

Abstract
In vivo gene manipulation is a cornerstone approach in modern physiology. Cre-Lox technology has been extensively used to delete genes and activate reporters in pancreatic β-cells, bringing new insight into the pathophysiology of diabetes. In all cases, it is important to understand the expression domain of the specific reporter-Cre combination in order to correctly interpret the data. In the case of targeted genes with significant expression and function in the brain, the use of Ins2 promoter driven Cre, commonly known as RIP-Cre, has been shown to confound data interpretation when appropriate controls are not present. The recent article from the Philipson group in Islets provides an important characterization of a new Cre-deleter model, referred to as MIP1-CreER, which employs the mouse Ins1 promoter. This Ins1 promoter, recapitulating the expression pattern of the endogenous Ins1 gene, does not drive significant transgene expression in the brain and therefore is highly specific for deleting genes or turning on reporters in the pancreatic β-cell. This model promises to be widely used in the field of islet biology. Here, I review recent developments in the area of in vivo gene modification and predict areas where such tools will be refined further.
Keywords: Cre-lox recombination, gene editing, islet cells, knockout mice, lineage tracing
The goal of modern medical research is to understand the mechanisms of disease in sufficient detail to intervene therapeutically. A critical component of this mission involves the understanding of the role of specific genes in the context of whole organism physiology. In the case of diabetes, many causal genes or genes that increase an individual's susceptibility to disease in the context of environmental factors are known, from both candidate gene approaches and genome-wide association studies.1 For example, increased risk for common and polygenic type 2 diabetes can be conferred by single nucleotide polymorphisms in at least 50 genes.1 Monogenic forms of diabetes, including the MODY category, have at least 10 known causal genes.1 The study of these gene-deficient patients and their families has provided critical insight into the essential pathways that mediate glucose homeostasis in man.2 However, such ‘knockout humans’ are generally very rare and, with this approach, it is not possible to gain sufficient insight into the effect of specific genes in common type 2 diabetes due to the small effect sizes of each gene and the genetic heterogeneity of the human population. Moreover, because invasive analysis and tissue-specific manipulations are virtually impossible in humans, a robust understanding of the physiological role of key genes requires the use of animal models. From a mechanistic standpoint, studies in genetically engineered rodents are more powerful than correlative and non-invasive human studies. While human studies are important in diabetes research, genetic manipulation of β-cells in small animal models, like mice, is required to understand mammalian glucose homeostasis and diabetes pathogenesis.
Many approaches have been taken to modulate gene expression in pancreatic islet cells, with the ultimate goal of understanding the control of glucose homeostasis. There are critical caveats and assumptions inherent to each approach. In the case of global germline knockout mice, gene deletion can be absolute (depending on the targeting strategy) and these models best mimic monogenic human diseases. However, the use of global knockout mice to identify specific tissues and developmental stages involved in a genetic pathway comes with clear caveats, given the interconnectivity across tissues. For human mutations that result in gain-of-function or change-of-function, germline knock-in mice are suitable models. Again, these models do not offer temporal or spatial control of gene function that is required to understand the relative roles of specific tissues or cell types.
In order to understand the specific function of a gene in a specific cell type, conditional gene manipulation must be used. This typically involves the use of a two-component system, involving a line of mice wherein the target gene has been modified to incorporate loxP sites and a second line of mice where Cre-recombinase is expressed in the domain of a tissue-specific gene promoter3,4 Complete knowledge of the domain within which Cre is expressed is critical for the proper interpretation of a Cre-Lox experiment. Magnuson has recently published a comprehensive review of Cre lines available for pancreas research.5 Here I will focus on the utility and caveats around β-cell models. Extensive research has shown that many widely used Cre mouse lines are not as tissue-specific as originally thought.5-7 In the field of pancreatic β-cell physiology, the first extensively used Cre driver line was the so-called RIP-Cre mouse,8 where RIP denotes short (∼0.6 kb) versions of the rat Ins2 promoter.5 Versions of this mouse line were produced by multiple laboratories (an up-to-date list has recently been published5). There have been reports of Cre toxicity with this model,9,10 but other groups have not reported similar observations,11 suggesting the possibility that such effects are strain- and/or environment-dependent. Care and adequate controls are essential in this realm of research. The rationale for Cre-only controls, to complement floxed controls and fully wildtype controls has recently been reviewed.12 Despite these caveats, these Cre-Lox models remain viable tools for stable tissue-selective gene deletion or reporter activation in cells with Ins2 promoter activity. In pancreatic β-cells, Ins2 promoters are active beginning at ∼E9.5, meaning that gene modulation encompasses a majority of developmental stages and the entire adult lifespan.
