

Abstract
The skin is colonized by an assemblage of microorganisms which, for the most part, peacefully coexist with their hosts. In some cases, these communities also provide vital functions to cutaneous health through the modulation of host factors. Recent studies have illuminated the role of anatomical skin site, gender, age, and the immune system in shaping the cutaneous ecosystem. Alterations to microbial communities have also been associated with, and likely contribute to, a number of cutaneous disorders. This review focuses on the host factors that shape and maintain skin microbial communities, and the reciprocal role of microbes in modulating skin immunity. A greater understanding of these interactions is critical to elucidating the forces that shape cutaneous populations and their contributions to skin homeostasis. This knowledge can also inform the tendency of perturbations to predispose and/or bring about certain skin disorders.
Keywords: Microbiome, Cutaneous immunity, Dermatology, Skin, Microbiota
Introduction
The skin is our primary interface to the external environment, supporting the growth of commensal microorganisms while impeding invasion by more pathogenic species. Culture-independent techniques that employ sequencing of marker genes, such as the bacterial-specific 16S ribosomal RNA (rRNA) gene, have begun to elucidate the community characteristics of these cutaneous microorganisms. In addition, these analyses have been used to inform elements of intrapersonal and interpersonal variability, as well as longitudinal dynamics of skin microbial communities. These studies have also led to investigations into the importance of host–microbe interactions, and their ability to shape the identity and composition of commensal relationships. This review will highlight these determinants as they pertain to a number of host factors. It will also address the role of microbiome–host interactions in certain skin disorders. While numerous microorganisms are thought to colonize the skin surface, we will emphasize the contribution of bacterial and fungal inhabitants. However, it is important to note that viruses, mites, and archaea are all capable of influencing residential populations of the skin.
Cutaneous architecture and biochemistry
To fully appreciate the microbial diversity of the skin, one must first understand the complex architecture and environment of this organ. As a critical barrier to the outside world, human skin is essential for activities such as thermoregulation, gas exchange, and hydration [1]. It also represents one of the body’s largest and most exposed organs with approximately 1.8 m2 of total surface area. The biogeography of the skin includes a number of planes, folds, and invaginations, each capable of maintaining a unique microenvironment. For this reason, microbial communities above the cool, desiccating skin surface often differ greatly from those found within shielded pores and follicles [2]. Different skin sites can also contribute to microbial heterogeneity through the production of various lipid- and water-based solutions. These determinants then work in concert with additional host factors and the external environment to shape an individual’s core microbiome.
Skin strata
Human skin consists of two main layers: the epidermis and the dermis (Fig. 1). As the most superficial layer, the epidermis contributes the majority of barrier functions while the dermis provides a structural framework made of fibrous and connective tissues. Underlying these strata is a layer of subcutaneous fat, which is critical for the protection of deeper tissues and bones.
Fig. 1.
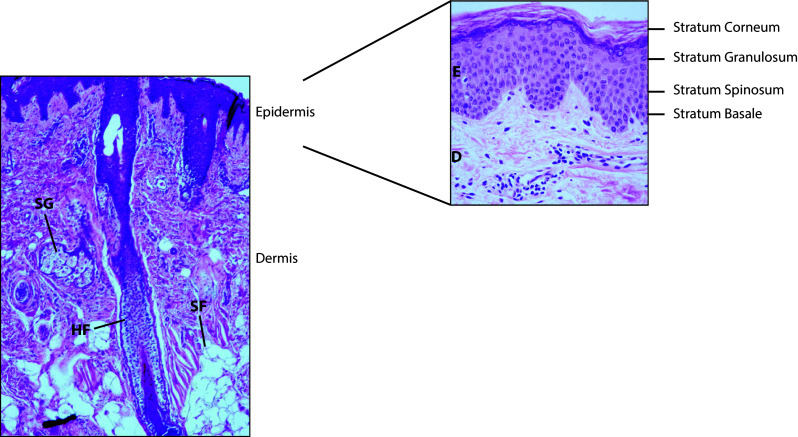
Skin structure and morphology. The skin can be divided into two main layers, the epidermis (E) and dermis (D), and underlying regions of subcutaneous fat (SF). Hair follicles (HF) extend from the skin surface into the dermis and are often associated with sebaceous glands (SG). The epidermis contains distinct layers of keratinocytes at varying stages of development. Basal stem cells are found at the stratum basale while daughter cells mature to populate the stratum spinosum, stratum granulosum, and upon terminal differentiation, the stratum corneum
As a continually self-renewing epithelium, the epidermis can be subdivided into four main strata, characterized by cells at varying stages of development (Fig. 1). The bottommost layer, the stratum basale, contains a single layer of undifferentiated stem cells that rest upon the epidermal basement membrane [3]. All keratinocytes originate from these basal cells, and they are essential for the regeneration of keratinocytes lost to terminal differentiation and desquamation [4]. During asymmetric cell division, these progenitor cells produce a subset of daughter cells that exit the cell cycle and separate from the basement membrane to form the stratum spinosum. In this layer, immature keratinocytes are characterized by abundant calcium-dependent desmosomes, which promote intercellular adhesion and resistance to mechanical stress [5]. As these cells continue to develop, they also flatten and initiate the formation of lamellar bodies and keratin filaments to support overall skin structure [5].
