

Abstract
Stress and pre-frontal cognitive dysfunction have key roles in driving smoking, however, there are no therapeutics for smoking cessation which attenuate the effects of stress on smoking and enhance cognition. Central noradrenergic pathways are involved in stress-induced reinstatement to nicotine and in the prefrontal executive control of adaptive behaviors. We used a novel translational approach employing a validated laboratory analogue of stress-precipitated smoking, fMRI, and a proof-of-concept treatment period to evaluate whether the noradrenergic α2a agonist, guanfacine (3mg/day) versus placebo (0mg/day) reduced stress-precipitated smoking in the laboratory, altered cortico-striatal activation during the Stroop cognitive-control task, and reduced smoking following a quit attempt. In nicotine-deprived smokers (n=33), stress versus a neutral condition significantly decreased the latency to smoke, and increased tobacco craving, ad-libitum smoking, and systolic blood pressure in placebo-treated subjects, and these effects were absent or reduced in guanfacine-treated subjects. Following stress, placebo-treated subjects demonstrated decreased cortisol levels whereas guanfacine-treated subjects demonstrated increased levels. Guanfacine, compared to placebo, altered prefrontal activity during a cognitive control task, and reduced cigarette use but did not increase complete abstinence during treatment. These preliminary laboratory, neuroimaging and clinical outcome data were consistent and complementary and support further development of guanfacine for smoking cessation.
Keywords: guanfacine, smoking cessation, stress, fMRI, Stroop, lapse, ad-lib smoking, craving
INTRODUCTION
Tobacco use is a leading cause of preventable morbidity and mortality worldwide, contributing to nearly 6 million deaths yearly (World Health Organization, 2014). All current FDA-approved smoking cessation medications target the nicotinic acetylcholine receptor system to some extent and attenuate nicotine-related reinforcement and withdrawal symptoms (De Biasi and Dani, 2011). However, most smokers using nicotinic acetylcholinergic agents fail to maintain long-term abstinence (Fiore et al, 2008), underscoring the need to identify novel compounds. Factors that maintain smoking and precipitate relapse are varied and complex, and the underlying biology has yet to be elucidated (Lester, 2011). Stress is a primary mechanism involved in the maintenance of and relapse to smoking (McKee et al, 2003), and targeting stress-related relapse for medications development is a critical, yet relatively unexplored strategy for smoking cessation.
Nicotine potently activates cortico-striatal-limbic pathways (Stein et al, 1998) and significant neuro-adaptations in stress responses with chronic nicotine exposure and withdrawal have been documented. Smokers demonstrate blunted cortisol and adrenocorticotropic hormone (ACTH) responses to stress during acute abstinence (al’Absi et al, 2005; McKee et al, 2011) and in response to nicotine (Mendelson et al, 2008). Stress-related alterations during acute abstinence are accompanied by increased tobacco craving and reduced control over smoking behavior (McKee et al, 2011). As attention and self-control are critical executive functions required to regulate stress arousal and craving states (Sinha, 2008), treatments that target reducing stress reactivity and improve self-control may optimize smoking cessation outcomes. Early abstinent smokers show reduced responses in anterior cingulate cortex (ACC) and prefrontal cortex (PFC) with functional magnetic resonance imaging (fMRI) during executive function and self-control tasks, while nicotine and nicotine cues increase such activations (Kober et al, 2010; Stein et al, 1998). Decrements in executive function and self-control in smokers may contribute to increased stress-induced tobacco craving and poor regulation of stress in smokers.
Noradrenergic transmission is involved in both stress-reactivity and PFC control of cognitive function. In animal models, increasing central noradrenergic activity pharmacologically or via foot-shock stress enhances reinstatement to drugs following extinction (Shaham et al, 2003), whereas reducing noradrenergic activity with α2-adrenergic agonists attenuates stress-induced relapse to drugs (Lê et al, 2005), including stress-induced reinstatement of nicotine-seeking behavior (Yamada and Bruijnzeel, 2011). Stress exposure impairs PFC functions in both animals and humans (Arnsten, 2009), and reduced PFC-based self-control may be one mechanism by which stress induces relapse to drug-seeking (Sinha 2008), including nicotine. Stress induces high levels of cyclic AMP (cAMP) that open potassium channels on dendritic spines near PFC network synapses, weakening PFC connections underlying working memory and behavioral inhibition (Arnsten, 2009). Conversely, the α2A-adrenergic agonist, guanfacine, inhibits cAMP production, which closes potassium channels, strengthens PFC network connections, increases PFC neuronal firing and improves PFC regulation of behavior (Arnsten, 2010). Targeting stress-related decrements in PFC function with guanfacine may improve self-control during stress and decrease stress-precipitated smoking relapse.
Guanfacine also improves executive functioning in non-stressed states. In monkeys, systemic guanfacine administration helped inhibit impulsive choices and wait for larger rewards, an important operation in achieving drug abstinence (Kim et al, 2012). Guanfacine also improved executive function deficits in adults with Attention-Deficit Hyperactivity Disorder (ADHD) (Taylor and Russo, 2001) and working memory deficits in adults and in those with mild traumatic brain injury (McAllister et al, 2011). The extended release formulation of guanfacine is FDA-approved for the treatment of ADHD. As PFC-based executive dysfunction exists in addictions (Everitt et al, 2008), strengthening PFC-based executive function more generally may also serve to improve smoking cessation outcomes.
In addition to attenuating stress reactivity and improving cognitive function, there is extensive evidence that noradrenergic function is also involved in the rewarding effects of drugs (Weinshenker et al, 2007) and drug withdrawal (Semenova and Markou, 2010). Noradrenergic agents decrease nicotine-evoked dopamine release in the nucleus accumbens (Forget et al, 2010; Villegier et al, 2007), reduce conditioned place preference to nicotine (Forget et al, 2009), attenuate nicotine withdrawal induced deficits in brain reward thresholds (Bruijnzeel et al, 2010), and reduce somatic signs of nicotine withdrawal in animals and humans (Bruijnzeel et al, 2010; Gourlay et al, 2004). Thus, there exist multiple possible mechanisms through which noradrenergic drugs may operate to influence human tobacco smoking.
The current study primarily tests whether guanfacine reduces stress-precipitated smoking and improves self-control in human subjects, while also investigating effects on PFC during a cognitive control task. Guanfacine, approved in 1986 to treat hypertension, is an α2 adrenergic agonist that is known to preferentially bind to the α2A subtype of norepinephrine receptors, which are highly concentrated in the prefrontal cortical regions. Non-selective α2-adrenergic agonists such as clonidine have demonstrated efficacy for smoking cessation (Gourlay et al, 2004), but are limited by possible orthostatic adverse events and sedation. Guanfacine is more selective for the α2A-adrenoceptor subtype (Uhlen and Wikberg, 1991), is less sedating (Arnsten et al, 1988), has fewer pre-synaptic actions (Engberg, 1991), and has a longer half-life compared to clonidine (PDR, 1994), potentially improving its clinical utility.
This study used a well-validated model of smoking-lapse which evaluates the ability to resist smoking following stress and neutral conditions (McKee et al, 2011). This task is similar to a delay discounting task in that subjects are required to resist smoking to earn larger sums of money. We hypothesized that guanfacine would attenuate stress effects and associated decrements in self-control by increasing the ability to resist smoking, attenuating stress-related increases in tobacco craving, and decreasing ad-libitum smoking. Medication effects on neural activity were assessed using functional magnetic resonance imaging (fMRI) while subjects performed a cognitive-control Stroop task. We hypothesized that guanfacine treatment would increase activation of the insula and ACC and decrease activation of the dorsolateral PFC (dlPFC). We based this hypothesis on prior findings that: 1) Stroop-related activation in the insula and ACC was associated with better treatment outcome amongst adolescent smokers (Krishnan-Sarin et al, 2013); and 2) Stroop-related activation in the dlPFC decreased in substance-dependent individuals during treatment (DeVito et al, 2012). Finally, we evaluated the effects of guanfacine on clinical outcomes during a brief (4-week) proof-of-concept treatment phase, hypothesizing that guanfacine would be associated with better treatment outcomes.
