

Abstract
Background
Human Papillomavirus (HPV) genotyping is an important approach to fight cervical cancer due to the relevant information regarding risk stratification for diagnosis and the better understanding of the relationship of HPV with carcinogenesis. This paper proposed two new feature extraction techniques, i.e. ChaosCentroid and ChaosFrequency, for predicting HPV genotypes associated with the cancer. The additional diversified 12 HPV genotypes, i.e. types 6, 11, 16, 18, 31, 33, 35, 45, 52, 53, 58, and 66, were studied in this paper.
In our proposed techniques, a partitioned Chaos Game Representation (CGR) is deployed to represent HPV genomes. ChaosCentroid captures the structure of sequences in terms of centroid of each sub-region with Euclidean distances among the centroids and the center of CGR as the relations of all sub-regions. ChaosFrequency extracts the statistical distribution of mono-, di-, or higher order nucleotides along HPV genomes and forms a matrix of frequency of dots in each sub-region. For performance evaluation, four different types of classifiers, i.e. Multi-layer Perceptron, Radial Basis Function, K-Nearest Neighbor, and Fuzzy K-Nearest Neighbor Techniques were deployed, and our best results from each classifier were compared with the NCBI genotyping tool.
Results
The experimental results obtained by four different classifiers are in the same trend. ChaosCentroid gave considerably higher performance than ChaosFrequency when the input length is one but it was moderately lower than ChaosFrequency when the input length is two. Both proposed techniques yielded almost or exactly the best performance when the input length is more than three. But there is no significance between our proposed techniques and the comparative alignment method.
Conclusions
Our proposed alignment-free and scale-independent method can successfully transform HPV genomes with 7,000 - 10,000 base pairs into features of 1 - 11 dimensions. This signifies that our ChaosCentroid and ChaosFrequency can be served as the effective feature extraction techniques for predicting the HPV genotypes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-015-0493-4) contains supplementary material, which is available to authorized users.
Keywords: HPV, Genotype, Chaos game representation, Singular value decomposition, Prediction
Background
Human Papillomavirus (HPV) is a small double-stranded and most common sexually transmitting DNA virus. At present, more than one hundred types of Human papillomavirus have been identified. They are differentiated by the genetic sequence of the outer capsid protein L1. Approximately forty types can infect the mucosal epithelium. They are categorized according to their epidemiologic association with cervical cancer. Infection with low risk HPV types such as types 6 and 11 can cause benign or low-grade cervical cell abnormalities and genital warts. In contrast, high risk HPV types such as 16 and 18 act as carcinogens that can lead to the development of cervical cancer and other anogenital cancers.
Cervical cancer is the second most common cancer significantly causing morbidity and mortality in women worldwide [1]. Persistent infection by high risk HPV is a necessary cause of this cancer. Especially, the most common high risk HPV types are 16 and 18, and approximately 70% of cervical cancer is due to infection by these genotypes [2]. Each genotype of HPV has a different risk level in the cervical cancer. Furthermore, there is a wide variation in genotype distribution in different regions around the world. To better understand the relationship of HPV with carcinogenesis, many countries have investigated the HPV infection among women with cytological status by HPV genotyping methods, as revealed in Switzerland [3], in Italy [4], in Cambodia [5], and in Romania [6].
HPV genotyping is necessary for managing effective medical treatment strategies to patients with persistent infection and for evaluating prevention strategies to individual patients to be immunized with type-specific HPV vaccines [7]. Currently, there are various kinds of HPV genotyping tests used for detecting the genotypes of Human Papillomavirus, in clinical laboratories. For example, PapilloCheck®;, PCR-RFLP, HPV genome sequencing, INNO-LiPA, Linear Array®; HPV Genotyping Test, etc. These methods detect the HPV genotypes from some regions of genomes. Even though these HPV genotyping tests are beneficial and employed for HPV diagnosis in patients nowadays, they have some limitations. To illustrate this aspect, the HPV genotypes are hardly detected in cases of inadequate samples or low amplification signals of some genotypes. Contamination with previously amplified material can lead to false positive results. Furthermore, mistaken classifications can be occurred through cross-reactivity among similar types in the tests based on hybridization [8].
To avoid these problems, some computational methods for identifying HPV types were developed [9-16]. Since discriminating whether the patients have been infected with the high risk types of Human papillomavirus is the most important and urgent aspect for diagnosis and treatment, multiple perspectives were proposed to focus on predicting the HPV risk types. For instance, Wang and Xiao [9] presented multitudinous physicochemical and statistical features from the protein sequences using Fuzzy K nearest neighbor classifier for the risk type prediction of Human papillomaviruses. They also further developed the better algorithm based on geometric moments of protein distance matrix images using a Fuzzy K nearest neighbor classifier [10]. In addition, classification of HPV risk types was also proposed through algorithms based on decision tree [11], text mining [12], genetic mining of DNA sequence structures [13], support vector machines [14], gap-spectrum kernels [15], and ensemble support vector machines with protein secondary structures [16].
While classifying the HPV into high and low risk types is the urgent aspect for diagnosis of the cancer as claimed by many researchers, the study on how to predict specific genotypes of the virus has not significantly focused. In fact, the identification of HPV genotypes infecting the patients is more essential than a rough classification of HPV risk types. To clarify this issue, HPV genotyping can provide more information regarding risk stratification. With the persistent infection, the risk of a precancerous lesion is in between 10% to 15% with HPV types 16 and 18 but below 3% for all other high risk types combined [2]. Furthermore, the relevant diagnosis with cost effectiveness can be done by selecting the virus types to be tested based on epidemiological and prevalence studies from a wide variation in the genotype distribution in different regions around the world. The diversity of virus types and the incidence of multiple infections have made it necessary to develop reliable methods to identify the different genotypes for epidemiological studies and medical treatment. HPV genotyping can make a great contribution to the following aspects: HPV diagnosis in case of single and multiple infection, more information regarding risk stratification, a better understanding of the relationship of HPV with carcinogenesis, and prevention of the cancer though the development of type-specific vaccines. Consequently, HPV genotyping has become an important approach to fight with cervical cancer. For these reasons, this research concentrated on the prediction of HPV genotypes.
Chaos Game Representation (CGR) was proposed as a unique and scale-independent representation for genomic sequences by Jeffrey [17]. It is an iterative mapping technique assigning each nucleotides in a DNA or amino acids in a protein to a unique coordinates in a 2-dimensional space. It can be viewed as a 2-dimensional image of distributed dots and captured in a form of 0-1 square matrix, where 1 represents a dot and 0 represents an empty coordinate. The distribution of positions has two properties of uniqueness and possibility to inverse a coordinate back to its corresponding nucleotide or amino acid [18]. Using graphic approaches to study biological systems can provide useful intuitive insights, as indicated by many previous studies on a series of important biological topics, such as DNA [19,20], RNA [21], genome [22-26], protein [27-35], drug metabolism systems [36], protein-protein interactions [37], analysis of protein sequence evolution [38]. Moreover, the cellular automaton graph has also been applied to study hepatitis B viral infections, HBV virus gene missense mutation, as well as represent complicated biological sequences and help to identify various protein attributes [39-41].