Time-controlled gene deletion in β-cells can be achieved with variants of this model wherein the Cre activity can be induced with a small molecule, usually the estrogen-related drug tamoxifen. These mice include the RIP-CreER strains that take advantage of the rat Ins2 promoter and Pdx1CreER strains (made by several groups) that utilize a fragment of the Pdx1 promoter that expresses in adult β-cells. It should be noted the widely used Pdx1CreER mouse generated by the Melton group was not reviewed in the article examining Cre-mediated excision in the brain,6 and several groups including ours have found an absence of broad hypothalamic excision using this model.13 An Ins2 Cre-ERT knock-in has recently been created and published.14 These systems work by using a Cre fusion protein that contains a modified version of the estrogen receptor that, in the absence of tamoxifen, prevents the Cre from entering the nucleus to cleave the loxP tagged DNA. Some technical challenges of this approach include spontaneous recombination in the absence of tamoxifen in older mice.15 Caveats associated with these complications can be circumvented by performing studies in younger mice and by employing multiple controls that take into account possible effects of Cre alone, tamoxifen alone, and spontaneous recombination. Another caveat that has been reported is the persistence of significant recombination, up to 2 weeks after the last high-dose tamoxifen pulse.16 Presumably, this can be alleviated by using lower doses and more sensitive ER systems.
Figure 1.
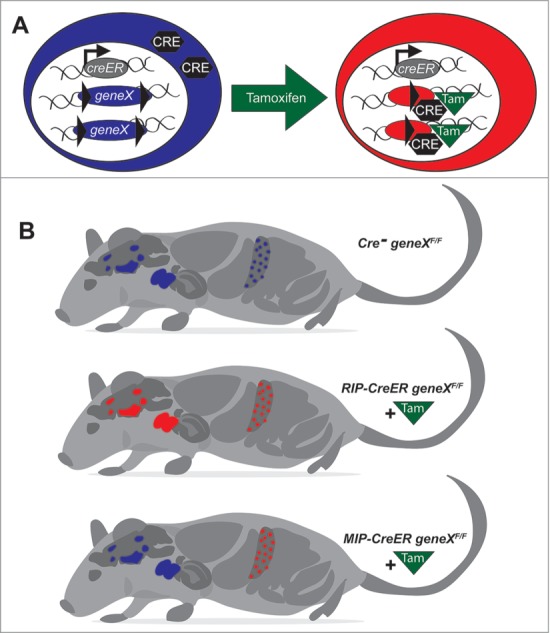
Schematic of Cre-LoxP mediated gene targeting in pancreatic β-cells and its relationship to the endogenous expression patterns of Insulin 1 and Insulin 2 in the mouse. In mice, Insulin 1 is present primarily in the pancreatic β-cells, whereas the ancestral Insulin 2 gene is expressed in the pancreatic β-cells, the thymus, and specific cell populations in the brain. Using a tamoxifen-sensitive system that causes Cre translocation to the nucleus (A), transgenic mice expressing Cre recombinase under the control of the mouse Ins1 promoter can selectively excise DNA sequences between LoxP sites in the pancreatic β-cells, without significant recombination in the brain (B).
