

Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers in western countries with a median survival of six months and an extremely low percentage of long term-surviving patients1. KRAS mutations represent a driver event of PDAC2, yet targeting mutant KRAS has proven challenging3. Targeting oncogene-driven signaling pathways, however, represents a clinically validated approach for several devastating diseases4,5. Still, despite significant tumor shrinkage, the frequency of relapse indicates that a fraction of tumor cells survives shut down of oncogenic signaling6, 7. Here, we explore the role of mutant KRAS in PDAC maintenance using a recently developed inducible mouse model of mutated KRAS2 (KRASG12D, herein KRas) in a p53LoxP/WT background. We demonstrate that a subpopulation of dormant tumor cells surviving oncogene ablation (hereafter, SCs (surviving cells)) and responsible for tumor relapse has features of cancer stem cells and relies on oxidative phosphorylation (OXPHOS) for survival. Transcriptomic and metabolic analyses of SCs reveal prominent expression of genes governing mitochondrial function, autophagy, lysosome activity, as well as strong reliance on mitochondrial respiration and decreased dependence on glycolysis for cellular energetics. Accordingly, SCs show high sensitivity to OXPHOS inhibitors, which can inhibit tumor recurrence. Our integrated analyses illuminate a therapeutic strategy of combined targeting of the KRAS pathway and mitochondrial respiration to manage pancreatic cancer.
Using our tetracycline-inducible KRas allele2 we controlled the expression of KRas in a temporal and pancreas-specific manner. Upon doxycycline withdrawal, we observed regression of pancreatic tumors within 2-3 weeks followed by relapse after 4-5 months, suggesting a fraction of tumor cells survived oncogene ablation. To investigate the impact of KRas ablation in detail, we transplanted cells from primary tumors subcutaneously into recipient mice fed with doxycycline. When tumors reached a diameter of 1 cm, doxycycline was withdrawn and lesions rapidly and apparently completely regressed (Fig.1a;EDfig.1a). However, analysis of residual scars detected epithelial remnants embedded in fibrotic tissue (Fig.1b;EDfig.1b,c). This phenotype was confirmed in vitro using a 3D-culture system in which cells from primary lesions were grown as spheres in semisolid medium. After doxycycline withdrawal (EDfig.1d,e), tumor spheres underwent regression due to apoptosis (EDfig.1f), and only a small population of dormant cells survived (EDfig.1d,g). Notably, upon KRas re-activation, SCs massively re-entered the cell cycle both in vitro and in vivo (Fig.1c;EDfig.1g,h) and rapidly reconstituted spheres and tumors, suggesting that subpopulations of cells differently addicted to KRas co-exist in pancreatic tumors.
Figure 1. Cells surviving oncogene ablation are enriched in tumorigenic cells.
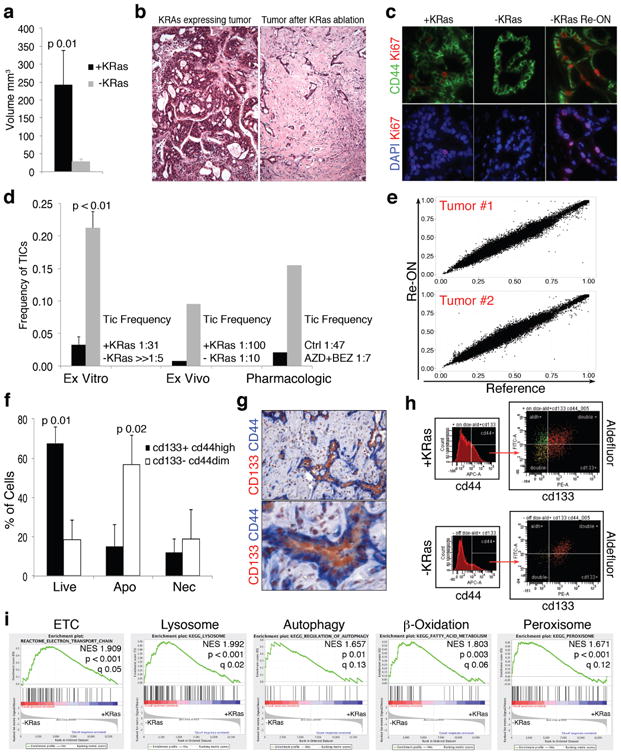
a, Tumor volume before/after KRas ablation (+/-KRas)(n=6). b, Histology depicting tumor remnants (10×). c, Immunofluorescence of KRas-expressing tumor (+KRas), regressed tumor (-KRas) and regressed tumors 48hs after KRas re-activation (-KRas Re-ON) for Ki67 (red), CD44 (green) and DAPI (blue)(40×). d, Limiting dilution transplantation, TIC frequency. Genetic model: +KRas (black) vs -Kras (grey) ex vitro (n=4) or ex vivo (n=2). Pharmacological down-regulation: control (black) vs treated spheres (grey, AZD8330+BEZ235) (n=2). e, Exome sequencing: allele frequencies after KRas re-activation in SCs (RE-ON) vs KRas-expressing cells (Reference) at 40383 and 44182 SNVs for 2 independent tumors. f, AnnexinV in spheres +/-KRas with respect to CD44/CD133 expression (n=3). g, IHC of -KRas tumors for CD44 (blue) and CD133 (red)(20-40×). h, Immunophenotyping of +/-KRas tumors for CD44/CD133/aldefluor. i, GSEA of pathways enriched in -KRas vs +KRas cells. Data are mean ± s.d.
To assess the tumorigenic potential of SCs, we isolated KRas-expressing cells and SCs from tumor spheres (ex vitro) and tumors (ex vivo) and transplanted them in limiting dilution into recipient mice. Surprisingly, SCs were highly enriched in tumor-initiating cells (TIC). Virtually all SCs ex vitro initiated tumors in mouse (TIC frequency ≫1:5 vs. 1:31 in KRas-expressing cells (p<0.001))(Fig.1d;ED fig.2a), and TIC frequency was similarly enriched in SCs ex vivo (1:10 vs 1:100 in KRas-expressing cells (p=0.003))(Fig.1d;ED fig.2b). Then to assess whether pharmacologic ablation of oncogenic pathways could mimic the genetic suppression of KRas we treated tumor spheres derived from a KRas constitutive mouse model8 with a combination of Mek1 (AZD8330) and a dual PI3K/mTOR (BEZ235) inhibitors (EDfig.2c). The treatment resulted in an enrichment of tumorigenic cells (TIC frequency 1:7 vs. 1:47 for treated vs. non-treated cells, respectively, p=0.01)(Fig.1d;EDfig.2d). Collectively, our data demonstrate that PDAC tumors are heterogeneous and a population of spherogenic and tumorigenic cells survives genetic and pharmacologic ablation of oncogenic pathways.
To exclude that SCs represent a more aggressive subclone of tumor cells, we performed exome sequencing of tumor cells during cycles of KRas activation-inactivation-reactivation (ON-OFF-ON cycles) and evaluated changes in the allelic frequency of single nucleotide variants (SNVs), a hallmark of clonal selection. Mutational profiles did not show any significant modification in allelic frequencies before versus after ON-OFF-ON cycles (Fig.1e;EDfig.2e), demonstrating that tumors after KRas reactivation are genetically identical to their primary counterparts. While these data formally exclude genetic clonal selection among SCs, epigenetically driven clonal selection of a more aggressive subclone remains possible.
To further characterize SCs, we examined expression of markers used to isolate cancer stem cells in human tumors9-11. We found that different subpopulations of tumor cells were differentially sensitive to KRas ablation; specifically, only CD133+CD44high cells avoided undergoing a massive apoptosis (Fig.1f;EDfig.1i). Consequently, tumor remnants in vivo are strongly positive for stem cell markers (Fig.1g,h;EDfig.2f,g). Together, the tumorigenicity and immunophenotypic similarity between SCs and previously identified human pancreatic cancer stem cells9-11 suggests SCs may possess cancer stem cell characteristics.
We next performed a transcriptomic analysis of cells isolated from tumor spheres. Gene Set Enrichment Analysis (GSEA) using Signaling Pathways c2.cp.v3.0 gene set revealed significant enrichment of genes involved in several metabolic pathways (e.g. mitochondrial electron transport chain (ETC), lysosome activity, autophagy, mitochondrial and peroxisomal β-oxidation) (Fig.1i;EDfig.3a-e), which suggested SCs might have increased mitochondrial activity. Indeed, Ppargc1a (PGC1a), a key regulator of mitochondrial biogenesis12, was increased at the mRNA and protein levels in SCs (Fig.2a;EDfig.4a), and we detected PGC1a accumulation in the nuclei of SCs in vivo (Fig.2c). Furthermore, SCs from tumor spheres stained intensely for MitoTracker Green, a marker of mitochondrial mass (EDfig.4b). These data were corroborated by increased expression of the mitochondrial marker, VDAC1, in SCs in vitro and in vivo (Fig.2b,d).
Figure 2. Surviving cells have more active mitochondria and impaired glycolysis.
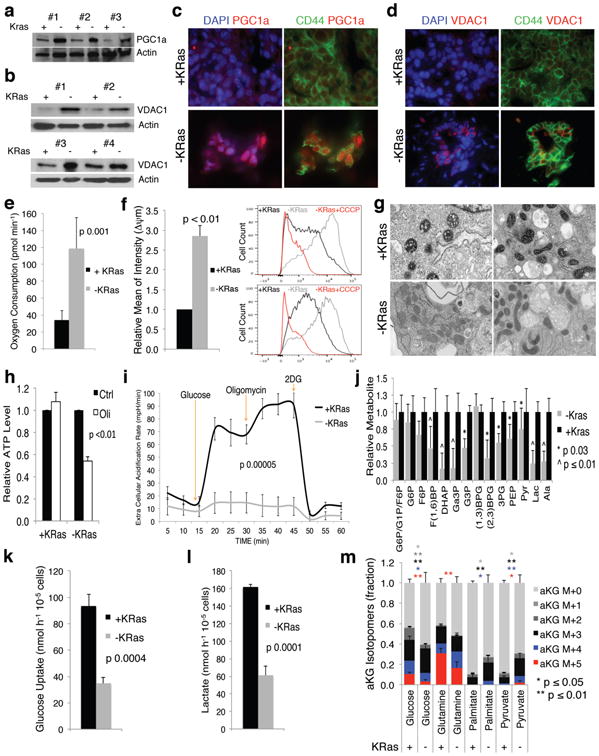
a-b, Immunoblot of +/-KRas cells probed with PGC1a (a) and VDAC1 (b) antibodies. c-d, In vivo immunofluorescence for CD44 (green), (c) PGC1a (red) and (d) VDAC1 (red) in +/-KRas tumors(60×). e, Oxygen consumption of +/-KRas cells (n=7). f, In vivo mitochondrial potential of +/-KRas tumors (n=3); representative flow-cytometry of two tumors, as control CCCP was added to acquired -KRas. g, Representative mitochondrial morphology in TEM (25000×). h, ATP levels of +/-KRas cells in response to oligomycin (Oli) or DMSO (Ctrl)(n=4). i, ECAR response of +/-KRas cells to glucose, oligomycin and 2DG (n=4). j, Fold change of glycolytic intermediates in +/-KRas cells (n=4). k-l, Glucose uptake (k) and lactate production (l) of +/-KRas cells (n=3). m, Isotopomer distribution for α-ketoglutarate following steady-state tracing with uniformly carbon-13-labeled substrates (n=3). Data are mean ± s.d.
