estimated by visual examination, and the plates were regularly stored at −80°C.
(c). Phenotypic characterization
After completion of the evolution experiment, single clones from 160 representative lineages were isolated by streaking onto Luria broth (LB) agar plates. These isolates were transferred to 96-well microtiter plates, incubated overnight and stored at −80°C for later analysis. All incubations were conducted in LB broth at 37°C without agitation. The phenotypic characterization measured susceptibility to a collection of chemicals, antibiotics and bacteriophage viruses. Assays were performed in triplicate after overnight growth of a replica of the frozen collection plates. These overnight cultures were 1 : 100 diluted and re-grown for 4 h. A 96 pin replicator was then used to transfer aliquots to square Petri dishes containing LB agar supplemented with varying concentrations of each specific agent. To examine the status of the AcrAB pump, the major pump extruding β-lactams, we estimated the MIC of three well-known substrates such as acriflavine, tetracycline and erythromycin (all purchased from Sigma-Aldrich) [35]. Altered-efflux mutants were identified using a conservative criterion: either the MIC of one them was elevated more than or equal to fourfold, or the MIC of at least two rose more than equal to twofold. To study alterations in porins OmpF and OmpC, the major route of entry of the antibiotic into the cell [36], we assessed resistance to the bacteriophages Tu1a and Tu1b, which use these porins, respectively, as their attachment site [69]. The resistance criterion was growth on a multiplicity of infection sufficient to prevent growth of the ancestor strain. As positive controls, Keio-derived knockout mutants of the genes acrR, ompF and ompC were routinely included in the aforementioned assays.
(d). Genetic characterization
PCR amplification and Sanger sequencing of the chromosomal bla gene was performed with oligonucleotides 5′-TGAACA TTCCGA AATGCG C-3′ and 5′-CCTTCG TTCACC GTCTTC A-3′. Regarding the plasmidic bla gene, pBRACI extraction and purification were performed using the Qiagen plasmid mini kit, and sequencing was performed with oligonucleotides 5′-GCTCAG GACTGG TCTAAC-3′ and 5′-CTTTGC GGTTAG ACTGGT C-3′. Amplification and sequencing of the ftsI gene was carried out with oligonucleotides 5′-GCCCAG CATGTT TCACAA GATG-3′ and 5′-CGAGCA GAGATG CTGCGA A-3′. The internal oligonucleotide 5′-CATCGT GCCCTA ACAACA ACC-3′ was also employed for sequencing, owing to the large size of the gene (approx. 1.8 kb). Sequencing services were provided by Secugen (www.secugen.es), sequence analysis were carried out with Ridom TraceEdit (www.ridom.de/traceedit) and sequence alignments were performed with MAFFT v. 6 (mafft.cbrc.jp/alignment/software).
Supplementary Material
Acknowledgements
We thank O. Tenaillon, J. Rodríguez-Beltrán, J. Poyatos, J. Rolff and O. Makarova for helpful comments on the manuscript, and L. López-Merino for priceless technical assistance.
Funding statement
This work was supported by predoctoral fellowship FI05/00569 to A.C. and grant nos. REIPI RD12/0015/0012 and FIS PI13/00063 to J.B. from Instituto de Salud Carlos III, Spain.