Singular value decomposition (SVD) is a matrix factorization technique with various applications. For instance, it can be used to solve underdetermined and overdetermined systems of linear equations, find inverse and the pseudo-inverse matrices, compute the matrix condition number and calculate the vector system orthogonality and orthogonal complement [42]. SVD is also applied to several areas in gene expression data and microarray data, such as analysis [43-46], search [47], image compression [48], gene extraction [42], and classification [49,50], etc. In this paper, we deployed SVD a tool to reduce the size of CGR into a smaller number of feature matrices without losing any knowledge from the original data. Therefore, a new feature extraction was proposed based on the combination of chaos game representation and singular value decomposition.
Due to the significance of HPV genotyping, the objective of this paper is to predict the HPV genotypes from their genomes, which is similar to the conventional methods of genome detection in clinical laboratories. The remaining sections of this paper are organized as follows. Section “Methods” describes for the methods used in this experiment, including collection of HPV data set, the proposed feature extraction techniques, predicting systems, and performance evaluation. Section “Results and discussion” illustrates the experimental results and discussion. Section “Conclusion” concludes the paper.
Methods
As realized by a series recent publications [51-58] in response to the call from [59], the following procedures to establish a really useful statistical predictor for a biological system were involved in our method: (i) construct or select valid benchmark data sets to train and test the predictor; (ii) formulate the biological samples with an effective mathematical expression that can truly reflect their intrinsic correlation with the predicted target; (iii) introduce or develop a powerful predicting algorithm (or engine); (iv) properly perform cross-validation tests to objectively evaluate the anticipated accuracy of the predictor; (v) establish a user-friendly web-server for the predictor accessible to the public. The detail of each procedure is discussed as follows.
HPV genome data from genotypes were collected and their features were extracted by our proposed feature extraction techniques, i.e. ChaosCentroid and ChaosFrequency, as inputs for classification. These features were divided into the training and testing sets by a 2-fold cross validation technique. Four different classification models were deployed to train and test the experimental data sets. Then, the prediction performance from the obtained results were evaluated and compared with other methods. Our proposed method consists of the following four main procedures, i.e. data collection, feature extraction, prediction, and performance evaluation.
Collection of HPV data set
To remove the homologous sequences from the benchmark data sets, a cut-off threshold of 25% was imposed in [60,61] to exclude those proteins from the benchmark data sets that are equal to or greater than 25% of sequence identity to any others in a same subset. However, in this study we did not use such a stringent criterion because the currently available data do not allow us to do so. Otherwise, the numbers of genomes for some subsets would be too few to have statistical significance.
HPV genotypes collected in this experiment are those important genotypes detectable by Linear Array®; HPV Genotyping Test. This HPV genotyping is a widely used qualitative test developed by Roche Molecular Diagnostics for detecting HPV genotypes associated with cervical cancer. The test can detect 37 high and low risk HPV genotypes, including those considered as a significant risk factor for HSIL progression to cervical cancer. To challenge the prediction, only HPV genotypes having genome diversity were concentrated in this experiment. Some of 37 genotypes containing few genomes were excluded. For this reason, only HPV genotypes 6, 11, 16, 18, 31, 33, 35, 45, 52, 53, 58 and 66 were involved. The genomes of these HPV genotypes were collected from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). The data set contains Human Papillomavirus genomes of 12 genotypes, including high, possible high, and low risk types. For each HPV genotype, the number of genomes as well as the minimum and maximum lengths are shown in Table 1.
Table 1.
The number of genomes, minimum and maximum genome lengths of HPV genotypes in the HPV data set
| HPV Genotypes | No. of genomes | Genome length (base pairs) | |
|---|---|---|---|
| Minimum | Maximum | ||
| 6 | 58 | 7954 | 8051 |
| 11 | 49 | 7931 | 10424 |
| 16 | 103 | 7881 | 7976 |
| 18 | 19 | 7824 | 7857 |
| 31 | 23 | 7878 | 7945 |
| 33 | 22 | 7830 | 7912 |
| 35 | 28 | 7820 | 7908 |
| 45 | 12 | 7841 | 7858 |
| 52 | 22 | 7933 | 7974 |
| 53 | 16 | 7856 | 7863 |
| 58 | 37 | 7814 | 7836 |
| 66 | 11 | 7816 | 7824 |
All viral genomes in this HPV data set were previously published and are publicly available on GenBank or NCBI databases. In addition, the genome names, NCBI access numbers, and HPV genotypes of all genomes in the HPV data set are properly cited in Additional file 1, and the HPV data set used in this experiment is also available in Additional file 2.
Detail of proposed feature extraction techniques
The following techniques, i.e. ChaosCentroid and ChaosFrequency, were proposed to extract the features from the chaos game representation of HPV genomes. To identify each genotype, the relations among subsets of HPV genomes must be clarified. These relations are actually the local features. Since the CGR captures the information of the whole genome data, extracting the global features from the CGR may not be efficient enough to distinguish the HPV genotypes. The local features hidden in various sub-regions of CGR must be more contemplated. In this work, we concentrate on extracting the local features rather than global features. The difference between ChaosCentroid and ChaosFrequency are the feature representation. HPV genomes contain A, C, G, and T nucleotides. Prior to the discussion of ChaosCentroid and ChaosFrequency, the detail of how to construct CGR is the following. Let x i and y i be the coordinates of nucleotide η i at the i th position in the nucleotide sequence. Algorithm 1 illustrates how to construct a CGR for capturing a given nucleotide sequence.
A CGR can be viewed as a square whose corners are at coordinates (-1,-1), (-1,1), (1,1), and (1,-1) representing nucleotides A, C, G, and T, respectively. Note that the size of CGR according to the coordinates of A, C, G, and T nucleotides is equal to 2×2 units. However, this unit size of original CGR is not appropriate for discussing our algorithm. Therefore, the geometrical structure and the physical size of our CGR are re-defined as follows. The size of CGR square is set to n×n and n∈R +. Its center is also located at the coordinates (0,0). Each corner of this square represents the same nucleotide as that of the original CGR. After Algorithm 1, CGR can be viewed as an image of distributed dots. Figure 1 shows some examples of CGR of HPV genotypes 6, 16, 18, and 31. Obviously, the number of dots in a CGR is equal to the number of nucleotides in a given sequence. Although this CGR image can be directly used in the prediction step, its computational time may be too high due to the large number of dots. Thus it is necessary to extract only those relevant features from this set of dots to reduce the computational time complexity in the prediction process. In this paper, we proposed two different features as the representation of CGR image. The first feature is called ChaosCentroid and the second one is called ChaosFrequency. The detail of each feature is the following.
Figure 1.