Several versions of the tamoxifen-inducible Cre have been published in the broader field, including a fusion protein containing two modified ER (MER) domains that reportedly exhibits fewer spontaneous recombination events.5 Another version, called the ERT2 system, employs a triple mutant version of the estrogen receptor that exhibits virtually no binding to endogenous estrogen at physiological concentrations, and therefore greater sensitivity and specificity for artificial estrogen receptor ligands such as tamoxifen.17 The ERT2 system should theoretically improve upon current models, although a complete set of controls should still be employed.
Rational Choice of Insulin Promoter or Knock-in Location for -Cell Targeting: Lessons from Insulin Biology
Understanding the consequences and complexities of transgenes driven by insulin promoters and alterations in the insulin locus requires some understanding of the biology of the insulin genes in mice. Insulins are ancient, conserved genes.18 Worms have 40 insulin-like genes expressed mostly in neurons.19 Flies have 7 insulin-like peptides, also in neurons, with diverse roles in growth and metabolism.20 There are 8 genes related to insulin in humans: an INSULIN gene, 2 IGFs, as well as insulin-like peptides and relaxins; they are expressed in a plethora of tissues.21 Mice and rats are unique in that the have 2 insulin genes, a second gene (Ins1) arising from a duplication event.22 The Ins2 gene is ancestral, equates to the INSULIN gene found in humans and is better characterized.22 Islets express almost twice as much Ins2 mRNA when compared to Ins1.22 It is probably the relative strength of the Ins2 promoter that led most investigators to employ Ins2 promoter constructs in transgenic mice.5 Insulin is expressed, albeit at much lower levels, in several tissues outside the pancreas. Specifically, Ins2/INSULIN is expressed in the pancreas, the thymus where it controls immune tolerance, and in certain regions of the brain where its function is unknown.23-33 We found clear, albeit modest, expression of Ins2 mRNA, but not Ins1 mRNA, in multiple brain regions of wildtype mice.23 Importantly, neither Ins2 mRNA expression nor insulin protein, were found in Ins2−/− mice, effectively controlling for qPCR or staining artifacts.23 We and others have described how mice with βGal knocked into the endogenous Ins2 locus, originally created in the 1990s, show clear expression in multiple regions of the brain.23 The recently published Ins2 Cre-ERT knock-in mouse14 would also be expected to produce Cre in the brain, as demonstrated by previous Ins2 lacZ knock-in and Ins2 diphtheria toxin knock-in mice.23,26,34 Thus, Cre expression in the brain while under the control of the Ins2 promoter6 or endogenous Ins2 gene elements reflects to at least some extent the endogenous expression domain of Ins2 in the brain,23 and is not necessarily entirely due to a poorly defined process of ‘leakiness’. The expression of Cre in the brain does not necessarily mean that a study is fatally flawed, just that complete controls are required, including controls for the effects of Cre alone, as well as detailed study of processes under central control. In some cases, the gene of interest will not be expressed or relevant in the Ins2 expressing central neurons. Conversely, the analysis of the endogenous expression domain of Ins1 in the adult mouse, which does not include significant expression in the intact brain, also explains why Ins1-based approaches are inherently more specific to the pancreas. In the accompanying paper,35 the authors report the characterization of a new Cre-deleter mouse model employing a large Ins1 promoter fragment that directs Cre expression to the pancreatic β-cells, but not the brain.6
Utilizing the Mouse Ins1 Promoter to Mitigate Brain Cre Expression
The need for a more β-cell specific Cre deleter model prompted the Philipson group to design the MIP1-CreERT mouse model using an 8.3 kb fragment of the Ins1 gene promoter, as described in the Tamarina publication.35 A long Ins1 promoter was also previously used to make the well-characterized MIP-GFP mice,36-38 as well as other mice designed for PET imaging and bioluminescence imaging.39 In this new publication, the authors used qRT-PCR to demonstrate that MIP1 does not drive creERT mRNA expression in a panel of non-pancreatic tissues. This complements previous studies demonstrating that this model does not drive significant Cre expression in the central nervous system (Fig. 1).6 Although a tiny number of GFP-positive can apparently be FACS separated the hypothalamus of MIP-GFP mice, they do not express Ins1 or Ins2 once immortalized and it is not clear what features of the resulting cells lines are recapitulated in vivo.40 Given previous reports of extra-pancreatic GFP expression in stressed MIP-GFP mice,41 a thorough, cell-level, immunohistochemical analysis of Cre and reporter gene expression (e.g., β-Gal) in all tissues will be required to confirm its absolute specificity to the islet β-cells.