METHODS AND MATERIALS
Design
A between-subject, double-blind, placebo-controlled design was used to compare guanfacine (3mg/day) to placebo (0mg/day). Following titration to steady-state medication levels, subjects (n=33) completed two laboratory sessions designed to model smoking lapse (stress vs neutral imagery, order counterbalanced), completed fMRI to assess cognitive control, and were then maintained on their randomized medication condition for an additional 4-week period. The quit day was scheduled following the fMRI session, and subjects were provided with weekly brief behavioral treatment. Medication was tapered after end of the treatment period (see Table 1).
Table 1.
Single Subject Timeline.
| DAY | Procedure |
|---|---|
| 1–21 | Titration to steady state medication levels. |
| 22 | Human laboratory session to model smoking lapse behavior (stress vs. neutral imagery, order counter-balanced). See Figure 1. |
| 24 | Human laboratory session to model smoking lapse behavior (stress vs. neutral imagery, order counter-balanced). See Figure 1. |
| 25 or 26 | fMRI session to evaluate attention and inhibitory control using Stroop task |
| 27 | Quit day |
| 34–53 | 1x weekly brief behavioral support during 4-week treatment period |
| 54–58 | Medication taper |
Note: Mean days between the two laboratory sessions = 2.24 days, SD=0.49; mean days from laboratory to fMRI session = 1.26 days, SD=0.47.
Participants
Eligible participants were 18–60 years of age, smoked ≥10 cigarettes/day for the past year, had urine cotinine levels ≥150 ng/ml, and were normotensive (sitting BP >90/60 and <160/100 mmHg). Subjects were excluded if they met criteria for current (past-6-month) Axis-I psychiatric disorders (excluding nicotine dependence) (First, 1996), were using illicit drugs (assessed by urine toxicology), had engaged in smoking-cessation treatment in the past six months, had medical conditions or used concurrent medicine that would contraindicate guanfacine use or smoking behavior assessed by physical exam (including electrocardiogram and basic blood chemistries). The study was approved by the Yale Human Investigations Committee. All subjects signed informed consent. Subjects were recruited from the community for a smoking laboratory study and during the consent process were informed that they could also participate in an imaging session and a brief treatment phase as part of the study. Following initial phone screening, 55 subjects completed eligibility screening and 50 were found eligible; a total of 33 subjects (17 guanfacine) completed the laboratory sessions. Of the 17 who did not complete, n=8 were non-starters (6 lost interest, 2 positive urine toxicology) and n=9 started medication but did not complete the study (4 were dismissed for failing to comply with experimental procedures, 4 lost interest, 1 positive urine toxicology). Following consent, 26 subjects expressed interest in the imaging session and were consented for the optional fMRI component, and 21 subjects arrived for and completed the fMRI session (9 guanfacine). Twenty-one subjects expressed interest in the treatment phase and 18 subjects (9 guanfacine) engaged in the treatment phase (3 lost interest). Subjects completing the laboratory sessions were primarily Caucasian, high-school-educated, and moderately nicotine-dependent (Fagerström Nicotine Dependence Test (FTND, Heatherton et al, 1991), with low levels of depressive symptomatology (Center for Epidemiological Studies-Depression Scale, CESD, Radloff, 1977). There were no significant differences in demographic or smoking behaviors across medication conditions (all p’s>.05; Table 2). Across the phases of the study (laboratory, imaging, treatment) the only difference in baseline characteristics was treatment motivation. Subjects consenting to treatment had significantly higher treatment motivation (i.e., Contemplation scores; range 1–10, Biener and Adams, 1991) (mean=7.50, SE=0.49), then those not consenting (mean=4.3, SE=0.54) and this did not differ by medication.
Table 2.
Demographics, smoking behavior, and vitals by medication condition.
| Mean, SD or n,% | Guanfacine (n=17) | Placebo (n=16) |
|---|---|---|
|
|
|
|
| Baseline | ||
|
|
|
|
| Age | 35.65, 11.34 | 36.13, 13.12 |
| Gender (male) | 11, 64.7% | 9, 56.3% |
| Race (Caucasian) | 13, 76.5% | 13, 81.3% |
| Education (high school) | 11, 64.7% | 8, 50% |
| Cigarettes per day | 19.84, 7.73 | 16.30, 7.41 |
| Carbon monoxide (ppm) | 30.65, 20.25 | 26.75, 13.20 |
| FTNDa | 6.18, 2.35 | 4.88, 2.13 |
| CESDb | 7.47, 6.88 | 4.56, 4.08 |
| Systolic BP (mmHg) | 119.86, 3.72 | 124.07, 14.39 |
| Diastolic BP (mmHg) | 75.43, 10.01 | 76.83, 9.38 |
| Heart rate (bpm) | 81.58, 12.84 | 80.13, 11.96 |
|
| ||
| Titrationc
| ||
| Cigarettes per day | 18.98, 7.93 | 15.63, 9.34 |
| Systolic BP (mmHg) | 109.07, 13.67 | 119.47, 12.81 |
| Diastolic BP (mmHg) | 64.00, 8.60 | 71.07, 8.06 |
| Heart rate (bpm) | 61.81, 11.18 | 69.97, 10.09 |
All baseline comparisons across medication conditions were not significant (p>.05).
Fagerström Test of Nicotine Dependence, FTND (Heatherton et al, 1991), range 1–10 for measure.
Center for Epidemiologic Studies Depression Scale, CES-D (Radloff, 1977), range 0–60.
Values collected at the end of titration.
Guanfacine Treatment
The medication condition was double-blind and placebo-controlled, and randomization was stratified by gender. Effective doses for the treatment of hypertension range from 2mg/day to 5mg/day with dose-dependent effects on blood pressure and adverse events (<http://www.drugs.com/pro/guanfacine.html>, accessed August 1, 2011). We evaluated 3mg/day immediate-release guanfacine due to previous work showing that this dosing significantly reduces nicotine craving in cocaine-dependent smokers, with minimal adverse events (Fox et al, 2012). Guanfacine was administered twice daily and titrated to steady-state levels over 21 days (0.5mg days 1–3, 1.5mg days 4–7, 2mg days 8–12, 2.5mg days 13–15, 3mg days 16–21). Subjects completing the treatment phase were maintained at their randomized dose for the additional 4-week period. Thereafter, subjects received a 5-day medication taper.
Laboratory Assessment of Stress-Precipitated Smoking Lapse
Procedures
Each subject completed two 6.5-hour laboratory sessions (stress vs. neutral imagery; Figure 1)
Figure 1.
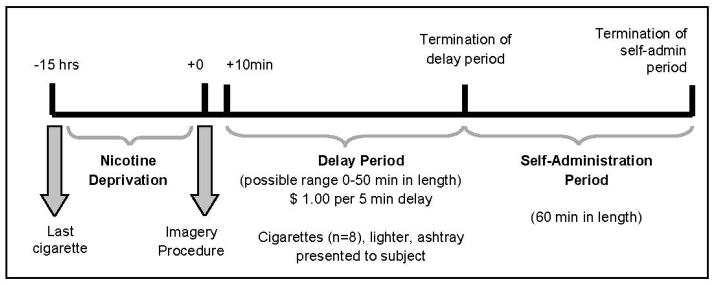
Timeline of laboratory procedures to evaluate smoking-lapse during stress versus neutral imagery. Note: Assessment of cortisol and ACTH occurred at −30 min, −15 min, +10 min, +20 min, +40 min, and +60 min from the imagery procedure and remained fixed regardless of when the termination of the delay period occurred. Assessments of craving, emotion, physiologic reactivity, and nicotine withdrawal occurred at −15min, +5min, and termination of the delay.
Baseline Assessment Period
Laboratory sessions started at 9:00am. Participants were instructed to smoke a final cigarette at 10:00pm the night before. Abstinence was confirmed with a carbon monoxide (CO) reading. An IV cannula was inserted to obtain blood samples. Baseline assessments of breath CO, breath alcohol, urine drug screens, urine pregnancy screen, and vital signs were obtained. Additional measures of emotion, tobacco craving, and nicotine withdrawal were obtained. Medication administration (1.5 mg or placebo) occurred at 10:00am. Participants were provided with a standardized lunch at 11:15am to control for time since last food consumption. From 10:00am to 12:30pm, subjects were able to watch television or read.