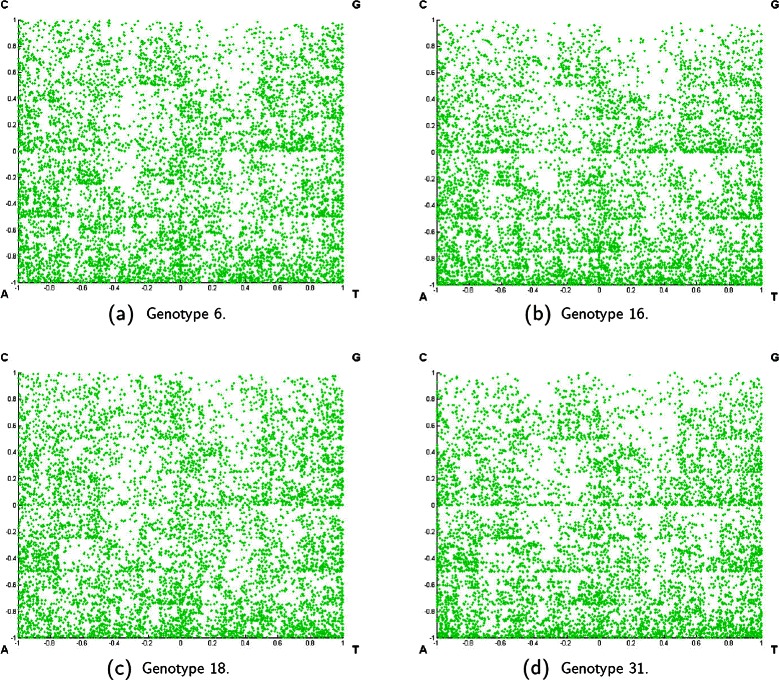
Chaos game representation (CGR) of HPV genotypes 6, 16, 18, and 31. (a) Genotype 6. (b) Genotype 16. (c) Genotype 18. (d) Genotype 31.

ChaosCentroid
According to [17], the k-th dot plotted on the CGR of a sequence corresponds to the first k-long initial sub-sequence of the sequence. Therefore, any visible pattern of the CGR corresponds to some pattern of the nucleotide sequence. CGR represents the global information of the nucleotide sequence. Partitioning the CGR into several sub-regions is implemented for revealing local information of the interested areas. If two dots are within the same quadrant, they correspond to sequences with the same last mononucleotide; if they are in the same sub-quadrant, the sequences have the same last dinucleotides; and so on. This can demonstrate the structure of the sequences yielding the dots. ChaosCentroid utilizes this biological significance by computing the centroid of the distributed dots of each sub-region. Therefore, the centroid, which can be converted to specific structure of the sequence, is represented as local information of the sub-region. For ChaosCentroid, the CGR is partitioned into equal sub-regions, where . This range is obtained by all possible numbers that can applied to the CGR. For instance, the CGR is not partitioned when , the CGR is partitioned into 4 equal sub-regions when , and so on. Furthermore, if the value of is greater than 11, some sub-regions does not contain any dots. So, 11 is the maximum value of in this experiment. For each of partitioned into the CGR, the centroid of each sub-region is computed first. Then all pairs of distances between the centroids and the center of CGR are computed and captured in a form of a matrix. This set of distances can be considered as the relation of information embedded in all sub-regions. However, the number of ChaosCentroids may be too large. Therefore, this matrix is decomposed by applying singular value decomposition (SVD) method to reduce information complexity. Finally, the diagonal elements from the -by- diagonal matrix of SVD are represented as the features of CGR and are subsequently used as the input vectors for prediction process. As a result, ChaosCentroid produces 11 formats of input vectors, i.e. the first format have 1 dimension, the second format have 2 dimensions, and so on. Extracting ChaosCentroid consists of the following steps, as illustrated in Algorithm 2. Additionally, Figure 2 shows an example of distances between the centroid of each sub-region and the center of CGR for HPV genotype 16 after being partitioned into sub-regions of size 2×2.
Figure 2.
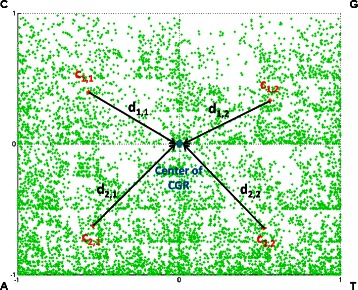
The distances between the centroids and the center of CGR for HPV genotype 16 after being partitioned into sub-regions of size 2×2.

ChaosFrequency
As elucidated in [20], the bias of distribution of different mono-, di-, tri-, or higher order nucleotides along the DNA/RNA sequences can generate different patterns in the CGR. This can be used as diagnostic patterns for different HPV genotypes. The CGRs of the HPV genomes of different genotypes tend to exhibit distinct patterns visually, as displayed in Figure 1. Thus, ChaosFrequency concentrates on the frequencies of sub-sequences occurred in the HPV genomes. Particularly, when is equal to 2k where k∈{1,2,3}, it represents the k-mer frequency occurred in the HPV sequences. Accordingly, the ratio between the number of dots in the sub-region and the total number of dots in the CGR are computed and represented as the feature of each sub-region. This ratio can be interpreted as the probability of distribution. Suppose each sub-region is of size g×g. After extracting the ChaosFrequency of each sub-region, the whole CGR be viewed as a matrix of size . This matrix is decomposed by SVD to extract the diagonal elements used as the feature of CGR. Likewise, this technique produces 11 formats of input vectors, in accordance with those of ChaosCentroid. The detail of this procedure is illustrated in Algorithm 3. Each sub-region is referred by its location according to the row and column after the partition of CGR. Let m i,j be the number of dots in sub-region at row i and column j. Suppose there are total M dots in CGR. Then we can calculate the probability of distribution as .

Predicting systems
To evaluate the performance of the proposed feature extraction techniques, the testing sets were fed to four different types of predicting systems. Each system has its own principle and criteria for predicting the corresponding HPV genotypes. The predicting systems are multi-layer perceptron neural network, radial basis function network, k-nearest neighbor technique, and fuzzy k-nearest neighbor technique. From 400 HPV genomes, one of 12 genotypes which are types 6, 11, 16, 18, 31, 33, 35, 45, 52, 53, 58, and 66 was identified. The detail of set-up for each predicting system in our experiments are as follows.
Multi-layer perceptron neural network
Each input pattern is the feature vector F obtained from Algorithms 2 and 3. Therefore, the numbers of input neurons are ranged from 1 to 11 according to the sizes of the feature vector F. The number of hidden neurons was empirically varied from 1 to 24 neurons to find the most suitable number. From the experiments, 16 hidden neurons are the best number of neurons for producing the best prediction of HPV genotypes. There are 12 output neurons, each of which corresponds to each HPV genotype. To make the testing efficient, the neuron 1 is for determining HPV genotype 6; neuron 2 for type 11; neuron 3 for type 16; neuron 4 for type 18; neuron 5 for type 31; neuron 6 for type 33; neuron 7 for type 35; neuron 8 for type 45; neuron 9 for type 52; neuron 10 for type 53; neuron 11 for type 58; and neuron 12 for type 66. Therefore, the network deployed in our experiments consists of an input layer with neurons, a hidden layer with 16 neurons, and an output layers with 12 neurons. Backpropagation learning rule was adopted to adjust the weights of the network during the training process. Mean squared normalized error function was used as a terminating criterion in the training process. In testing procedure, the predict HPV genotype is determined by this equation. Let o i be the output value of output neuron i.
| (1) |
argtype is the mapping from neuron index to its corresponding HPV genotype previously defined.
Radial basis function network
After finding the optimal spread distances for the prediction, the spread of radial basis function (RBF) is set to 0.4 for ChaosCentroid and 0.1 for ChaosFrequency. The same network structure of multi-layer perceptron was adopted for this RBF network. The determination in Equation (1) of HPV genotypes for multi-layer perceptron was used in this RBF predicting system.