References
- 1.De Visser JAGM, Cooper TF, Elena SF. 2011. The causes of epistasis. Proc. R. Soc. B 278, 3617–3624. ( 10.1098/rspb.2011.1537) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinreich DM, Watson RA, Chao L. 2005. Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evol. Int. J. Org. Evol. 59, 1165–1174. ( 10.1111/j.0014-3820.2005.tb01769.x) [DOI] [PubMed] [Google Scholar]
- 3.Bridgham JT, Carroll SM, Thornton JW. 2006. Evolution of hormone-receptor complexity by molecular exploitation. Science 312, 97–101. ( 10.1126/science.1123348) [DOI] [PubMed] [Google Scholar]
- 4.Franke J, Klözer A, de Visser JAGM, Krug J. 2011. Evolutionary accessibility of mutational pathways. PLoS Comput. Biol. 7, e1002134 ( 10.1371/journal.pcbi.1002134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kvitek DJ, Sherlock G. 2011. Reciprocal sign epistasis between frequently experimentally evolved adaptive mutations causes a rugged fitness landscape. PLoS Genet. 7, e1002056 ( 10.1371/journal.pgen.1002056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lalić J, Elena SF. 2012. Magnitude and sign epistasis among deleterious mutations in a positive-sense plant RNA virus. Heredity 109, 71–77. ( 10.1038/hdy.2012.15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salverda MLM, Dellus E, Gorter FA, Debets AJM, van der Oost J, Hoekstra RF, Tawfik DS, de Visser JAGM. 2011. Initial mutations direct alternative pathways of protein evolution. PLoS Genet. 7, e1001321 ( 10.1371/journal.pgen.1001321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weinreich DM, Delaney NF, Depristo MA, Hartl DL. 2006. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312, 111–114. ( 10.1126/science.1123539) [DOI] [PubMed] [Google Scholar]
- 9.Poelwijk FJ, Tănase-Nicola S, Kiviet DJ, Tans SJ. 2011. Reciprocal sign epistasis is a necessary condition for multi-peaked fitness landscapes. J. Theor. Biol. 272, 141–144. ( 10.1016/j.jtbi.2010.12.015) [DOI] [PubMed] [Google Scholar]
- 10.Stern DL. 2013. The genetic causes of convergent evolution. Nat. Rev. Genet. 14, 751–764. ( 10.1038/nrg3483) [DOI] [PubMed] [Google Scholar]
- 11.de Visser JAGM, Krug J. 2014. Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 15, 480–490. ( 10.1038/nrg3744) [DOI] [PubMed] [Google Scholar]
- 12.LeClerc JE, Li B, Payne WL, Cebula TA. 1996. High mutation frequencies among Escherichia coli and salmonella pathogens. Science 274, 1208–1211. ( 10.1126/science.274.5290.1208) [DOI] [PubMed] [Google Scholar]
- 13.Oliver A, Cantón R, Campo P, Baquero F, Blázquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1253. ( 10.1126/science.288.5469.1251) [DOI] [PubMed] [Google Scholar]
- 14.Sniegowski PD, Gerrish PJ, Lenski RE. 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387, 703–705. ( 10.1038/42701) [DOI] [PubMed] [Google Scholar]
- 15.Pal C, Maciá MD, Oliver A, Schachar I, Buckling A. 2007. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 450, 1079–1081. ( 10.1038/nature06350) [DOI] [PubMed] [Google Scholar]
- 16.Notley-McRobb L, Pinto R, Seeto S, Ferenci T. 2002. Regulation of mutY and nature of mutator mutations in Escherichia coli populations under nutrient limitation. J. Bacteriol. 184, 739–745. ( 10.1128/JB.184.3.739-745.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tenaillon O, Toupance B, Nagard HL, Taddei F, Godelle B. 1999. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics 152, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canfield GS, Schwingel JM, Foley MH, Vore KL, Boonanantanasarn K, Gill AL, Sutton MD, Gill SR. 2013. Evolution in fast forward: a potential role for mutators in accelerating Staphylococcus aureus pathoadaptation. J. Bacteriol. 195, 615–628. ( 10.1128/JB.00733-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez-Rojas A, Couce A, Blazquez J. 2010. Frequency of spontaneous resistance to fosfomycin combined with different antibiotics in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 54, 4948–4949. ( 10.1128/AAC.00415-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perron GG, Hall AR, Buckling A. 2010. Hypermutability and compensatory adaptation in antibiotic-resistant bacteria. Am. Nat. 176, 303–311. ( 10.1086/655217) [DOI] [PubMed] [Google Scholar]