We functionally validated our findings by measuring respiratory capacity. SCs had a four-fold increase in oxygen consumption rate (OCR) compared to KRas-expressing cells (118 vs. 33 pmol min-1, respectively, p=0.001;Fig.2e). Likewise, both in vivo and in vitro, mitochondria of SCs either genetic or pharmacological selected were more hyperpolarized (Fig.2f;EDfig.4c-f) and generated more reactive oxygen species (ROS)(EDfig.4g,h), indicating a more active electron transport chain (ETC). We also detected morphological differences in mitochondria using transmission electron microscopy (TEM)(Fig.2g). Because transmembrane mitochondrial potential regulates the mitochondrial permeability transition pore (lower potential=lower threshold for pore opening) and because cells positive for stem cell markers in KRas-expressing tumors have hyperpolarized mitochondria (EDfig.4i,j), the higher mitochondrial potential per se could explain why cells positive for stem cell markers are less prone to KRas ablation-mediated apoptosis. Altogether, our data support that altered metabolic and mitochondrial functions are critical features of SCs.
SCs and KRas-expressing cells were next exposed to mitochondrial stressors. Treatment with oligomycin, a Fo-ATPase inhibitor of Complex V, significantly reduced mitochondrial respiration in both populations (EDfig.5a). Conversely, normalization to basal respiration revealed different effects of the uncoupler FCCP (EDfig.5b), indicating that the mitochondria of SCs function near their maximal rate and have minimal spare respiratory capacity. Despite similar overall responses to Complex V inhibition, ATP levels of SCs were dramatically reduced upon oligomycin treatment compared to KRas-expressing cells (Fig.2h), suggesting a deficit in energy compensatory mechanisms. Indeed, KRas-expressing cells exposed to oligomycin strongly upregulated their extracellular acidification rate (ECAR) and lactate production, a compensation that did not occur in SCs (Fig.2i;EDfig.5c,e), confirming that SCs failed to increase glycolysis after OXPHOS inhibition in a manner sufficient to rescue the defects in ATP production.
To comprehensively assess differences in metabolism, we performed a metabolomic analysis using a LC-MS/MS based platform2, 13 revealing that several metabolic pathways were deregulated in SCs (EDfig.5d). Consistent with the above findings, glycolytic intermediates downstream of phospho-fructose kinase (PFK) were significantly less abundant in SCs versus Kras-expressing cells (Fig.2j). The impaired glycolysis of SCs was independently validated by measuring glucose uptake and lactate production in vitro (Fig.2k,l;EDfig.6a) and in vivo (EDfig.6b). The ratio between lactate and glucose in spent media supports even stronger this difference ([Lactate]/[Glucose]: KRas-expressing cells 16.9, SCs 0.9). Importantly, SCs surviving pharmacological ablation of KRas showed a similar phenotype (EDfig.6c,d).
We also detected decreased abundance of metabolic intermediates specific to the tricarboxylic acid (TCA) cycle in SCs (EDfig.5f). We used carbon-13 labeled primary metabolic substrates to trace their contribution to central carbon metabolism. After 36-hour labeling, SCs relied less on glucose and glutamine and more on pyruvate and palmitate to generate TCA cycle intermediates and branching metabolites (Fig.2m;EDfig.7e-h). This is consistent with reports describing the mutant KRas-mediated activation of anabolic glucose and glutamine metabolism in PDAC2,14-16. It is also worth noting that SCs had lower levels of high-energy metabolites (EDfig.5g)(compatible with less anabolic, dormant cells) and increased total glutathione (EDfigs.5g,8h). Importantly, any effect of cell cycle fluctuations on metabolic processes was excluded by our experimental design, as comparisons were made between quiescent SCs and quiescent fully formed KRas-expressing spheres (EDfig.1g). In fact we demonstrated that sphere formation is a dynamic and regulated process in which tumor cells expressing KRas exit the cell cycle when tumor spheres are fully formed. Thus, observed metabolic alterations can appropriately be attributed to an autonomous metabolic program.
The lower energy levels and a lack of energetic compensatory mechanisms in response to mitochondrial stressors in SCs suggested that treatment with OXPHOS inhibitors might impact their survival. As expected, even a short, 24-hour exposure to oligomycin completely abrogated the ability of SCs to reform tumor spheres upon KRas re-expression, whereas KRas-expressing cells were unaffected (Fig.3a;ED fig.8a). Similar effects were observed with other OXPHOS inhibitors, though with less dramatic effects relative to Complex V inhibitors (EDfig.8b). To determine whether OXPHOS inhibition synergized with KRas ablation in vivo, we induced tumor regression for two weeks via doxycycline withdrawal, treated mice with oligomycin (0.5mg/kg daily,IP) or vehicle, and subsequently re-induced KRas and tumor relapse (EDfig.8c-e). While mice treated with vehicle relapsed immediately (median survival 15 days, maximal survival 19 days), one fourth of oligomycin-treated mice survived >60 days (p <0.0001)(Fig.3b;EDfig.8f). Because OXPHOS inhibitors induce ROS17 and SCs have elevated basal mitochondrial ROS levels (Fig.S4g; ED fig.8g), we excluded that the observed synthetic lethality of oligomycin was caused by oxidative stress (EDfig.8h-k).
Figure 3. OXPHOS inhibition specifically targets surviving cells.
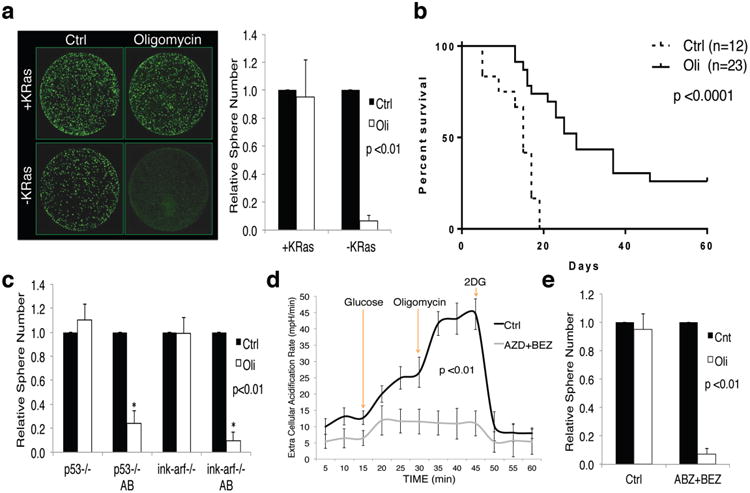
a, Effect of oligomycin (Oli) and DMSO (Ctrl) on spherogenic potential of +/-KRas cells (n=8), representative calcein staining. b, Kaplan-Meier overall survival after KRas reactivation in mice bearing regressed tumors treated two weeks with oligomycin or vehicle. c, Effects of oligomycin and DMSO on spherogenic potential of cells treated (AB) or not with AZD8330/BEZ235 in p53-/- and ink-arf-/- backgrounds (n=3). d, ECAR response of human tumor spheres treated (AZD+BEZ) and untreated (Ctrl) to glucose, oligomycin and 2DG (n=3). e, Effect of oligomycin and DMSO on spherogenic potential of human treated and untreated tumor cells (n=3). Data are mean ± s.d.
Oligomycin treatment also strongly abrogated the spherogenic potential of SCs treated with AZD8330+BEZ235 independently of their p53 status (Fig.3c).
To explore the therapeutic potential of inhibiting OXPHOS in human tumors, we derived spheres from early passage patient-derived xenografts18. Similar to the mouse model, combination treatment with AZD8330 and BEZ235 for one week strongly decreased sphere formation (EDfig.9a-c), sparing cells endowed with spherogenic potential, increased mitochondrial potential (EDfig.9d) and, most importantly, unable to up-regulate ECAR in response to OXPHOS inhibition (Fig.3d). As with mouse cells, short exposure of human SCs to oligomycin dramatically decreased their spherogenic potential (Fig.3e). Similarly, transient genetic down-regulation of TFAM (a key regulator of mitochondrial transcription19) and TUFM (a GTPase that delivers amino-acylated tRNAs to mitochondrial ribosomes20) decreased the spherogenic potential of human cells surviving combined AZD8330+BEZ235 treatment (EDfig.9e-h). Altogether, these data highlight mitochondrial respiration as an attractive, druggable target that may effectively eradicate SCs in PDAC.
The low energy state of SCs suggested that these cells may be experiencing metabolic stress. We thus measured AMPK phosphorylation at Thr172, an established indicator of metabolic stress. Surprisingly, AMPK phosphorylation was lower in SCs versus Kras-expressing cells (Fig.4a); however, marked induction of AMPK phosphorylation was observed specifically in SCs upon oligomicyn treatment, stressing again the importance of ETC activity for their energy production (Fig.4b;EDfig.9i).
Figure 4. Surviving cells are not in metabolic stress and activate autophagy.