The authors provide evidence that the MIP1-CreERT model is physiologically normal. Specifically, no significant effects of Cre induction with tamoxifen were found on glucose homeostasis in either male or female mice under the conditions tested.35 Intracellular Ca2+ responses of isolated whole islets to glucose, a robust index of β-cell function and health,42 also appeared normal in this model. Islet architecture was grossly normal,35 although morphometric counting of β-cells and α-cells and analysis at more ages and under stress conditions may be required to fully eliminate an effects of Cre to β-cell health. Moreover, it will be important in future studies to assess the β-cell function of this model under diabetic conditions.
The work by Tamarina et al. demonstrates for the first time that a target gene, in this case β-catenin, can be robustly down-regulated using the MIP1-CreERT deleter mouse line,35 although MIP-Cre has previously been used to activate a target gene in β-cells.43 It is expected that there will be some caveats with this model, as there always are with any model. For example, the expression level of Cre might be expected to be slightly less than in Ins2-driven models, given the relative weakness of the Ins1 promoter. Subsequent studies were unable to show complete protein ablation of GLP-1 receptors using this MIP1-CreERT deleter mouse,44 which impacts the interpretation of those studies. We and others have shown that some Ins1 promoter constructs may only be robustly activated in a subset of mature β-cells.45,46 While this caveat apparently did not come into play in the Tamarina studies,35 less than complete gene ablation is something that groups using this model should carefully examine using qPCR and imaging of a lineage-tracing reporter in littermate controls.
Future Directions and Perspectives
The field of genetic engineering in pancreatic β-cells is clearly entering an exciting new phase, with a range of available and improved models. It will be a challenge for the field to keep track of each model, in part, due to the shifting nomenclature. Ideally, the field would move to a nomenclature used in publications where each of the strains has a unique identifier. For example, Cre-deleter mice could have information on the promoters used built into their name (e.g., mIns1−xxx-xxxbp CreERT).
The field looks forward to additional Cre-deleter lines for pancreatic β-cell research. In addition to transgenic approaches, which can come with caveats related to insertion sites, groups are employing alternative approaches including bacterial artificial chromosomes, as well as knock-in/gene editing. A very recent article reports on the creation of an Ins1-based Cre driver using bacterial artificial chromosome transgenesis.47 Multiple efforts under way to produce additional Cre and CreERT2 knock-in alleles that replace Ins1 in the endogenous insulin gene locus. Such mice would have the advantage of near ideal specificity for mature β-cells. While such Ins1 knock-in mice would necessarily have only 3 of 4 insulin alleles, we and others have shown that mice have normal glucose homeostasis even when lacking 2 insulin alleles.23 It should be noted that the Ins1 promoter is generally considered to be slightly weaker than the Ins2 promoter. However, with the sensitivity of the Cre-lox system, one might not expect this to be a serious problem. It is also expected that investigators may begin to use multiple site-specific recombinases, such as Flp and Dre,5 which may permit double lineage tracing and other complex experimental designs, once suitable target alleles become available. Expanded use of a hybrid approach, where Cre or similar recombinases are delivered using viruses to mice with conditional alleles, is also expected. Indeed, studies have shown that adeno-associated viruses, such as AAV8 and AAV6, are particularly effective at delivering genes to islets in vivo.48-51 This approach obviates the need for tamoxifen, which may be less than ideal in some experiments.