- 21.Miller JH. 1996. Spontaneous mutators in bacteria: insights into pathways of mutagenesis and repair. Annu. Rev. Microbiol. 50, 625–643. ( 10.1146/annurev.micro.50.1.625) [DOI] [PubMed] [Google Scholar]
- 22.Couce A, Guelfo JR, Blázquez J. 2013. Mutational spectrum drives the rise of mutator bacteria. PLoS Genet. 9, e1003167 ( 10.1371/journal.pgen.1003167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindgren PK, Marcusson LL, Sandvang D, Frimodt-Møller N, Hughes D. 2005. Biological cost of single and multiple norfloxacin resistance mutations in Escherichia coli implicated in urinary tract infections. Antimicrob. Agents Chemother. 49, 2343–2351. ( 10.1128/AAC.49.6.2343-2351.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salverda MLM, De Visser JAGM, Barlow M. 2010. Natural evolution of TEM-1 β-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 34, 1015–1036. ( 10.1111/j.1574-6976.2010.00222.x) [DOI] [PubMed] [Google Scholar]
- 25.Blazquez J, Morosini MI, Negri MC, Baquero F. 2000. Selection of naturally occurring extended-spectrum TEM beta-lactamase variants by fluctuating beta-lactam pressure. Antimicrob. Agents Chemother. 44, 2182–2184. ( 10.1128/AAC.44.8.2182-2184.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schenk MF, Szendro IG, Krug J, de Visser JAGM. 2012. Quantifying the adaptive potential of an antibiotic resistance enzyme. PLoS Genet. 8, e1002783 ( 10.1371/journal.pgen.1002783) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schenk MF, Szendro IG, Salverda MLM, Krug J, de Visser JAGM. 2013. Patterns of epistasis between beneficial mutations in an antibiotic resistance gene. Mol. Biol. Evol. 30, 1779–1787. ( 10.1093/molbev/mst096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denamur E, Matic I. 2006. Evolution of mutation rates in bacteria. Mol. Microbiol. 60, 820–827. ( 10.1111/j.1365-2958.2006.05150.x) [DOI] [PubMed] [Google Scholar]
- 29.Covarrubias L, Cervantes L, Covarrubias A, Soberón X, Vichido I, Blanco A, Kupersztoch-Portnoy YM, Bolivar F. 1981. Construction and characterization of new cloning vehicles. V. Mobilization and coding properties of pBR322 and several deletion derivatives including pBR327 and pBR328. Gene 13, 25–35. ( 10.1016/0378-1119(81)90040-8) [DOI] [PubMed] [Google Scholar]
- 30.Cupples CG, Miller JH. 1989. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl Acad. Sci. USA 86, 5345–5349. ( 10.1073/pnas.86.14.5345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seier T, Padgett DR, Zilberberg G, Sutera VA, Toha N, Lovett ST. 2011. Insights into mutagenesis using Escherichia coli chromosomal lacZ strains that enable detection of a wide spectrum of mutational events. Genetics 188, 247–262. ( 10.1534/genetics.111.127746) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lovett ST, Hurley RL, Sutera VA, Aubuchon RH, Lebedeva MA. 2002. Crossing over between regions of limited homology in Escherichia coli: RecA-dependent and RecA-independent pathways. Genetics 160, 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vidmar JJ, Cupples CG. 1993. MutY repair is mutagenic in mutT- strains of Escherichia coli. Can. J. Microbiol. 39, 892–894. ( 10.1139/m93-133) [DOI] [PubMed] [Google Scholar]