Upon further maturation, keratinocytes progress upwards to populate the stratum granulosum, so-named for the presence of prominent keratohyalin granules. These vesicles contain filaggrin, keratin filaments, loricrin, and involucrin—all necessary components for the hydration and structure of mature epidermal tissue [5]. Keratinocytes of the stratum granulosum are also held together by a number of extracellular tight junction proteins including claudins and occludins, which are essential to epidermal barrier function [6]. During the terminal stages of differentiation, cells of the granular layer compress and anucleate to form the stratum corneum. At this stage, keratinocytes then become known as corneocytes for their highly cornified cellular envelopes. These protein-enriched cells are also held together by keratins, corneodesmosomes, and a lipid-enriched extracellular matrix to provide a strong physical barrier that is resistant to mechanical stress, UV damage, and permeation [7].
Appendages
In addition to these strata, the skin is also characterized by a number of appendages that can extend beyond the epidermis into the dermis. These include sebaceous glands, hair follicles, and sweat glands. Sebaceous glands specialize in the secretion of sebum, an oily, lipid-rich substance that provides skin flexibility and waterproofing. Most sebaceous glands are also connected to hair follicles to form pilosebaceous units that concentrate on the face and upper body [8]. Pilosebaceous follicles support an array of niche-specific microorganisms that can thrive in anoxic environments rich in sebum-derived lipids [9]. These lipids can then be metabolized into free fatty acids by bacterial commensals, which contribute to the acidic pH of the skin [10]. Importantly, while the number and distribution of sebaceous glands remain relatively constant throughout life, their size and activity fluctuate widely depending on age and hormone levels [8]. It is thus not surprising that puberty marks a defining period in skin development characterized by the elevated production of sebum and sebum-related products, as well as the subsequent growth of lipophilic skin microbial inhabitants [11].
Sweat glands, another critical appendage of the epidermis, can be divided into two major types: apocrine and eccrine. Like sebaceous glands, apocrine sweat glands release oily secretions into upper hair follicles and are especially active during puberty. Apocrine sweat is composed of a milieu of proteins, lipids and steroids [12]. Apocrine glands are also more sparsely distributed, often localized to especially pileous regions such as the axillae and perineum [12].
Eccrine sweat glands, in contrast, are widely distributed throughout the body with high concentrations at the forehead, axillae, palms, and soles [13]. They are also the only gland with direct access to the skin surface, and as such, continuously bathe the epidermis in a water- and salt-based sweat solution. These secretions are critical to thermoregulation and hydration, and also contribute to the relatively acidic pH of skin surfaces.
In all, the dissemination and activity of epidermal appendages provide essential roles for the human body. By creating habitats with unique levels of moisture, pH and nutrients, they also represent specialized niches that can promote the growth of distinct microbial communities. This then contributes to the unique stratification of bacterial populations at skin sites throughout the body.
Host factors and the skin microbiota
Topographical variability
The site specificity of the skin microbiota has been borne out in multiple experiments analyzing unique topographical locations of the skin (Fig. 2). For example, a study of 20 distinct body sites representing sebaceous, moist, and dry physiological environments found that Propionibacterium and Staphylococcus species dominated sebaceous skin sites including the face and upper body [14]. By contrast, Corynebacteria, β-Proteobacteria, and Staphylococcus were the major genera at moist sites such as the axilla, antecubital fossae (inner elbow), and popliteal fossae (inner knee). Dry sites including the forearm and buttock were found to be more variable, supporting the growth of numerous phylotypes including β-Proteobacteria, Corynebacteria, and Flavobacteriales.
Fig. 2.