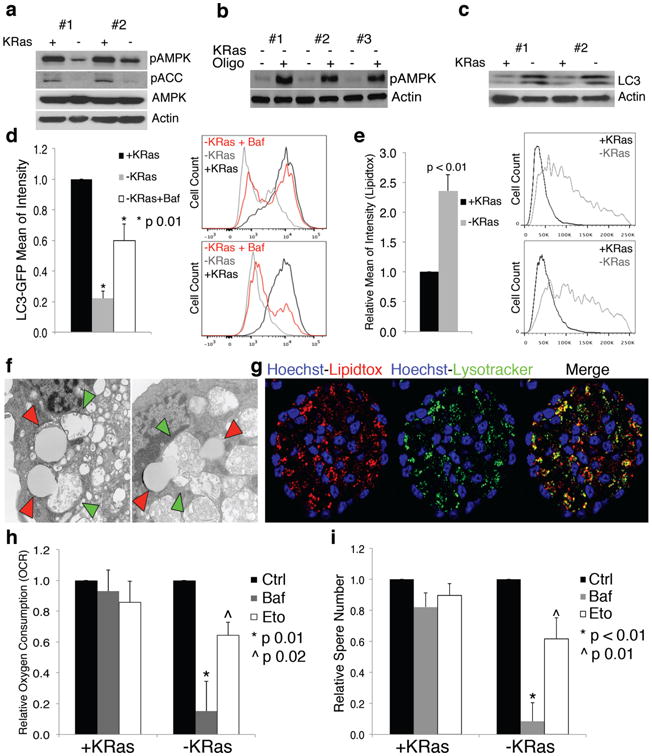
a, Immunoblot of Thr172-phosphorylated and total AMPK, Ser79-phospho-acetyl-coA-carboxylase (pACC) and β-actin in +/-KRas cells. b, Immunoblot of -KRas cells treated or not with oligomycin probed with anti-Thr172-phospho-AMPK and β-actin antibodies. c, Immunoblot of LC3 and β-actin in +/-KRas cells. d, Autophagic flux of +/-KRas cells stably expressing GFP-LC3. Bafilomycin treatment rescues GFP (-KRas+Baf) (n= 3); representative flowcytometry of two tumors. e, Lipid droplet quantification in +/-KRas cells (n=4); representative flowcytometry of two tumors. f, Fusion between lipid droplets (red arrowheads) and autophagosomes (green arrowheads) in -KRas cells (TEM-25000×). g, Confocal microscopy for lipid droplets (Lipidtox-red), lysosomes (Lysotracker-green), Hoechst (blue) in -KRas spheres. h, Oxygen consumption of +/-KRas cells pretreated with bafilomycin, etomoxir or vehicle (n= 4). i, Effect of bafilomycin and etomoxir on spherogenic potential of +/-KRas cells (n=6). Data are mean ± s.d.
To further elucidate the metabolic mechanisms active in SCs, we investigated other targets identified by transcriptomic analysis, including autophagy and β-oxidation. Autophagy and lysosome activity, two of the most activated pathways based on GSEA (Fig.1i), have a significant role in the metabolism of SCs. The autophagic marker microtubule-associated protein light-chain 3 (LC3) was abundantly expressed in SCs (Fig.4c), and lysosomes and autophagosomes at various stages of evolution were abundant (EDfig.10a). To investigate the activation of autophagic flux, we transduced KRas-expressing tumor spheres with LC3-GFP fusion protein. Cells stably expressing LC3-GFP were maintained in the presence or absence of doxycycline and subsequently analyzed by flow cytometry. As shown in Fig.4d, SCs had dramatically reduced GFP signal (80%) compared to KRas-expressing cells, indicating GFP was quenched upon autolysosome formation, as also confirmed by the treatment with bafilomycin, a vacuolar H+-ATPase inhibitor (Fig.4d).
Upon TEM analysis, we observed increased content of cytoplasmic lipid droplets in SCs (EDfig.10b) that we quantified with neutral lipid-specific dyes (Fig.4e). Importantly, and unique to SCs, we observed unambiguous evidence of microlipophagy21, the fusion of lipid droplets with autophagosomes (Fig.4f), which we confirmed by demonstrating strong co-localization of lipid droplets and lysosomes upon staining in SCs (Fig.4g). Consistent with observations that SCs catabolize more endogenous protein and fatty acid, tracing metabolic experiments illustrate that more carbon in central metabolism is unaccounted for from labeled substrates in SCs versus KRas-expressing cells (EDfig.7e-h). To determine whether autophagy and microlipophagy may represent a strategy for SCs to maintain their energetic balance22,23, we explored their contribution in maintaining mitochondrial activity by measuring OCR in cells pre-treated with autophagic and β-oxidation inhibitors. Neither inhibitor effected OCR in KRas-expressing cells, while treatment with either bafilomycin or etomoxir both markedly reduced OCR in SCs (85% and 35%, respectively)(Fig.4h). Importantly, both inhibitors also induced metabolic stress specifically in SCs (EDfig.9j,k) and dramatically affected both their spherogenic potential (decrease >90% and 39% respectively, p ≤0.01)(Fig.4i) and survival (EDfig.9l). We therefore conclude that autophagy and microlipophagy are as critical as ETC activity for the survival of SCs.
For the first time, we isolated and characterized tumor cells surviving the genetic and pharmacologic ablation of oncogenic pathways, demonstrating they have features of TICs and a distinct metabolic profile. Unlike highly glycolytic tumor cells depending on lactate production to regenerate NAD+ to support continued glycolysis for their anabolic metabolism (Warburg effect), SCs are less glycolytic and more dependent on mitochondrial respiration. Their dependence on OXPHOS for energy production is consistent with recent reports describing that both normal and leukemic stem cells rely on mitochondrial respiration20,24-26. Importantly their strong dependence of ETC activity on autophagic and catabolic processes makes SCs more resistant to nutrient deprivation and environmental stressors. However, their inability to increase compensatory fluxes (namely, glycolysis) following OXPHOS inhibition confers extreme sensitivity to inhibition of mitochondrial function that dramatically impacts their spherogenic and tumorigenic potential. Our findings hold important implications for tumor treatment, paving the way for targeting OXPHOS in association with oncogenic pathway inhibitors to eradicate SCs and prevent tumor relapse in pancreatic cancer.
Methods
Mice
The inducible tetO-LSL-KRasG12D, ROSA26-LSL-rtTA, p53LoxP/WT, p48-Cre mouse model was generated as described2. After weaning, mice were fed doxycycline (2g/l) in drinking water supplemented with sucrose (20g/l) and monitored for tumor development. Constitutive LSL-KRasG12D, p53LoxP/LoxP, Pdx1-Cre and LSL-KRasG12D, Ink4a/ArfLoxP/LoxP, Pdx1-Cre mice were generated as previously reported8. All animals were kept in FVB/C57Bl6 mix or pure FVB backgrounds in a pathogen-free facility at the University of Texas MD Anderson Cancer Center. All manipulations were performed under IACUC-approved protocols.
Antibodies, plasmids and chemical reagents
Primary antibodies used for flow cytometry, immunofluorescence, immunohistochemistry and immunoblotting were: CD44 (eBioscience, IM7), CD133 (eBioscience, 13A4; Abcam, 165118), phospho-p44/42 (Cell Signaling, D10), VDAC1 (Abcam, ab15895), PGC1a (Abcam, ab54481), BrdU (Abcam, ab82421), Ras (05-516, Millipore), LC3A/B (Cell Signaling, 4108), Phospho-AMPKα (Thr172) (Cell Signaling, 40H9), AMPKα (Cell Signaling, D63G4), TFAM (Cell Signaling, D5C8), TUFM (Abcam, ab175199), β-Actin (sc-1615, Santa Cruz). Plasmids: pBABEpuro GFP-LC327 (Addgene plasmid 22405), pLKO TFAM shRNAs (TRCN0000016093 5′- CCGG-CGTGAGTATATTGATCCAGAA-CTCGAG-TTCTGGATCAATATACTCACG-TTTTT-3′; TRCN0000016095 5′-CCGG-GTAAGTTCTTACCTTCGATTT-CTCGAG-AAATCGAAGGTAAGAACTTAC-TTTTT-3′) and pLKO TUFM shRNAs (TRCN0000280863 5′- CCGG-GCTCACCGAGTTTGGCTATAA-CTCGAG-TTATAGCCAAACTCGGTGAGC-TTTTTG-3′; TRCN0000280864 5′-CCGG-GAGGACCTGAAGTTCAACCTA-CTCGAG-TAGGTTGAACTTCAGGTCCTC-TTTTTG-3′) (Sigma), Tet-inducible pLKO was provided by Institute for Applied Cancer Science at MDACC. Chemical reagents: oligomycin A (Sigma), buthioninesulphoximine, (Sigma), 4-hydroxynonenal (Cayman Chemical), α-tocopherol (Sigma), n-acetylcysteine (Sigma), tetrakis (Calbiochem), bafilomycin (Sigma), etomoxir (Sigma), dicyclohexylcarbodiimide (Sigma), venturicidin A (Sigma), rotenone (Seahorse), antimycin (Seahorse), doxycycline (Research Product International), AZD8330-AZD6244-BEZ235 (Selleckchem).
Tumor culture
Patient-derived xenograft tumors were generated transplanting subcutaneously in mice small tumor fragments isolated directly from surgical specimens according to Kim et al18. Patient-derived samples were obtained from consented patients under IRB-approved protocol LAB07-0854 chaired by J.B.F. Cells derived from early passage patient-derived xenograft tumors (F1-F2) and primary mouse tumors were kept in culture as spheres in semi-solid media for <15-16 passages. Briefly, after explant, tumors were digested at 37° C for one hour (Collagenase IV-Dispase 4 mg/ml; Invitrogen). Single-cell suspensions were plated in stem cell medium (SCM) MEBM (Lonza) supplemented with 2 mM glutamine (Invitrogen), B27 (Invitrogen), 20 ng/ml hEGF (PeproTech), 20 ng/ml hFGF (PeproTech), 5 µg/ml h-Insulin (Roche), 0.5 µM hydrocortisone (Sigma), 100 µM β-mercaptoethanol (Sigma), 4µg/ml heparin (Sigma). Methocult M3134 (Stemcell Technologies) was added to SCM (final concentration 0.8%) to keep tumor cells growing as clonal spheres versus aggregates. Fully formed tumor spheres were collected and digested with 0.05% trypsin (Gibco) to single cells and re-plated in culture. KRas inducible system: for maintenance, tumor cells were kept in doxycycline (2 µg/ml). For experiments, cells were plated in the presence or absence of doxycycline to select SCs. After 8 days in culture, tumor spheres or SCs were collected, washed and processed for experiments.
