

Abstract
Wiskott-Aldrich Syndrome (WAS) is an inherited immunodeficiency caused by mutations in the gene encoding WASP, a protein regulating the cytoskeleton. Hematopoietic stem/progenitor cell (HSPC) transplants can be curative but, when matched donors are unavailable, infusion of autologous HSPCs modified ex vivo by gene therapy is an alternative approach. We used a lentiviral vector encoding functional WASP to genetically correct HSPCs from three WAS patients and re-infused the cells after reduced-intensity conditioning regimen. All three patients showed stable engraftment of WASP-expressing cells and improvements in platelet counts, immune functions, and clinical score. Vector integration analyses revealed highly polyclonal and multi-lineage haematopoiesis resulting from the gene corrected HSPCs. Lentiviral gene therapy did not induce selection of integrations near oncogenes and no aberrant clonal expansion was observed after 20–32 months. Although extended clinical observation is required to establish long-term safety, lentiviral gene therapy represents a promising treatment for WAS.
Introduction
Wiskott-Aldrich Syndrome (WAS) is an X-linked primary immunodeficiency characterized by infections, microthrombocytopenia, eczema, autoimmunity and lymphoid malignancies (1, 2). The disorder is caused by mutations in the WAS gene, which codes for WASP, a protein that regulates the cytoskeleton. WASP-defective immune cells display alterations in proliferative responses after activation, cell migration, immunological synapsis formation and cytotoxicity (3–5). Allogeneic hematopoietic stem/progenitor cell (HSPC) transplantation can be curative, but it is often associated with significant morbidity and mortality, particularly in the absence of fully matched donors (6–8).
For patients without matched donors, an alternative therapeutic strategy is the infusion of autologous HSPC that have been genetically corrected ex vivo. This gene therapy approach has been successful in more than 50 patients affected by primary immunodeficiencies, including 10 WAS patients treated with HSPC transduced with a γ-retroviral vector encoding a functional WAS gene (9–15). Gene therapy combined with a reduced intensity conditioning regimen proved to be effective and safe in patients with Severe Combined Immunodeficiency (SCID) due to adenosine deaminase (ADA) deficiency, who were followed up to 13 years after treatment (9, 15, 16). In contrast, despite the initial clinical benefit, gene therapy with γ-retroviral transduced HSPC was associated with development of leukemia or myelodysplasia in patients with SCID-X1, Chronic Granulomatosis Disease, and WAS (14, 17–20). These adverse events were ascribed to vector insertion sites (ISs) near specific proto-oncogenes, leading to their trans-activation by enhancer/promoter sequences within the long-terminal repeat (LTR) of the retroviral vector (10–12, 21–23). In the case of WAS, characterization of ISs over the first two years of follow-up revealed a highly skewed insertion profile in vivo, resulting in expansion of clones with insertions in proto-oncogenes such as LMO2 (12), some of which progressed to leukemias (14, 24). The possibility of vector-driven leukemogenesis is a particular concern for WAS patients, who are cancer-prone (1).
Lentiviral vectors with self-inactivating (SIN) LTRs integrate efficiently in HSPC, allow robust transgene expression from a promoter of choice inserted within the vector and could potentially be safer for gene therapy applications (24–26). Lentiviral-based HSPC gene therapy combined with full conditioning has been used to treat three patients with adrenoleukodystrophy (ALD) (27) and one patient with β-thalassemia (28), resulting in 10–15% progenitor cell marking with therapeutic benefit. Although a relative expansion of a clone harboring an insertion in the HMGA2 gene was observed in the β-thalassemia patient (28), no aberrant clonal proliferation has been reported for the lentiviral-based trials up to 5 years after treatment (27, 29).
We developed a SIN lentiviral vector coding for human WASP under the control of a 1.6 kb reconstituted WAS gene promoter (LV-w1.6W) (3). The use of this endogenous promoter ensures that the transgene is expressed in a physiological manner (4), restoring WASP expression and function in human and murine WAS cells (3, 30–34). Its moderate enhancer activity combined with the SIN LTR design reduces the risk of insertional mutagenesis (35), as shown by in vitro transformation assays (36) and preclinical in vivo studies in WASP-deficient mice (34, 37). These data provided the rationale for a phase I/II clinical trial in which LV-w1.6W was used as a gene therapy vector for treatment of patients with WAS (38).
Results
Lentiviral transduction of HSPC and infusion of gene-corrected cells into patients pretreated with reduced intensity conditioning
Three children with WAS, who had been shown by genotyping to carry severe mutations in the X-linked WAS gene and who did not have compatible allogeneic donors, were enrolled in the phase I/II clinical trial (Table 1). All patients suffered from recurrent infections, eczema, bleeding, and thrombocytopenia, with a disease score ranging from 3 to 4 (39) (Table 1). Autologous bone BM derived CD34+ cells were collected, transduced twice with purified LV-w1.6W vector using an optimized protocol (fig. S1) (34) and re-infused intravenously back into the patients three days after collection. The vector and genetically modified cells were characterized extensively for quality and safety (tables S1 to S3, figs. S1 and S2).
Table 1.
Characteristics and treatment of the three WAS patients.
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Infectious manifestations | Recurrent ENT | Pneumonias, Colitis arthritis/cellulitis, URTI, UTI | Pneumonia with respiratory distress, URTI, otitis |
| Pathogens | VZV, CMV, HSV, EBV | CMV, HSV6, candida | Pneumocystis jirovecii, CMV |
| Thrombocytopenia manifestations | Skin petechiae | Skin petechiae, GI bleeding | Skin petechiae, GI bleeding, epistaxis |
| Eczema | Moderate-severe | Moderate-severe | Severe |
| Other | Developmental disorder, allergy | Failure to thrive, ⇑Inflammatory indexes/vasculitis, hepatosplenomegaly | GE reflux/food adversion (fed by naso-gastric tube), allergy |
| WAS mutation | Exon 10: C>T 995 (R321X) | IVS10del11nt | 37C>T (R13X) |
| WASP expression | <5% | <5% | <5% |
| Zhu score | 3 | 4 | 4 |
| Age at treatment (y) | 5.9 | 1.6 | 1.1 |
| Infused CD34+ cells (×106/kg) | 3.66 (BM) + 5.25 (MPB) | 14.1 | 10.2 |
| Vector copies/genome | 1.9 (BM)−1.4 (MPB) | 2.4 | 2.8 |
| Transduction efficiency (CFC) | 92% (BM)−88% (MPB) | 97% | 100% |
| Follow-up (mo) | 32 | 23 | 20 |
| Current clinical conditions | A&W No eczema No major bleeding/petechiae Off IVIG |
A&W No eczema No major bleeding/petechiae |
A&W No eczema No major bleeding/petechiae |
ENT, ear nose throat, URTI; upper respiratory tract infection; UTI, urinary tract infection; GI, gastrointestinal. GE, gastroesophageal. IVIG, intravenous immunoglobulins. WASP expression analysis was performed on PB lymphocytes by FACS. Patient 1 also received G-CSF mobilized peripheral blood (MPB)-derived CD34+ cells, previously collected as back-up, to achieve the target HSPC dose. F.U., follow-up. A&W, alive and well. Y, years. Mo, months.
Vector-specific Q-PCR on individually collected colony-forming cells (CFC) revealed high gene transfer efficiency – i.e., 88 to 100%. Average vector copy number (VCN) per genome measured in bulk cultured CD34+ cells was 2.3 ± 0.6 (Table 1). WASP expression in the transduced CD34+ cells was confirmed by immunoblotting (fig. S3). Before HSPC reinfusion, patients received a reduced intensity conditioning regimen, consisting of anti-CD20 mAb, busulfan (7.6–10.1 mg/Kg i.v. targeted for weight and pharmacokinetics) and fludarabine (60 mg/m2), designed to achieve significant depletion of endogenous HSPC and immune cells with limited toxicity (see Materials and Methods). The mean infused CD34+ cell dose was 11.07 ± 2.70 × 106 cells/kg (Table 1 and table S4). No adverse reactions were observed after the infusions.
Engraftment of lentiviral-transduced cells and restoration of WASP expression
All three WAS patients showed robust and multi-lineage engraftment of gene-corrected cells in BM and peripheral blood (PB), persisting up to the latest time point analyzed (30 months after gene therapy). Gene-marking in PB granulocytes peaked in the first month after treatment and then stabilized at a VCN range of 0.4–0.9 (Fig. 1A). Similar marking levels were seen in PB monocytes (Fig. 1A) as well as in granulocytic and megakaryocytic cells, CD34+ cells (Fig. 1B) and erythroid cells (fig. S4) collected from the BM. The in vivo engraftment of lentiviral-transduced HSPC was confirmed throughout the follow-up and persisted at high levels (Pt1 34%, Pt2 26%, Pt3 48%) 1 year after treatment as shown by vector-specific PCR on BM CFC (Fig. 1C). Based on these data, we estimated that each transduced progenitor cell contained on average 1 to 1.6 copies of the vector. Transduced B and natural killer (NK) cells were detectable 1 month after gene therapy and increased over time, with PB-derived B cells showing 1.5–2 fold higher VCN as compared to BM B cells (Fig. 1A, B). Transduced T cells appeared 3–6 months after gene therapy and reached the highest VCN as compared to other lineages (VCN range 1.0–2.4) (Fig. 1A).