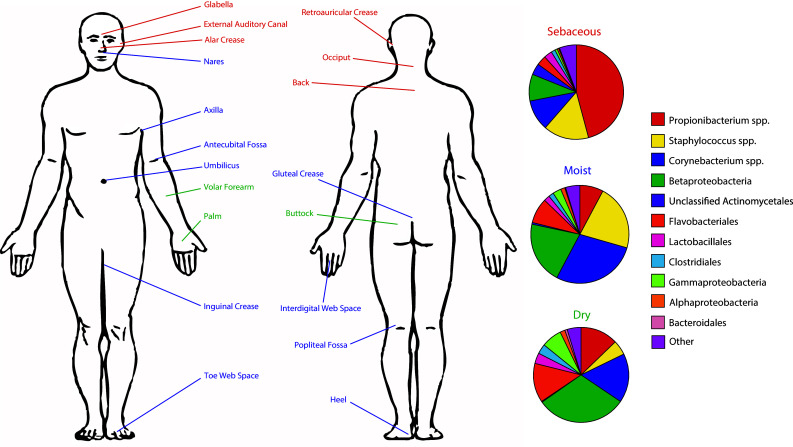
Regional variation of skin microbial communities. The cutaneous microbiota varies according to body site and is strongly influenced by differences in cutaneous physiological environments. Each pie chart represents the mean bacterial community of a given biogeographic region. Sebaceous (red), moist (blue), and dry (green) regions are highlighted. Data from Grice et al. [13]
Upon more in-depth analyses, it was revealed that the sites richest in bacterial operational taxonomic units (OTUs; a sequence-level proxy for designating species) were often dry regions such as the forearm, while sebaceous sites including the upper back and retroauricular crease (behind the ear) were home to fewer bacterial phylotypes. In addition, sebaceous regions were consistently lower in bacterial evenness as measured by the relative distribution of sequences among OTUs. Interpersonal variation (differences between individuals) was found to be greater than intrapersonal variation (differences within individuals) over time. This suggests that individuality and body site physiology are both strong determinants of bacterial community membership and structure.
Similarly to above, Costello et al. [15] observed that temporal intrapersonal variability was less pronounced than interpersonal variability between individuals. These studies also confirmed that spatial intrapersonal variability (e.g. variability in microbiomes of distinct body sites such as forehead, arm, and umbilicus) was even greater than interpersonal variability at the same skin site. As such, although individual microbial populations of the skin are often more similar to themselves in regard to symmetry and time, these likenesses appear to breakdown when comparing separate biogeographic regions.
In accordance with Grice et al., this group also found high levels of Propionibacterium at sebaceous sites on the face, and greater diversity at areas such as the popliteal fossa, forearm, and palm. Moreover, it was shown that the variation of these sites remained relatively constant over time, as the palm and forearm were both consistently more diverse than the forehead at four separate collection periods.
The influence of body site in regard to overall community structure was also tested by inoculating bacteria from foreign sites onto new areas of the skin. These studies observed a relative flexibility in forearm community membership, while the forehead microbiota rapidly returned to a population resembling its native state. This suggests that host factors may vary in their ability to promote bacterial colonization, especially at sebaceous sites with strong environmental biases.
Whereas these studies sought to compare multiple body sites, additional research has focused on individual skin regions. These studies largely complement one another, providing greater insight into the contribution of topography to skin microbial communities. For example, studies performed on the human forearm have illustrated relatively high degrees of bacterial diversity, although this population is consistently dominated by Propionibacterium, Corynebacterium, Staphylococcus, Streptococcus, and Acinetobacter [14–17]. While these major taxa appear throughout the literature, however, it appears that their relative contributions to the forearm community can fluctuate greatly.
The same can be said of the palmar region, which is frequently exposed to new surfaces and environments—while major phylotypes such as Propionibacteria, Streptococcaceae, and Staphylococcaceae are consistently observed, a great amount of variability exists in regard to their absolute numbers and proportions [14, 15, 18, 19]. Thus, it appears that certain exposed regions including the palm and forearm are less restricted in overall community membership and highly susceptible to temporal variability.
By contrast, other regions, including those with high sebaceous gland activity, are much more exclusive. For example, the forehead harbors fewer bacterial species and is largely dominated by Propionibacterium [14, 15, 17, 20]. This observation is congruent among multiple studies, and as such, represents a relatively consistent trend. Whether this effect is inherent to the lipid-rich environment of the forehead, or whether Propionibacterium can successfully restrict membership alone is currently unknown. Regardless, this region appears largely invariant compared to more diverse sites of the skin, and thus represents a more stable overall community structure.
While compelling, the stratification illustrated by certain dry and sebaceous sites is by no means absolute, as multiple sites of the skin are characterized by intermediate diversity with both dominant and transient taxa [14, 15]. Therefore, further research will be necessary to determine the role of intrinsic host factors and extrinsic microbial traits as they pertain to skin bacterial communities.