In summary, the models described herein are forming an increasingly varied and powerful toolkit that will allow investigators to tailor studies to test highly complex and specific hypotheses. The combined use of multiple models to answer a given question will give the field increased confidence in the underlying biology. The rapid increase in new mouse models for β-cell genetic engineering will provide new insights into β-cell biology and accelerate diabetes research.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The author thanks Tim Kieffer, Jake Kushner, Ernesto Bernal-Mizrachi, and Dan Luciani for helpful discussions, and Michael Bround for the technical illustration.
Funding
Related work in the Johnson laboratory is supported by grants from the JDRF and the Stem Cell Network.
References
- 1. Groop L,, Pociot F. Genetics of diabetes—Are we missing the genes or the disease? Mol Cell Endocrinol 2014; 382:726-39; PMID: 23587769; http://dx.doi.org/10.1016/j.mce.2013.04.002 [DOI] [PubMed] [Google Scholar]
- 2. Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011; 34:1878-84; PMID: 21788644; http://dx.doi.org/10.2337/dc11-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis 2000; 26:99-109; PMID: 10686599; http://dx.doi.org/10.1002/(SICI)1526-968X(200002)26:2<99::AID-GENE1>3.0.CO;2-B [PubMed] [Google Scholar]
- 4. Orban PC, Chui D, Marth JD. Tissue- and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci USA 1992; 89:6861-5; PMID: 1495975; http://dx.doi.org/10.1073/pnas.89.15.6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Magnuson MA, Osipovich AB. Pancreas-specific Cre driver lines and considerations for their prudent use. Cell Metab 2013; 18:9-20; PMID: 23823474; http://dx.doi.org/10.1016/j.cmet.2013.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, et al. Conditional gene targeting in mouse pancreatic Beta-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 2010; 59:3090-8; PMID: 20802254; http://dx.doi.org/10.2337/db10-0624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Song J, Xu Y, Hu X, Choi B, Tong Q.. Brain expression of Cre recombinase driven by pancreas-specific promoters. Genesis 2010; 48:628-34; PMID: 20824628; http://dx.doi.org/10.1002/dvg.20672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999; 96:329-39; PMID: 10025399; http://dx.doi.org/10.1016/S0092-8674(00)80546-2 [DOI] [PubMed] [Google Scholar]
- 9. Magnuson MA, Burlison JS. Caveats and considerations for performing pancreas-specific gene manipulations in the mouse. Diabetes Obes Metab 2007; 9(Suppl 2):5-13; PMID: 17919173; http://dx.doi.org/10.1111/j.1463-1326.2007.00771.x [DOI] [PubMed] [Google Scholar]
- 10. Lee JY, Ristow M, Lin X, White MF, Magnuson MA, Hennighausen L. RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J Biol Chem 2006; 281:2649-53; PMID: 16326700; http://dx.doi.org/10.1074/jbc.M512373200 [DOI] [PubMed] [Google Scholar]
- 11. Fex M, Wierup N, Nitert MD, Ristow M, Mulder H. Rat insulin promoter 2-Cre recombinase mice bred onto a pure C57BL6J background exhibit unaltered glucose tolerance. J Endocrinol 2007; 194:551-5; PMID: 17761894; http://dx.doi.org/10.1677/JOE-07-0161 [DOI] [PubMed] [Google Scholar]
- 12. Harno E, Cottrell EC, White A. Metabolic pitfalls of CNS Cre-based technology. Cell Metab 2013; 18:21-8; PMID: 23823475; http://dx.doi.org/10.1016/j.cmet.2013.05.019 [DOI] [PubMed] [Google Scholar]