In vitro treatments
For drug treatments, KRas-expressing spheres or surviving tumor cells after 8 days of doxycycline withdrawal were collected, washed, digested with trypsin and repeatedly counted (Countless, Invitrogen). Similar procedures were used for spheres derived from human tumors and from the KRas constitutive mouse model except that surviving cells were selected treating tumor spheres with AZD8330 (10 nM) and BEZ235 (100 nM) for 1 week. Equal numbers of live KRas-expressing cells and surviving tumor cells (AZD8330+BEZ235 treated cells for the constitutive KRas and human tumors) were treated with oligomycin (200 nM for 24hs), venturicidin (500 nM for 24hs), DCCD (1 µM for 24hs), bafilomycin (50 nM for 48hs) or etomoxir (100 µM for 48hs). For OCR measurement and western-blot experiments, cells were treated with bafilomycin and etomoxir for 24 and 6 hours, respectively. To test the effects of ROS on spherogenic potential, cells were treated with buthionine-sulphoximine (100 µM) to deplete GSH or pretreated with α-tocopherol (100 µM), n-acetylcysteine (1 mM) or Mn-tetrakis (50 µM) before oligomycin treatment. After drug wash out, treated cells were re-plated for 7 days (on doxycycline when using the KRas-inducible system). The number of tumor spheres (spherogenic potential) was quantified using ImageXpress Velos Laser Scanning Cytometer (Molecular Devices) upon calcein staining (Molecular Probes).
Tumor transplantation, transplantation in limiting dilution and in vivo drug treatments
Tumor cells isolated from in vivo tumors or from in vitro spheres were digested to single cells (see Tumor culture). Usually, 104-105 tumor cells were used for routine transplantation, instead for transplantation in limiting dilution were used 103, 102 or 10 tumor cells. Tumor cells were suspended in stem cell medium (SCM, see Tumor culture) and Matrigel (BD Biosciences, 356231) (1:1 dilution) and injected subcutaneously into the flank of 6- to 8-week-old female immunodeficient mice (NCR-NU, Taconic). If cells were derived from KRas-inducible tumors, mice were injected with doxycycline (3 mg/kg, IP) at the time of transplantation and then fed with doxycycline in drinking water. Tumor-initiating cell (TIC) frequencies were determined by Poisson statistical analysis using L-Calc software (Stemcell Technologies). For BrdU incorporation experiments, mice were injected IP with 1 mg of BrdU 3 times (every 8 hours) after 24hs or 48hs of KRas reactivation before being sacrificed. For In vivo oligomycin treatment, mice were transplanted with tumor cells and fed with doxycycline in drinking water (+Dox) until tumors reached 1 cm in diameter. At that time, doxycycline was withdrawn (-Dox) and, after 2 weeks, when tumors were almost completely regressed, mice were injected with oligomycin (0.5 mg/kg, IP, Sigma) or vehicle, 5 days a week, for two weeks. After treatment, doxycycline was added back to drinking water and mice were monitored for tumor relapse. Genetically identical and age matched recipient mice were used for transplantation experiments and were randomly allocated for treatment with oligomycin upon tumor regression. Experimenters were not blinded to the experimental groups in evaluating treatment outcome.
For pharmacologic inhibition of KRas pathways using small molecule inhibitors, mice bearing tumors derived from the KRas constitutive system were treated with a combination of AZD6244 and BEZ235 (100 mg/kg and 40 mg/kg respectively, per oral gavage daily) for at least 7 days. Tumor volume was calculated using the formula: V=l2*L/2 (l length; L width). All manipulations were performed under IACUC-approved protocols.
Isolation of tumor cells
To isolate pure populations of tumor cells from transplanted tumors, we took advantage of dim-high ubiquitous expression of CD44 in pancreatic tumor cells (EDfig.2f,g). Single-cell suspensions of digested tumors were stained with anti-CD44 biotinylated antibody (eBioscience, IM7). Positive cells were then purified using Easy Sep Biotin Selection Kit (Stemcell Technologies) according to manufacturer's instructions. Counterstaining of isolated cells with anti-CD45 and CD31 antibodies and fluorochrome-conjugated streptavidin were used to check the purity by flowcytometry.
shRNA expression and gene down-regulation
shRNAs against TFAM (TRCN0000016093; TRCN0000016095) and TUFM (TRCN0000280863; TRCN0000280864) (Sigma) were cloned in a Tet-inducible pLKO vector provided by Institute for Applied Cancer Science at MDACC (for sequences see also antibodies, plasmids and chemical reagents). Human tumor spheres were transduced with viral particles and selected with puromycin. Upon selection, tumor spheres were treated or not with a combination of Mek/Pi3K inhibitors (10 nM AZD83330 plus 100 nM BEZ235) for one week. On the second day after beginning the combination drug treatment, doxycycline was added to the culture to induce shRNA expression and was maintained for 5 days. Then tumor cells were collected, washed, and replated to evaluate their spherogenic potential. Down regulation of the target was evaluated by western blot at 72hs after shRNA induction.
Flow cytometry, cell-sorting, immunohistochemistry, immunofluorescence, immunoblotting analysis and pull-down assay
Flowcytometry and cell-sorting
Single cells isolated from tumors or spheres were stained with primary antibodies after blocking with 10% BSA and 5% rat serum. Aldefluor (Stemcell Technologies) and AnnexinV (eBioscience) staining were performed according to manufacturer's instructions. To study the cell cycle of tumor spheres, BrdU Flow Kit (BD Pharmingen) was used according to datasheet specifications. Mitochondrial potential in vitro was measured using MitoProbe DilC1(5) Assay Kit for Flow Cytometry (Molecular Probes) according to specifications, and CCCP treatment was used for controls. In vivo mitochondrial potential was evaluated according to Zheng et al28. Briefly, mice bearing KRas-expressing or regressed tumors (inducible model or pharmacologically treated with AZD6244+BEZ235) were injected with 25 nmol/kg of TMRE (Molecular Probes) as a tail vein bolus. After one hour, mice were sacrificed and tumors explanted, digested (as described above in tumor culture adding 10% FBS to the digestion mix) and stained for CD44. Samples were kept on ice and immediately acquired gating CD44-positive cells. The same approach and timeline were used to evaluate glucose uptake in vivo, injecting 25 µmol/kg 2NBDG (Molecular Probes) as a tail vein bolus according to Ytoh et al29. For in vitro experiments, 2NBDG was used at a concentration of 10 µM in complete stem cell medium containing 2 mM glucose29. Cells were incubated for 6 hours then washed and analyzed by flow cytometry. MitoTracker Green and Deep Red (Molecular Probes) were utilized to measure mitochondrial mass. ROS were evaluated using MitoSOX red (Molecular Probes) and induced by 4-hydroxynonenal treatment (10 µM) for positive controls. LipidTox deep red (Molecular Probes) was used for quantifying lipid droplet content. All staining procedures were performed according to manufacturer's protocols. DAPI (Invitrogen) was used to stain DNA content or to exclude dead cells depending on the experiment. For measuring autophagic flux, KRas-expressing cells were transduced with pBABEPuro GFP-LC327 and, upon selection, doxycycline was withdrawn for 8 days. Mean of fluorescence of surviving and matched KRas-expressing cells was quantified and surviving tumor cells treated for 24 hs with bafilomycin (50nm) were used as a control. Gating strategies to exclude doublets and dead cells (DAPI) were always employed. After staining, samples were acquired using a BD FACSCantoII flow cytometer or sorted using BD Influx cell sorter. Data were analyzed by BD FACSDiva or FlowJo (Tree Star).
ImmunoHistoChemistry/ImmunoFluorescence
Tumor samples were fixed in 4% formaldehyde for 2 to 4 hours on ice, moved in 70% ethanol for 12 hours, and then embedded in paraffin (Leica ASP300S). After cutting (Leica RM2235), baking and deparaffinization, slides were treated with Citra-Plus Solution (BioGenex) according to specifications. For IHC staining, endogenous peroxidases were inactivated by 3% hydrogen peroxide. Non-specific signals were blocked using 3% BSA, 10% goat serum and 0.1% triton. Tumor samples were stained with primary antibodies. For BrdU detection, samples were digested on slides for 1 hour at 37° C with DNAse I (300 µg/ml) before staining. For IHC, ImmPress and ImmPress-AP (Vector Lab) were used as secondary antibodies and Nova RED, Vector BLUE and DAB were used for detection (Vector Lab). Images were captured with a Nikon DS-Fi1 digital camera using a wide-field Nikon EclipseCi microscope. For immunofluorescence, secondary antibodies conjugated with Alexa488 and 555 (Molecular Probes) were used. Images were captured with a Hamamatsu C11440 digital camera, using a wide-field Nikon EclipseNi microscope. LipidTox, Lysotracker, MitoTracker, CellRox and Hoechst 33342 (Molecular Probes) were used on live spheres and cells at the concentrations suggested by manufacturer's protocols and images were acquired using a Nikon high-speed multiphoton confocal microscope A1 R MP.
Immunoblotting
Protein lysates were resolved on 5-15% gradient polyacrylamide SDS gels and transferred onto PVDF membranes according to standard procedures. Membranes were incubated with indicated primary antibodies, washed, and probed with HRP-conjugated secondary antibodies. The detection of bands was carried out upon chemiluminescence reaction followed by film exposure.
Ras pull-down assay
The amount of active Ras was evaluated by detecting the fraction of Ras protein that co-precipitated with RAF kinase. Cell lysates from KRas-expressing cells and cells surviving KRas ablation were co-incubated with RAF-linked agarose beads for 2 hours. After incubation, beads were collected, washed and boiled for 5 minutes in the presence of laemmli loading buffer supplemented with 10% 2-mercapthoethanol and ultimately loaded onto SDS-PAGE gels. The detection of the active fraction of RAS was carried out using standard western blot procedures with anti-Ras antibody.
Transmission electron microscopy
TEM was performed at the UT MDACC High Resolution Electron Microscopy Facility. Samples were fixed with a solution containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.3, for 1 hour. After fixation, the samples were washed and treated with 0.1% Millipore-filtered cacodylate buffered tannic acid, post-fixed with 1% buffered osmium tetroxide for 30 min, and stained en bloc with 1% Millipore-filtered uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol, infiltrated and embedded in LX-112 medium. The samples were polymerized in a 60 C oven for 2 days. Ultrathin sections were cut in a Leica Ultracut microtome (Leica, Deerfield, IL), stained with uranyl acetate and lead citrate in a Leica EM Stainer and examined in a JEM 1010 transmission electron microscope (JEOL, USA, Inc., Peabody, MA) at an accelerating voltage of 80 kV. Digital images were obtained using an AMT Imaging System (Advanced Microscopy Techniques Corp, Danvers, MA).