Personalized Imagery Procedure
Exposure to stress and neutral imagery used personalized guided-imagery methods (McKee et al, 2011; Sinha, 2009). In a prior session, stress-imagery scripts were developed by having subjects identify and describe in detail highly stressful experiences occurring within the last 6 months. Only situations rated as 8 or greater (1=’not at all stressful’ and 10=’the most stress they recently felt in their life’) were accepted as appropriate for script development. A neutral-relaxing script was developed from subjects’ descriptions of personal neutral/relaxing situations. Scripts were developed by a PhD-level clinician and audiotaped for presentation during the laboratory sessions. Each script was approximately 5min in length. During the laboratory session at 12:55pm, subjects listened to scripts (stress or neutral) via headphones. See Supplementary Materials for additional details.
Delay Period
At 1:10pm, participants were presented with a tray containing 8 cigarettes of their preferred brand, a lighter, and an ashtray. Participants were instructed that they could commence smoking at any point over the next 50min. However, for each 5min block of time they delayed or ‘resisted’ smoking, they would earn $1 for a maximum of $10. Time when subjects announced they wanted to smoke (range 0–50min) was recorded.
Smoking Self-Administration Period
The ad-lib smoking-session duration was 60min and started once participants decided to end the delay period (or delayed for 50min). Participants were provided with 8 cigarettes of their preferred brand. Participants were instructed to ‘smoke as little or as much as you wish’. Subjects were discharged at 3:15pm.
Assessments
Primary measures included the length of the delay period (i.e., time to initiate smoking), number of cigarettes smoked during the ad-lib period, and tobacco craving (Questionnaire of Smoking Urges-Brief; Cox et al, 2001). Other measures were collected but not included in this report. Additional measures are described below.
Mood & Nicotine Withdrawal
The Differential Emotion Scale (DES), a 30-item self-report questionnaire, was used to assess current emotional state for positive (e.g., happy, joy) and negative (e.g., sadness, anger) emotion states (VAS scale, range 1–100; Izard, 1972). DSM-IV symptoms of nicotine withdrawal were assessed with the 8-item Minnesota Nicotine Withdrawal Scale (MNWS; Hughes and Hatsukami, 1986). Instructions were worded to assess current symptoms of withdrawal (range 0–32).
Physiologic measures
A pulse sensor was attached to the subject’s forefinger to obtain a measure of pulse rate. Blood pressure was measured using a Critikon Dynamap.
Cortisol & ACTH
Four mls of blood were collected at each timepoint to assess plasma ACTH and cortisol (see Supplementary Materials for processing methods).
Guanfacine Levels
Five mls of blood were collected at the start of each laboratory session to evaluate plasma-trough-guanfacine levels (see Supplementary Materials for processing methods)
Timing of Assessments
Tobacco craving, emotion ratings, physiologic reactivity, and nicotine withdrawal were assessed pre-imagery, post-imagery (prior to the presentation of cigarette cues), end of delay, and +30min and +60min during the ad-lib-smoking period. ACTH and cortisol levels were assessed −30, −15, +5, +20, +40, +60min post-imagery.
Statistical Analysis
Multivariate analysis of variance were used to examine effects of medication condition by time (week 1, week 2, week 3) on cigarettes per day, systolic BP, diastolic BP, and heart rate during the titration period. Multivariate analyses of variance were used to examine within-subject effects of imagery condition (stress, neutral) by medication condition (guanfacine, placebo) on the primary outcomes of the length of the delay period and the number of cigarettes smoked during the ad-lib period. Multivariate analyses of variance were used to examine outcomes of tobacco craving, HPA-axis levels, emotion ratings, physiologic reactivity, and nicotine withdrawal within imagery condition (stress, neutral), within time (pre-imagery, post-imagery), and by medication condition (guanfacine, placebo). The post-imagery timepoint occurred prior to the start of the smoking lapse task. Post-hoc analyses examined differences in stress vs. neutral imagery change scores (differences pre- to post-imagery) within each medication group. According to the predefined analytical plan, age, sex, baseline cigarettes per day, FTND scores, and CES-D scores were evaluated as potential covariates (or as a between-subjects variable in the case of sex), and were retained if they reduced residual variance, or were otherwise excluded.
fMRI
Stroop
The fMRI Stroop color-word interference task has been described previously (DeVito et al, 2012; Krishnan-Sarin et al, 2013). The task involves frequent exposure of subjects to matched color-word pairs and infrequent exposure to mismatched color-word pairs (pseudo-randomly presented every 13th to 16th presentation) during fMRI. As in prior studies (Jastreboff et al, 2013), an hour prior to fMRI, subjects were given the opportunity to smoke a cigarette so that they were not in acute tobacco intoxication or withdrawal during fMRI. Subjects completed two practice runs to gain familiarity with the task and then then participated in five runs each of 168 sec. Each run includes seven incongruent stimuli and 105 stimuli total. Subjects received instructions in all cases to name silently the color rather than read the word. This procedure of silent naming has been used successfully by our group to minimize subject motion during fMRI (Brewer et al, 2008; Leung et al, 2000; Potenza et al, 2003). Task performance was assessed outside of the scanner immediately following fMRI. During the presentation of mismatched color-word pairs, performance of this task involves conflict monitoring and cognitive control, and specifically the inhibition of the pre-potent response to read the word (Botvinick et al, 2001; Carter et al, 2000; Kerns et al, 2004). The fMRI design is event-related and analyses focus on activation changes related to incongruent and congruent stimuli (Brewer et al, 2008; Potenza et al, 2003).
Imaging Data Acquisition
Images were obtained using a 3-T Siemens Trio MRI system equipped with a standard quadrature head coil, using T2*-sensitive gradient-recalled single shot echo planar pulse sequence. Subjects were positioned in the coil and head movements were restrained using foam pillows. Functional, blood oxygen level dependent (BOLD) images were acquired with a single-shot gradient-echo echo planar imaging (EPI) sequence. Twenty-five 4mm axial slices parallel to the AC-PC line (1mm skip) were acquired with TR = 1,500msec, TE = 27msec, flip angle = 60 degrees, field of view = 220×220mm, matrix = 64×64. In addition, a high-resolution 3D Magnetization Prepared Rapid Gradient Echo (MPRAGE) image was acquired for each subject (TR=2530 ms; TE =3.34 ms; flip angle = 7 degrees; slice thickness=1mm; field of view=256 × 256mm; matrix=256 × 256).
Imaging Data Preprocessing
Functional images underwent preprocessing using SPM5 (Wellcome Trust Center for Neuroimaging) following published methods (Kober et al, 2010), including the following: slice scan-time correction to the middle slice of each volume; realignment of all functional images to the first image of the first functional scan; co-registration of the anatomical image and the average of these realigned functional images; co-registration of all functional images using parameters obtained from co-registration of the mean image; application of the SPM Unified Segmentation process to the anatomical scan, using prior information from the ICBM Tissue Probabilistic Atlas and estimation of non-linear warping parameters (Ashburner and Friston, 2005); warping the functional images to the Montreal Neurological Institute (MNI) template space, followed by smoothing of functional images using a 6mm Gaussian kernel.
Imaging Data Analysis
First-level robust regression was performed on each participant’s preprocessed images, using the standard general linear model but with iteratively reweighted least squares using the bisquare weighting function for robustness (Kober et al, 2010), as implemented in MATLAB 7.3 (Mathworks, Natick, MA; robust.m). Motion parameters and high-pass filter parameters were added as additional regressors of no interest. Activity during congruent and incongruent trials was estimated as percent signal change from baseline. Next, a second-level, random effects analysis was performed to compare activity between condition and between groups, using NeuroElf (NeuroElf.net). Findings were Family-Wise-Error-corrected for multiple comparisons at p<.05 using Monte-Carlo simulation.
Treatment Phase Evaluating Medication Effects on Clinical Outcomes
Procedures
The quit day was scheduled within a week of completing the laboratory and fMRI sessions. Participants attended weekly appointments to receive brief behavioral support (15–20 min), and to complete research assessments (adverse events, timeline followback to assess cigarette use, CO levels, mood ratings, craving, and withdrawal). Following standard clinical guidelines (Fiore et al, 2008), basic behavioral support was provided by a PhD-level clinician following the Mayo Clinic’s manual “Smoke-Free and Living It” (Mayo Clinic, 2000).