K-nearest neighbor technique
In this technique, the determination of HPV genotypes depends upon the value of k nearest neighbors measured by Euclidean distance. For any tested feature vector, the HPV genotype of its nearest neighbor is assigned as the HPV genotype of the tested feature vector. Empirically, it was found that k=1 gave the best performance.
Fuzzy K-nearest neighbor technique
Fuzzy k-nearest neighbor technique was proposed by James M. Keller, Michael R. Gray, and James A. Givens [62]. It is a special variation of the k-nearest neighbor technique family. The algorithm of fuzzy k-nearest neighbor assigns class membership to a sample vector rather than assigning the vector to a particular class. An advantage is that no arbitrary assignments are made by the algorithm. Additionally, membership values of the vector should provide a level of assurance to accompany the resultant classification. In this technique, we set k to 1.
Performance evaluation
Among the independent statistical accuracy testing methods for predicted results such as sub-sampling (e.g., 2, 5 or 10-fold cross-validation) test and jackknife test, jackknife test was deemed the most objective that can always yield a unique result for a given benchmark data set, as elucidated in [59] and demonstrated by Equations 28, 29 and 30 in [59]. Therefore, the jackknife test has been increasingly used and widely recognized by investigators to test the power of various prediction methods (see, e.g., [63-73]). Although jackknife is widely used, its computational time is rather high. However, to reduce the computational time, we adopted the 2-fold cross-validation in this experiment to deal with the parameter optimization. Therefore, the reported prediction performance was obtained by the combination of both validating sets.
In this experiment, we adopted Equation 11 of [52] to formulate the set of four metrics, including Sensitivity(Sen), Specificity(Spec), Accuracy(Acc), and Matthew’s Correlation Coefficient(MCC), for evaluating the prediction performance. The formulation of the four metrics is defined by the following equations.
| (2) |
where N + is the total number of HPV genomes of the investigated genotype whereas the number of HPV genomes of the investigated genotype that is incorrectly predicted as the other genotypes; N − the total number of HPV genomes of the other genotypes that are not investigated whereas the number of HPV genomes of the other genotypes that is incorrectly predicted as the investigated genotype. The investigated HPV genotype is 6, 11, 16, 18, 31, 33, 35, 45, 52, 53, 58, or 66. For example, if the investigated genotype is 6, N + is the total number of HPV genomes of genotype 6, while N − is the total number of the genomes of the other genotypes, excluding genotype 6.
According to Equation 2, the prediction performance can be evaluated in a meaningful explanation, as follows. The sensitivity is used for evaluating the performance of the predicting systems in identifying the investigated genotype. When , none of HPV genomes of the investigated genotype was incorrectly predicted as the other genotypes, so the sensitivity is 1. In contrast, while , all HPV genomes of the investigated genotype were incorrectly predicted as the other genotypes, so the sensitivity is 0. The specificity is used for evaluating the performance of the systems in excluding the other genotypes. When , none of HPV genomes of the other genotypes was incorrectly predicted as the investigated genotype, so the specificity is 1; while , all HPV genomes of the other genotype were incorrectly predicted as the investigated genotype, so the specificity is 0. The accuracy is used for evaluating the performance of the systems in classifying the investigated genotype and the other genotypes. When , none of HPV genomes of the investigated genotype and none of HPV genomes of the other genotypes was incorrectly predicted, so the accuracy is 1; while and all HPV genomes of the investigated genotype and all HPV genomes of the other genotypes were incorrectly predicted, so the accuracy is 0. Typically, the Matthew’s Correlation Coefficient (MCC) is used for measuring the quality of binary classification. When , none of HPV genomes of the investigated genotypes and none of HPV genomes of the other genotypes was incorrectly predicted, so MCC is 1; when and , MCC is 0 meaning no better than random prediction; When and , MCC is -1 indicating total disagreement between prediction and observation.
However, the set of metrics in Equation 2 is valid only for single-label systems. For multi-label systems whose existence has become more frequent in system biology [61,74] and system medicine [67,75], a completely different set of metrics as defined in [76] is needed.
Results and discussion
The value of variable in Algorithms 2 and 3 was set from 1 to 11. The performance of HPV genotype prediction was separately summarized according to each predicting system and two feature extracting schemes. The obtained results are the following.
Results from multi-layer perceptron neural network
The results of the HPV genotype prediction gained by ChaosCentroid and by ChaosFrequency feature extraction with the predicting system based on multi-layer perceptron neural network are summarized in Tables 7 and 8, respectively, of Additional file 3. The results were reported according to different values of . It is rather remarkable when .
When , the number of sub-regions of CGR is equal to one. Thus there is only one centroid computed by ChaosCentroid and the probability of distribution of CGR computed by ChaosFrequency is equal to one. The overall performance of ChaosFrequency is much lower than those of ChaosCentroid. ChaosFrequency gain 0% of sensitivity and 100% of specificity in all genotypes, excepting genotype 16. It implies that the features of all genomes extracted by ChaosFrequency are totally predicted to genotype 16. In contrast, ChaosCentroid can obtain high performance metrics, including accuracy, sensitivity, specificity, and Matthew’s Correlation Coefficient in almost all genotypes. This is because a centroid is computed from the coordinates of every dots. It is obvious that different HPV genotypes must have different distribution of dots and centroids. So, predicting HPV genotypes with high performance from these centroids is possible. But in case of ChaosFrequency, the probability of distribution of every HPV genotype is equal. This makes the feature of each HPV genotype indistinguishable.
However, when the value of is greater than one, the local information regarding the frequency of sub-sequence among nucleotides in each sub-region is brought out and the performance is increased in proportion to the value of . It is noticeable that there is no significant difference between the overall performance obtained from ChaosCentroid and ChaosFrequency when . In addition, we can conclude that, to achieve high performance of prediction, the local information of each sub-region is more relevant than global information.
Results from radial basis function network
The results of the HPV genotype prediction gained by ChaosCentroid and by ChaosFrequency feature extraction with the predicting system based on radial basis function network are summarized in Tables 9 and 10, respectively, of Additional file 3. According to the results, the performance values obtained by this predicting system are unstable among input dimensions. This is because this experiment set only one optimal spread distance, which gain the maximum average accuracy of all dimensions, for each predicting system of ChaosCentroid and ChaosFrequency, respectively. In fact, it is possible that each input dimension has its own proper spread distance, and one value of spread distance can not fit for all dimensions. In addition, it is noticeable that ChaosFrequency with RBF at 4-dimensional input can achieve the best performance with minimum input dimension. The overall performance trend obtained from this predicting system is similar to those of multi-layer perceptron. But the peformance from multi-layer perceptron is significantly higher than the performance from radial basis function.
Results from K-nearest neighbor technique
The results of the HPV genotype prediction gained by ChaosCentroid and by ChaosFrequency feature extraction with the predicting system based on k-nearest neighbor technique are summarized in Tables 11 and 12, respectively, of Additional file 3. The experimental results have shown the high performance of prediction. Therefore, it can imply that, in each sub-region, the structure of sequence in a form of centroid by ChaosCentroid and the statistical distribution of mono-, di-, or higher order nucleotides in a form of frequency by ChaosFrequency, are closed to each other in the same genotype. The overall performance trend obtained from this predicting system is similar to those of multi-layer perceptron. But the performance from this predicting system is slightly higher than the performance of multi-layer perceptron.