Recent studies have also begun to elucidate the topographical diversity of fungal communities on human skin [21]. Specifically, it was shown that Malassezia predominated at core body and arm sites, but that discrete signatures could be observed at the species level. For example, the face was dominated by Malassezia restricta while the back, occiput (back of neck), and inguinal crease (groin) were all characterized by higher levels of Malassezia globosa.
In contrast to these areas, regions of the foot such as the plantar heel, toenail, and toe-web space were all defined by significantly greater amounts of fungal diversity. While Malassezia was still detected in all samples, subjects were also colonized by relatively high proportions of Aspergillus and Epicoccum. Interestingly, regional localization was found to be the strongest determinant of fungal community membership as feet, arms, the head, and torso all formed distinct communities regardless of physiological environment. This suggests that while bacterial populations are subject to factors such as sebum content and hydration, fungal communities are more flexible in resource utilization, a less surprising realization given their pronounced evolutionary differences.
Gender
The contribution of gender to skin microbial diversity likely arises as a downstream effect of male and female steroid production [13]. For example, it is thought that androgen expression and identity are both critical to sex-defined differences in skin thickness [22, 23]. Males also exhibit increased levels of sebaceous and sweat gland activity compared to females, a trait that strongly contributes to differences in skin surface biochemistries [24, 25]. Even the presence or absence of body hair could presumably result in alternative microenvironments with the potential to support the growth of niche-specific microorganisms. Interestingly, mixed results have been observed in regard to gender and pH. While some studies have detected a more acidic pH in female skin, others have demonstrated no differences [26–29]. This suggests that variation in male and female physiologies has the potential to influence microbial communities, but that certain factors likely contribute to skin habitats more strongly than others.
With this in mind, a recent study that sampled the palmar regions of male and female undergraduate students observed significantly different bacterial communities on the skin surface in regard to gender [18]. While no taxa were specific to either sex, there were marked differences in the relative abundances of numerous bacterial groups. For example, Propionibacterium and Corynebacterium were 37 and 80 % more abundant in men, respectively, along with a trend towards higher levels of Staphylococcus. By contrast, Enterobacteriales, Moraxellaceae, Lactobacillaceae, and Pseudomonadaceae were all over 150 % more abundant in females. Women were also found to harbor significantly greater levels of alpha diversity, a metric that defines “within” sample diversity and is often measured by numbers of OTUs, their evenness, and their degree of phylogenetic difference.
In contrast to these results, a study of healthy Chinese undergraduates showed no significant differences between the palmar bacterial communities of men and women [19]. However, higher relative abundances of distinct taxa such as Propionibacterium, Corynebacterium, and Staphylococcus were once again observed in male subjects while Lactobacillus was over-represented in females. Interestingly, Enhydrobacter and Deinococcus also made up a large portion of female hand communities, while Fierer et al. found no such contribution in either sex. This suggests that geographical or cultural aspects may also play a large role in diversifying skin microbial communities, a concept that has been supported by a number of additional reports as well [30, 31].
In a study comparing the skin microbiota at varying developmental stages, males and females between the ages of 2 and 40 were swabbed at the antecubital and popliteal fossae, the volar forearm, and the nares [32]. In all, no significant differences were observed between the bacterial communities of males and females regardless of age group. Moreover, a study comparing the levels of Propionibacterium and coagulase negative Staphylococcus in middle-aged men and women found no significant differences at the forehead, cheek, upper chest, or back [33]. However, it was found that males harbored greater total amounts of the fungi Malassezia.
Studies have also examined the human axilla, upper buttock, forehead, and forearm as potential sites of gender variability. Interestingly, the bacterial communities of the axillary vault were found to stratify into two main groups, those colonized predominantly by Staphylococcus and those with high relative abundances of Corynebacterium [34]. While not absolute, female subjects were generally found within the Staphylococcus cluster whereas males were more often associated with the Corynebacterium cluster. Analysis of the upper buttock also exhibited a strong effect of gender with males illustrating relatively high proportions of Corynebacterium, Dermacoccus, Streptococcus, and Finegoldia while females displayed elevated levels of Lactobacillus, Propionibacterium, Staphylococcus, and Enhydrobacter [35]. Despite these distinctions, there were no significant differences between genders when taking the entire microbial community into account, suggesting that individualized signatures were still the best indicators of variability.
On the forehead, males and females were found to harbor differences in overall bacterial diversity [17]. However, when accounting for the use of make-up, significant variability between these groups was no longer detected. In contrast, microbial diversity of the forearm was significantly different between men and women at both the genus and species level.
In all, it appears that gender may contribute to microbial community structure, but that the importance of this factor likely varies in a site-dependent manner. As male and female physiology differs throughout the body, it is not surprising that the contribution of gender to microbial communities is also inconstant. More detailed studies will be necessary to determine the importance of potential driving factors, as no studies to date have measured microbial populations and biochemical signatures in concert.