Statistical Analysis
The primary outcomes were cigarettes/day and retention over the 4-week treatment period. Complete abstinence and percent days abstinent (arcsine transformed) were also calculated. The effect of guanfacine on cigarettes/day was evaluated using linear mixed models with a conservative approach for missing values. Missing values were left as missing versus imputing baseline values based on the assumption that missing subjects had returned to baseline levels of smoking. Cigarettes/day represented the dependent variable, medication was included as a between-subjects explanatory variable, and time represented a within-subjects factor. Covariates were handled similarly to what was described for the laboratory sessions. Treatment and time interactions were modeled and followed by appropriate post-hoc tests. Similar analyses were conducted for weekly measures of CO levels, positive mood ratings, negative mood ratings, craving, and withdrawal.
RESULTS
Human Laboratory Analogue Evaluating Medication Effects on Stress-Precipitated Smoking Lapse
Medication Compliance and Adverse Events
Compliance was 100% as assessed by a riboflavin marker (Del Boca et al, 1996) and pill counts. Plasma-trough-guanfacine levels collected at the start of each laboratory session did not differ (stress condition mean=4.25 ng/ml, SE=0.44; neutral condition mean=3.97 ng/ml, SE=0.34, p>.05 paired comparison). Levels were consistent with those expected following steady state dosing (Sokin and Heel, 1986). Adverse events were assessed twice weekly during titration and once weekly during treatment (Levine and Schooler, 1986). Common adverse events associated with guanfacine are reported (Table 3). All were rated as minimal or mild. No subject discontinued or required dosing adjustment due to adverse events.
Table 3.
Relative frequencies of treatment emergent adverse events commonly associated with guanfacine (3mg/day) versus placebo during the 3-week titration period and the 4-week treatment phase.
| Adverse Event | 3-week Titration Period | 4-week Treatment Phase | ||
|---|---|---|---|---|
| Guanfacine | Placebo | Guanfacine | Placebo | |
|
|
|
|
||
| Dry Mouth | 88.2%* | 18.8% | 88.9%* | 0% |
| Drowsiness | 47.1% | 18.8% | 11.1% | 0% |
| Dizziness | 6.3% | 24.5% | 11.1% | 0% |
| Headache | 29.4% | 25.0% | 33.3% | 11.1% |
| Impotence | 0% | 6.3% | 0% | 0% |
| Constipation | 23.5% | 6.3% | 11.1% | 0% |
| Fatigue | 47.1%* | 6.3% | 11.1% | 11.1% |
All events were rated as minimal or mild on five point scale (0=absent, 1=minimal, 2=mild, 3=moderate, 4=severe). A chi-square comparison across adverse events revealed higher incidence of dry mouth during the titration and treatment phases, and higher incidence of fatigue during the titration period only (*p<.05).
Titration Phase
Cigarettes per day did not significantly change by medication during the titration period. Systolic blood pressure significantly decreased following guanfacine administration (main effect of medication; F1,27=4.74, p<.05), whereas diastolic blood pressure (p=.10) and heart rate (p=.11) demonstrated reductions following guanfacine administration that did not reach statistical significance (Table 2).
Latency to Smoking, Ad-libitum Smoking, Craving, and Withdrawal
As expected, there was a significant medication-by-imagery-condition interaction on time to resist smoking (F1,28=4.45, p<.05; Cohen d=0.80, large effect) indicating that stress reduced smoking resistance in the placebo group, and this effect was eliminated in the guanfacine group (p<.05; Figure 2A). Once subjects started smoking, there was a significant medication-by-imagery interaction on cigarettes smoked during the 60-minute ad-libitum period (F1,28=4.42, p<.05, Cohen d=0.79, large effect; Figure 2B). Stress increased the number of cigarettes smoked in the placebo group (p<.05), and this effect was absent in the guanfacine group. For tobacco craving, there was a significant medication–by-imagery-condition interaction on tobacco craving (F1,29=4.54, p<.05, Cohen d=0.80, large effect; Figure 2C). Post-hoc analysis demonstrated that stress versus neutral imagery increased tobacco craving in the placebo group (pre to post-imagery), and this effect was reduced in the guanfacine group (p<.05). Further, stress-related increases in tobacco craving were greater in the placebo versus guanfacine-treated subjects (p<.05). Additional analyses demonstrated that baseline levels in craving did not differ across imagery or medication groups (grand mean = 41.92, SE=4.52), and did not differ by the end of the self-administration session (grand mean = 17.38, SE=2.66). There were no main effects of medication on latency, smoking, or craving. There were no effects of medication or imagery condition on nicotine withdrawal ratings. At the start of the laboratory session, subjects demonstrated withdrawal symptoms consistent with overnight deprivation (guanfacine mean=4.24, SE=1.06, placebo mean=5.75, SE=1.09).
Figure 2.
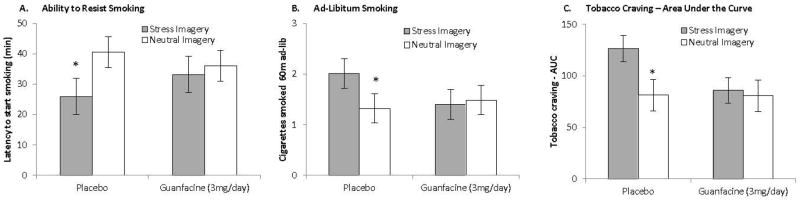
Stress increases the ability to resist smoking (2A), ad-libitum smoking (2B), and tobacco craving (2C) during a human laboratory experiment and these effects are absent or reduced in guanfacine-treated subjects (guanfacine n=17; placebo n=16). (A): Stress (versus neutral imagery) reduced the ability to resist smoking in the placebo group, and this effect was absent in the guanfacine group. *p<.05 stress vs. neutral imagery within medication group. (B): Stress increased the number of cigarettes smoked in the placebo group, and this effect was absent in the guanfacine group. *p<.05 stress vs. neutral imagery within medication group. (C): Stress (versus neutral imagery) increased tobacco craving (change from pre to post-imagery) in the placebo group, and this effect was reduced in the guanfacine group. During the stress session, the increase in craving was greater in the placebo vs. guanfacine group. (*p<.05 stress vs. neutral imagery within medication group; and guanfacine vs. placebo within stress imagery). NOTE: The post-imagery timepoint occurs prior to the smoking lapse task.
Physiologic Measures
Systolic blood pressure demonstrated a significant main effect of medication (F1,31=18.72, p<.0005), with lower values in the guanfacine group (guanfacine mean=101.37 mmHg, SE=2.28; placebo mean=115.53 mmHg, SE=2.35). While stress increased systolic blood pressure in the placebo group (p<.05), the overall interaction across medication, imagery, and time (pre-post imagery) was not significant (p>.05). Post-hoc analysis demonstrated that placebo-treated but not guanfacine-treated subjects demonstrated an increase in systolic blood pressure during stress vs. neutral imagery sessions (Figure 3A). Diastolic blood pressure demonstrated a main effect of medication (F1,31=15.04, p<.001), with lower values in the guanfacine group (guanfacine mean=57.92 mmHg, SE=1.71; placebo mean=67.44 mmHg, SE=1.76). Heart rate demonstrated a trend for a main effect of medication (F1,31=3.20, p<.10), with lower values in the guanfacine group (guanfacine mean=59.06 bpm, SE=2.40; placebo mean=65.23 bpm, SE=2.48). There were no significant interactions across medication or imagery condition for diastolic blood pressure or heart rate.
Figure 3.

Stress increases systolic blood pressure in placebo but not guanfacine-treated subjects (3A) and stress increases cortisol response in guanfacine but not placebo-treated subjects (3B). (A): Effect of medication (guanfacine vs placebo) and imagery (stress vs neutral) on systolic blood pressure during the smoking-lapse paradigm(guanfacine n=17; placebo n=16). Main effect of guanfacine on decreasing systolic blood pressure. Post-hoc analysis of change (pre to post imagery) demonstrated that stress vs. neutral imagery increased systolic blood pressure in placebo but not guanfacine-treated subjects (*p<.05 stress versus neutral imagery within medication). (B): Effect of medication (guanfacine vs placebo) and imagery (stress vs neutral) on cortisol levels during the smoking-lapse paradigm (guanfacine n=13; placebo n=14). Post-hoc analysis of change (pre to post-imagery) demonstrated that cortisol levels differed in guanfacine across stress and neutral imagery sessions but not in placebo-treated subjects (*p<.05 stress versus neutral imagery within medication) and differed across medication groups within stress condition (*p<.05 guanfacine vs. placebo within stress). NOTE: The post-imagery timepoint occurs prior to the smoking lapse task.