Fig. 1. Engraftment of transduced cells and WASP expression after gene therapy.
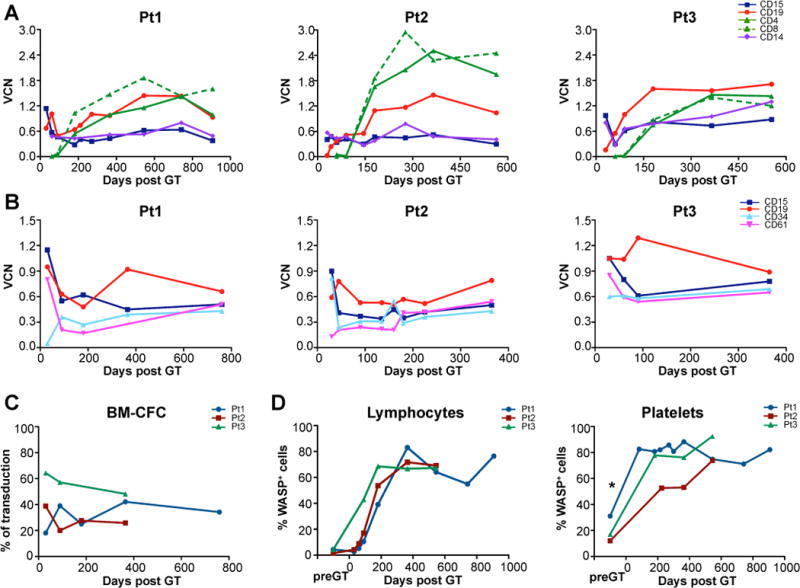
Vector copy number (VCN) per genome was evaluated by qPCR at different time points (up to 2.5 years) after gene therapy (GT) in CD15+ granulocytes, CD19+ B cells, CD4+ and CD8+ T cells, CD14+ monocytes, CD34+ progenitors and CD61+ (megakaryocytic lineage), purified either from peripheral blood (A) or bone marrow (B) in Patient 1 (left), 2 (center) and 3 (right). (C) Percentage of vector-positive bone marrow colony-forming cells (BM CFC) evaluated by PCR analyses on individual colonies derived from ex vivo purified CD34+ cells after gene therapy. (D) WAS protein expression measured by cytofluorimetric analysis at different time points after gene therapy in patient lymphocytes (left) and platelets (right). *WASP expression was measured after transfusion of donor platelets. Transgene expression was confirmed by immunoblot analysis of peripheral blood mononuclear cells, untransformed T-cell lines and a EBV-transformed B cell line (Fig. S3).
WASP expression, as measured by flow cytometry, progressively increased over time in PB lymphocytes and platelets in all three patients (Fig. 1D). WAS protein was present in a large proportion of T cells (CD4+ and CD8+), NK cells and monocytes at mean expression level comparable to normal donors, whereas in B cells and platelets its expression level was lower than controls (fig. S5). Interestingly, the proportion of WASP+ cells was higher in PB-derived platelets as compared to their BM-derived counterparts (fig. S6), suggesting a preferential migration or selective survival advantage of gene-corrected platelets in the periphery.
Clinical and immunological improvement after gene therapy
All treated children are clinically well with post-treatment follow-up of 20–32 months (Table 1). After conditioning, patients experienced severe neutropenia (neutrophil counts <500/ul) lasting from 12 to 19 days, followed by normalization of neutrophil counts. None of the patients experienced mucositis or other chemotherapy-related toxicity. Serious adverse events occurred in Pt2 and Pt3 within the first 2–6 months of gene therapy. These events were mainly of infectious origin, likely favored by the underlying clinical conditions and immune deficiency typical of the early post-transplant period. Both patients fully recovered from them (see Materials and Methods). No abnormal cellular expansion was detected in BM and PB by immunophenotypic, morphologic, and karyotypic analyses. Assays to detect anti-HIV Gag p24 antibodies and replication-competent lentivirus in patients’ blood were negative at all times of analysis.
In all three children, symptoms of WAS showed significant improvement. Pre-treatment eczema resolved between 6 and 12 months after gene therapy and did not recur (Table 1). Beginning 6 months after gene therapy, the frequency and severity of infections progressively decreased and cytomegalovirus replication was well-controlled, allowing withdrawal of anti-infectious prophylaxis in Pt1 and Pt3. Pt1 discontinued i.v. immunoglobulin supplementation 6 months after gene therapy, with positive antibody responses after vaccination.
Platelet counts improved significantly during the first year after gene therapy (Fig. 2A), as assessed by a mixed linear model applied to the serially repeated sampling (p-value=0.03). Although these counts never reached normal values, the platelet volumes normalized after gene therapy (MPV range: 8.3–9.2 fl, normal values: 7.4–10.9 fl). Following discontinuation of platelet transfusions (1–7 months after gene therapy), none of the patients experienced bleeding or skin manifestations of thrombocytopenia, with the exception of occasional grade 1 gastrointestinal bleeding in Pt2 (Fig. 2B). Moreover, all patients tested negative for a large panel of autoantibodies 1 year after gene therapy. The overall clinical improvement resulted in a reduced disease severity score in all patients (Fig. 2C).
Fig. 2. Clinical features and immune function of WAS patients after gene therapy.
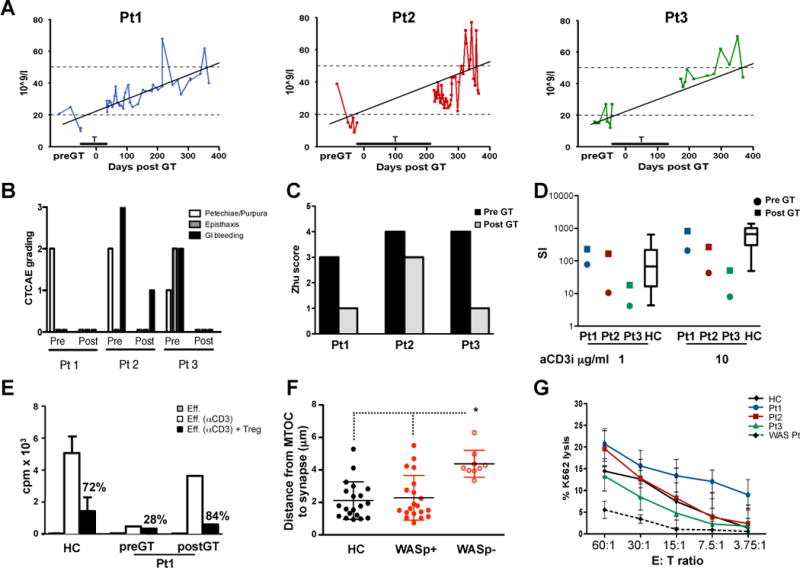
(A) Platelet counts before and 1 year after gene therapy. Kinetics of platelet counts during the first year of follow-up, analyzed by a mixed linear model applied to the individual repeated counts. Platelet transfusions (T) are indicated by an horizontal bar. (B) Summary of bleeding events. Each of the 3 categories [skin manifestations (petechiae/purpura), epistaxis, GI bleeding] was given an arbitrary score: 0, absent; 1, moderate; 2, severe (with the total score ranging between 0 and 6). Patients were evaluated after gene therapy following discontinuation of platelet transfusions (see Materials and Methods). (C) Disease score (Zhu score) evaluated pre- and post-gene therapy (39). (D) TCR-driven proliferation in PB mononuclear cells from WAS patients (before and 1 year after gene therapy) or healthy controls (HCs) (Boxplot with mean, 1st and 4th quartile, 5th and 95th percentile, n=15); SI. Stimulation index. (E) Treg-mediated suppression of effector T cells pre-gene therapy and 1 year post-gene therapy in Pt1, in comparison to healthy controls (mean ± SD, n=9). Eff: unstimulated effector cells. Eff.(a-CD3): effector cells stimulated with allogeneic accessory cells and soluble anti-CD3 mAb. Treg cells (sorted for CD4+/CD25high/CD127low/neg cells), were added to effector cells at 1:1 ratio and proliferation was measured by 3H-thymidine incorporation. Percentages indicate inhibition of proliferation. (F) Formation of NK immunological synapse. Distance of the microtubule organizing center (MTOC) from the immunological synapses in NK cells contacting K562 target cells. Analyses were performed by confocal microscopy in Pt1, 18 months after gene therapy. Patient NK cells were identified as positive or negative for WASP expression by anti-WASP antibody. (G) NK cell cytotoxic activity. Shown is the percent lysis of K562 human erythroleukemia cells by patient-derived PB mononuclear cells, as measured in a chromium release assay.
Lymphocyte subpopulation counts decreased after the conditioning regimen but progressively returned to normal levels for age within 6–12 months after treatment (fig. S7). In all patients, proliferative responses to anti-CD3 mAb increased after treatment and reached the range of age-matched controls (Fig. 2D). Similarly, the proliferative capacity of T-cell lines from Pt1 and Pt3 greatly improved after gene therapy (fig. S8). T cell receptor (TCR) Vβ profiling revealed that all three WAS patients displayed a broader polyclonal repertoire after gene therapy, in agreement with the polyclonal reconstitution observed by ISs analysis (see below) (fig. S9).