Results from Fuzzy K-nearest Neighbor Technique
The results of the HPV genotype prediction gained by ChaosCentroid and by ChaosFrequency feature extraction with the predicting system based on fuzzy k-nearest neighbor technique are summarized in Tables 13 and 14, respectively, of Additional file 3. The overall performance trend obtained from this predicting system is similar to those of multi-layer perceptron. Additionally, the overall performance of this predicting system is slightly higher than the performance of multi-layer perceptron but it is statistically equal to the performance of k-nearest neighbor technique due to setting the same value of k.
Comparative results with Related Method
NCBI viral genotyping tool [77] is a web-based tool for identifying the genotype of a viral sequence. It works by sliding a window along the query sequence and processing each window/sequence segment separately. Each segment is compared to a set of reference sequences using BLAST, which returns the similarity scores for the local alignments. The reference sequence genotype that matches the query with the highest similarity score is assigned to the query segment. The process is repeated for the next window until the whole length of the query sequence has been covered. The results from all windows are combined. If the same genotype is assigned to most segments, then the query sequence is considered the genotype. This tool is a web-based resource that provides a reliable method based on alignment. Then, this experiment adopted this tool for identifying genotypes of the viral genomes in the HPV data set. To evaluate the prediction performance, the result obtained by this genotyping tool were compared with the best results obtained by the proposed ChaosCentroid and ChaosFrequency feature extraction techniques with all predicting systems, as illustrated in Tables 2, 3, 4, 5 and 6.
Table 2.
Best results of the HPV genotype prediction based on the features extracted by ChaosCentroid and by ChaosFrequency with multi-layer perceptron neural network
| HPV Genotypes | ChaosCentroid | ChaosFrequency | ||||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | Sensitivity | Specificity | MCC | Accuracy | Sensitivity | Specificity | MCC | |
| 6 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 11 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 16 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 18 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 31 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 33 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 35 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 45 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 52 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 53 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 58 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 66 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
Table 3.
Best results of the HPV genotype prediction based on the features extracted by ChaosCentroid and by ChaosFrequency with radial basis function network
| HPV Genotypes | ChaosCentroid | ChaosFrequency | ||||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | Sensitivity | Specificity | MCC | Accuracy | Sensitivity | Specificity | MCC | |
| 6 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 11 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 16 | 99.50 | 99.03 | 99.66 | 0.99 | 100.00 | 100.00 | 100.00 | 1.00 |
| 18 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 31 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 33 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 35 | 99.50 | 96.43 | 99.73 | 0.96 | 100.00 | 100.00 | 100.00 | 1.00 |
| 45 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 52 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 53 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 58 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 66 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
Table 4.
Best results of the HPV genotype prediction based on the features extracted by ChaosCentroid and by ChaosFrequency with k-nearest neighbor technique
| HPV Genotypes | ChaosCentroid | ChaosFrequency | ||||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | Sensitivity | Specificity | MCC | Accuracy | Sensitivity | Specificity | MCC | |
| 6 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 11 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 16 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 18 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 31 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 33 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 35 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 45 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 52 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 53 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 58 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 66 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
Table 5.
Best results of the HPV genotype prediction based on the features extracted by ChaosCentroid and by ChaosFrequency with fuzzy k-nearest neighbor technique
| HPV Genotypes | ChaosCentroid | ChaosFrequency | ||||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | Sensitivity | Specificity | MCC | Accuracy | Sensitivity | Specificity | MCC | |
| 6 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 11 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 16 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 18 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 31 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 33 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 35 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 45 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 52 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 53 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 58 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
| 66 | 100.00 | 100.00 | 100.00 | 1.00 | 100.00 | 100.00 | 100.00 | 1.00 |
Table 6.
Results of the HPV genotype prediction obtained by NCBI viral genotyping tool
| HPV Genotypes | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| 6 | 100.00 | 100.00 | 100.00 | 1.00 |
| 11 | 100.00 | 100.00 | 100.00 | 1.00 |
| 16 | 100.00 | 100.00 | 100.00 | 1.00 |
| 18 | 100.00 | 100.00 | 100.00 | 1.00 |
| 31 | 100.00 | 100.00 | 100.00 | 1.00 |
| 33 | 100.00 | 100.00 | 100.00 | 1.00 |
| 35 | 100.00 | 100.00 | 100.00 | 1.00 |
| 45 | 100.00 | 100.00 | 100.00 | 1.00 |
| 52 | 100.00 | 100.00 | 100.00 | 1.00 |
| 53 | 100.00 | 100.00 | 100.00 | 1.00 |
| 58 | 100.00 | 100.00 | 100.00 | 1.00 |
| 66 | 100.00 | 100.00 | 100.00 | 1.00 |
The experimental results have shown that all methods, excepting ChaosCentroid with radial basis function network, can achieve the best performance of the four metrics, including accuracy, sensitivity, specificity, and Matthew’s Correlation Coefficient, in predicting the HPV genotypes of the data set. It demonstrated that both of the proposed techniques and the NCBI genotyping tool can be used to predict the genotypes of HPV genomes. Even though there is no significance between the proposed techniques and the NCBI genotyping tool, some issues should be considered.
The NCBI genotyping tool provides a reliable method based on homology searching sequence alignment procedure. The limitation of alignment is that it is difficult to identify or classify the protein or DNA sequences in the case that they does not have a significant sequence homology. Besides, the alignment with multiple sequences will take time consuming and only one query sequence at a time can be processed by this tool. So, this method is not appropriate for large scale tasks.
In contrast, the proposed techniques, i.e. ChaosCentroid and ChaosFrequency, are based on Chaos game representation, which provides a unique and scale-independent representation of DNA sequences through the statistical distribution of mono-, di-, tri-, or higher order nucleotides along DNA sequences. An advantage of CGR over alignment is that it has the potential to reveal the evolutionary and/or functional relationships between the sequences having no significant homology, as elucidated in [35]. Furthermore, it does not require prior knowledge of consensus sequences, nor does it involve exhaustive searches for sequences in databases. The limitation of CGR is that it takes a computational time to generate the representations from DNA sequences. Nevertheless, this experiment utilized the singular value decomposition to reduce the size of CGR into a smaller number of feature matrices so the computational time in the prediction process was also reduced. From the experimental results, it have shown that the proposed ChaosCentroid and ChaosFrequency, which are based on chaos game representation and singular value decomposition, can successfully extract the characteristic parameters of HPV genotypes for the prediction.
Since user-friendly and publicly accessible web-servers represent the future direction for developing practically more useful models, simulated methods, or predictors [78-80], we may make efforts in our future work to provide a web-server for the method presented in this paper.
Conclusion
This paper proposed two new feature extraction techniques, i.e. ChaosCentroid and ChaosFrequency, based on chaos game representation and singular value decomposition for predicting HPV genotypes from nucleotide sequences in HPV genomes. Both extracting techniques concentrate on the local information among nucleotides. For the sub-regions in CGR, ChaosCentroid pays attention to capture the structures of the sequences in a form of centroids, while ChaosFrequency focuses on capture the distribution of sub-sequences in a form of frequencies. Four different predicting systems, i.e. multi-layer perceptron neural network, radial basis function network, K-nearest neighbor technique, and fuzzy K-nearest neighbor technique, were deployed. From the experiment, we found that the features extracted by our proposed feature extraction techniques are significant and independent of the predicting systems. The comparative results demonstrated no significance between our proposed techniques and the NCBI viral genotyping tool. In addition, local information is more important than global information in order to achieve high performance of prediction.