DNA, RNA, cDNA and qPCR
DNA and RNA were extracted using DNeasy Blood and Tissue Kit (Qiagen) and RNeasy Mini Kit (Qiagen) according to technical specifications. A mix of random hexamers and oligo(dT) were applied for cDNA synthesis using SuperScript III First-Strand-Synthesis System (Invitrogen). For qPCR, 10 ng of DNA or cDNA was amplified with EXPRESS SYBR GreenER qPCR SuperMix (Invitrogen) using a Startagene Mx3005p thermal-cycler. Primers used for mitochondrial and lipid metabolic gene amplification are shown in EDfig.10c. β-actin (F-GACGGCCAGGTCATCACTAATTG, R-AGGAAGGCTGGAAAAGAGCC), 28S (F-TCATCAGACCCCAGAAAAGG, R-GATTCGGCAGGTGAGTTGTT) and β2-microglobulin (F-ATTCACCCCACTGAGACTG, R-TGCTATTTCTTTCTGCGTGC) were used as house-keeping genes for normalization. Expression of genes involved in the ETC, mitochondria and autophagy was evaluated using Qiagen commercial arrays: RT2 Profiler PCR Array Mouse Mitochondrial Energy Metabolism, Mouse Mitochondria and Mouse Autophagy.
Expression profiling and data analysis
Gene expression profiling was performed at the Dana-Farber Cancer Institute Microarray Core Facility. RNA isolated from KRas-expressing and surviving tumor cells was hybridized on a Gene Chip Mouse Genome 430 2.0 Array (Affymetrix). Raw data (CEL files) were pre-processed using a robust multi-array analysis (RMA) and analyzed with GSEA30 using Signaling Pathways c2.cp.v3.0 and TFT c3.tft.v3.0 symbols gene sets. Complete profiles are available at GEO at GSE58307.
Exome sequencing
Two independent tumor spheres expressing KRas were collected, digested to single cells, and replated on two plates in the presence of doxycycline (ON DOX, +KRas) for one week. Spheres from one plate were collected and snap frozen for DNA extraction (REFERENCE), while spheres from the other plate were plated OFF doxyclycline (-KRas) for one week prior to re-addition of doxycycline to re-express KRas. After one week, reformed spheres were collected and snap frozen for DNA extraction (RE-ON). Genomic DNA was extracted by phenol-chloroform and further purified from contamination on columns (Qiagen). The exonic DNA regions were captured using Nimblegen SeqCap EZ Mouse Exome kit. The DNA sequences recovered were processed through a standard SNP calling pipeline. Reads were aligned using BWA following removal of duplicates, realignment and recalibration (using Broad's Genome Analysis Toolkit or GATK). Further SNPs were called using GATK's Unified Genotyper and annotated using Annovar. For the analysis we considered SNVs which were 1) called in both samples, and 2) had a minimum coverage of 200 in both, providing enough confidence to compare allelic frequencies between the sample pairs. Roughly 12% (40383/335076) and 13% (44182/339398) of the common SNVs for Tumor #2 and Tumor #1, respectively, satisfied the coverage threshold of 200.
Metabolomics
For metabolomic analyses, surviving and KRas-expressing spheres (grown as reported above in Tumor Culture), were collected after 8 days of culture when both KRas-expressing spheres and surviving tumor cells were confirmed to have exited the cell cycle (to avoid the confounding effects of proliferation on metabolism) (EDfig.1g). The day before collection, medium was changed and spheres re-plated in fresh medium. After 24 hours, cells were collected by centrifugation, washed three times and samples were then immediately lysed in methanol:water (80:20) at dry-ice temperature. The quantity of the metabolite fraction analyzed was adjusted to the corresponding protein concentration calculated upon processing a parallel sample. Metabolite fractions were processed and analyzed by targeted LC-MS/MS via selected reaction monitoring (SRM), as described2,13,15. Processed data were analyzed in Cluster 3.0 and TreeViewer. The analysis was performed on four independent tumors in biological triplicate
Carbon-13 metabolic tracing
Cells were prepared in a manner identical to that for routine metabolomic profiling (see above), except that media formulations were adjusted to account for isotopically labeled substrates. Specifically, surviving and KRas-expressing cells were plated in RPMI medium (reconstituted with growth factors) devoid of glucose, glutamine, pyruvate, palmitate and supplemented with one of the four carbon-13-labeled substrates where the remaining substrates were unlabeled (glucose 10 mM, glutamine 2 mM, pyruvate 1 mM and palmitate 75 µM). After 24 hours of incubation with labeled substrates, cells were collected and replated again in fresh media supplemented with carbon-13-labeled substrates for another 12 hours to minimize the effects of cellular uptake on the concentration of different substrates. After 36 total hours of labeling, samples were collected by centrifugation and immediately lysed in methanol:water (80:20) at dry-ice temperature. As for metabolomics, the quantity of the metabolite fraction analyzed was adjusted to the corresponding protein concentration from a sample processed in parallel. The analysis was performed for each of the four substrates on three independent tumors in biological triplicate. Data were collected and processed as above, as described2,13,15.
Oxygen consumption, glycolytic capacity, glucose uptake, lactate production, ATP and glutathione assays
Oxygen consumption and glycolytic capacity
Tumor spheres and surviving cells were digested to single cells and spun into XF96 Cell Culture Microplates (Seahorse Bioscience) previously treated with Cell-Tak (BD Biosciences) immediately before the experiment. To measure OCR and the response to OXPHOS inhibition, we plated cells in complete stem cell medium (see Tumor culture) prepared with MEBM lacking NaHCO3 (Lonza) and supplemented with 5 mM pyruvate (Sigma). Oligomycin, FCCP, antimycin and rotenone (XF Cell Mito Stress Test Kit, Seahorse Bioscience) were sequentially injected at a final concentration of 1 µM. To measure ECAR, we plated tumor cells in base assay medium (D5030, Sigma) adjusted to pH 7.4. Glucose (10 mM final concentration, Sigma), oligomycin (1 µM final concentration) and 2-deoxy-glucose (100 mM final concentration) were sequentially injected (XF Glycolysis Stress Test Kit, Seahorse Bioscience). Specifically for the experiment in EDfig.5c, ECAR was measured in complete stem cell media to evaluate the glycolytic reserve of tumor cells in a nutrient-rich environment. Oligomycin and 2DG were sequentially injected at a final concentration of 1 µM and 100 mM, respectively. Experiments were run using a XF96 analyzer and raw data were normalized to metabolically active cells, evaluated as Hoechst 33342 positive/Propidium Iodide-negative, by an Operetta High-Content Imaging System (PerkinElmer) immediately after each experiment.
Glucose uptake and lactate production
To measure glucose uptake, we used two different approaches: by flow cytometry using the fluorescent glucose analog 2NBDG (see above Flowcytometry and cell-sorting) and by YSI 2950 Biochemistry Analyzer (YSI Life Sciences). For YSI, the same number of surviving cells and KRas-expressing cells was plated in triplicate in a 96-well plate in 200 µl of complete stem cell medium. After 24 hours, the medium was collected and glucose and lactate concentrations evaluated by YSI analyzer, using unconditioned fresh medium as a reference. Lactate production in response to oligomycin treatment (200 nM) was independently measured by Lactate Kit (Trinity Biotech) to directly evaluate the concentration of lactic acid in stem cell medium 6 to 12 hours after tumor cell plating.
ATP and glutathione assays
ATP production of tumor cells in response to 6 to 12 hours of oligomycin treatment (200 nM) was measured using Cell Titer Glo (Promega) following manufacturer's instructions. For glutathione, cells were lysed by sonication in 1X GSH MES buffer (Cayman Chemical) and debris pelleted by high-speed centrifugation. Samples were deproteinized by adding vol:vol of meta-phosphoric acid (1 mg/ml) followed by centrifugation and pH equilibration by the addition of triethanolamine. Glutathione content was evaluated through an enzymatic recycling method by using a commercially available kit (Cayman Chemical). PHERAStar plus microplate reader (BMG Labtech) was used to measure luminescence and absorbance.
Statistical Analysis
In vitro and in vivo data are presented as the mean ± s.d. (standard deviation). Statistical analyses were performed using a two-tailed Student's t-test after the evaluation of variance. Results from survival experiments were analyzed with a Gehan-Breslow-Wilcoxon test and expressed as Kaplan-Meier survival curves.
Extended Data
Extended Data Figure 1. Oncogene ablation leads to tumor regression in vitro and in vivo.
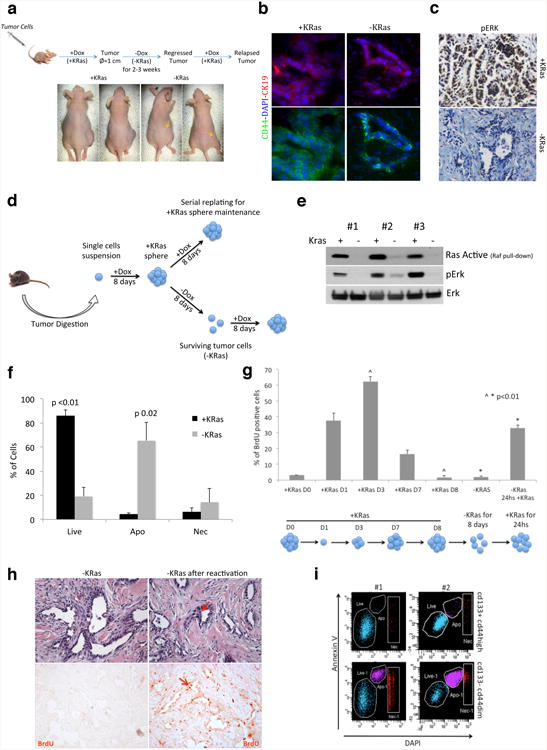
a, In vivo experimental scheme. Tumor cells isolated from primary tumors or tumor spheres were injected in nude mice fed with doxycycline in drinking water (+Dox). When mice developed tumors, doxycycline was withdrawn (-Dox) and tumors underwent a complete macroscopic regression after 2-3 weeks (arrows indicate regressed tumors). In residual lesions few tumor cells remain quiescent for months and they can quickly reform tumors upon KRas reactivation (+Dox). b-c, Tumor expressing KRas (+KRas) and tumor remnants after regression (-KRas) are positive for ductal epithelial marker CK19 (b) (40×). Tumor expressing KRas (+KRas) and epithelial remnants after tumor regression (-KRas) were stained for phosphorylated-p42/44 (pErk). No signal is detected in surviving cells (c) (20×). d, In vitro experimental scheme. After digestion to a single cells suspension, tumor cells isolated from primary tumors were plated in stem cell medium in presence of doxycycline (+Dox, +KRas). Spherogenic cells form tumor spheres (+KRas) that can be maintained by serial replating in presence of doxycycline. Upon doxycycline withdrawal (-Dox) tumor spheres undergo involution and only a minority of cells survives the ablation of KRas (surviving cells, -KRas). Surviving cells readily reform tumor spheres upon re-activation of KRas (+Dox). e, The amount of active Ras in KRas expressing cells (+KRas) and surviving cells (-KRas) has been evaluated in three independent tumor spheres by detecting the fraction of Ras protein that co-precipitates with Raf kinase. Total lysates were probed with anti-phospho-p42/44 (pErk), total p42/44 (Erk) antibodies. f, AnnexinV staining in tumor spheres after 3 days +/-KRas (n=3). g, Sphere formation is a regulated process and tumor cells enter and exit cell cycle. BrdU incorporation (pulse of 3 hours) has been evaluated at different time points during sphere formation and regression. KRas expressing fully formed spheres (Day 0 and 8) are quiescent. Upon sphere dissociation and replating (D0), spherogenic cells enter cell cycle (D1) and tumor cells continue to grow till day 3-4, when spheres reach their maximal S-phase. Then tumor cells gradually exit the cell cycle and become quiescent (D8). After doxycycline withdrawing (-KRas) tumor spheres undergo involution and surviving cells remain quiescent till KRas is re-expressed (-KRas 24hs +KRas) and spheres are reformed. Ruling out the effect of the cell cycle, transcriptomic and metabolomic characterizations have been done matching quiescent surviving tumor cells to quiescent fully formed KRas expressing spheres at D8 (n=3). h, HE and IHC of regressed tumors after three 8-hour pulses of BrdU show that epithelial remnants in regressed tumors after KRas ablation (-KRas) are completely quiescent (left panels). 48 hours after KRas reactivation (doxycycline IP injection) tumor cells re-enter massively the cell cycle (right panels). Red arrows indicate mitotic cells (20×). i, Representative AnnexinV staining with respect to CD133 and CD44 after three days of KRas ablation, two independent tumors are represented. Data are mean ± s.d.