Age
The human skin begins to develop in utero during the first trimester of gestation, and by 34 weeks, a well-defined stratum corneum has formed [36]. In the weeks leading up to delivery, the epidermis further matures, and begins to resemble a competent adult-like barrier by week 40 [37]. Upon birth, the skin undergoes a number of rapid changes as it acclimates to a dry, gaseous climate very much at odds with its former aqueous environment. During this time, the skin is characterized by quantal bursts of improved barrier function that persist for multiple weeks postnatal delivery [38]. Development then continues during the first year, after which point infant skin begins to resemble that of mature adults [39].
During maturation, infant skin is defined by a thin layer of corneocytes that are, on average, much smaller than adult corneocytes [40]. In addition, infant skin contains lower lipid content resulting in an epidermal barrier with higher water levels and increased permeability [39, 41]. Neonates are also born with a relatively alkaline skin pH that remains less acidic than adult skin for the first two years of life [42].
All of these developmental features likely contribute to the differences seen between adult and infant bacterial communities. For example, Staphylococcus species, which are known to predominate at moist body sites on the adult epidermis, have been found at significantly higher levels on neonatal skin. In fact, a recent study of the infant microbiota observed that Staphylococcus and Streptococcus species could account for up to 40 % of skin bacterial populations during the first six months of life, before giving way to a more diverse community [43]. Interestingly, site specificity also began to appear within the first few months of life. Staphylococcus, Streptococcus, Corynebacterium, and Propionibacterium were all found to predominate at the arm and forehead of infant skin while the buttock was colonized by both gut- and skin-associated taxa such as Clostridium, Staphylococcus, Streptococcus, and Ruminococcus. This suggests that as the skin matures, it becomes more adept at influencing resident bacterial communities at certain body sites.
Additional experiments have also examined the route of delivery as a direct contributor to the human skin microbiota [44]. These analyses have shown that vaginally born neonates harbor skin bacterial communities very similar to those found in the vagina. This includes an abundance of both Lactobacillus and Prevotella. In contrast, babies born by Cesarean section were colonized by common skin residents such as Acinetobacter, Bacillales, Micrococcineae, and Staphylococcus. Interestingly, this study also found that babies born through conventional methods displayed skin bacterial communities most similar to their mother’s microbiota, while babies born by Cesarean section were no more similar to their own mother than any other subject. As such, while an initial vertical transmission of the bacterial microbiota existed in vaginally delivered neonates, no such transmission occurred in babies delivered by Cesarean section. Rather it appears that incidental exposures, likely provided by hospital staff and environmental surfaces, were the greatest contributors to microbial communities in these subjects.
While the initial inhabitants of infant skin can vary greatly depending upon age and delivery mode, their microbiomes appear to stabilize over time, reaching an adult-like community at sexual maturity. A study employing Tanner staging to distinguish between children and adults found that the microbiota of subjects within Tanner stages 1, 2, and 3 segregated significantly from that of individuals at stages 4 and 5 [32]. Similarly to above, it was also shown that higher levels of Proteobacteria and Firmicutes such as Streptococcaceae distinguished the microbiota of younger cohorts, while adolescents/post-adolescents were dominated by Propionibacterium and Corynebacterium. This particular result corresponds well with the developmental milestones reached at higher Tanner stages including elevated hormone levels and increased sebaceous gland activity, as both factors promote the growth of more lipophilic microorganisms [45].
Interestingly, it has also been shown that the common fungal commensal Malassezia colonizes neonate skin during the birthing process [46]. At day 0 following delivery, Malassezia DNA was successfully detected in 24 of 27 subjects, and by day 30 approximately 104 residents were estimated by qPCR. While the specific distribution of Malassezia residents differed greatly in newborns compared to their mothers, these rates stabilized to a level very near that of adulthood by day 30.
Overall, these results suggest that the skin and its microbial inhabitants develop together over time. While the physiological and biochemical attributes of the skin contribute a great deal to microbial diversity, this niche also represents a blank slate with the potential to accommodate a vast array of microbial organisms. For this reason, further research will be necessary to fully elucidate the dynamic nature of age-related succession.
It may also be necessary to revise the long-held belief that most fetuses develop in a sterile environment. Recent evidence suggests that bacteria can be reproducibly isolated from newborn meconium and umbilical cords of healthy, full-term neonates [47, 48]. Enterococcus faecium has also been isolated from newborn meconium and amniotic fluid following oral inoculation of pregnant mice, and fluorescent in situ hybridization can be used to visualize 16S rRNA-containing species deep within human fetal membranes [47–49]. A recent study of the placental microbiome also reported a diverse community of bacterial species characterized by increased levels of Proteobacteria [50]. In addition, both Gram-positive and Gram-negative intracellular bacteria have been detected in over a quarter of placental basal plate samples [51]. These findings are in stark contrast to the notion that newborns are not exposed to microorganisms until birth, and these microbes could contribute to the initial inoculum present on newborn epidermis.