Acknowledgements
The authors express their gratitude to the Thailand Research Fund (TRF) for supporting this research under the Golden Jubilee Scholarship.
Abbreviations
- HPV
Human papillomavirus
- LSIL
Low grade squamous intraepithelial lesion
- HSIL
High grade squamous intraepithelial lesion
- CGR
Chaos game representation
- SVD
Singular value decomposition
- MLP
Multi-layer perceptrons
- RBF
Radial basis function
- KNN
K-Nearest neighbor
- FKNN
Fuzzy K-Nearest neighbor
Additional files
Genome names, NCBI access numbers, and HPV genotypes of all genomes in the HPV data set.
HPV data set.
Results of the HPV genotype prediction based on the features extracted by ChaosCentroid and ChaosFrequency with all predicting systems.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CL and YP participated in the design of the feature extractions and contributed in the refinement of the work. WT participated in the design of the feature extractions, collected the HPV data set, implemented the feature extractions, evaluated the prediction performance, and drafted the manuscript. All authors participated in revising the manuscript. All authors read and approved the final manuscript.
Contributor Information
Watcharaporn Tanchotsrinon, Email: Watcharaporn.T@Student.chula.ac.th.
Chidchanok Lursinsap, Email: lchidcha@gmail.com.
Yong Poovorawan, Email: Yong.P@chula.ac.th.
References
- 1.Sheng J, Zhang WY. Identification of biomarkers for cervical cancer in peripheral blood lymphocytes using oligonucleotide microarrays. Chin Med J. 2010;123:1000–5. [PubMed] [Google Scholar]
- 2.Abreu ALP, Souza RP, Gimenes F, Consolaro MEL. A review of methods for detect human papillomavirus infection. Virol J. 2012;9:262. doi: 10.1186/1743-422X-9-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobec M, Bannwart F, Kilgus S, Kaeppeli F, Cassinotti P. Human papillomavirus infection among women with cytological abnormalities in switzerland investigated by an automated linear array genotyping test. J Med Virol. 2011;83:1370–6. doi: 10.1002/jmv.22126. [DOI] [PubMed] [Google Scholar]
- 4.Rossi PG, Chini F, Bisanzi S, Burroni E, Carillo G, Lattanzi A, et al. Distribution of high and low risk hpv types by cytological status: a population based study from italy. Infect Agents Cancer. 2011;6:2. doi: 10.1186/1750-9378-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Couture MC, Page K, Stein ES, Sansothy N, Sichan K, Kaldor J, et al. Cervical human papillomavirus infection among young women engaged in sex work in phnom penh, cambodia: prevalence, genotypes, risk factors and association with hiv infection. BMC Infect Dis. 2012;12:166. doi: 10.1186/1471-2334-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ursu RG, Onofriescu M, Nemescu D, Iancu LS. HPV prevalence and type distribution in women with or without cervical lesions in the northeast region of romania. Virol J. 2011;8:558. doi: 10.1186/1743-422X-8-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SH, Vigliotti VS, Vigliotti JS, Pappu S. Routine human papillomavirus genotyping by dna sequencing in community hospital laboratories. Infect Agents Cancer. 2007;2:11. doi: 10.1186/1750-9378-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carvalho NO, Castillo DM, Perone C, Januario JN, Melo VH, Filho GB. Comparison of hpv genotyping by type-specific pcr and sequencing. Mem Inst Oswaldo Cruz. 2010;105(1):73–8. doi: 10.1590/S0074-02762010000100011. [DOI] [PubMed] [Google Scholar]
- 9.Wang P, Xiao X. Proceedings of 5th International Conference on Bioinformatics and Biomedical Engineering: 10-12 May 2011; Wuhan, China. USA: IEEE; 2011. Predicting the risk type of human papillomaviruses based on sequence-derived features. [Google Scholar]
- 10.Xiao X, Wang P. Proceedings of 2011 International Conference on Electronics, Communications and Control: 9-11 September 2011; Ningbo. USA: IEEE; 2011. A new approach using geometric moments of distance matrix image for risk type prediction of human papillomaviruses. [Google Scholar]
- 11.Park S, Hwang S, Zhang B. Proceedings of 4th International Conference on Intelligent Data Engineering and Automated Learning: 21-23 March 2003; Hong Kong, China. Germany: Springer Berlin Heidelberg; 2003. Classification of the risk types of human papillomavirus by decision trees. [Google Scholar]
- 12.Park S, Hwang S, Zhang B. Classification of human papillomavirus (hpv) risk type via text mining. Genomics Informatics. 2003;1(2):80–6. [Google Scholar]
- 13.Eom J, Park S, Zhang B. Proceedings of the 11th International Conference on Neural Information Processing: 22-25 November 2004; Calcutta, India. Germany: Springer Berlin Heidelberg; 2004. Genetic mining of DNA sequence structures for effective classification of the risk types of human papillomavirus (HPV) [Google Scholar]
- 14.Kim S, Zhang B. Proceedings of the 2006 International Conference on Applications of Evolutionary Computing: 10-12 April 2006; Budapest, Hungary. Germany: Springer Berlin Heidelberg; 2006. Human papillomavirus risk type classification from protein sequences using support vector machines. [Google Scholar]
- 15.Kim S, Eom J. Proceedings of Third International Symposium on Neural Networks: 28 May - 1 June 2006; Chengdu, China. Germany: Springer Berlin Heidelberg; 2006. Prediction of the human papillomavirus risk types using gap-spectrum kernels. [Google Scholar]
- 16.Kim S, Kim J, Zhang B. Ensembled support vector machines for human papillomavirus risk type prediction from protein secondary structures. Comput Biol Med. 2009;39:187–93. doi: 10.1016/j.compbiomed.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Jeffrey HJ. Chaos game representation of gene structure. Nucleic Acids Res. 1990;18:2163–70. doi: 10.1093/nar/18.8.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almeida JS, Carrico JA, Maretzek A, Noble PA, Fletcher M. Analysis of genomic sequences by chaos game representation. Bioinformatics. 2001;17:429–37. doi: 10.1093/bioinformatics/17.5.429. [DOI] [PubMed] [Google Scholar]
- 19.Lu J, Hu X, Liu X, Shi F. Proceedings of 5th International Conference on Bioinformatics and Biomedical Engineering: 10-12 May 2011; Wuhan. USA: IEEE; 2011. Predicting thermophilic nucleotide sequences based on chaos game representation features and support vector machine. [Google Scholar]
- 20.Dutta C, Das J. Mathematical characterization of chaos game representation: New algorithms for nucleotide sequence analysis. J Mol Biol. 1992;228:715–29. doi: 10.1016/0022-2836(92)90857-G. [DOI] [PubMed] [Google Scholar]
- 21.Xiao Q, Zhou J, Shi L. Proceedings of Sixth International Conference on Natural Computation: 10-12 August 2010; Yantai, Shandong. USA: IEEE; 2010. A novel 3D graphical representation of RNA secondary structures based on chaos game representation. [Google Scholar]