- 34.Poole K. 2004. Resistance to beta-lactam antibiotics. Cell. Mol. Life Sci. 61, 2200–2223. ( 10.1007/s00018-004-4060-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okusu H, Ma D, Nikaido H. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 178, 306–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikaido H, Rosenberg EY, Foulds J. 1983. Porin channels in Escherichia coli: studies with beta-lactams in intact cells. J. Bacteriol. 153, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davin-Regli A, Bolla J-M, James C, Lavigne J-P, Chevalier J, Garnotel E, Molitor A, Pages J-M. 2008. Membrane permeability and regulation of drug ‘influx and efflux’ in enterobacterial pathogens. Curr. Drug Targets 9, 750–759. ( 10.2174/138945008785747824) [DOI] [PubMed] [Google Scholar]
- 38.Nishino K, Yamaguchi A. 2001. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 183, 5803–5812. ( 10.1128/JB.183.20.5803-5812.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu EW, Aires JR, McDermott G, Nikaido H. 2005. A periplasmic drug-binding site of the acrb multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J. Bacteriol. 187, 6804–6815. ( 10.1128/JB.187.19.6804-6815.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaara M. 1993. Antibiotic-supersusceptible mutants of Escherichia coli and Salmonella typhimurium. Antimicrob. Agents Chemother. 37, 2255–2260. ( 10.1128/AAC.37.11.2255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orencia MC, Yoon JS, Ness JE, Stemmer WP, Stevens RC. 2001. Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat. Struct. Biol. 8, 238–242. ( 10.1038/84981) [DOI] [PubMed] [Google Scholar]
- 42.Hedge PJ, Spratt BG. 1985. Resistance to beta-lactam antibiotics by re-modelling the active site of an E. coli penicillin-binding protein. Nature 318, 478–480. ( 10.1038/318478a0) [DOI] [PubMed] [Google Scholar]
- 43.Hedge PJ, Spratt BG. 1985. Amino acid substitutions that reduce the affinity of penicillin-binding protein 3 of Escherichia coli for cephalexin. Eur. J. Biochem. FEBS 151, 111–121. ( 10.1111/j.1432-1033.1985.tb09075.x) [DOI] [PubMed] [Google Scholar]
- 44.Sun S, Selmer M, Andersson DI. 2014. Resistance to β-lactam antibiotics conferred by point mutations in penicillin-binding proteins PBP3, PBP4 and PBP6 in Salmonella enterica. PLoS ONE 9, e97202 ( 10.1371/journal.pone.0097202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ubukata K, Shibasaki Y, Yamamoto K, Chiba N, Hasegawa K, Takeuchi Y, Sunakawa K, Inoue M, Konno M. 2001. Association of amino acid substitutions in penicillin-binding protein 3 with beta-lactam resistance in beta-lactamase-negative ampicillin-resistant Haemophilus influenzae. Antimicrob. Agents Chemother. 45, 1693–1699. ( 10.1128/AAC.45.6.1693-1699.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen-Distèche M, Fraipont C, Buddelmeijer N, Nanninga N. 1998. The structure and function of Escherichia coli penicillin-binding protein 3. Cell. Mol. Life Sci. 54, 309–316. ( 10.1007/s000180050157) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tavío MM, Aquili VD, Vila J, Poveda JB. 2013. Resistance to ceftazidime in Escherichia coli associated with AcrR, MarR, and PBP3 mutations and overexpression of sdiA. J. Med. Microbiol. 63, 56–65. ( 10.1099/jmm.0.063727-0) [DOI] [PubMed] [Google Scholar]
- 48.Efron B, Thisted R. 1976. Estimating the number of unseen species: how many words did Shakespeare know? Biometrika 63, 435–447. ( 10.2307/2335721) [DOI] [Google Scholar]
- 49.Dabernat H, Delmas C, Seguy M, Pelissier R, Faucon G, Bennamani S, Pasquier C. 2002 Diversity of β-lactam resistance-conferring amino acid substitutions in penicillin-binding protein 3 of Haemophilus influenzae. Antimicrob. Agents Chemother 46, 2208–2218. ( 10.1128/AAC.46.7.2208-2218.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weigand MR, Sundin GW. 2012. General and inducible hypermutation facilitate parallel adaptation in Pseudomonas aeruginosa despite divergent mutation spectra. Proc. Natl Acad. Sci. USA 109, 13 680–13 685. ( 10.1073/pnas.1205357109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dettman JR, Rodrigue N, Melnyk AH, Wong A, Bailey SF, Kassen R. 2012. Evolutionary insight from whole-genome sequencing of experimentally evolved microbes. Mol. Ecol. 21, 2058–2077. ( 10.1111/j.1365-294X.2012.05484.x) [DOI] [PubMed] [Google Scholar]