Extended Data Figure 2. Transplantation in limiting dilution and characterization of epithelial remnants.
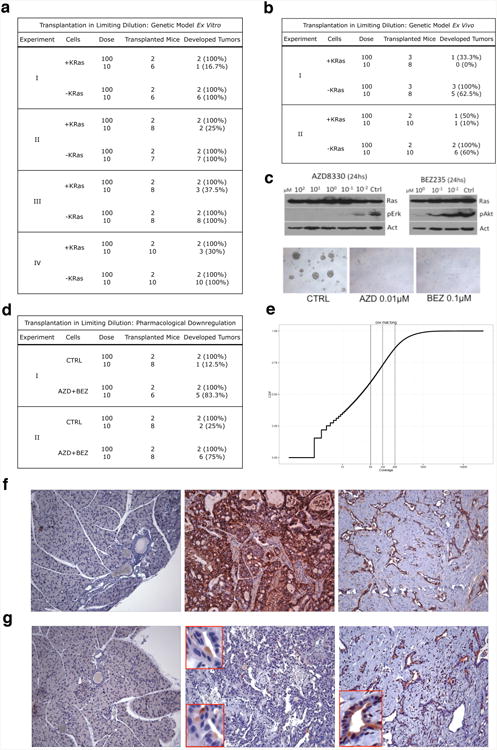
a-d, Transplantation in limiting dilution. Experiments, number of transplanted mice and percentage of developed tumor are shown. a, Limiting dilution experiments using tumor spheres (+KRas) and surviving cells (-KRas) (genetic model ex vitro). b, Limiting dilution experiments using cells isolated from KRas expressing (+KRas) and regressed tumors (-KRas) (genetic model ex vivo). c, Upper panels: immunoblots of tumor spheres treated with different concentrations of Mek1 (AZD8330) and a dual PI3K/mTOR (BEZ235) inhibitors probed with anti-phospho-p42/44 (pErk), phospho-Akt (pAkt), pan- Ras (Ras) and β-actin (Act) antibodies. Lower panels: Effects of AZD8330 (AZD 0.01µM) and BEZ235 (BEZ 0.1µM) treatment for 1 week on tumor sphere formation, some cells, as single or in small clusters, are able to survive the treatment (5×). d, Limiting dilution experiments using cells surviving pharmacologic downregulation of oncogenic pathways (AZD+BEZ, combination of AZD8330 and BEZ235) and control cells (CTRL). e, The plot shows the cumulative distribution of coverage at all the SNVs called by Unified Genotyper (across samples). f, CD44 is expressed during tumorigenesis in mouse: no positive cells are detected in normal pancreas (left panel), KRas expressing tumors express high level of CD44 (middle panel), epithelial remnants in regressed tumors maintain their positivity for CD44 (right panel)(10×). g, Validation of CD133 (ab16518) in IHC: this antibody does not recognize cells and ductal structures in normal pancreas (left panel), a small population of cells is stained by ab16518 in KRas expressing tumors (middle panel), epithelial remnants in regressed tumors are strongly positive for CD133 (right panel) (10×). At higher magnification (red boxes) is possible to appreciate the classical polarized pattern of CD133.
Extended Data Figure 3. QPCR validation of pathways enriched in surviving cells.
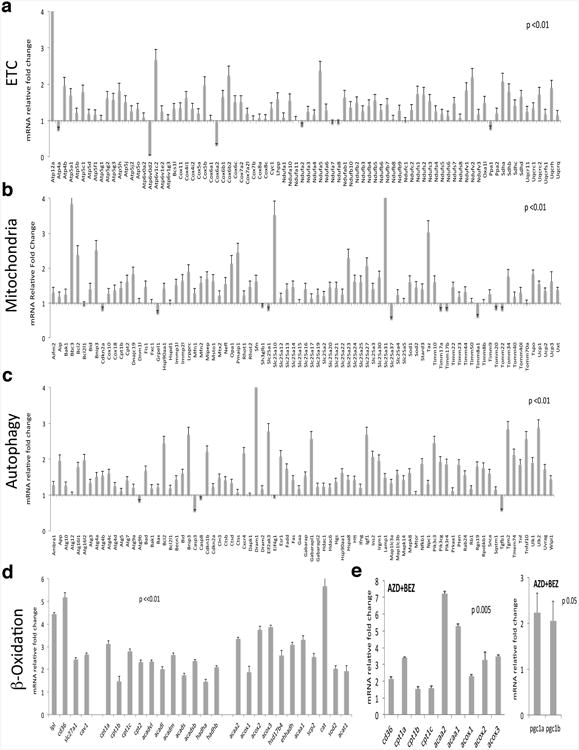
mRNA fold change in surviving cells normalized to KRas expressing cells. a, Genes involved in electron transport chain (ETC) (n=5). b, Genes involved in the biogenesis and function of mitochondria (Mitochondria). Genes encoding proteins of the autophagic molecular machinery and its key regulators (n=5) (c) and (β-Oxidation (n=5) (d). e, mRNA fold change in cells surviving AZD8330/BEZ235 treatment (AZD+BEZ) versus controls (n=3). Data are mean ± s.d.
Extended Data Figure 4. Surviving cells have more active mitochondria.
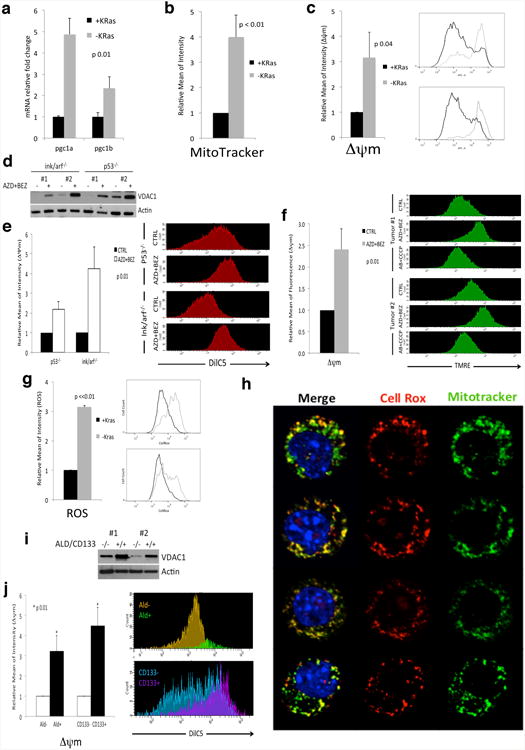
a, mRNA fold change of PGC1 genes in -KRas versus +KRas cells (n=5). b, Quantification of MitoTracker Green staining in +KRas and -KRas cells (n=3). c, Mitochondrial membrane potential (Δψm) of +KRas and -KRas cells (n=4); representative flow-cytometry analysis of two tumors. d, Immunoblot of two independent tumor spheres derived from different genetic backgrounds (ink/arf-/- and p53-/-) treated or not with AZD8330 and BEZ235 (AZD+BEZ) for 7 days and probed with anti-VDAC1 (VDAC1) and (β-actin (Actin) antibodies. e, Cells surviving AZD+BEZ treatment have higher mitochondrial transmembrane potential (Δψm) than untreated cells (CTRL) (n=3); representative flow-cytometry analysis is reported. f, Mice bearing tumors have been treated (AZD+BEZ) or not (CTRL) with a combination of AZD6244 and BEZ235 for one week. Upon tail vein injection of a bolus of TMRE tumors were explanted and analyzed by flow-cytometry for their mitochondrial potential (Δψm) upon gating on CD44+ DAPI- cells (n=3). A representative flow-cytometry analysis of two different tumors is reported, AB+CCCP samples represent reacquisition of AZD+BEZ samples after incubation with CCCP for 5 minutes. g, ROS production in +KRas and -KRas cells (n=3); representative flow-cytometry analysis of two tumors. h, Live confocal imaging of surviving cells stained for mitochondria (MitoTracker Green), ROS (CellRox-Red) and DNA (Hoechst). The vast majority of signal generated by ROS colocalizes with mitochondria. i, Immunoblot of Aldefluor/CD133 double positive and double negative cells sorted from two independent tumors probed with anti-VDAC1 (VDAC1) and (β-actin (Actin) antibodies. j) KRas expressing cells positive for aldefluor (Ald+) and CD133 (CD133+) have higher mitochondrial transmembrane potential (Δψm) than tumor cells that do not express the same markers (Ald- and CD133-) (n=3); a representative flow-cytometry analysis is reported. Data are mean ± s.d.
Extended Data Figure 5. OCR, ECAR and metabolomics.