Because regulatory T (Treg) cell function is defective in WAS patients (32, 40), we evaluated the suppressive ability of Treg cells (VCN: 1.37) sorted from Pt1. As shown in Fig. 2E, the in vitro suppressive function of Treg cells was restored after gene therapy. This is consistent with the finding that CD4+/CD25high cells from both Pt1 and Pt3 express WASP after gene therapy (fig. S10). It is known that WASP-deficient NK cells show an altered immune function (41). We found that WASP-expressing, but not WASP-deficient, NK cells isolated from Pt1 after gene therapy, functioned normally in immunological synapse formation (Fig. 2F, fig. S11). The improved NK cell function of all three patients resulted in normal cytotoxic activity after gene therapy (Fig. 2G).
Multi-lineage and polyclonal engraftment of gene-corrected HSPC, assessed by longitudinal integration site profiling
To monitor the clonal contribution of gene-corrected cells to hematopoiesis in the three patients, we performed a high-throughput IS analysis (42, 43) on multiple lineages and time points up to 18 months after infusion of transduced HSPCs (see Supplementary Text, Figures S12–S46 and Tables S8–S25). Linear amplification-mediated (LAM) PCR and next-generation sequencing (42, 43) detected >2,400,000 IS sequences that were mapped to 33,363 unique chromosomal positions in the patients: 11,137 from Pt1, 10,889 from Pt2, and 10,337 from Pt3.
The relative proportion of sequencing reads corresponding to each specific ISs, as collected from BM CD34+ cells and PB myeloid, B and T cells, was used as a surrogate of clonal repertoire of gene-corrected cells and safety readout (Fig. 3; fig. S12–S14). We found that, beginning 1 month after gene therapy, the vast majority of ISs from all lineages and timepoints were well below 5% with a few occasionally reaching up to 15–20% of the total reads retrieved from a lineage (Fig. 3A). By monitoring the most frequent ISs in each lineage, we found that highly represented insertion sites were different at each timepoint and fluctuated over time for up to 6–12 months after gene therapy (Fig. 3B). At later timepoints, in line with the stabilization of hematopoietic output, we detected a persistent multi-lineage contribution of several ISs, some of which were highly represented (Fig. 3B), but remained below 10% of all the reads belonging to ISs retrieved from the PB cells in the one-year interval (Fig. 3A, fig. S14). It is possible that HSPC activity in Pt2 was affected by the occurrence of bacterial sepsis 6 months after gene therapy. At that time, a group of ISs marking T-cells, B-cells and myeloid cells became relatively more abundant, possibly revealing the stress-induced activation of a limited pool of progenitors.
Fig. 3. Long-term polyclonal engraftment of gene-corrected HSPC, assessed by longitudinal integration site profiling.
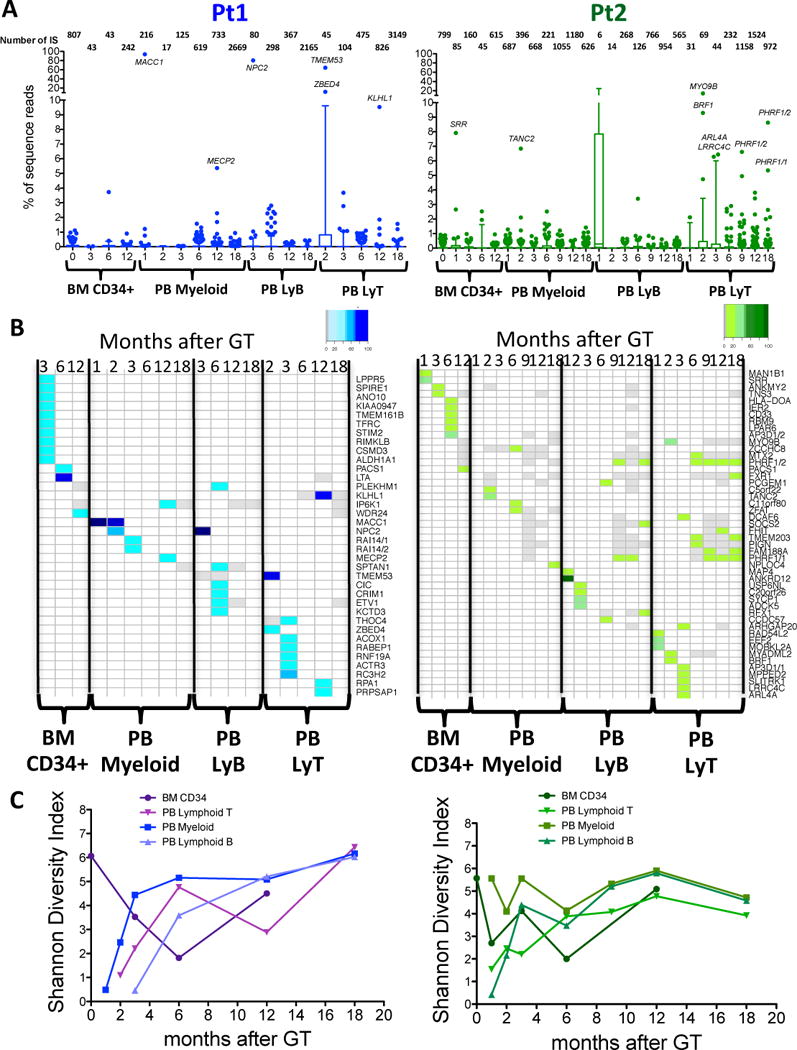
(A) Distribution of sequence reads in different lineages at different timepoints. Each boxplot shows the distribution of sequence reads in two WAS patients in the different lineages. Percentages of sequence reads for each IS are calculated over the total number of sequence reads from the same source (BM or PB) and timepoint. X-axis indicates months after gene therapy, Y-axis percentage of sequence reads. IS outliers (over 95 percentile of ISs dataset from same timepoint) are shown as dots. The total number of ISs is shown on top of each boxplot for the relative lineage and timepoint. Genes proximal to IS representing more than 5% of sequence reads from same source and timepoint are reported on top of outlier dots. (B) Top contributing ISs inside each lineage in BM CD34+ cells, PB Myeloid B and T cells in two WAS patients. Percentages of sequence reads for each IS are calculated over the total number of sequence reads from the same lineage. Top IS shown in the heatmaps account for more than 5% of sequence reads from their lineage. Each column corresponds to a timepoint (months after gene therapy), and each row to a given top ISs based on sequence reads. Color intensity on the heatmaps represents for each IS the percentages of relative sequence reads on total sequence reads from same lineage and timepoint, ranging from white (IS not detected at that timepoint) to gray (IS detected at percentages lower than 5% of sequence reads from that given timepoint) to gradient of colors (IS detected at more than 5% of sequence reads from that given timepoint). (C) Diversity of ISs in different lineages and timepoint. For each ISs dataset from the same lineages and timepoints of panel A and B a Shannon diversity index was calculated and plotted.
The Shannon diversity index was used to assess the overall clonal repertoire composition of in vivo derived BM CD34+ and myeloid cells. In the first months after treatment, clonal diversity was lower then measured in the in vitro transduced cell population, but later increased and stabilized at 12–18 months (Fig. 3C). As predicted by the slower dynamics of thymic reconstitution, T cells showed different kinetics with increasing diversity from 3 to 6 months and stabilization to levels similar to other lineages at 9–18 months after gene therapy.
We next compared ISs retrieved from BM CD34+ cells, myeloid, and lymphoid lineages in each patient, applying stringent analytical filters to take into account sample impurity and cross-contaminations. This analysis unveiled a group of ISs that was shared among these datasets (Fig. 4A). The shared ISs in CD34+ cells and myeloid/lymphoid lineages increased from 18.2% to 27.2% at 1 year after gene therapy in Pt1. A higher proportion of integrations shared with other lineages (44.1%) was detected in BM CFC from Pt1. A substantial fraction of the ISs retrieved from BM CD34+ cells or CFC at 1 year after gene therapy (Fig. 4B) could be traced back to earlier follow up samples of mature cells, pointing to the efficient engraftment of self-renewing gene-marked HSPCs.
Fig. 4. Multi-lineage engraftment and activity of gene-corrected HSPC.