- 13. Luciani DS, White SA, Widenmaier SB, Saran VV, Taghizadeh F, Hu X, Allard MF, Johnson JD. Bcl-2 and Bcl-xL suppress glucose signaling in pancreatic beta-cells. Diabetes 2013; 62:170-82; PMID: 22933114; http://dx.doi.org/10.2337/db11-1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura K, Minami K, Tamura K, Iemoto K, Miki T, Seino S. Pancreatic beta-cells are generated by neogenesis from non-beta-cells after birth. Biomed Res 2011; 32:167-74; http://dx.doi.org/10.2220/biomedres.32.167 [DOI] [PubMed] [Google Scholar]
- 15. Liu Y, Suckale J, Masjkur J, Magro MG, Steffen A, Anastassiadis K, Solimena M. Tamoxifen-independent recombination in the RIP-CreER mouse. PLoS One 2010; 5:e13533; PMID: 21063464; http://dx.doi.org/10.1371/journal.pone.0013533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reinert RB, Kantz J, Misfeldt AA, Poffenberger G, Gannon M, Brissova M, Powers AC. Tamoxifen-induced Cre-loxP recombination is prolonged in pancreatic islets of adult mice. PLoS One 2012; 7:e33529; PMID: 22470452; http://dx.doi.org/10.1371/journal.pone.0033529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun 1997; 237:752-7; PMID: 9299439; http://dx.doi.org/10.1006/bbrc.1997.7124 [DOI] [PubMed] [Google Scholar]
- 18. Ferrannini E, Galvan AQ, Gastaldelli A, Camastra S, Sironi AM, Toschi E, Baldi S, Frascerra S, Monzani F, Antonelli A, et al. Insulin: new roles for an ancient hormone. Eur J Clin Invest 1999; 29:842-52; http://dx.doi.org/10.1046/j.1365-2362.1999.00536.x [DOI] [PubMed] [Google Scholar]
- 19. Narasimhan SD, Yen K, Tissenbaum HA. Converging pathways in lifespan regulation. Curr Biol 2009; 19:R657-66; PMID: 19674551; http://dx.doi.org/10.1016/j.cub.2009.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Broughton S, Partridge L. InsulinIGF-like signalling, the central nervous system and aging. Biochem J 2009; 418:1-12; PMID: 19159343; http://dx.doi.org/10.1042/BJ20082102 [DOI] [PubMed] [Google Scholar]
- 21. Lok S, Johnston DS, Conklin D, Lofton-Day CE, Adams RL, Jelmberg AC, Whitmore TE, Schrader S, Griswold MD, Jaspers SR. Identification of INSL6, a new member of the insulin family that is expressed in the testis of the human and rat. Biol Reprod 2000; 62:1593-9; PMID: 10819760; http://dx.doi.org/10.1095/biolreprod62.6.1593 [DOI] [PubMed] [Google Scholar]
- 22. Hay CW, Docherty K. Comparative analysis of insulin gene promoters: implications for diabetes research. Diabetes 2006; 55:3201-13; PMID: 17130462; http://dx.doi.org/10.2337/db06-0788 [DOI] [PubMed] [Google Scholar]
- 23. Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, Botezelli JD, Asadi A, Hoffman BG, Kieffer TJ, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metabolism 2012; 16:723-37; PMID: 23217255; http://dx.doi.org/10.1016/j.cmet.2012.10.019 [DOI] [PubMed] [Google Scholar]
- 24. Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J 2009; 28(18):2812-24; http://dx.doi.org/10.1038/emboj.2009.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giddings SJ, Chirgwin J, Permutt MA. Evaluation of rat insulin messenger RNA in pancreatic and extrapancreatic tissues. Diabetologia 1985; 28:343-7; PMID: 2412922; http://dx.doi.org/10.1007/BF00283141 [DOI] [PubMed] [Google Scholar]
- 26. Lamotte L, Jackerott M, Bucchini D, Jami J, Joshi RL, Deltour L. Knock-in of diphteria toxin A chain gene at Ins2 locus: effects on islet development and localization of Ins2 expression in the brain. Transgenic Res 2004; 13:463-73; PMID: 15587270; http://dx.doi.org/10.1007/s11248-004-9587-x [DOI] [PubMed] [Google Scholar]