Immune system
The host immune system and the skin microbiota are in constant communication as each works to establish a steady equilibrium. This is not surprising given the intimate contact made between the two. In fact, it is thought that as many as 107 bacteria/cm2 colonize the epidermis at any given time [52]. Although the vast majority of these microorganisms inhabit the stratum corneum, recent evidence has shown that bacterial species may also reside within deeper layers of the epidermis and dermis [35, 53]. For this reason, it is essential for hosts to control the cutaneous immune response, and tailor it to a given threat, as persistent activation against resident skin bacteria could lead to chronic inflammatory disorders.
To perform this function, the skin is equipped with a number of professional innate and adaptive immune cells including multiple dendritic and T cell subsets (Fig. 3). Keratinocytes also provide support through the expression of Toll- and Nod-like receptors and the secretion of antimicrobial peptides, proinflammatory cytokines, and chemokines [54]. Even melanocytes can assist in the overall immune response by recognizing and responding to specific foreign antigens [55].
Fig. 3.
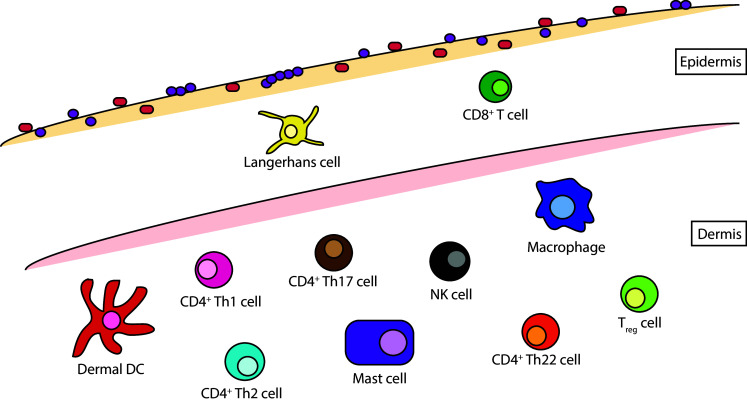
Major skin immune cell subsets. Human skin is characterized by an array of innate and adaptive immune cells. In the epidermis, this includes Langerhans dendritic cells and CD8+ T cells. The dermis is home to a more varied population of innate dermal dendritic cells, NK cells, and mast cells, as well as adaptive CD4+ Th1, Th2, Th17, and Th22 cells
While all of these cells play a crucial role in epidermal barrier function, Langerhans cells (LCs) are thought to act as the key initiators of cutaneous immunity by sampling the upper strata for microbial antigens and presenting these peptides to adaptive immune cells [56, 57]. However, the exact role of these specific dendritic cells has recently come into question, as many of the tasks previously attributed to LCs, such as cross-presentation, may actually be performed in vivo by a separate subset of myeloid cells known as dermal dendritic cells [58, 59]. Regardless of subtype, it appears that dendritic cells are crucial to mediating the initial response to barrier disruption. Upon antigen uptake, these cells travel to cutaneous-draining lymph nodes where they present foreign peptides to naïve T cells. These T cells then become activated and imprinted with skin-specific homing markers such as cutaneous leukocyte antigen (CLA), CCR4, CCR8, and CCR10 [60–63]. The ligands for these receptors are expressed at low levels during steady state, but they can be upregulated during inflammation, allowing for the recruitment of effector T cells to the skin epithelium. Upon antigen clearance, these mature T cells differentiate into resident or effector memory T cell subsets. Resident memory CD8+ T cells are then thought to remain within the epidermis while effector memory CD4+ T cells traffic to more distal sites of the skin [64, 65].
While this pathway has been established in response to infection, less information exists with regard to the skin’s response to commensal microorganisms. Specifically, it is currently unclear how the immune system can differentiate between pathogenic and non-pathogenic species, especially when considering the close proximity of keratinocytes, melanocytes, and LCs to conserved microbial antigens. A recent paper sheds some light on this debate by suggesting that LCs may perform separate roles depending on the state of epidermal tissue [66]. This group found that upon insult, resident LCs were crucial for the activation of resident memory T cells. However, at steady state, these cells promoted a homeostatic balance through the activation and preservation of regulatory T cells. While it is proposed that these regulatory T cells are important for the maintenance of self-tolerance, this process could also regulate the host immune response to resident skin microorganisms and inhibit excess inflammation.