Cortisol & ACTH
For cortisol levels, the multivariate interaction of medication by imagery (stress, neutral) by time (pre and post-imagery) was significant (F1,26=7.19, p<.02). Post-hoc analysis examining change scores (pre- to post-imagery) of imagery condition by guanfacine or placebo finds that cortisol levels were greater following stress for guanfacine-treated subjects (p<.05), but did not differ for placebo-treated subjects. Following stress, cortisol levels decreased in the placebo-treated subjects and increased in the guanfacine-treated subjects (p<.05) (Figure 3B). Additional analyses demonstrated that baseline levels of cortisol did not differ across imagery or medication groups (grand mean = 10.00 ng/ml, SE=0.86), but differed by imagery condition 60 minutes following the imagery manipulation (F1,25=6.49, p<.02; stress imagery mean=11.45 ng/ml, SE=1.42, neutral imagery=9.46, SE=1.21). It should be noted that the majority of subjects had commenced smoking by the +60 cortisol timepoint, which increased cortisol levels. ACTH levels did not demonstrate significant effects of medication or imagery condition, although examination of mean values demonstrated that guanfacine tended to alter ACTH values in an opposite pattern to cortisol.
Manipulation Check on Stress and Neutral Imagery
Significant time-by-imagery-condition interactions were observed for positive (F1,31=53.93, p<.0005) and negative (F1,31=18.12, p<.0005) mood ratings. Following stress imagery, positive mood decreased and negative mood increased. There were no effects of medication on mood ratings.
fMRI Session
Stroop Performance
Overall reaction times were similar to those in our prior published work (Congruent reaction time (RT) mean = 580.40ms, SD = 62.18ms; Incongruent RT mean = 816.11ms, SD= 112.15ms) (DeVito et al, 2012; Krishnan-Sarin et al, 2013). A repeated measures ANOVA with condition as a within-subjects factor and group as a between-subjects factor revealed the anticipated effect of condition (incongruent vs. congruent; (F1,19=139.67, p<.001). There was no effect of guanfacine on Stroop RT (F1,19=.01, p=.9) and no interactive effects. An independent sample t-test revealed no medication differences with respect to accuracy of responses (t19=.52, p=.61).
fMRI
CO levels did not significantly differ across guanfacine (mean=27.33, SE=5.76) and placebo (mean=29.83, SE=4.99) groups at the start of the scanning session. Consistent with prior studies (Leung et al, 2000; Potenza et al, 2003), incongruent versus congruent stimuli presentation (“Stroop Effect”) was associated with activation of ventrolateral and dorsolateral PFC, insula, striatum, thalamus, anterior cingulate/dorsomedial PFC and parietal cortex (Figure S1, Supplementary Material). In the guanfacine relative to placebo group, increased activation was observed in the anterior cingulate, sensorimotor cortex, ventromedial PFC (vmPFC), insula and middle and superior temporal gyri, and decreased activation was observed predominantly in the dorsolateral PFC (dlPFC) and parietal cortex (Table 4; Figure 4; Figure S2, Supplementary Material).
Table 4.
Brain regions showing significant group differences in Stroop effect.
| Regions of Activation | R/L | Peak Coordinates
|
||||
|---|---|---|---|---|---|---|
| x | y | z | k | Max Statistic | ||
| Guanfacine (Inc>Con) > Placebo (Inc>Con) | ||||||
| Superior Temporal Gyrus/Posterior Insula | R | 69 | −15 | 12 | 113 | 4.64 |
| Superior Temporal Gyrus/Posterior Insula | L | −42 | −21 | 18 | 239 | 4.56 |
| Superior and Middle Temporal Gyrus/Mid Insula | L | −42 | −6 | −27 | 176 | 4.29 |
| Mid Cingulate/Post Central Gyrus/Sensorimotor | R | 24 | −21 | 45 | 107 | 3.81 |
| Ventromedial PFC | R | 12 | 45 | −9 | 116 | 3.78 |
| Placebo (Inc>Con) > Guanfacine (Inc>Con) | ||||||
| Superior/Middle Frontal Gyrus | L | −18 | 45 | 24 | 230 | 4.10 |
| Superior Parietal | L | −42 | −54 | 42 | 231 | 3.87 |
| Superior/Middle Frontal Gyrus | R | 36 | 33 | 42 | 214 | 3.81 |
| Superior Temporal/Thalamus/Caudate | R | 30 | −42 | 21 | 105 | 3.57 |
| Superior Parietal | R | 42 | −42 | 36 | 104 | 3.50 |
Results are whole-brain family-wise-error-corrected at p<0.05. L = left; R = right; k = number of 3×3×3 voxels; Max statistic = T value at peak voxel. Xyz coordinates are in MNI space.
Figure 4.

Guanfacine alters prefrontal activation to the incongruent stimuli in a Stroop task during the fMRI session (guanfacine n=9; placebo n=12). Guanfacine also attenuated demand on dorsal attention networks during the task. During this Stroop task, participants see color words (e.g., BLUE) presented in either congruent (blue) or incongruent color fill (green). Participants are asked to silently name the color fill while ignoring the word itself, and their ability to do so is thought to reflect attention, conflict resolution, and inhibitory control during incongruent stimuli presentations. Percent-signal-change values during incongruent (red) and congruent trials (green) are displayed in regions that showed significant between-group difference in the incongruent> congruent contrast in whole brain analysis (family-wise-error corrected p<.05). Figure shows increased activity in (A): Dorsal Cingulate/Sensorimotor area (SMA), (B): ventromedial prefrontal cortex (vmPFC), and (C): Superior Temporal Gyrus (STG)/Insula. Attenuated activity is shown in (D): Superior Parietal, and (E): Dorsal Prefrontal Cortex (dPFC).
Treatment Phase
During the 4-week treatment phase, guanfacine decreased cigarette use (estimate of fixed effect for medication, t46=5.15, p<.0005; Cohen d=1.52, large effect; Figure 5A) and improved treatment retention (z=2.65, p<.01; Figure 5B). No subject achieved complete abstinence for the 4-week phase, however, there was a trend towards increased percent days abstinent in the guanfacine (24%) compared to the placebo group (1%) (p=.10). Measures of CO levels, positive mood ratings, negative mood ratings, craving, and withdrawal did not demonstrate medication effects during the treatment phase.
Figure 5.

Guanfacine reduces cigarette use and improves retention during a brief treatment phase (guanfacine n=9; placebo n=9). (A): Effect of medication (guanfacine vs placebo) on cigarette use during baseline (mean 30 day average), titration (mean of final titration week), and the 4-week treatment period (4-week average adjusted for baseline). Guanfacine significantly reduces cigarette use during the treatment phase (*p<.0005). (B): Effect of medication (guanfacine vs placebo) on retention in treatment during the 4-week quit attempt. Guanfacine significantly increases retention during the treatment phase (*p<.01).
DISCUSSION
Laboratory, neuroimaging, and clinical outcome data were consistent and complementary. Results suggest that in overnight nicotine deprived daily cigarette smokers, guanfacine reduced stress-precipitated lapse by increasing the ability to resist smoking in the laboratory, increased ventromedial prefrontal brain activity during the cognitive control task, and decreased smoking during a brief treatment phase. The α2A-agonist guanfacine may ameliorate stress responses through multiple inter-related mechanisms, including reductions in norepinephrine and dopamine release (Jentsch et al, 1998) and strengthening PFC network connections via post-synaptic α2A inhibition of cAMP-sensitive potassium channel signaling in PFC neurons (Arnsten, 2010). Guanfacine may also “replace” nicotine’s enhancing effects at α7-nicotinic receptors in the PFC, which are permissive for NMDA receptor actions (Yang et al, 2013). Consistent with these ideas, guanfacine increased self-control in the laboratory (i.e., increased the latency to start smoking), reduced stress-related increases in tobacco craving, and decreased smoking behavior in the laboratory and during short-term treatment.