- 27. Deltour L, Leduque P, Blume N, Madsen O, Dubois P, Jami J, Bucchini D. Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proc Natl Acad Sci USA 1993; 90:527-31; PMID: 8421685; http://dx.doi.org/10.1073/pnas.90.2.527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005; 435:220-3; PMID: 15889095; http://dx.doi.org/10.1038/nature03523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pugliese A, Zeller M, Fernandez A Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet 1997; 15:293-7; PMID: 9054945; http://dx.doi.org/10.1038/ng0397-293 [DOI] [PubMed] [Google Scholar]
- 30. Devaskar SU, Singh BS, Carnaghi LR, Rajakumar PA, Giddings SJ. Insulin II gene expression in rat central nervous system. Regul Pept 1993; 48:55-63; PMID: 8265817; http://dx.doi.org/10.1016/0167-0115(93)90335-6 [DOI] [PubMed] [Google Scholar]
- 31. Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm 1998; 105:423-38; PMID: 9720972; http://dx.doi.org/10.1007/s007020050068 [DOI] [PubMed] [Google Scholar]
- 32. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease–is this type 3 diabetes? J Alzheimers Dis 2005; 7:63-80; PMID: 15750215. [DOI] [PubMed] [Google Scholar]
- 33. Hrytsenko O, Wright JR Jr, Morrison CM, Pohajdak B. Insulin expression in the brain and pituitary cells of tilapia (Oreochromis niloticus). Brain Res 2007; 1135:31-40; PMID: 17196948; http://dx.doi.org/10.1016/j.brainres.2006.12.009 [DOI] [PubMed] [Google Scholar]
- 34. Duvillie B, Cordonnier N, Deltour L, Dandoy-Dron F, Itier JM, Monthioux E, Jami J, Joshi RL, Bucchini D. Phenotypic alterations in insulin-deficient mutant mice. Proc Natl Acad Sci USA 1997; 94:5137-40; PMID: 9144203; http://dx.doi.org/10.1073/pnas.94.10.5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tamarina NA, Roe MW, Philipson L. Characterization of mice expressing Ins1 gene promoter driven CreERT recombinase for conditional gene deletion in pancreatic beta-cells. Islets 2014; 6; http://dx.doi.org/10.4161/isl.27685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hara M, Wang X, Kawamura T, Bindokas VP, Dizon RF, Alcoser SY, Magnuson MA, Bell GI. Transgenic mice with green fluorescent protein-labeled pancreatic beta-cells. Am J Physiol Endocrinol Metab 2003; 284:E177-83; PMID: 12388130 [DOI] [PubMed] [Google Scholar]
- 37. Huang YC, Gaisano HY, Leung YM. Electrophysiological identification of mouse islet alpha-cells: from isolated single alpha-cells to in situ assessment within pancreas slices. Islets 2011; 3:139-43; PMID: 21623173; http://dx.doi.org/10.4161/isl.3.4.16166 [DOI] [PubMed] [Google Scholar]
- 38. Yang YH, Vilin YY, Roberge M, Kurata HT, Johnson JD. Multiparameter screening reveals a role for Na(+) channels in cytokine-induced beta-cell death. Mol Endocrinol 2014; 28:406-17; PMID: 24438339; http://dx.doi.org/10.1210/me.2013-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McGirr R, Hu S, Yee SP, Kovacs MS, Lee TY, Dhanvantari S. Towards PET imaging of intact pancreatic beta cell mass: a transgenic strategy. Mol Imaging Biol 2011; 13:962-72; PMID: 20924688; http://dx.doi.org/10.1007/s11307-010-0435-5 [DOI] [PubMed] [Google Scholar]
- 40. Wang ZC, Wheeler MB, Belsham DD. Isolation and immortalization of MIP-GFP neurons from the hypothalamus. Endocrinology 2014; 155:2314-9; PMID: 24617526; http://dx.doi.org/10.1210/en.2013-2128 [DOI] [PubMed] [Google Scholar]