With this in mind, various groups have explored the direct interactions of skin inhabitants with keratinocytes and the immune system. For example, the common skin commensal bacterium Staphylococcus epidermidis has been found to activate TLR2 signaling and the production of antimicrobial peptides and proinflammatory cytokines, augmenting the immune response to both group A Streptococcus and HPV infection [67–69]. The TLR2 ligand lipoteichoic acid has also been shown to reduce TLR3-mediated inflammation in keratinocytes and promote the induction of cathelicidin-producing mast cells [70, 71]. Interestingly, this effect does not appear to extend to macrophages, dendritic cells, or mouse endothelial cells, as exposure in these cell types results in an inflammatory response that is equal to or greater than that of epidermal keratinocytes. Therefore, a division of labor may exist within the cutaneous epithelium in which only certain cells can promote inflammation, a finding supported by the differential expression of Toll-like receptors at distinct layers of the epidermis [72].
Our lab and others have also focused on the relationship between host immunity and skin bacterial residents to identify key members of this host–microbe interaction network. By treating mice with a C5aR antagonist, we have shown that disruptions to the complement pathway can lead to significant changes in skin community structure including an increase in Actinobacteria and a decrease in Firmicutes [73]. We also observed a significant decrease in bacterial diversity (defined as the number of OTUs and their evenness), upon treatment, as well as a reduction in the overall number of bacterial OTUs. In addition, the expression of antimicrobial peptides, cytokines, chemokines, cell adhesion molecules, and pattern recognition receptors was all reduced in antagonist-treated mice, along with decreased levels of immune cell infiltration. This suggests that complement proteins may act to induce and/or maintain stable levels of these effectors, and that alterations to this balance can significantly shape skin microbial populations. The expression of complement genes in the skin of germ-free and conventionally raised mice was also compared to determine the importance of bacterial stimulation to complement gene expression. In the absence of bacterial colonization, we observed significantly lower expression of over 30 genes related to complement activation and binding, indicating that both the skin and its resident microorganisms are capable of influencing the identity of their respective interaction partners.
The ability of the immune system to shape bacterial communities has also been observed by comparing the skin microbiota of healthy and immunocompromised mice [74]. Here, it was found that healthy mice were colonized by an abundance of Proteobacteria including Acinetobacter, Escherichia/Shigella, and Acidovorax while immunodeficient mice were dominated by Firmicutes, especially those of the Staphylococcus genus. This difference was borne out in diversity metrics as well, with healthy mice displaying a significantly greater degree of variation when compared to immunodeficient mice.
Importantly, a recent study of humans with primary immunodeficiencies (PIDs) shows that this effect is not limited to murine models [75]. PID patients were defined by increases in microbial permissiveness to atypical microorganisms such as the opportunistic pathogen Serratia marcescens. Depending on the specific PID, patients were also characterized by decreases in site specificity, interpersonal variation, and longitudinal stability, suggesting a generalized dysbiosis caused by alterations to the host immune response. Paradoxically, these changes did not result in significant alterations to microbial diversity, however, indicating that site-specific restraints in humans may still control overall community structure.
Work has also compared the adaptive immune systems of germ-free (GF) and specific pathogen-free (SPF) mice to determine the importance of commensal bacteria to cutaneous immunity [76]. This study found that skin bacterial residents influence T cell number and function, as GF mice had higher levels of Foxp3+ regulatory T cells and lower amounts of the cytokines IFN-γ and IL-17A. Importantly, this effect on IL-17A could be rescued by monocolonization with the skin commensal bacterium Staphylococcus epidermidis. These results were also extended to infection by the parasite Leishmania major. In this model, GF mice were unable to mount a robust immune response to L. major while monoassociation with S. epidermidis could restore protection in an IL-17A-dependent manner. IL-1α expression was essential for this response, as neutralization of this cytokine impaired the restoration of IL-17A signaling. As such, it appears that IL-1 signaling pathways are enhanced by the skin microbiota, and that this response can promote overall skin immune fitness.
A more recent report supports this finding by confirming the ability of T cells to shape skin bacterial communities [77]. Adoptive transfer of T cells from WT mice into Rag1 −/− mice resulted in the rapid proliferation of both CD4+ and CD8+ T cells within skin-draining lymph nodes, consistent with a memory immune response to skin bacterial antigens. The number of live bacteria and 16S rRNA bacterial sequences was also higher in Rag1 −/− compared to WT mice, and the transfer of T cells from WT to immunodeficient mice resulted in a steady decline of these markers. This response was abrogated in the absence of IL-17A and IFN-γ, while B cell deficient mice mirrored WT phenotypes, suggesting that certain T cell profiles are essential for the recognition and control of skin bacterial residents.