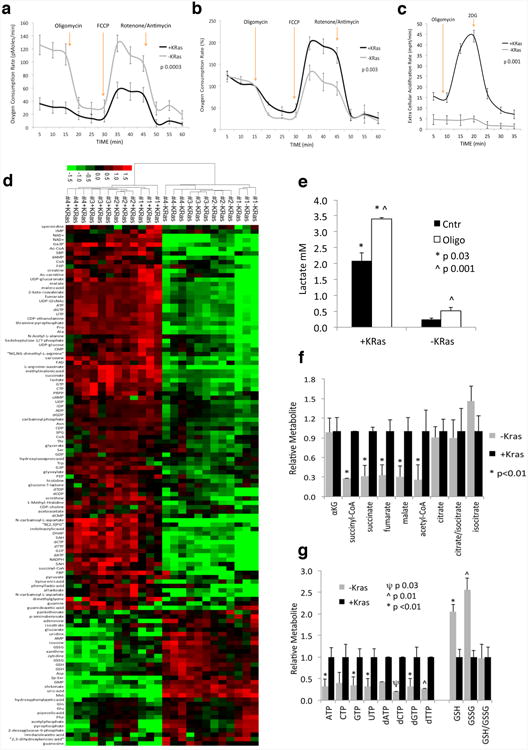
a, Oxygen consumption rate (OCR) of KRas expressing (+KRas) and surviving (-KRas) cells in response to oligomycin, FCCP and rotenone/antimycin (n=4). b, Same as in (a) but normalized to basal respiration of +KRas and -KRas cells. c, Extracellular acidification (ECAR) response of +KRas and -KRas cells to oligomycin and 2-deoxy-D-glucose (2DG). The experiment has been carried out in complete stem cell media to evaluate the glycolytic reserve of tumor cells in a nutrient rich environment. d, Metabolome analysis for +/-KRas cells; unsupervised hierarchical clustering and heat map of significantly (p<0.05) deregulated metabolites (n=4). e, Lactate production of +KRas and -KRas cells in response to oligomycin (Oligo) or DMSO (Ctrl) treatment (n=3). f, Fold change of TCA cycle intermediates in +KRas versus -KRas cells (αKG, α-ketoglutarate) (n=4). g, Fold change of nucleotide triphosphates and deoxynucleotide triphosphates, glutathione (GSH) and glutathione disulfide (GSSG) in -KRas versus +KRas cells (n=4). Data are mean ± s.d.
Extended Data Figure 6. Surviving cells in vitro and in vivo have an impaired glucose uptake.
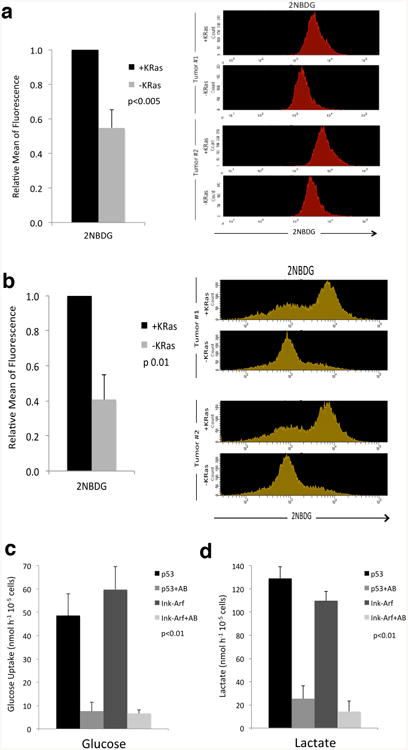
a, KRas expressing cells (+KRas) and surviving cells (-KRas) were incubated with 2NBDG for 6 hours then analyzed by flow-cytometry (n=3); a representative flow-cytometry analysis of spheres derived from two different tumors is reported. b, Mice bearing KRas expressing tumors (+KRas) and three-week regressed tumors (-KRas) were injected with a tail vein bolus of 2NBDG. After one hour tumors were explanted and analyzed by flow-cytometry upon gating on CD44+ DAPI- cells (n=3); a representative flow-cytometry analysis of two different tumors is reported. c-d, Tumor spheres derived from different genetic backgrounds (ink/arf-/- and p53-/-) were treated (+AB) or not with AZD8330 and BEZ235 for 7 days then plated in fresh stem cell medium. After 24hs medium was collected and analyzed by YSI analyzer for glucose uptake (c) and lactate production (d) (n=3). Data are mean ± s.d.
Extended Data Figure 7. Fuel carbon contribution to TCA cycle and TCA branch metabolites.
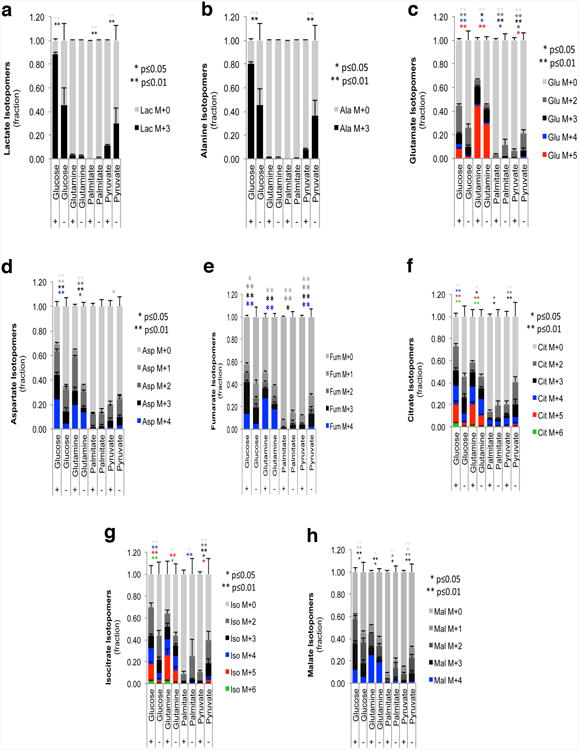
a-h, Isotopomer distribution for lactate (a), alanine (b), glutamate (c), aspartame (d), fumarate (e), citrate (f), isocitrate (g) and malate (h) in KRas expressing (+) and surviving cells (-) following steady-state tracing (36hs labeling) with uniformly carbon-13-labeled substrates: glucose, glutamine, palmitate and pyruvate (n=3). Data are mean ± s.d.
Extended Data Figure 8. Differential sensitivity of tumor cells to OXPHOS inhibition.
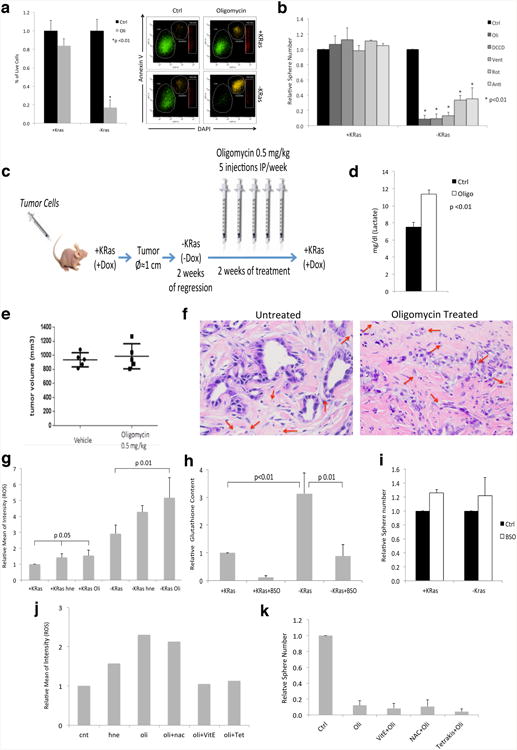
a, AnnexinV staining of cells treated with oligomycin 200nM (Oli) for 24hs shows a significant decrease in viability in surviving cells (-KRas). On the contrary control cells expressing KRas (+KRas) are minimally affected (n=3); a representative flow-cytometry analysis is reported. b, Effect of oligomycin (Oli), dicyclohexylcarbodiimide (DCCD), veturicidin (Vent), rotenone (Rot), antimycin (Anti) and DMSO (Ctrl) on spherogenic potential of KRas expressing (KRas+) and surviving tumor cells (KRas-) (n=4). c, In vivo treatment experimental scheme: mice were transplanted with tumor cells and fed with doxycycline in drinking water (+KRas, +Dox) until they developed tumors of 1 cm in diameter. Then doxycycline was withdrawn (-KRas, -Dox) and after 2 weeks, when tumors were regressed, mice were treated with oligomycin (0.5mg/kg, IP) or vehicle for 5 days a week, for two weeks. After treatment, KRas was re-induced (+Dox) and mice were monitored for tumor relapse. d, One dose of oligomycin (0.5mg/kg, IP) is sufficient to increase lactate concentration in plasma of treated mice after 4hs from injection (Oligo: oligomycin; Ctrl: vehicle) (n=4). e, Tumor volume of KRas expressing tumors treated with either vehicle or oligomycin 0.5mg/kg, 5 days a week, for two weeks. Treatment has started when tumors reached 5mm of diameter (5 mice per group). f, Surviving cells after treatment with oligomycin show signs of degeneration and epithelial remnants change their morphology. Red arrows indicate the presence of capillaries (red blood cells) indicating regressed tumors are vascularized (40×). g, Oligomycin (Oli) induces ROS production in KRas expressing cells (+KRas) and surviving cells (-KRas). Its effect is even stronger than that of positive control 4-hydroxynonenal (hne) (n=3). h, Glutathione levels in KRas expressing cells (+KRas) and surviving tumor cells (-KRas) before and after buthionine sulphoximine (BSO) treatment. Glutathione is increased in surviving cells and BSO treatment is effective in reducing its level (n=3). i, Effect of glutathione depletion on spherogenic potential of KRas expressing (+KRas) and surviving (-KRas) cells (n=3). j, ROS production in surviving cells after treatment with 4-hydroxynonenal (hne) and oligomycin (oli) in presence or absence of antioxidants: α-tocopherol (vitE), n-acetylcysteine (nac) and tetrakis (Tet) (n=2). k, Effect of oligomycin on spherogenic potential of surviving cells pretreated with antioxidants (n=4). Data are mean ± s.d.
Extended Data Figure 9. Effect of mitochondrial downregulation in human tumor spheres and metabolic stress mediated by inhibition of autophagy.