- 52.Tenaillon O, Rodríguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS. 2012. The molecular diversity of adaptive convergence. Science 335, 457–461. ( 10.1126/science.1212986) [DOI] [PubMed] [Google Scholar]
- 53.Albert TJ, et al. 2005. Mutation discovery in bacterial genomes: metronidazole resistance in Helicobacter pylori. Nat. Methods 2, 951–953. ( 10.1038/nmeth805) [DOI] [PubMed] [Google Scholar]
- 54.Toprak E, Veres A, Michel J-B, Chait R, Hartl DL, Kishony R. 2012. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet. 44, 101–105. ( 10.1038/ng.1034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong A, Rodrigue N, Kassen R. 2012. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genet. 8, e1002928 ( 10.1371/journal.pgen.1002928) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maharjan RP, Ferenci T. 2013. Epistatic interactions determine the mutational pathways and coexistence of lineages in clonal Escherichia coli populations. Evolution 67, 2762–2768. ( 10.1111/evo.12137) [DOI] [PubMed] [Google Scholar]
- 57.Lunzer M, Miller SP, Felsheim R, Dean AM. 2005. The biochemical architecture of an ancient adaptive landscape. Science 310, 499–501. ( 10.1126/science.1115649) [DOI] [PubMed] [Google Scholar]
- 58.da Silva J, Coetzer M, Nedellec R, Pastore C, Mosier DE. 2010. Fitness epistasis and constraints on adaptation in a human immunodeficiency virus type 1 protein region. Genetics 185, 293–303. ( 10.1534/genetics.109.112458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gavrilets S. 1997. Evolution and speciation on holey adaptive landscapes. Trends Ecol. Evol. 12, 307–312. ( 10.1016/S0169-5347(97)01098-7) [DOI] [PubMed] [Google Scholar]
- 60.Poyart C, Mugnier P, Quesne G, Berche P, Trieu-Cuot P. 1998. A novel extended-spectrum TEM-type β-lactamase (TEM-52) associated with decreased susceptibility to moxalactam in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 42, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barlow M, Fatollahi J, Salverda M. 2009. Evidence for recombination among the alleles encoding TEM and SHV β-lactamases. J. Antimicrob. Chemother. 63, 256–259. ( 10.1093/jac/dkn475) [DOI] [PubMed] [Google Scholar]
- 62.Pozzi C, et al. 2012. Methicillin resistance alters the biofilm phenotype and attenuates virulence in Staphylococcus aureus device-associated infections. PLoS Pathog. 8, e1002626 ( 10.1371/journal.ppat.1002626) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okabe T, Yamazaki Y, Shiotani M, Suzuki T, Shiohara M, Kasuga E, Notake S, Yanagisawa H. 2010. An amino acid substitution in PBP-3 in Haemophilus influenzae associate with the invasion to bronchial epithelial cells. Microbiol. Res. 165, 11–20. ( 10.1016/j.micres.2008.03.003) [DOI] [PubMed] [Google Scholar]
- 64.Blázquez J. 2003. Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 37, 1201–1209. ( 10.1086/378810) [DOI] [PubMed] [Google Scholar]
- 65.Baba T, et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 20060008 ( 10.1038/msb4100050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia Coli and related bacteria. New York, NY: CSHL Press. [Google Scholar]
- 67.Hegreness M, Shoresh N, Hartl D, Kishony R. 2006. An equivalence principle for the incorporation of favorable mutations in asexual populations. Science 311, 1615–1617. ( 10.1126/science.1122469) [DOI] [PubMed] [Google Scholar]
- 68.Lee SW, Edlin G. 1985. Expression of tetracycline resistance in pBR322 derivatives reduces the reproductive fitness of plasmid-containing Escherichia coli. Gene 39, 173–180. ( 10.1016/0378-1119(85)90311-7) [DOI] [PubMed] [Google Scholar]
- 69.Datta DB, Arden B, Henning U. 1977. Major proteins of the Escherichia coli outer cell envelope membrane as bacteriophage receptors. J. Bacteriol. 131, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.