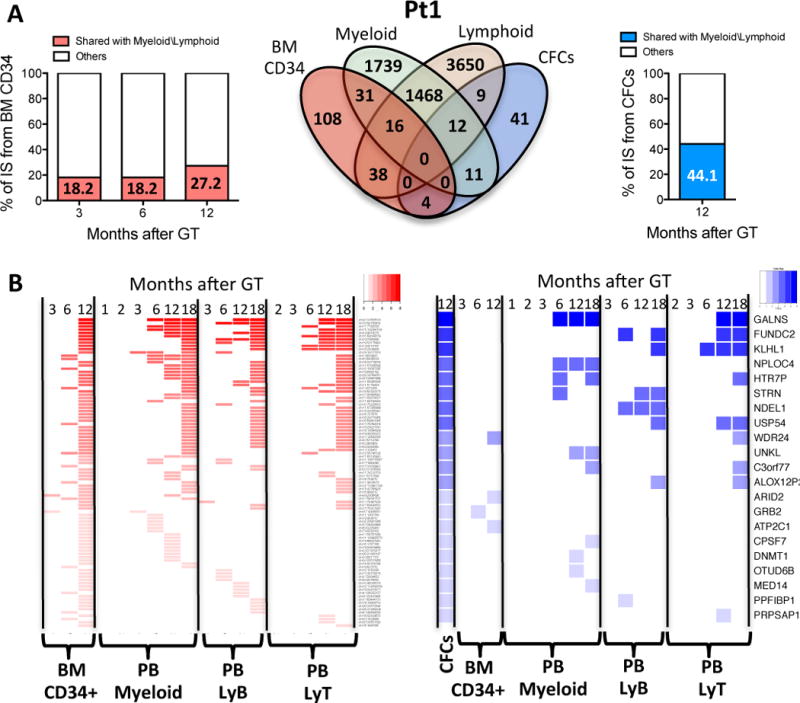
(A) Multi-lineage detection of identical IS. Venn diagram show overlaps among CD34+, myeloid, lymphoid cells and CFCs ISs datasets from Pt1. Column graphs show percentage of CD34+ cells and CFC ISs from Pt1 shared with myeloid\lymphoid lineages (red and blue portion of column respectively) at different months after gene therapy. (B) Detection of shared ISs over time. Heatmaps show CD34+ cells and CFCs ISs at different timepoint shared with the 4 lineages of panel A–C. Each column shows a lineage and a timepoint and each row a shared ISs belonging to CD34+ cells (red) or CFCs (blue). The intensity of colors indicates degree of ISs detection in multiple lineages and timepoints from highly shared ISs (high intensity of red and blue) to ISs shared with a single lineage and timepoint (light red or blue)
Our data allowed an estimation of the number of transduced HSPC contributing to hematopoiesis in vivo. Vector integrations were analyzed in two independent samplings (12 and 18 months after gene therapy) of purified PB myeloid cells, which are short-lived and provide a readout of stem cell output. Using the Petersen/Schnabel model estimator of population size (44), we calculated a theoretical minimal number of about 6,300 and 1,700 transduced active stem cells in Pt1 and Pt2 respectively (table S25) which corresponds to about 1.5–2.5 in 1×105 infused CD34+ cells.
Comparison of lentiviral versus γ-retroviral “integrome” in gene therapy of WAS
We compared the integration profile of LV-w1.6W vector with 11,294 unique ISs retrieved from two WAS patients enrolled in the γ-retroviral gene therapy trial (12) collected over a similar time frame and analysed through the same mapping pipeline to obtain a comparable dataset (Table S10). The genomic insertional pattern of LV-w1.6W vector was similar in all three patients and fit the classical distribution of lentivirus showing a strong tendency to integrate within transcriptional units (fig. S15B) and cluster in gene-rich regions (Fig. 5A, fig. S15A–B). No major difference was observed in the genomic distribution of LV-w1.6W in the in vitro vs patients derived samples (fig. S16A). By contrast, the profile found in the cells from patients treated with γ-retroviral gene therapy was very different, showing a skewed profile towards transcription start sites (fig. S15A).
Fig. 5. Comparative analysis of vector integration sites in patients treated by gene therapy with lentiviral or γ-retroviral vectors.
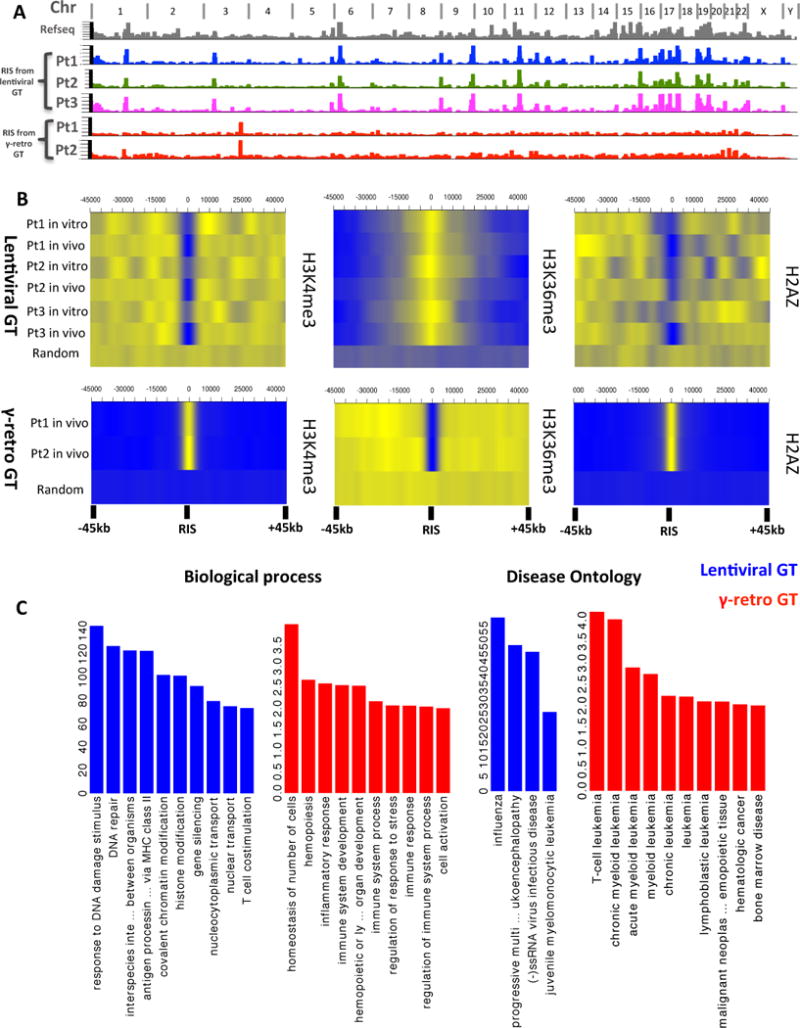
(A) Genomic distribution of ISs from five patients with WAS, treated by lentiviral (n=3) or γ-retroviral (n=2) vectors. Chromosome names are reported on top of the graph. Refseq Genes and ISs frequency distributions are shown in bins of 1Mbp (grey, colored/red columns respectively). (B) Chromatin modifications surrounding ISs. Probability density distributions of histone modifications mapped on CD34+ cells in a +/−45kb window surrounding IS are shown as heatmaps. Color intensity show under (blue) or over (yellow) representation of each given histone modification as compared to random in silico generated reference. Each row represents an in vitro or in vivo ISs dataset from the patients treated with lentiviral or γ-retroviral vectors. (C) Gene Ontology of genes proximal to ISs. Biological process and Diseases significantly associated to the functions of genes proximal to ISs in lentiviral and γ-retro gene therapy are represented as blue and red bars, respectively. The values on y-axis show the fold enrichment scale for gene ontology categories.
As reported in previous studies (45, 46), vector integration site selection could be influenced by the chromatin status of target cells during transduction/infection. Thus, we analyzed the density distribution of histone modifications mapped in CD34+ cells (47) on a 100kb window surrounding the ISs from the two cohorts of patients (Fig. 5B, fig. S20). The probability density map showed that certain histone modifications were equally preferred (H3K4me1) or avoided (H3K9me3 and H3K27me3) by lentivirus and retrovirus integrations. In addition, no major differences were observed between in vitro and in vivo lentivirus datasets. By contrast, other chromatin features such as H3K4me3, H3K36me3 and H2AZ, marking respectively transcription start sites, coding, and enhancer/promoter regions of actively expressed genes, were present at differential intensity around ISs of the two vectors (Fig. 5B). Of note, the concomitant low density of H3K4me3 and H2AZ seems to provide an optimal chromatin environment for lentivirus insertions (fig. S17 to 19) and help explaining the different genomic distribution observed for retroviruses.
We also analyzed the ontological categories of the genes targeted by vector integration sites in the datasets from the two patient cohorts by using the GREAT (Genomic Regions Enrichment of Annotations Tool) software (48) (Fig. 5C). A broad spectrum of biological categories including antigen presentation, response to virus, chromatin modification and gene silencing was represented among the lentivirus targeted genes. In contrast, the retroviral insertions were more often proximal to genes related to hematopoietic system maintenance as well as to hematological diseases. Of note, the vast majority of gene categories targeted by ISs in our LV-w1.6W patients were also targeted in the previous ALD gene therapy trial (27) (fig. S21, S22). Thus, differently from retroviruses, LV-w1.6W has a lower probability to interfere with the expression of genes involved in transformation of hematopoietic cells due to its wider genomic distribution.
We next assessed the presence of common insertion sites (CIS) to study potential insertional hotspots at the time of transduction and define surrogate markers of in vivo selection of clones. CIS were detected using a canonical statistical method that assumes a random distribution of vector integrations (49) and a novel analysis for CIS discovery based on genome-wide Grubbs test for outliers (for details on both methods see Supplementary Text). The highest ranking order WAS lentiviral CIS were KDM2A (targeted by 125 integrations), PACS1 (117) and TNRC6C (94) (table S23) which are well known hotspots in preclinical studies of lentiviral vectors (50, 51) and in the ALD lentiviral gene therapy trial (27) and have not been associated with clonal expansion (Fig. 6A)(Fig. S40). In contrast, γ-retroviral vector was found clustered in vivo in the MECOM (MDS-EVI1) and LMO2 loci (Fig. 6A), hosting 67 and 15 of 11,294 integrations, respectively (12). In the current LV-w1.6W gene therapy trial, these two genes were hit only by 10 and 5 out of 33,363 integrations, respectively (Fig. 6B). Additionally, the relative sequence reads associated to these ISs (0.44% and 0.05% of reads from the same timepoint and sample source for MECOM and LMO2, respectively) were below the average of all other insertions and these specific integrants were not consistently detected overtime. These observations were confirmed for insertions near other genes previously involved in clonal dominance like CCND2 and PRDM16 (Fig. S15C). The HMGA2 locus was hit in both datasets (Fig. S15C) but in contrast to what was reported in the β-thalassemia patient (28), we did not observe over-representation of these integrations during followup. Together, these results indicate the lack of evident clonal expansions associated to CIS in proximity of known proto-oncogenes, suggesting that the insertional hotspots found in our trial are the product of vector integration biases at the time of transduction rather than in vivo genetic selection of ISs conferring a selective growth advantage.