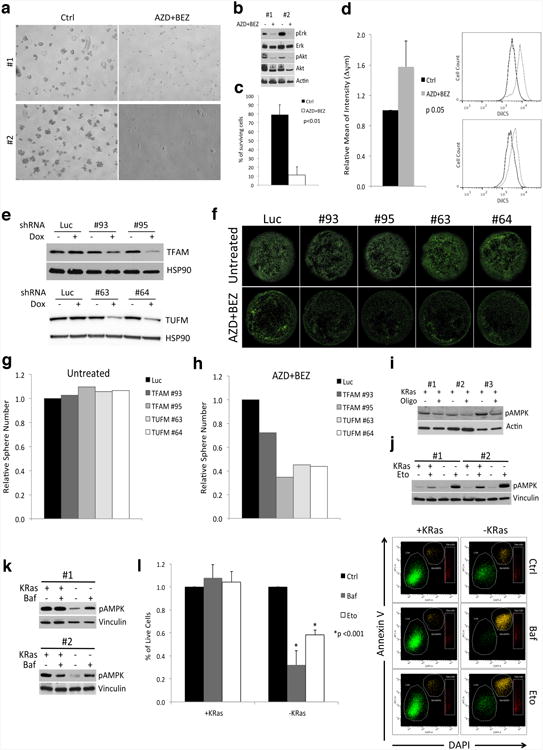
a, Effects of the combination of AZD8330 and BEZ235 (AZD+BEZ) on human tumor spheres. Some cells, usually doublets, are able to survive the treatment (5×). b, Immunoblots of human tumor spheres treated or not with the AZD+BEZ probed with anti-phospho-p42/44 (pErk), total-Erk (Erk), phospho- Akt (pAkt), Akt and (β-actin (Actin) antibodies, two independent tumors were reported. c, Annexin V staining of treated (AZD+BEZ) and control (Ctrl) cells after 4 days of treatment (n=3). d, Mitochondrial transmembrane potential (Δψm) of untreated (Ctrl) and treated (AZD+BEZ) human spheres with AZD8330/BEZ235 for 7 days (n=3), representative flow-cytometry analysis of two tumors. e-h, TFAM and TUFM were downregulated using two inducible shRNAs each (TFAM: #93, #95; TUFM: #63, #64) in human spheres expressing KRas (untreated) and cells surviving one week treatment with AZD8330 and BEZ235 (AZD+BEZ), after 5 days of shRNA induction cells were replated for evaluating their spherogenic capacity. e, Immunoblots of tumor spheres after 72hs of shRNA induction (+Dox) probed with anti-TFAM, TUFM and HSP90 antibodies, f, representative calcein staining after spheres replating. g-h, Effects of downregulation of TFAM and TUFM on spherogenic potential of untreated and treated cells respectively, data represent the average of two independent human tumors. i, Immunoblot of KRas expressing cells treated or not with oligomycin 200nM (Oligo, +/-) probed with anti-Thr172-phospho-AMPK and actin antibodies. Immunoblots of +KRas and -KRas cells treated with: j, etomoxir (Eto, 100 µM for 6hs) and k, bafilomycin (Baf, 50nM for 24hs) probed with anti-Thr172-phospho-AMPK and vinculin antibodies. l, AnnexinV staining of cells treated for 48hs with bafilomycin 50nM (Baf) and etomoxir 100µM (Eto) clearly shows a significant decrease in viability in surviving cells (-KRas). Controls cells expressing KRas (+KRas) are not affected (n=3); representative dot-plots are reported. Data are mean ± s.d.
Extended Data Figure 10. Cells surviving oncogene ablation are engorged with autophagosomes and lysosomes and contain lipid droplets.
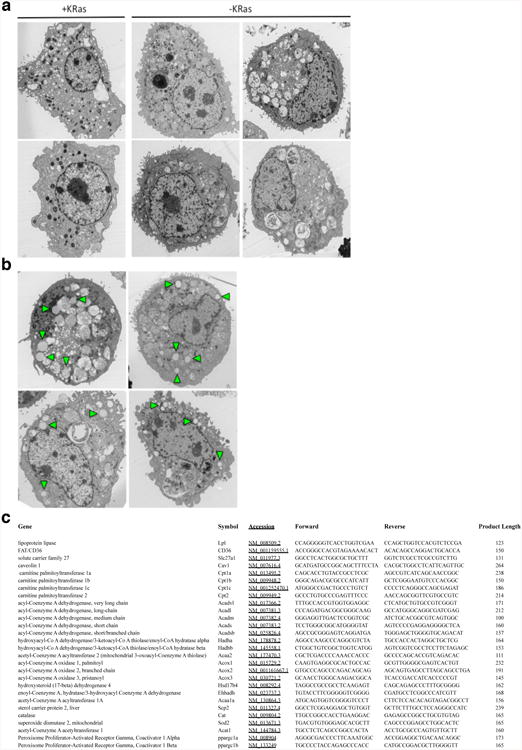
a, Surviving cells (-KRas) have the cytoplasm full of phagosomes and autophagosomes, a feature absent in KRas expressing cells (+KRas) (TEM 7500×). b, Surviving cells are characterized by the presence of several lipid droplets (arrowheads) in the cytoplasm (TEM 7500×). c, Primers used for amplification of mitochondrial and lipid metabolic genes.
Acknowledgments
We thank Ajit Divakaruni, Jay Dunn, Craig Smith, Katy McGirr and David Ferrick at Seahorse Bioscience for their support; Trang Tieu for vector cloning and Jeff Kovacs for YSI; James D. Lechleiter for the in vivo TMRE protocol; Hector Sandoval, Carlo Tacchetti, Di Francesco Maria Emilia, Joe Marszalek and Philip Jones for discussions and suggestions; Kenneth Jr. Dunner and High Resolution Electron Microscopy Facility at MDACC for TEM (Cancer Center Core Grant CA16672); Walter N. Hittelman and Center for Targeted Therapy for sharing confocal microscope; the Dana-Farber Cancer Institute Microarray Core Facility for affymetrix and the MDACC Sequencing and Microarray Facility (SMF) funded by NCI Grant CA016672(SMF) for exome sequencing; The MDACC Flow Cytometry and Cellular Imaging (FCCI) Core Facility Supported by Grant NCI#P30CA16672 for flow-cytometers and FACS. Debnath Jayanta for providing GFP-LC3 constructs; Bastianella Perrazzona, Usha Varadarajan and Robert Dewan for lab management and Shan Jiang for assistance in maintenance of mouse colonies. A.V. is thankful to Agnese Fantino, Sonia Rapi, Vito Giuliani and Pietro Viale for their continuous support.
This study was supported by grants from the Hirshberg Foundation for Pancreatic Cancer Research to A.V., Harvard Stem Cell Institute to R.A.D. and A.V., Sheikh Ahmed Center for Pancreatic Cancer Research to G.F.D. T.P.H. and A.V., American Italian Cancer Foundation to G.F.D., NIH P01CA117969 to R.A.D., NIH/NCI P01CA120964 to J.M.A., The Viragh Family Foundation to J.B.F.; C.A.L. is a Pancreatic Cancer Action Network-AACR Pathway to Leadership Fellow.
Footnotes
Author Contributions: A.V., P.G., R.A.D. and G.F.D. designed the studies, interpreted the data and wrote the manuscript; A.V., P.G., H.Y., N.S., M.M., A.C., T.G., V.G. performed the experiments; C.A.L. was responsible for metabolomics and C13 tracing experiments; S.S. was responsible for CNV and bioinformatics analysis; M.K.A, F.M., S.C., L.N., G.G., A.K.D., A.K., W.Y., E.B., Y.K., T.P.H., A.K., H.W., J.B.F. contributed essential reagents and resources; M.Y., J.M.A. helped with the metabolomics analysis; F.M., Y.A.W., L.C.C. assisted in data interpretation; A.K.D. edited the manuscript.
Author Disclosures: L.C.C. owns equity in, receives compensation from, and serves on the Board of Directors and Scientific Advisory Board of Agios Pharmaceuticals. Agios Pharmaceuticals is identifying metabolic pathways of cancer cells and developing drugs to inhibit such enzymes in order to disrupt tumor cell growth and survival.
References
- 1.NCI/SEER. 2012 http://seer.cancer.gov/statfacts/html/pancreas.html - incidence-mortality.
- 2.Ying H, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nature reviews Molecular cell biology. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kantarjian H, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. The New England journal of medicine. 2002;346:645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- 5.Flaherty KT, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. The New England journal of medicine. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quintas-Cardama A, Kantarjian H, Cortes J. Imatinib and beyond--exploring the full potential of targeted therapy for CML. Nature reviews Clinical oncology. 2009;6:535–543. doi: 10.1038/nrclinonc.2009.112. [DOI] [PubMed] [Google Scholar]
- 7.Jang S, Atkins MB. Which drug, and when, for patients with BRAF-mutant melanoma? The lancet oncology. 2013;14:e60–69. doi: 10.1016/S1470-2045(12)70539-9. [DOI] [PubMed] [Google Scholar]
- 8.Bardeesy N, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermann PC, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell stem cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Li C, et al. Identification of pancreatic cancer stem cells. Cancer research. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 11.Kim MP, et al. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PloS one. 2011;6:e20636. doi: 10.1371/journal.pone.0020636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. The American journal of clinical nutrition. 2011;93:884S–890. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nature protocols. 2012;7:872–881. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaglio D, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Molecular systems biology. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vizan P, et al. K-ras codon-specific mutations produce distinctive metabolic phenotypes in NIH3T3 mice [corrected] fibroblasts. Cancer research. 2005;65:5512–5515. doi: 10.1158/0008-5472.CAN-05-0074. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Schubert DR. The specificity of neuroprotection by antioxidants. Journal of biomedical science. 2009;16:98. doi: 10.1186/1423-0127-16-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim MP, et al. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nature protocols. 2009;4:1670–1680. doi: 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, et al. Mammalian transcription factor A is a core component of the mitochondrial transcription machinery. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16510–16515. doi: 10.1073/pnas.1119738109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skrtic M, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes & development. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang S, et al. Pancreatic cancers require autophagy for tumor growth. Genes & development. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lagadinou ED, et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell stem cell. 2013;12:329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samudio I, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. The Journal of clinical investigation. 2010;120:142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Molecular biology of the cell. 2008;19:797–806. doi: 10.1091/mbc.E07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng W, Talley Watts L, Holstein DM, Wewer J, Lechleiter JD. P2Y1R-initiated, IP3R-dependent stimulation of astrocyte mitochondrial metabolism reduces and partially reverses ischemic neuronal damage in mouse. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:600–611. doi: 10.1038/jcbfm.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itoh Y, Abe T, Takaoka R, Tanahashi N. Fluorometric determination of glucose utilization in neurons in vitro and in vivo. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24:993–1003. doi: 10.1097/01.WCB.0000127661.07591.DE. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]