- 41. Kojima H, Fujimiya M, Matsumura K, Nakahara T, Hara M, Chan L. Extrapancreatic insulin-producing cells in multiple organs in diabetes. Proc Natl Acad Sci USA 2004; 101:2458-63; PMID: 14983031; http://dx.doi.org/10.1073/pnas.0308690100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luciani DS, Misler S, Polonsky KS. Ca2+ controls slow NAD(P)H oscillations in glucose-stimulated mouse pancreatic islets. J Physiol 2006; 572:379-92; PMID: 16455690; http://dx.doi.org/10.1113/jphysiol.2005.101766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kaihara KA, Dickson LM, Jacobson DA, Tamarina N, Roe MW, Philipson LH, Wicksteed B.. beta-Cell-specific protein kinase A activation enhances the efficiency of glucose control by increasing acute-phase insulin secretion. Diabetes 2013; 62:1527-36; PMID: 23349500; http://dx.doi.org/10.2337/db12-1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith EP, An Z, Wagner C, Lewis AG, Cohen EB, Li B, Mahbod P, Sandoval D, Perez-Tilve D, Tamarina N, et al. The role of beta cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metab 2014; 19:1050-7; PMID: 24836562; http://dx.doi.org/10.1016/j.cmet.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Szabat M, Luciani DS, Piret JM, Johnson JD. Maturation of adult beta-cells revealed using a Pdx1insulin dual-reporter lentivirus. Endocrinology 2009; 150:1627-35; PMID: 19095744; http://dx.doi.org/10.1210/en.2008-1224 [DOI] [PubMed] [Google Scholar]
- 46. Szabat M, Pourghaderi P, Soukhatcheva G, Verchere CB, Warnock GL, Piret JM, Johnson JD. Kinetics and genomic profiling of adult human and mouse beta-cell maturation. Islets 2011; 3:175-87; PMID: 21633187; http://dx.doi.org/10.4161/isl.3.4.15881 [DOI] [PubMed] [Google Scholar]
- 47. Hasegawa Y, Daitoku Y, Mizuno S, Tanimoto Y, Mizuno-Iijima S, Matsuo M, Kajiwara N, Ema M, Oishi H, Miwa Y, et al. Generation and characterization of Ins1-cre-driver C57BL6N for exclusive pancreatic beta cell-specific Cre-loxP recombination. Exp Anim 2014; 63:183-91; PMID: 24770644; http://dx.doi.org/10.1538/expanim.63.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riedel MJ, Gaddy DF, Asadi A, Robbins PD, Kieffer TJ. DsAAV8-mediated expression of glucagon-like peptide-1 in pancreatic beta-cells ameliorates streptozotocin-induced diabetes. Gene Ther 2010; 17:171-80; PMID: 19865180; http://dx.doi.org/10.1038/gt.2009.143 [DOI] [PubMed] [Google Scholar]
- 49. Rehman KK, Trucco M, Wang Z, Xiao X, Robbins PD. AAV8-mediated gene transfer of interleukin-4 to endogenous beta-cells prevents the onset of diabetes in NOD mice. Mol Ther 2008; 16:1409-16; PMID: 18560422; http://dx.doi.org/10.1038/mt.2008.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prasad KM, Yang Z, Bleich D, Nadler JL. Adeno-associated virus vector mediated gene transfer to pancreatic beta cells. Gene Ther 2000; 7:1553-61; PMID: 11021593; http://dx.doi.org/10.1038/sj.gt.3301279 [DOI] [PubMed] [Google Scholar]
- 51. Montane J, Bischoff L, Soukhatcheva G, Dai DL, Hardenberg G, Levings MK, Orban PC, Kieffer TJ, Tan R, Verchere CB. Prevention of murine autoimmune diabetes by CCL22-mediated Treg recruitment to the pancreatic islets. J Clin Invest 2011; 121, 3024-8; http://dx.doi.org/10.1172/JCI43048 [DOI] [PMC free article] [PubMed] [Google Scholar]