Fig. 6. Common insertion sites and oncogenic hits in lentiviral vs γ-retroviral gene therapy.
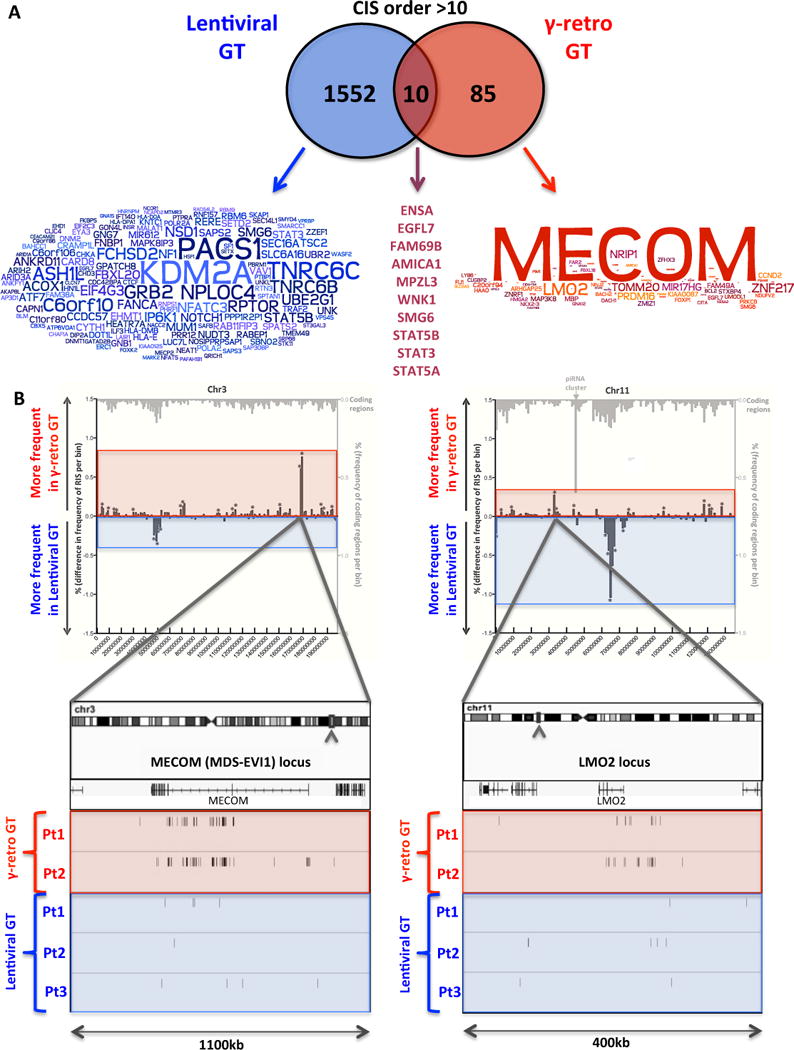
(A) Overlaps among CIS genes in lentiviral and γ-retroviral gene therapy. Genes proximal to CIS of order >10 are reported in the overlapping circles for lentiviral and γ-retro gene therapy (blue and red, respectively). Word clouds show the intensity of ISs clustering in each of the CIS gene (the bigger the gene name the higher the number of ISs inside or in the proximity of that gene). The names of the 6 CIS genes detected in both gene therapy trials are reported on the right at the intersection between the circles. (B) Incidence of ISs at two oncogenes in patients treated with lentiviral or γ-retroviral gene therapy. The comparative frequency distributions of insertions from lentiviral and γ-retroviral gene therapy in bins of 2kb are shown for chromosome 3 (left panel) and 11 (right panel). Mirror graph shows for each bin more frequent insertions in γ-retroviral gene therapy (columns above X-axis) and more frequent insertions in lentiviral gene therapy (columns below X-axis) (*p<0.05 Fisher exact test). Light grey columns on top show transcriptional units frequency for the same bins. Boxes on bottom show a detail of MECOM and LMO2 loci where ISs from the three lentiviral and two retroviral gene therapy patients are shown as sticks below the gene transcript. Raw IS data from the patients treated with the γ-retroviral vector were generated by Boztug et al (12) and re-processed through our informatic pipeline (Table SOM3).
Discussion
We have shown that infusion of autologous HSPC transduced with a lentiviral vector encoding a functional WASP gene resulted in robust and stable multi-lineage engraftment in three WAS patients who had been pre-treated with a reduced intensity preparative regimen. The gene corrected blood cells expressed WASP under control of a reconstituted WAS promoter. The patients showed improved immune function and amelioration of clinical manifestations of the disease, including protection from bleeding and severe infections as well as resolution of eczema.
In this trial, the in vivo gene marking of hematopoiesis was superior (≈25–50% of BM progenitors and mature PB myeloid cells) to that described in other lentiviral-based gene therapy trials for ALD and β-thalassemia (27, 28), which used myeloablative preparative regimens. The use of highly purified and homogenous batches of lentiviral vectors, optimized culture conditions and vector exposure (34), and the infusion of cells shortly after transduction, likely account for the robust engraftment. In a parallel trial using the same procedures to treat patients with metachromatic leukodystrophy (MLD) (52), in vivo gene marking of HSPC was even higher (45–80%). The preparative conditioning regimen in the latter trial was more intensive, suggesting that chemotherapy regimens can be modulated based on the engraftment threshold required for therapeutic benefit. Notably, stable engraftment of marked progenitor cells in vivo was higher in this lentiviral vector trial than in the previous WAS trial based on γ-retrovirus vector (3–22%) (12). A selective advantage of lymphoid cells carrying a functional WAS gene (12), was observed in both clinical trials in line with previous studies in the murine disease model (12, 30, 53, 54).
Although gene therapy did not normalize platelet counts in the patients the counts did increase significantly and the platelets were of normal size, improvements that were sufficient to protect patients from bleeding and related disorders. Considering that the BM must contain a mixed population of WASP-positive and WASP-negative megakaryocytes and only platelets with normal levels of WASP were detected in peripheral blood, it is conceivable that a threshold level of WASP is required for platelets to exit the BM and/or to survive in the periphery. It is noteworthy that platelet counts in WAS patients treated by lentiviral gene therapy are in the range measured in patients with mixed donor/host chimerism after successful allogeneic BM transplant (7). A more intense conditioning regimen or the use of a lentiviral vector with a stronger promoter might result in a greater number of gene-corrected platelets. However, these options should be carefully balanced with the higher risk of toxicity, as reported for the conditioning used in allogeneic BM transplant, as well as of transactivation and insertional oncogenesis associated with strong enhancer/promoters, such as those of γ-retroviral vectors (55).
A consistent multi-lineage re-capture of ISs 12 months after gene therapy indicated the active contribution of gene-corrected long-term HSPC to hematopoiesis. Our results lead us to propose that a takeover of hematopoietic output by long-term HSPC occurs at a defined time window after gene therapy, generating a diverse clonal repertoire in the blood progeny. The high proportion of ISs found in clonogenic BM progenitors that are shared with mature lineages validates IS-based tracking of stem cell dynamics and the colony assay as a representative surrogate readouts of in vivo HSPC activity. The calculated minimal number of 1,700–6,300 transduced active stem cells engrafted in vivo is consistent with data from the parallel MLD trial (52) and with estimates that repopulating HSPC account for 1 out of 104–105 infused CD34+ cell population (56), considering the impact of cell manipulation and the potentially limited BM homing after transplant.
The study of ISs from WAS patients treated with lentiviral or retroviral vector (12) allowed the first comparative study of the safety and clinical performance of two different vectors in the context of the same disease background. Our analyses expand previous observations that chromatin conformation and histone modifications differently impact the genomic distribution of retroviral vectors (45, 57), with the identification of H3K4me3 and H2AZ as two major chromatin determinants of differential insertions between lentiviruses and retroviruses. Differently from the retroviral gene therapy trial, where clonal enrichment of ISs targeting oncogenes like MECOM, CCND2 and LMO2 occurred already at early follow-up, oncogenic ISs were not overrepresented in our patients, in agreement with previous preclinical and clinical studies (27, 34, 51). Many genes and related categories targeted by lentivirus in our WAS patients were also hit in the parallel MLD trial (52) and in a previous ALD lentiviral trial (27) where no clonal dominance or leukemia have been reported up to 5 years after treatment (14). These findings are particularly relevant considering the high risk of lymphomagenesis described in WAS patients. The overall tendency of LV-w1.6W to distribute over megabase-wide areas and a broader spectrum of gene classes, together with the lower transformation potential of our SIN LTR-based construct likely make lentiviral vector less prone to aberrantly interact with proto-oncogenes and induce genotoxicity. Collectively, although a definitive conclusion on safety must await the long-term observations, these data suggest that lentiviral-based HSPC gene therapy is safer than retroviral gene therapy.