Using a validated human laboratory analogue of smoking-lapse which targets the first instance of smoking during a quit attempt (McKee et al, 2012), we demonstrated that personalized stress imagery decreased the ability to resist smoking, and increased tobacco craving and subsequent cigarette smoking in placebo-treated subjects, supporting prior findings (McKee et al, 2011). The effect of stress on these outcomes was eliminated or reduced in subjects who received guanfacine. Importantly, the magnitude of these effects was large and suggests that guanfacine potently counteracted known stress-effects on smoking behavior. Our human laboratory findings extend preclinical results demonstrating that α2 adrenergic agonists attenuate stress-precipitated relapse behavior (Lê et al, 2005; Yamada & Bruijnzeel, 2011). Associations between stress and tobacco relapse episodes are well documented (McKee et al, 2003; Shiffman and Waters, 2004), and we have hypothesized that stress promotes ongoing use and relapse by increasing craving and decreasing self-control for rewarding substances in addicted individuals (Sinha, 2008). The finding of relatively diminished activation of dlPFC and parietal cortices in the guanfacine versus placebo groups in the absence of between-medication-group differences in Stroop performance suggests that guanfacine may facilitate function of executive control circuitry by promoting more efficient processing in these regions. The current findings on reduced smoking and craving response also extend preclinical results documenting that noradrenergic agents attenuate both nicotine taking (Forget et al., 2010) and nicotine seeking (Forget et al., 2010; Yamada & Bruijnzeel, 2011). As documented with varenicline, pharmacological targets which effectively address both consumption and craving have the potential to be highly effective treatments (see Rollema et al, 2007).
Consistent with the laboratory findings, guanfacine increased reductions in cigarette use from baseline by 70%, and increased retention to 100% during the brief treatment period. These results, while preliminary, compare favorably to clonidine which has been found to increase quit rates by 63% (Gourlay et al, 2004). It is possible that guanfacine’s specificity for the α2A-adrenoceptor subtype (Uhlen, 1991) in comparison to clonidine which is non-specific for α2 receptors, may be mediating differences for effects on smoking behavior. However, effects on complete abstinence, CO levels, mood, craving, and withdrawal were not demonstrated, which may have been due, in part, to the sample recruited for this study. While subjects agreed to engage in a treatment phase as part of the protocol and had reasonably high treatment motivation, they were initially recruited for a laboratory study. In future work it will be important to determine specific mechanisms underlying guanfacine’s efficacy in clinical trial evaluations recruiting smokers specifically interested in smoking cessation.
During the titration period guanfacine significantly reduced systolic blood pressure, and also tended to reduce diastolic blood pressure and heart rate. Mean values of blood pressure were approaching definitions outlined for hypotension, suggesting that smokers with existing hypotension may not be appropriate to treat with guanfacine. Future work should investigate whether lower doses of guanfacine would be efficacious for smoking cessation while minimizing reductions in blood pressure, heart rate, and other orthostatic adverse events.
Stress-precipitated changes in cortisol levels demonstrated reductions in placebo-treated subjects, whereas guanfacine-treated subjects demonstrated increased levels. When compared to non-smokers, smokers typically demonstrate a blunted hypothalamic-pituitary-adrenal-axis activation in response to stress, and this blunted response has been associated with smoking relapse (al’Absi, 2005). No significant effects of imagery condition or medication were observed for ACTH levels. These findings suggest that guanfacine may normalize the cortisol response to stress in overnight-deprived daily smokers. Chronic nicotine alters corticotrophin releasing factor (CRF) and noradrenergic signaling in stress-reactive extrahypothalamic circuits such as the amygdala and the bed nucleus of the stria terminalis (BNST; Buczek et al, 1999), regions involved in regulation of the HPA axis and cortisol responses (Bornstein et al., 2008). The chronic nicotine related alterations in these circuits may contribute to blunted cortisol responses to stress and to nicotine in daily smokers. Guanfacine has been shown to have its effects on stress-induced nicotine reinstatement via effects on the amygdala and the BNST (Yamada and Bruijnzeel, 2011), which may in turn affect amygdala and BNST efferents to the HPA axis and to autonomic regulation. However, it should be noted that this hypothesis regarding the normalization of blunted cortisol response is speculative as we did not include a non-smoker control group to evaluate the strength of the stress manipulation used in the current study.
Neuroimaging results demonstrated altered activation in brain areas associated with attention and inhibitory control, consistent with findings that guanfacine treatment is associated with improvement in attention and PFC-based executive functioning tasks (Kim et al, 2012). Guanfacine’s enhancement of Stroop-related activation in the anterior cingulate, ventromedial PFC (vmPFC), and bilateral temporal gyri/insula, while decreasing activity in dlPFC and parietal cortices, suggests improved engagement of PFC-based cognitive and impulse-control systems and decreased reliance on dorsal attentional circuits while attending to incongruent versus congruent stimuli. A parsimonious explanation for this pattern of neural activation is that guanfacine may increase efficiency within dorsal attention networks engaged during Stroop performance, and further study is needed to examine this possibility directly. Interestingly, amongst treatment-seeking adolescent smokers, Stroop-related activations in the insula and anterior cingulate positively correlated with treatment-related reductions in cotinine levels (Krishnan-Sarin et al, 2013); thus, these and the current findings indicate a relationship between increased Stroop-related insula and anterior-cingulate activation and better treatment outcome for smokers. However, insula lesions have been associated with smoking cessation (Naqvi et al, 2007) and insula activation to smoking cues associated with smoking slips (Janes et al, 2010), suggesting a context-dependent mechanism for the relationship between insula activation and smoking. A similar pattern of Stroop-related increased vmPFC activation and diminished dlPFC activation as observed presently was previously associated with improved outcome in cocaine dependence (Brewer et al, 2008). Relatively diminished activation of dorsal PFC was also observed following behavioral treatment for substance abuse (DeVito et al, 2012).
Additional limitations to those already mentioned include a modest sample size and attrition. Although the sample size and rates of attrition were comparable to other laboratory studies of stress in smokers and medication effects on our primary outcomes were robust, it will be important to replicate findings. We did not collect data on time of last dose of guanfacine/placebo during the fMRI session, although this concern might be mitigated by the scanning being performed at steady-state dosing levels of guanfacine. Stress was not specifically measured during the treatment phase, although measures of negative mood (including anxiety and distress) did not demonstrate significant findings.
Overall, results point to guanfacine as a potential pharmacotherapy for smoking cessation. Using a novel translational approach, we report for the first time that guanfacine significantly reduced smoking lapse and craving using a well-validated laboratory analog of stress-precipitated smoking, altered brain activity during a cognitive-control task, and reduced smoking and improved retention during a subsequent treatment period. Our findings are consistent with preclinical results that guanfacine attenuates stress-related relapse and rescues decrements in self-control, and support further development of guanfacine as a potential pharmacotherapy for smoking cessation.
Supplementary Material
Acknowledgments
FUNDING
Supported by NIH grants P50DA033945, RL1DA024857, R21DA033597, R01DA035001, UL1DE019586, PL1DA024859, PL1DA024860, UL1RR024139, RL1AA017539, K12DA000167, P20DA027844, R01DA014241, and the Connecticut Department of Mental Health and Addictions Services.
Footnotes
DECLARATION OF CONFLICT OF INTEREST
Dr. Arnsten and Yale receive royalties from the sales of Intuniv (extended-release guanfacine) for the treatment of pediatric ADHD. They do not receive royalties from the sales of generic guanfacine which is used to treat adults. Generic guanfacine was used in the current study. All other authors have no disclosures to report.
Dr. Sofuoglu is supported by the Veterans Administration Mental Illness Research, Education and Clinical Center (MIRECC). Dr. Sofuoglu served as an expert witness on behalf of Pfizer in lawsuits related to varenicline.
Dr. Potenza has received financial support or compensation for the following: Dr. Potenza has consulted for and advised Ironwood, Lundbeck, Shire and iNSYS; has received research support from the National Institutes of Health, Mohegan Sun Casino, the National Center for Responsible Gaming, and Psyadon pharmaceuticals; has participated in surveys, mailings or telephone consultations related to drug addiction, impulse control disorders or other health topics; has consulted for law offices and gambling entities on issues related to impulse control disorders; provides clinical care in the Connecticut Department of Mental Health and Addiction Services Problem Gambling Services Program; has performed grant reviews for the National Institutes of Health and other agencies; has guest-edited or edited journals or journal sections; has given academic lectures in grand rounds, CME events and other clinical or scientific venues; and has generated books or book chapters for publishers of mental health texts.
Drs. McKee, Kober, Sofuoglu, Picciotto, Weinberger, Ashare, and Sinha reported no conflicts of interest with this study.