- 22.Deschavanne PJ, Giron A, Vilain J, Fagot G, Fertil B. Genomic signature: characterization and classification of species assessed by chaos game representation of sequences. Mol Biol Evol. 1999;16:1391–9. doi: 10.1093/oxfordjournals.molbev.a026048. [DOI] [PubMed] [Google Scholar]
- 23.Tavassoly I, Tavassoly O, Rad MSR, Dastjerdi NM. Proceedings of Frontiers in the Convergence of Bioscience and Information Technologies: 11-13 October 2007; Jeju City. USA: IEEE; 2007. Three dimensional chaos game representation of genomic sequences. [Google Scholar]
- 24.Yu Z-G, Shi L, Xiao Q-J, Anh V. Proceedings of the 2nd International Conference on Bioinformatics and Biomedical Engineering: 16-18 May 2008; Shanghai. USA: IEEE; 2008. Chaos game representation of genomes and their simulation by recurrent iterated function systems. [Google Scholar]
- 25.Nair VV, Vijayan K, Gopinath DP, Nair AS. Proceedings of 2010 Second International Conference on Machine Learning and Computing: 9-11 February 2010; Bangalore. USA: IEEE; 2010. ANN based classification of unknown genome fragments using chaos game representation. [Google Scholar]
- 26.Messaoudi I, Oueslati AE, Lachiri Z. Proceedings of 2012 6th International Conference on Sciences of Electronics, Technologies of Information and Telecommunications: 21-24 March 2012; Sousse. USA: IEEE; 2012. Genomic data visualization. [Google Scholar]
- 27.Yu Z-G, Anh V, Lau KS. Chaos game representation of protein sequences based on the detailed HP model and their multifractal and correlation analyses. J Theor Biol. 2004;226:341–8. doi: 10.1016/j.jtbi.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Yang JY, Yu ZG, Anh V. Clustering structure of large proteins using multifractal analyses based on 6-letters model and hydrophobicity scale of amino acids. Chaos, Solitons Fractals. 2009;40:607–20. doi: 10.1016/j.chaos.2007.08.014. [DOI] [Google Scholar]
- 29.Yang JY, Peng ZL, Yu ZG, Zhang RJ, Anh V, Wang D. Prediction of protein structural classes by recurrence quantification analysis based on chaos game representation. J Theor Biol. 2009;257:618–26. doi: 10.1016/j.jtbi.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 30.Hu XH, Xia JB, Niu XH, Ma X, Song CH, Shi F. Proceedings of 3rd International Conference on Bioinformatics and Biomedical Engineering: 11-13 June 2009; Beijing. USA: IEEE; 2009. Chaos game representation for discriminating thermophilic from mesophilic protein sequences. [Google Scholar]
- 31.Nana L, Xiaohui N, Feng S, Xuehai H. Proceedings of 3rd International Conference on Bioinformatics and Biomedical Engineering: 11-13 June 2009; Beijing. USA: IEEE; 2009. Subcellular locations prediction of proteins based on chaos game representation. [Google Scholar]
- 32.Song C, Shi F. Proceedings of International Conference on Future BioMedical Information Engineering: 13-14 December 2009; Sanya. USA: IEEE; 2009. Subcellular location of apoptosis proteins based on chaos game representation. [Google Scholar]
- 33.Yu Z-G, Xiao Q-J, Shi L, Yu Z-W, Anh V. Chaos game representation of functional protein sequences, and simulation and multifractal analysis of induced measures. Chinese Phys B. 2010;19:068701. doi: 10.1088/1674-1056/19/6/068701. [DOI] [Google Scholar]
- 34.Olyaee M, yaghubi M. Proceedings of Fourth Asia International Conference on Mathematical/Analytical Modelling and Computer Simulation: 26-28 May 2010; Bornea. USA: IEEE; 2010. Improved protein structural class prediction based on chaos game representation. [Google Scholar]
- 35.Basu S, Pan A, Dutta C, Das J. Chaos game representation of proteins. J Mol Graphics Modell. 1997;15:279–89. doi: 10.1016/S1093-3263(97)00106-X. [DOI] [PubMed] [Google Scholar]
- 36.Chou KC. Graphic rule for drug metabolism systems. Curr Drug Metab. 2010;11:369–78. doi: 10.2174/138920010791514261. [DOI] [PubMed] [Google Scholar]
- 37.Zhou GP. The disposition of the lzcc protein residues in wenxiang diagram provides new insights into the protein-protein interaction mechanism. J Theor Biol. 2011;284:142–8. doi: 10.1016/j.jtbi.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu ZC, Xiao X. 2D-MH: a web-server for generating graphic representation of protein sequences based on the physicochemical properties of their constituent amino acids. J Theor Biol. 2010;267:29–34. doi: 10.1016/j.jtbi.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 39.Xiao X, Chou KC. Using pseudo amino acid composition to predict protein attributes via cellular automata and others approaches. Curr Bioinf. 2011;6:251–60. doi: 10.2174/1574893611106020251. [DOI] [Google Scholar]
- 40.Xiao X, Wang P. Cellular automata and its applications in protein bioinformatics. Curr Protein Pept Sci. 2011;12:508–19. doi: 10.2174/138920311796957720. [DOI] [PubMed] [Google Scholar]
- 41.Xiao X, Wang P. GPCR-2L: predicting g protein-coupled receptors and their types by hybridizing two different modes of pseudo amino acid compositions. Mol Biosystems. 2011;7:911–9. doi: 10.1039/C0MB00170H. [DOI] [PubMed] [Google Scholar]
- 42.Alshalalfa M, Alhajj R. Proceedings of 8th IEEE International Conference on BioInformatics and BioEngineering: 8-10 October 2008; Athens. USA: IEEE; 2008. Combining singular value decomposition and t-test into hybrid approach for significant gene extraction from microarray data. [Google Scholar]
- 43.Duan ZH, Liou LS, Shi T, DiDonato JA. Proceedings of the 2003 IEEE Bioinformatics Conference: 11-14 August 2003. USA: IEEE; 2003. Application of singular value decomposition and functional clustering to analyzing gene expression profiles of renal cell carcinoma. [Google Scholar]
- 44.Tomfohr J, Lu J, Kepler TB. Pathway level analysis of gene expression using singular value decomposition. BMC Bioinf. 2005;6:225. doi: 10.1186/1471-2105-6-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger JA, Hautaniemi S, Mitra SK, Astola J. Jointly analyzing gene expression and copy number data in breast cancer using data reduction models. IEEE/ACM Trans Comput Biol Bioinf. 2006;3:2–16. doi: 10.1109/TCBB.2006.10. [DOI] [PubMed] [Google Scholar]
- 46.Baty F, Rudiger J, Miglino N, Kern L, Borger P, Brutsche M. Exploring the transcription factor activity in high-throughput gene expression data using RLQ analysis. BMC Bioinf. 2013;14:178. doi: 10.1186/1471-2105-14-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aghili SA, Sahin OD, Agrawal D, Abbadi AE. Proceedings of the Fourth IEEE Symposium on Bioinformatics and Bioengineering: 19-21 May 2004. USA: IEEE; 2004. Efficient filtration of sequence similarity search through singular value decomposition. [Google Scholar]
- 48.Peters TJ, Smolikova-Wachowiak R, Wachowiak MP. Proceedings of the 29th Annual International Conference of the IEEE EMBS Cite Internationale: 22-26 Aug. 2007; France. USA: IEEE; 2007. Microarray image compression using a variation of singular value decomposition. [DOI] [PubMed] [Google Scholar]