Materials and Methods
Clinical protocol and patients
A phase I/II clinical trial of HSPC gene therapy for WAS was initiated on April 2010, after authorization by Istituto Superiore di Sanità on March 15th 2010 and by San Raffaele Scientific Institute Ethics Committee on April 2nd 2010 (Eudract # 2009-017346-32). The study promoter is HSR-TIGET, San Raffaele Hospital, Italy and the financial sponsor of the study is Telethon Foundation. The medicinal product received Orphan Drug Designation (ODD) by the European Medicines Agency (EMA) (EU/3/12/998) and FDA (ODD#10-3043).
Male children with WAS suffering from a severe clinical condition (Zhu clinical score ≥ 3) or severe mutation/absent WASP expression, without a suitable matched donor for allogeneic transplant or ineligible for HSPC transplantation (because of age >5 years or clinical features), were eligible for the study. The parents of all subjects provided written informed consent for experimental treatment. Details on clinical study can be found at ClinicalTrials.gov (# NCT01515462). Biological samples were obtained from WAS patients, healthy children or adults, with approval of the San Raffaele Scientific Institute’s Ethics Committee and consent from parents or subjects.
Lentiviral vector
The lentiviral vector used in this clinical trial is a 3rd generation SIN vector derived from HIV, named pCCLsin.cPPT.hw1.6.hWAS.WPREmut (abbreviated as LV-w1.6W), and it has been previously described (3)(58). This is a pseudotyped vector made by a core of HIV-1 structural proteins and enzymes, the envelope of the Vesicular Stomatitis Virus (VSV) and a genome containing HIV-1 cis-acting sequences, no viral genes and one expression cassette for the WAS transgene. In the cassette, the endogenous 1.6 KB promoter of WAS controls the expression of WAS cDNA. The three vector components (core, envelope and genome) are transiently expressed in vector producer cells by four different constructs: two core packaging constructs, the envelope construct and the transfer vector construct. Only the vector construct is transferred and integrated into the target cells.
Clinical lots of vectors were produced under Good Manufacturing Practice (GMP) conditions by MolMed S.p.a. (Milan, Italy), a certified GMP facility. Vectors were produced by large scale process based on transient quadritransfection in 293T cells followed by purification through endonuclease treatment, anion exchange chromatography with gradient elution, gel filtration, resuspension in serum free media, 0.22 μm filtration and aseptic filling (fig. S1). Each lot was characterized in terms of titer, potency, purity and safety aspects (table S1 and S2).
CD34+ cell gene transfer
Patients’ CD34+ cell manipulation and transduction were performed at MolMed S.p.A as detailed in fig. S2. Patient’s BM was diluted, stratified with Lymphoprep (Axis Shield, Oslo, Norway) and centrifuged in order to collect mononuclear cells (MNC). CD34+ cell positive selection from BM MNC was performed using immunomagnetic beads (CliniMACS, Miltenyi Biotec, Bergisch-Gladbach, Germany) and an immunomagnetic enrichment device. For mobilised PB (MPB) leukapheresis CD34+ cells were processed using the CliniMACS device. Two independent lots of transduced cells were prepared for Pt1 (one for BM and one for MPB). The number of CD34+ cells collected from the BM and used for transduction ranged from 4.4 to 14.5 × 106/kg (table S3). MPB derived CD34+ cells in Pt1 were 6.5 × 106/kg after thawing on day-3 (table S3). Purified CD34+ cells were seeded on VueLife bags (American Fluoroseal Corp., Gaithersburg, MD, USA) at 1 × 106 cells/ml in serum-free CellGro SCGM Medium (Cell Genix Technologies, Freiburg, Germany) in the presence of cells culture grade Stem Cell Factor (SCF) 300 ng/ml (Amgen Inc., Thousand Oaks, CA, USA), FLT3-L 300 ng/ml, Thrombopoietin (TPO) 100 ng/ml and IL-3 60 ng/ml (all from Cell Genix Technologies). Following 24 hour of pre-stimulation, cells were transduced with LV-w1.6W at 100 multiplicity of infection (MOI) for 2 hits of transduction with a wash period of 12–14 hours between the vector exposures. Transduced cells were tested for immune phenotype, sterility, endotoxin, mycoplasma, large T antigen, E1A DNA, Large T antigen DNA, clonogenic content, transduction efficiency, and replication competent lentivirus (table S4). Cells were resuspended in saline and transferred in a syringe for infusion. An aliquot of cells was cultured in IMDM, 10% FBS (Cambrex, East Rutherford, NJ, USA) with the same cytokines at 20 ng/ml concentration and harvested after 15 days to perform proviral integration evaluation by qPCR, WASP expression measurement, and integration profile analysis.
Rationale for conditioning regimen
Allogeneic HSPC transplant for WAS patients requires usually high dose myeloablative and immune suppressive regimen to deplete host bone marrow stem cells and prevent rejection/graft versus host disease. Previous studies in the context of ADA-SCID gene therapy indicated that a reduced dose chemotherapy regimen with busulfan at 25% of standard dose was sufficient to achieve significant engraftment of gene corrected cells while reducing conditioning-related toxicity (9, 15, 16). Since WAS requires higher levels of stem cell correction with respect to SCID, our chemotherapy regimen is based on administration of both busulfan and fludarabine as depleting agents for endogenous HSPC. The dose of busulfan and fludarabine are approximately 50% and 30% of the ones employed in standard allogeneic transplantation. Fludarabine was also used to break the homeostasis in the compartment of early lymphoid progenitors and to favour the establishment of a pool of corrected naïve T cells in the periphery, since the selective advantage for WASP+ cells is thought to take place mainly through peripheral expansion rather than during thymic differentiation (59, 60). Anti-CD20 mAb was introduced as depleting agent for B cells and particularly of autoreactive cells, thereby facilitating the engraftment and expansion of gene corrected B cells expressing WAS. In addition, anti-CD20 was used as pre-emptive treatment for lympoproliferative disorder due to EBV, which represents a high risk factor for the development of lymphoma in WAS patients.
Patient’s treatment
In Pt1, mobilization of peripheral blood stem cells after G-CSF stimulation was performed to collect back up HSPC and store an adequate HSPC dose for subsequent transduction and reinfusion in case of need. In Pt2 and Pt3, due to their younger age, only a BM back up was performed. A central venous catheter was implanted in all patients.
On day-3 autologous BM was collected from iliac crests under general anesthesia. A reduced intensity-conditioning regimen was then administered before reinfusion of the autologous-engineered HSPC. It consisted of iv Busulfan (bodyweight-based and area under the curve (AUC) adjusted-dose) (range: 0.8–1.2 mg/kg/dose), administered consecutively in 8 doses every 6 hours from days −3 to day −1 and iv Fludarabine (30 mg/sqm/day) on days −3 and −2. The actual doses and AUC of Busulfan received by each patient are summarized in table S5. A single dose of anti-CD20 mAb (Rituximab, 375 mg/sqm) was administered on day −22. Transduced autologous LV-w1.6W CD34+ cells, manufactured as indicated above, were infused i.v. in 20 minutes. Patients received anti-bacterial, anti-fungal, anti-Pneumocistis Jirovecii and anti-viral prophylaxes according to local standards. The treatment was administered at the Pediatric Immunology and BM Transplantation Unit and patients were maintained hospitalized in isolation for 40, 70 and 43 days, respectively.
Patients’ features and clinical course after GT
The clinical features before treatment and the current clinical conditions of the first 3 patients treated are summarized in Table 1. Pt1 was a 5.7 year-old boy lacking an HLA-compatible donor, with clinical history of recurrent, frequent ENT and viral infections, skin petechiae and moderate-severe eczema (Zhu score: 3). His clinical course after treatment was uneventful. The second and third patients (Pt2 and Pt3) were younger and had a Zhu score of 4, due to history of severe infections, severe eczema, and GI bleeding. Pt2 was affected by a colitis associated with human herpesvirus 6 (HHV-6) and CMV infection as well as persistently elevated inflammatory indexes and vasculitis-like skin manifestations. Pt3 had important feeding problems, with severe gastroesophageal (GE) reflux and food adversion, and was fed by naso-gastric tube. Pt2 developed an autoimmune thrombocytopenia, which is frequently observed after allogeneic transplant and was treated with intravenous immunoglobulins and anti-CD20 mAb. The patient experienced a Gram negative sepsis complicated by a disseminated Intravascular Coagulation and Acute Respiratory Distress Syndrome (ARDS), for which he received steroids for about 5.5 months. Pt3 clinical course was characterized by respiratory infections related to aspiration due to the underlying GE reflux, including an episode of interstitial aspiration pneumonia, complicated by ARDS, and requiring steroid treatment for about 2 months. Both patients recovered well from these events without sequelae. No immune suppressive treatment was administered to the patients subsequently. Pt1 responded to vaccination with recombinant antigens with production of specific antibodies and in vitro proliferative responses to tetanus toxoid.