References
- al’Absi M, Hatsukami D, Davis G. Attenuated adrenocorticotropic responses to psychological stress are associated with early smoking relapse. Psychopharmacology (Berl) 2005;181:107–117. doi: 10.1007/s00213-005-2225-3. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. The use of alpha2A adrenergic agonists for the treatment of attention-deficit/hyperactivity disorder. Expert Rev Neurother. 2010;10:1595–1605. doi: 10.1586/ern.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT. Stress signaling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Cai JX, Goldman-Rakic PS. The alpha 2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: Evidence for alpha-2 receptor subtypes. J Neurosci. 1988;8:4287–4298. doi: 10.1523/JNEUROSCI.08-11-04287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Steere JC, Hunt RD. The contribution of alpha-2 noradrenergic mechanisms to prefrontal cortical cognitive function: Potential significance to Attention Deficit Hyperactivity Disorder. Arch Gen Psychiatry. 1996;53:448–455. doi: 10.1001/archpsyc.1996.01830050084013. [DOI] [PubMed] [Google Scholar]
- Biener L, Adams D. The Contemplation Ladder: Validation of a measure of readiness to consider smoking cessation. Health Psychology. 1991;10:360–365. doi: 10.1037//0278-6133.10.5.360. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Podell DM, Arnsten AFT. Noradrenergic alpha-2 receptor agonists reverse working memory deficits induced by the anxiogenic drug, FG7142, in rats. Pharmacol Biochem Behav. 2000;67:397–403. doi: 10.1016/s0091-3057(00)00306-3. [DOI] [PubMed] [Google Scholar]
- Botvinick M, Braver TS, Barch DM, Carter CS, Cohen JD. Conflict monitoring and cognitive control. Psychol Rev. 2001;108:624–652. doi: 10.1037/0033-295x.108.3.624. [DOI] [PubMed] [Google Scholar]
- Brewer J, Worhunsky P, Carroll K, Rounsaville B, Potenza M. Pretreatment brain activation during Stroop task is associated with outcomes in cocaine-dependent patients. Biol Psychiatry. 2008;64:998–1004. doi: 10.1016/j.biopsych.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW, Bishnoi M, van Tuijl IA, Keijzers KF, Yavarovich KR, Pasek TM, Ford J, Alexander JC, Yamada H. Effects of prazosin, clonidine, and propranolol on the elevations in brain reward thresholds and somatic signs associated with nicotine withdrawal in rats. Psychopharmacology (Berl) 2010;212:485–99. doi: 10.1007/s00213-010-1970-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczek Y, Lê AD, Wang A, Stewart J, Shaham Y. Stress reinstates nicotine seeking but not sucrose solution seeking in rats. Psychopharmacology (Berl) 1999;144(2):183–188. doi: 10.1007/s002130050992. [DOI] [PubMed] [Google Scholar]
- CDC . Smoking-attributable mortality, years of potential life lost, and productivity losses---United States, 2000--2004. MMWR Morb Mortal Wkly Rep. 2008;57(45):1226–1228. [PubMed] [Google Scholar]
- Carter CS, Macdonald AM, Botvinick MM, Ross LL, Stenger VA, Noll D. Parsing executive processes: Strategic vs. evaluative functions of the anterior cingulate cortex. Proc Natl Acad Sci U S A. 2000;97:1944–1948. doi: 10.1073/pnas.97.4.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LS, Tiffany ST, Christen AG. Evaluation of the brief questionnaire of smoking urges (QSU-brief) in laboratory and clinical settings. Nicotine Tob Res. 2001;3:7–16. doi: 10.1080/14622200020032051. [DOI] [PubMed] [Google Scholar]
- De Biasi M, Dani JA. Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci. 2011;34:105–130. doi: 10.1146/annurev-neuro-061010-113734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Boca FK, Kranzler HR, Brown J, Korner PF. Assessment of medication compliance in alcoholics through UV light detection of a riboflavin tracer. Alcohol Clin Exp Res. 1996;20:1412–1417. doi: 10.1111/j.1530-0277.1996.tb01142.x. [DOI] [PubMed] [Google Scholar]
- DeVito EE, Worhunsky PD, Carroll KM, Rounsaville BJ, Kober H, Potenza MN. A preliminary study of the neural effects of behavioral therapy for substance use disorders. Drug Alcohol Depend. 2012;122(3):228–235. doi: 10.1016/j.drugalcdep.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engberg G, Eriksson E. Effects of alpha-2-adrenoceptor agonists on locus coeruleus firing rate and brain noradrenaline turnover in EEDQ-treated rats. Naunyn-Schmiedeberg Arch Pharmacol. 1991;343:472–477. doi: 10.1007/BF00169548. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Belin D, Economidou D, Pelloux Y, Dalley JW, Robbins TW. Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philos Trans R Soc Lond B Biol Sci. 2008;363(1507):3125–3135. doi: 10.1098/rstb.2008.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore MC, Jaén CR, Baker TB, Bailey WC, Benowitz NL, Curry SJ, et al. Treating Tobacco Use and Dependence: 2008 Update. U.S. Department of Health and Human Services; Rockville, MD: 2008. [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JB. Structured Clinical Interview for DSM-IV Axis I Disorders--Patient Edition. Biometrics Research Department, New York State Psychiatric Institution; New York: 1996. [Google Scholar]
- Forget B, Hamon M, Thiébot MH. Involvement of alpha1-adrenoceptors in conditioned place preference supported by nicotine in rats. Psychopharmacology (Berl) 2009;205:503–15. doi: 10.1007/s00213-009-1559-7. [DOI] [PubMed] [Google Scholar]
- Forget B, Wertheim C, Mascia P, Pushparaj A, Goldberg SR, Le Foll B. Noradrenergic alpha1 receptors as a novel target for the treatment of nicotine addiction. Neuropsychopharmacology. 2010;35:1751–60. doi: 10.1038/npp.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox HC, Seo D, Tuit K, Hansen J, Kimmerling A, Morgan PT, et al. Guanfacine effects on drug craving, stress arousal and prefrontal activation in cocaine dependent individuals: A preliminary investigation. J Psychopharmacol. 2012;26(7):957–972. doi: 10.1177/0269881111430746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. The prefrontal landscape: implications of functional architecture for understanding human mentation and the central executive. Philos Trans R Soc Lond B Biol Sci. 1996;351:1445–1453. doi: 10.1098/rstb.1996.0129. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Rasmusson AM, Bunney SB, Roth RH. Role of the amygdala in the coordination of behavioral, neuroendocrine and prefrontal cortical monoamine responses to psychological stress in the rat. J Neurosci. 1996;16:4787–4798. doi: 10.1523/JNEUROSCI.16-15-04787.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlay SG, Stead LF, Benowitz NR. Clonidine for smoking cessation. Cochrane Database Syst Rev. 2004;(3):Art. No.: CD000058. doi: 10.1002/14651858.CD000058.pub2. Updated 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heatherton TF, Kozlowski LT, Frecker RC, Fagerström KO. The Fagerström Test for Nicotine Dependence: a revision of the Fagerström Tolerance Questionnaire. Br J Addict. 1991;86:1119–1127. doi: 10.1111/j.1360-0443.1991.tb01879.x. [DOI] [PubMed] [Google Scholar]
- Hughes JR, Hatsukami DK. Signs and symptoms of tobacco withdrawal. Arch Gen Psychiatry. 1986;43:289–294. doi: 10.1001/archpsyc.1986.01800030107013. [DOI] [PubMed] [Google Scholar]
- Izard CE. Patterns of emotions: A new analysis of anxiety and depression. Academic Press; London: 1972. [Google Scholar]
- Jastreboff AM, Sinha R, Lacadie C, Small DM, Sherwin RS, Potenza MN. Neural correlates of stress- and food-cue-induced food craving in obesity: Association with insulin levels. Diabetes Care. 2013;36:394–402. doi: 10.2337/dc12-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes AC, Pizzagalli DA, de Richardt SB, Frederick B, Chuzi S, Pachas G, et al. Brain reactivity to smoking cues prior to smoking cessation predicts ability to maintain tobacco abstinence. Biol Psychiatry. 2010;67(8):722–729. doi: 10.1016/j.biopsych.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch JD, Wise A, Katz Z, Roth RH. Alpha-noradrenergic receptor modulation of the phencyclidine- and delta9-tetrahydrocannabinol-induced increases in dopamine utilization in rat prefrontal cortex. Synapse. 1998;28:21–26. doi: 10.1002/(SICI)1098-2396(199801)28:1<21::AID-SYN3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Kerns J, Cohen JD, MacDonald AW, III, Cho RY, Stenger VA, Carter CS. Anterior cingulate conflict monitoring and adjustments in control. Science. 2004;303:1023–1026. doi: 10.1126/science.1089910. [DOI] [PubMed] [Google Scholar]