- 49.Hu P, Bull SB, Jiang H. Gene network modular-based classification of microarray samples. BMC Bioinf. 2012;13(Suppl 10):17. doi: 10.1186/1471-2105-13-S10-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holec M, Klema J, Zelezny F, Tolar J. Comparative evaluation of set-level techniques in predictive classification of gene expression samples. BMC Bioinf. 2012;13(Suppl 10):15. doi: 10.1186/1471-2105-13-S10-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan YN, Xiao X, Min JL. iNR-Drug: predicting the interaction of drugs with nuclear receptors in cellular networking. Int J Mol Sci. 2014;15:4915–37. doi: 10.3390/ijms15034915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo SH, Deng EZ, Xu LQ, Ding H, Lin H, Chen W, et al. iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics. 2014;30:1522–9. doi: 10.1093/bioinformatics/btu083. [DOI] [PubMed] [Google Scholar]
- 53.Liu B, Zhang D, Xu R, Xu J, Wang X. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics. 2014;30:472–9. doi: 10.1093/bioinformatics/btt709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu WR, Xiao X. iRSpot-TNCPseAAC: identify recombination spots with trinucleotide composition and pseudo amino acid components. Int J Mol Sci. 2014;15:1746–66. doi: 10.3390/ijms15021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen W, Feng PM, Lin H. iSS-PseDNC: identifying splicing sites using pseudo dinucleotide composition. Biomed Res Int. 2014;2014:623149. doi: 10.1155/2014/623149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiu WR, Xiao X, Lin WZ. iMethyl-PseAAC: identification of protein methylation sites via a pseudo amino acid composition approach. BioMed Res Int. 2014;2014:947416. doi: 10.1155/2014/947416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ding H, Deng EZ, Yuan LF, Liu L. iCTX-Type: a sequence-based predictor for identifying the types of conotoxins in targeting ion channels. BioMed Res Int. 2014;2014:286419. doi: 10.1155/2014/286419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y, Wen X, Shao XJ, Deng NY. iHyd-PseAAC: predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int J Mol Sci. 2014;15:7594–610. doi: 10.3390/ijms15057594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition (50th anniversary year review) J Theor Biol. 2011;273:236–47. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chou KC, Shen HB. Review: Recent progresses in protein subcellular location prediction. Anal Biochem. 2007;370:1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 61.Chou KC, Wu ZC, Xiao X. iLoc-Hum: using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol Biosyst. 2012;8:629–41. doi: 10.1039/C1MB05420A. [DOI] [PubMed] [Google Scholar]
- 62.Keller JM, Gray MR, Givens JA. A fuzzy k-nearest neighbor algorithm. IEEE Trans Syst Man Cybernet. 1985;SMC-15:580–5. doi: 10.1109/TSMC.1985.6313426. [DOI] [Google Scholar]
- 63.Lin WZ, Fang JA, Xiao X. iLoc-Animal: a multi-label learning classifier for predicting subcellular localization of animal proteins. Mol Biosystems. 2013;9:634–44. doi: 10.1039/c3mb25466f. [DOI] [PubMed] [Google Scholar]
- 64.Chou KC, Cai YD. Prediction and classification of protein subcellular location: sequence-order effect and pseudo amino acid composition. J Cell Biochem. 2003;90:1250–60. doi: 10.1002/jcb.10719. [DOI] [PubMed] [Google Scholar]
- 65.Min JL, Xiao X, Chou KC. iEzy-Drug: A web server for identifying the interaction between enzymes and drugs in cellular networking. BioMed Res Int. 2013:701317. [DOI] [PMC free article] [PubMed]
- 66.Xiao X, Min JL, Wang P. iCDI-PseFpt: identify the channel-drug interaction in cellular networking with pseaac and molecular fingerprints. J Theor Biol. 2013;337:71–9. doi: 10.1016/j.jtbi.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 67.Xiao X, Wang P, Lin WZ. iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal Biochem. 2013;436:168–77. doi: 10.1016/j.ab.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 68.Kong L, Zhang L, Lv J. Accurate prediction of protein structural classes by incorporating predicted secondary structure information into the general form of chou’s pseudo amino acid composition. J Theor Biol. 2014;344:12–8. doi: 10.1016/j.jtbi.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 69.Mondal S, Pai PP. Chou’s pseudo amino acid composition improves sequence-based antifreeze protein prediction. J Theor Biol. 2014;356:30–5. doi: 10.1016/j.jtbi.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 70.Hajisharifi Z, Piryaiee M, Mohammad Beigi M, Behbahani M, Mohabatkar H. Predicting anticancer peptides with chou’s pseudo amino acid composition and investigating their mutagenicity via ames test. J Theor Biol. 2014;341:34–40. doi: 10.1016/j.jtbi.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 71.Chou KC, Cai YD. Prediction of membrane protein types by incorporating amphipathic effects. J Chem Inf Model. 2005;45:407–13. doi: 10.1021/ci049686v. [DOI] [PubMed] [Google Scholar]
- 72.Liu B, Xu J, Lan X, Xu R, Zhou J, Wang X, et al. iDNA-Prot ∣dis: Identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS ONE. 2014;9:106691. doi: 10.1371/journal.pone.0106691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu Y, Wen X, Wen LS, Wu LY. iNitro-Tyr: prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS ONE. 2014;9:105018. doi: 10.1371/journal.pone.0105018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chou KC, Wu ZC, Xiao X. iLoc-Euk: a multi-label classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins. PLoS One. 2011;6:18258. doi: 10.1371/journal.pone.0018258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen L, Zeng WM, Cai YD. Predicting anatomical therapeutic chemical(atc) classification of drugs by integrating chemical-chemical interactions and similarities. PLoS ONE. 2012;7:35254. doi: 10.1371/journal.pone.0035254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chou KC. Some remarks on predicting multi-label attributes in molecular biosystems. Mol Biosyst. 2013;9:1092–100. doi: 10.1039/c3mb25555g. [DOI] [PubMed] [Google Scholar]
- 77.Rozanov M, Plikat U, Chappey C, Kochergin A, Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004;32:654–9. doi: 10.1093/nar/gkh419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu R, Zhou J, Liu B, He Y, Zou Q, Wang X, Chou K-C. Identification of dna-binding proteins by incorporating evolutionary information into pseudo amino acid composition via the top-n-gram approach. J Biomolecular Struct Dynamics. 2014. doi:10.1080/07391102.2014.968624. [DOI] [PubMed]
- 79.Qiu WR, Xiao X, Lin WZ. iUbiq-Lys: Prediction of Lysine Ubiquitination Sites in Proteins by Extracting Sequence Evolution Information Via a Grey System Model. in press. [DOI] [PubMed]
- 80.Lin SX, Lapointe J. Theoretical and experimental biology in one. J Biomed Sci Eng. (JBiSE) 2013;6:435–42. doi: 10.4236/jbise.2013.64054. [DOI] [Google Scholar]