Laboratory studies
CFC assay
CFC assay was performed, immediately after transduction or on ex vivo BM samples according to the manufacturer’s procedure in Methocult medium (Stem Cell Technologies, Vancouver, Canada). At day 14, colonies were scored to determine number and type of colonies, singly picked and analysed by qPCR to evaluate the percentage of transduction.
Flow-cytometric analysis
Surface staining of transduced CD34+ cells was performed with anti-CD34 (8G12) and anti-CD45 (HI30) monoclonal antibodies (BD Biosciences, San Jose, CA). Cells from PB of patients and healthy donors, purified by standard density gradient technique (Lymphoprep), were stained for the expression of surface markers CD3 (SK7), CD4 (SK3), CD8 (SK1) CD19 (SJ25C1), CD14 (MΦP9), CD56 (NCAM16.2), CD25 (2A3), CD41 (HIP8) (BD Biosciences) and of intracytoplasmatic expression of WASP. Detection of WASP was performed after permeabilization (Cytofix/Cytoperm kit, BD Biosciences) by two lots of rabbit anti-human WASP polyclonal antibodies generated against WASP peptides (one of them was a kind gift of Dr. H. Ochs) and followed by staining with a secondary Alexa 488 or 647 conjugated goat anti-rabbit antibodies. A rabbit anti-goat Alexa 488 or 647 conjugated tertiary antibodies (all from Invitrogen, Carlsbad, CA, USA) were used. For analyses of TCR repertoire, 24 different TCR Vβ specificities were analysed by flow cytometric analysis by I0Test Beta Mark kit (Immunotech, Marseilles, France) according to the manufacturer’s procedure. Cells were acquired using a FACSCantoII (BD Biosciences) and analysed with Flow Jo Software (Tree Star Inc., Ashland, OR, USA).
Western Blot Analysis
Western Blot was performed as previously described (3, 58), using anti-hWASP (H250; Santa Cruz Biotechnologies, Santa Cruz, CA, USA), anti-hGAPDH (Chemicon, Temecula, CA, USA) antibodies, followed by secondary HRP-coupled antibodies (DAKO A/S, Glostrup, Denmark).
Purification of peripheral blood and bone marrow lineages
CD3, CD4, CD8, CD14, CD15, CD19, CD56 cells were purified from MNC from PB. CD3, CD15, CD19, CD34, CD56, CD61, Glycophorin were purified from BM derived MNCs by positive selection with immunomagnetic beads according to the manufacturer’s procedure (Miltenyi Biotec, Bergisch-Gladbach, Germany).
Determination of Vector Copy Number by Real-time PCR
In order to evaluate the number of lentiviral vector copies integrated per genome, a quantitative PCR was performed using specific primer and probes for human Telomerase and lentiviral vector, as described previously (58). A reference standard was obtained from serially diluted transduced human T-cell lines carrying 1 copy of integrated lentiviral vector. Results of integrated vector copies were normalized for the number of evaluated genomes. As negative control, samples of untransduced cells were used. All the reactions were performed according to the manufacturer’s instructions and analysed with an ABI PRISM 7900 sequence detection system (Applied Biosystem, Foster City, CA).
Vector integration analyses and bioinformatics
These methods are described extensively in the Supplementary material section.
In vitro suppression assays
Suppression assays were performed as previously described (61). Briefly, CD4+CD25highCD127−/low Treg cells and CD4+CD25− effector T cells were isolated from MNC by FACS sorting. Cells from healthy subjects were used as control. 15 × 103 CD4+CD25− effector T cells were stimulated by CD3-depleted APCs and 1 μg/ml of soluble anti-CD3 mAbs (Orthoclone OKT3; Janssen-Cilag). Suppressive activity of nTreg cells was assessed by co-culture of effector T cells with nTreg cells at 1 to1 ratio. Proliferation was evaluated by 16 hours liquid scintillation counting of 3H-thymidine (Amersham Biosciences) incorporation after 96 hours of stimulation. Cpm: counts per minutes.
Analysis of the NK cell immunological synapse
NK cell immunological synapse were analysed as previously described (62). In brief, NK cells were co-cultured with K562 target cells for 30 minutes to permit the formation of conjugates. F-actin and WASP accumulation, as well as perforin and MTOC polarization were measured to determine synapse maturity. The distance of the MTOC from the synapse was measured using the Velocity software package (Improvision-Perkin Elmer, Lexington, MA). Mean distances of the MTOC to the immunological synapse are a measure of synapse maturity and were compared by using Mann-Whitney Rank Sum Test.
Chromium release cytotoxicity assay
The ability of NK cells to lyse MHC devoid target cells was performed using a standard Chromium release cytotoxicity assay (63). Briefly, K562 target cells were labelled with 60 μCi of Na2(51CrO4) (Perkin Elmer Inc., Waltham, MA, USA) per 106 cells for 2hr at 37°C and resuspended at used concentration. Effectors MNC were isolated and co-incubated at serial diluted effectors to target (E:T) ratios of 60:1, 30:1, 15:1, 7.5:1, 3.75:1, in duplicates in a 96 well v-bottom plate (Corning Costar, NY, USA) for 4 hrs at 37°C in the atmosphere of 5% CO2. Radioactivity was measured in the culture supernatants by Canberra Packard Cobra II Auto Gamma Counter (Canberra Packard, Canberra, Canada). Percent specific target cell lysis was calculated as followed: (experimental.cpm-spontaneous.cpm)/(maximal.cpm-spontaneous.cpm)x100. For spontaneous release, targets were cultured with medium alone and for maximum release in 3% (v/v) Triton X-100 (Sigma, St. Louis, MO) in PBS.
T cell-response to anti-CD3
T cell-response to anti-CD3 was performed as previously described (64). Briefly, 0.1 × 106 PBMC were seeded into a round-bottom 96-well plate in IMDM medium containing 5% human AB serum (Lonza, Basel, Switzerland) in presence of coated 1 or 10 μg/ml of anti-CD3 (Orthoclone OKT3, Janssen-Cilag). Proliferation was assessed by 3H-thymidine (Amersham Biosciences) incorporation after 72 or 96 hours of stimulation. Data were expressed as the maximal count per minute (cpm) and stimulation index (SI, calculated as cpm/background cpm). For proliferation assay on untrasformed T cell lines, overnight starved T cells were stained with 2 μM CFSE and 0.1 × 106 cells were cultured in a round-bottom 96-well plate coated with different concentration (from 0.01 to 10 μg/ml) of anti-CD3. Proliferation was assessed after 72 hours evaluating CFSE dilution on a FACSCantoII and data were analysed with Flow Jo Software.
Supplementary Material
Acknowledgments
This work was supported by Fondazione Telethon (TIGET core grant to AA, MGR, LN, EM, and AV), the European Commission (CLINIGENE LSHB-CT2006-018933 to MRG and AA; CELL-PID HEALTH-F5-2010-261387 to AA; GA 222878 PERSIST and ERC, Advanced Grant 249845 TARGETING GENE THERAPY to LN) and Ministero della Salute (Ricerca Finalizzata, 005/RF-2009-1485896 to AA and AV; Progetto Giovani Ricercatori GR-2007-684057 to EM). We thank all medical staff of the HSR-TIGET Pediatric Clinical Research Unit, Pediatric Immunohematology, and Bone Marrow Transplant Unit; R. Fiori, P. Silvani, E. Zoia, A. Mandelli and A. Moscatelli for patient care; L. Dupre for initiating the project and conducting preclinical studies; R. Chiesa and B. Cappelli for initial contributions to the clinical trial; M. Gabaldo for support with project management; M. Bonopane and G. Tomaselli for clinical trial management; P. Massariello and other Molmed staff for patient cell manipulation; H. Ochs and L. Notarangelo for the anti-WASP antibody; G. Royal, L. King and F. Govani, for technical help on 454-pyrosequencing and sequence data management; J. Appleby and A. Chuang for advice and support; and F. Pasinelli for continuous support to this project. We are indebted to the patients and their families for their commitment and endurance. LN is an inventor of several patents on lentiviral vector technology that are owned by the Salk Institute and Cell Genesis and are licensed to Lentigen. LN is entitled to receive royalties from one of these patents (#U.S. Patent No. 6,013,516 with Salk Institute).