- Kim S, Bobeica I, Gamo NJ, Arnsten AFT, Lee D. Effects of α-2A adrenergic receptor agonist on time and risk preference in primates. Psychopharmacology (Berl) 2012;219:363–375. doi: 10.1007/s00213-011-2520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kober H, Mende-Siedlecki P, Kross E, Weber J, Mischel W, Hart CL, et al. A prefrontal-striatal pathway underlies the cognitive regulation of craving. Proc Natl Acad Sci U S A. 2010;107:14811–14816. doi: 10.1073/pnas.1007779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Balodis IM, Kober H, Worhunsky PD, Liss T, Xu J, et al. An exploratory pilot study of the relationship between neural correlates of cognitive control and reduction in cigarette use among treatment-seeking adolescent smokers. Psychol Addict Behav. 2013;27(2):526–532. doi: 10.1037/a0032479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê AD, Harding S, Juzytsch W, Funk D, Shaham Y. Role of alpha-2 adrenoceptors in stress-induced reinstatement of alcohol seeking and alcohol self-administration in rats. Psychopharmacology (Berl) 2005;179:366–373. doi: 10.1007/s00213-004-2036-y. [DOI] [PubMed] [Google Scholar]
- Lester RA. Cognitive mechanisms underlying relapse to nicotine. Rev Neurosci. 2011;22:467–470. doi: 10.1515/RNS.2011.038. [DOI] [PubMed] [Google Scholar]
- Leung H, Skudlarski P, Gatenby JC, Peterson BS, Gore JC. An event-related functional MRI study of the Stroop color word interference task. Cereb Cortex. 2000;10:552–560. doi: 10.1093/cercor/10.6.552. [DOI] [PubMed] [Google Scholar]
- Levine J, Schooler NR. SAFTEE: A technique for the systematic assessment of side effects in clinical trials. Psychopharmacol Bull. 1986;22:343–381. [PubMed] [Google Scholar]
- Mayo Clinic. Smoke Free and Living It. Mayo Clinic; Rochester, MN: 2000. [Google Scholar]
- McAllister TW, McDonald BC, Flashman LA, Ferrell RB, Tosteson TD, Yanofsky NN, et al. Alpha-2 adrenergic challenge with guanfacine one month after mild traumatic brain injury: Altered working memory and BOLD response. Int J Psychophysiol. 2011;82:107–114. doi: 10.1016/j.ijpsycho.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee SA, Maciejewski PK, Falba T, Mazure CM. Sex differences in the effects of stressful life events on changes in smoking status. Addiction. 2003;98:847–855. doi: 10.1046/j.1360-0443.2003.00408.x. [DOI] [PubMed] [Google Scholar]
- McKee SA, Sinha R, Weinberger AH, Sofuoglu M, Harrison EL, Lavery M, et al. Stress decreases the ability to resist smoking and potentiates smoking intensity and reward. J Psychopharmacol. 2011;25(4):490–502. doi: 10.1177/0269881110376694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee SA, Weinberger AH, Shi J, Tetrault J, Coppola S. Developing and validating a human laboratory model to screen medications for smoking cessation. Nicotine Tob Res. 2012;14(11):1362–1371. doi: 10.1093/ntr/nts090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson JH, Goletiani N, Sholar MB, Siegel AJ, Mello NK. Effects of smoking successive low-and high-nicotine cigarettes on hypothalamic-pituitary-adrenal axis hormones and mood in men. Neuropsychopharmacology. 2008;33(4):749–760. doi: 10.1038/sj.npp.1301455. [DOI] [PubMed] [Google Scholar]
- Naqvi NH, Rudrauf D, Damasio H, Bechara A. Damage to the insula disrupts addiction to cigarette smoking. Science. 2007;315(5811):531–534. doi: 10.1126/science.1135926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PDR. Physicians Desk Reference. Medical Economics Data: Medical Economics Data; 1994. [Google Scholar]
- Potenza MN, Leung H-C, Blumberg HP, Peterson BS, Skudlarski P, Lacadie C, et al. An fMRI stroop study of ventromedial prefrontal cortical function in pathological gamblers. Am J Psychiatry. 2003;160:1990–1994. doi: 10.1176/appi.ajp.160.11.1990. [DOI] [PubMed] [Google Scholar]
- Radloff LS. The CES-D scale: A self-report depression scale for research in the general population. Appl Psychol Meas. 1977;1:385–401. [Google Scholar]
- Roozendaal B, McGaugh JL. Memory modulation. Behav Neurosci. 2011;125:797–824. doi: 10.1037/a0026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollema H, Coe JW, Chambers LK, Hurst RS, Stahl SM, Williams KE. Rationale, pharmacology and clinical efficacy of partial agonists of alpha4beta2 nACh receptors for smoking cessation. Trends Pharmacol Sci. 2007;28:316–25. doi: 10.1016/j.tips.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Shalev U, Lu L, de Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- Shiffman S, Waters AJ. Negative affect and smoking lapses: A prospective analysis. J Consult Clin Psychol. 2004;72(2):192–201. doi: 10.1037/0022-006X.72.2.192. [DOI] [PubMed] [Google Scholar]
- Semenova S, Markou A. The alpha2 adrenergic receptor antagonist idazoxan, but not the serotonin-2A receptor antagonist M100907, partially attenuated reward deficits associated with nicotine, but not amphetamine, withdrawal in rats. Eur Neuropsychopharmacol. 2010;20:731–46. doi: 10.1016/j.euroneuro.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R. Chronic stress, drug use, and vulnerability to addiction. Ann N Y Acad Sci. 2008;1141:105–130. doi: 10.1196/annals.1441.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R. Modeling stress and drug craving in the laboratory: Implications for addiction treatment development. Addict Biol. 2009;14:84–98. doi: 10.1111/j.1369-1600.2008.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin EM, Heel RC. Guanfacine: A review of its pharmacodynamic and pharmacokinetic properties, and therapuetic efficacy in the treatment of hypertension. Drugs. 1986;31:301–336. doi: 10.2165/00003495-198631040-00003. [DOI] [PubMed] [Google Scholar]
- Stein EA, Pankiewicz J, Harsch HH, Cho J-K, Fuller SA, Hoffmann RG, et al. Nicotine-induced limbic cortical activation in the human brain: A functional MRI study. Am J Psychiatry. 1998;155:1009–1015. doi: 10.1176/ajp.155.8.1009. [DOI] [PubMed] [Google Scholar]
- Taylor FB, Russo J. Comparing guanfacine and dextroamphetamine for the treatment of adult attention-deficit/hyperactivity disorder. J Clin Psychopharmacol. 2001;21:223–228. doi: 10.1097/00004714-200104000-00015. [DOI] [PubMed] [Google Scholar]
- Uhlen S, Wikberg JES. Delineation of rat kidney alpha 2A and alpha 2B-adrenoceptors with [3H]RX821002 radioligand binding: computer modeling reveals that guanfacine is an alpha-2A-selective compound. Eur J Pharmacol. 1991;202:235–243. doi: 10.1016/0014-2999(91)90299-6. [DOI] [PubMed] [Google Scholar]
- Villégier AS, Lotfipour S, Belluzzi JD, Leslie FM. Involvement of alpha1-adrenergic receptors in tranylcypromine enhancement of nicotine self-administration in rat. Psychopharmacology (Berl) 2007;193:457–65. doi: 10.1007/s00213-007-0799-7. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, Schroeder JP. There and back again: a tale of norepinephrine and drug addiction. Neuropsychopharmacology. 2007;32:1433–51. doi: 10.1038/sj.npp.1301263. [DOI] [PubMed] [Google Scholar]
- World Health Organization. [Accessed May 31, 2014];2014 http://www.who.int/campaigns/no-tobacco-day/2014/event/en/
- Yamada H, Bruijnzeel AW. Stimulation of α2-adrenergic receptors in the central nucleus of the amygdala attenuates stress-induced reinstatement of nicotine seeking in rats. Neuropharmacology. 2011;60:303–311. doi: 10.1016/j.neuropharm.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.