References and Notes
- 1.Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. Curr Opin Hematol. 2008;15:30. doi: 10.1097/MOH.0b013e3282f30448. [DOI] [PubMed] [Google Scholar]
- 2.Catucci M, Castiello MC, Pala F, Bosticardo M, Villa A. Autoimmunity in Wiskott-Aldrich Syndrome: An Unsolved Enigma. Front Immunol. 2012;3:209. doi: 10.3389/fimmu.2012.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dupre L, et al. Lentiviral vector-mediated gene transfer in T cells from Wiskott-Aldrich syndrome patients leads to functional correction. Mol Ther. 2004;10:903. doi: 10.1016/j.ymthe.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Bosticardo M, Marangoni F, Aiuti A, Villa A, Roncarolo MG. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood. 2009;113:6288. doi: 10.1182/blood-2008-12-115253. [DOI] [PubMed] [Google Scholar]
- 5.Ochs HD, Filipovich AH, Veys P, Cowan MJ, K N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15:84. doi: 10.1016/j.bbmt.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Ozsahin H, et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008;111:439. doi: 10.1182/blood-2007-03-076679. [DOI] [PubMed] [Google Scholar]
- 7.Moratto D, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an international collaborative study. Blood. 2011;118:1675. doi: 10.1182/blood-2010-11-319376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin CR, et al. Outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome. Bone Marrow Transplant. 2012 Mar 19;47:1428. doi: 10.1038/bmt.2012.31. [DOI] [PubMed] [Google Scholar]
- 9.Aiuti A, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 10.Hacein-Bey-Abina S, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363:355. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaspar HB, et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2011;3:97ra79. doi: 10.1126/scitranslmed.3002715. [DOI] [PubMed] [Google Scholar]
- 12.Boztug K, et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med 363. 2010:1918. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaspar HB, et al. How I treat ADA deficiency. Blood. 2009;114:3524. doi: 10.1182/blood-2009-06-189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seymour LW, Thrasher AJ. Gene therapy matures in the clinic. Nat Biotechnol. 2012 Jul;30:588. doi: 10.1038/nbt.2290. [DOI] [PubMed] [Google Scholar]
- 15.Candotti F, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012 Nov 1;120:3635. doi: 10.1182/blood-2012-02-400937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aiuti A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 17.Howe SJ, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. The Journal of clinical investigation. 2008 Sep;118:3143. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hacein-Bey-Abina S, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 20.Cavazzana-Calvo M, Fischer A, Hacein-Bey-Abina S, Aiuti A. Gene therapy for primary immunodeficiencies: Part 1. Current opinion in immunology. 2012 Oct;24:580. doi: 10.1016/j.coi.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 21.Wang GP, et al. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood. 2010;115:4356. doi: 10.1182/blood-2009-12-257352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deichmann A, et al. Insertion sites in engrafted cells cluster within a limited repertoire of genomic areas after gammaretroviral vector gene therapy. Mol Ther. 2011;19:2031. doi: 10.1038/mt.2011.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biasco L, Baricordi C, Aiuti A. Retroviral Integrations in Gene Therapy Trials. Mol Ther. 2012;20:709. doi: 10.1038/mt.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corrigan-Curay J, et al. Challenges in vector and trial design using retroviral vectors for long-term gene correction in hematopoietic stem cell gene therapy. Mol Ther. 2012;20:1084. doi: 10.1038/mt.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12:301. doi: 10.1038/nrg2985. [DOI] [PubMed] [Google Scholar]
- 26.Montini E, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 27.Cartier N, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 28.Cavazzana-Calvo M, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartholomae C. Assessing the integration profile of lentiviral vectors in gene therapy for X-Adrenoleukodystrophy (abstract) Hum Gene Ther. 2012;23:A3. [Google Scholar]
- 30.Dupre L, et al. Efficacy of gene therapy for Wiskott-Aldrich syndrome using a WAS promoter/cDNA-containing lentiviral vector and nonlethal irradiation. Hum Gene Ther. 2006;17:303. doi: 10.1089/hum.2006.17.303. [DOI] [PubMed] [Google Scholar]
- 31.Charrier S, et al. Lentiviral vectors targeting WASp expression to hematopoietic cells, efficiently transduce and correct cells from WAS patients. Gene Ther. 2007;14:415. doi: 10.1038/sj.gt.3302863. [DOI] [PubMed] [Google Scholar]
- 32.Marangoni F, et al. WASp regulates suppressor activity of human and murine CD4(+)CD25(+)FOXP3(+) natural regulatory T cells. J Exp Med. 2007;204:369. doi: 10.1084/jem.20061334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bosticardo M, et al. Lentiviral-mediated gene therapy leads to improvement of B-cell functionality in a murine model of Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2011;127:1376. doi: 10.1016/j.jaci.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 34.Scaramuzza S, et al. Preclinical Safety and Efficacy of Human CD34(+) Cells Transduced With Lentiviral Vector for the Treatment of Wiskott-Aldrich Syndrome. Mol Ther. 2012 Feb 28; doi: 10.1038/mt.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montini E, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119:964. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Modlich U, et al. Insertional Transformation of Hematopoietic Cells by Self-inactivating Lentiviral and Gammaretroviral Vectors. Mol Ther. 2009 Aug 11; doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marangoni F, et al. Evidence for long-term efficacy and safety of gene therapy for Wiskott-Aldrich syndrome in preclinical models. Mol Ther. 2009;17:1073. doi: 10.1038/mt.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aiuti A, Bacchetta R, Seger R, Villa A, Cavazzana-Calvo M. Gene therapy for primary immunodeficiencies: Part 2. Curr Opin Immunol. 2012;24:585. doi: 10.1016/j.coi.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 39.Zhu Q, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype. Blood. 1997;90:2680. [PubMed] [Google Scholar]
- 40.Maillard MH, et al. The Wiskott-Aldrich syndrome protein is required for the function of CD4(+)CD25(+)Foxp3(+) regulatory T cells. J Exp Med. 2007;204:381. doi: 10.1084/jem.20061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orange JS, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci USA. 2002;99:11351. doi: 10.1073/pnas.162376099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt M, et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat Methods. 2007;4:1051. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- 43.Paruzynski A, et al. Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat Protoc. 2010;5:1379. doi: 10.1038/nprot.2010.87. [DOI] [PubMed] [Google Scholar]
- 44.Chao A. An overview of closed capture-recapture models. J Agric Biol Environ Stat. 2001;6:158. [Google Scholar]
- 45.Biasco L, et al. Integration profile of retroviral vector in gene therapy treated patients is ng to gene expression and chromatin conformation of target cell. EMBO Mol Med. 2011;3:89. doi: 10.1002/emmm.201000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome research. 2007 Aug;17:1186. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cui K, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abel U, et al. Analyzing the number of common integration sites of viral vectors–new methods and computer programs. PLoS One. 2011;6:e24247. doi: 10.1371/journal.pone.0024247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Biffi A, et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332. doi: 10.1182/blood-2010-09-306761. [DOI] [PubMed] [Google Scholar]
- 51.Cattoglio C, et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood. 2007;110:1770. doi: 10.1182/blood-2007-01-068759. [DOI] [PubMed] [Google Scholar]
- 52.Biffi A, et al. Therapeutic Benefit in Metachromatic Leukodystrophy by Lentiviral Hematopoietic Stem Cell Gene Therapy. Science. 2013 doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 53.Westerberg LS, et al. Wiskott-Aldrich syndrome protein (WASP) and N-WASP are critical for peripheral B-cell development and function. Blood. 2012;119:3966. doi: 10.1182/blood-2010-09-308197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bosticardo M, et al. Lentiviral-mediated gene therapy leads to improvement of B-cell functionality in a murine model of Wiskott-Aldrich syndrome. The Journal of allergy and clinical immunology. 2011 Jun;127:1376. doi: 10.1016/j.jaci.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 55.Astrakhan A, et al. Ubiquitous high-level gene expression in hematopoietic lineages provides effective lentiviral gene therapy of murine Wiskott-Aldrich syndrome. Blood. 2012;119:4395. doi: 10.1182/blood-2011-03-340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bystrykh LV, Verovskaya E, Zwart E, Broekhuis M, de Haan G. Counting stem cells: methodological constraints. Nat Methods. 2012;9:567. doi: 10.1038/nmeth.2043. [DOI] [PubMed] [Google Scholar]
- 57.Cattoglio C, et al. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood. 2010;116:5507. doi: 10.1182/blood-2010-05-283523. [DOI] [PubMed] [Google Scholar]
- 58.Scaramuzza S, et al. Preclinical Safety and Efficacy of Human CD34(+) Cells Transduced With Lentiviral Vector for the Treatment of Wiskott-Aldrich Syndrome. Molecular therapy : the journal of the American Society of Gene Therapy. 2012 Feb 28; doi: 10.1038/mt.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamaguchi K, et al. Mixed chimera status of 12 patients with Wiskott-Aldrich syndrome (WAS) after hematopoietic stem cell transplantation: evaluation by flow cytometric analysis of intracellular WAS protein expression. Blood. 2002 Aug 15;100:1208. doi: 10.1182/blood-2002-01-0211. [DOI] [PubMed] [Google Scholar]
- 60.Wada T, et al. Second-site mutation in the Wiskott-Aldrich syndrome (WAS) protein gene causes somatic mosaicism in two WAS siblings. The Journal of clinical investigation. 2003 May;111:1389. doi: 10.1172/JCI15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sauer AV, et al. Alterations in the adenosine metabolism and CD39/CD73 adenosinergic machinery cause loss of Treg cell function and autoimmunity in ADA-deficient SCID. Blood. 2012;119:1428. doi: 10.1182/blood-2011-07-366781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Orange JS, et al. IL-2 induces a WAVE2-dependent pathway for actin reorganization that enables WASP-independent human NK cell function. The Journal of clinical investigation. 2011 Apr;121:1535. doi: 10.1172/JCI44862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heo DS, et al. Evaluation of tetrazolium-based semiautomatic colorimetric assay for measurement of human antitumor cytotoxicity. Cancer Res. 1990;50:3681. [PubMed] [Google Scholar]
- 64.Trifari S, et al. Defective Th1 cytokine gene transcription in CD4+ and CD8+ T cells from Wiskott-Aldrich syndrome patients. J Immunol. 2006;177:7451. doi: 10.4049/jimmunol.177.10.7451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.