In all, these results indicate that the immune system and skin microbiota are in constant communication, and that each is necessary to promote homeostasis at the skin surface. However, these interactions appear to vary greatly depending on the specific immune cell subset and signaling pathway, and perhaps even the conditions in which mice are housed. Indeed one group recently reported no differences between the skin microbiota of healthy and immunocompromised mice, although variation is readily detectable when comparing the mice within different experimental groups [78]. As such, further research will be necessary to describe the intimate relationship between hosts and bacterial inhabitants, and to determine the key players of this particular host–microbe interaction network.
Host–microbiome interactions in cutaneous disease
Many cutaneous disorders are caused by, or associated with, overt microbial infection. Here we focus on three of these disorders: acne vulgaris, psoriasis and atopic dermatitis. While complex in etiology, these conditions are thought to involve both microbial and host components. In addition, studies of these diseases have included deep sequencing approaches as a means to elucidate the contribution of skin microbial communities to disease pathology. As such, these disorders represent a model system to study the interactions of host factors and bacterial residents as they pertain to disruptions in skin homeostasis.
Acne
Acne vulgaris is one of the most prevalent skin diseases in the world, representing a financial burden of over 3 billion dollars per year in the United States alone [79]. Despite this figure and studies showing that acne can affect approximately 80 % of adolescents and young adults [80], relatively little is known with regard to the events underlying this disorder. In particular, it remains unclear whether: (1) comedone formation is the cause or effect of inflammation in pilosebaceous follicles, (2) which immune cells and cytokines drive the overall inflammatory response, and (3) the specific role of skin microbial residents such as Propionibacterium acnes.
Over the past decade, a number of groups have begun to address these questions, outlining a multifactorial process driven, in large part, by increases in androgen production during puberty. This increase in hormone signaling activates sebaceous gland activity and induces epithelial hyperproliferation and keratinization [81]. These changes can then promote the colonization and growth of Propionibacterium acnes, and contribute to the chronic inflammation seen in affected pilosebaceous follicles.
Multiple in vitro studies have demonstrated the ability of P. acnes to increase the expression of key inflammatory cytokines such as IL-1α, IL-1β, IL-6, IL-8, IL-12, and TNF-α by human sebocytes, keratinocytes, and monocytes [82–84]. The presence of infiltrating CD4+ T cells has also been observed by a number of groups, suggesting that the recruitment of these cells could promote inflammation within acne lesions [85–87].
Recently, a number of independent reports confirmed the ability of P. acnes to upregulate the production of IL-1β through the activation of the NLRP3 inflammasome [88–90]. Higher expression levels of NLRP3 and caspase-1 were observed in the areas surrounding acne lesions and both markers co-localized with infiltrating tissue macrophages [88, 90]. Mice challenged with P. acnes also showed increased expression of caspase-1 and IL-1β, while NLRP3 knockout mice displayed a significant decrease in these inflammatory markers [89, 90]. In sebocytes, this activity was dependent upon reactive oxygen species and P. acnes protease activity, while monocytes required bacterial uptake, potassium efflux, and reactive oxygen species [88–90]. This information, coupled with studies showing increased expression of TLR-2 on acne-localized macrophages [83], suggests a mechanism by which monocytes are recruited to early acne lesions, and then activated by P. acnes to induce a more robust inflammatory response.
Recent studies have also demonstrated the ability of P. acnes to stimulate Th17 differentiation and activity. These reports have shown that IL-17-expressing cells often localize to affected pilosebaceous follicles and are elicited by the production of IL-1β, IL-6, and TGF-β [91]. In addition, P. acnes-reactive Th17 cells were isolated from the blood of acne patients at higher frequencies than those of healthy subjects [92]. Two commonly employed dermatologic acne treatments, all-trans retinoic acid and 1,25-dihydroxyvitamin D3, were also found to downregulate P. acnes-induced IL-17 mRNA and protein expression [91]. Together these results suggest that CD4+ Th17 cells may be key mediators of the chronic inflammation found within moderate-to-severe acne lesions, and that modulation of these cells could resolve certain aspects of P. acnes-induced pathology.
While convincing, these results do not address the fact that P. acnes is a common skin inhabitant regardless of acne phenotype. Rather, reconciliation with this observation has come in the form of more detailed experiments describing the specific localization and genetic signatures of individual P. acnes clones. These studies have shown that pilosebaceous follicles are more frequently colonized by P. acnes in affected, compared to unaffected, individuals [93, 94]. This bacterium is also found more commonly as macrocolonies within acne lesions in contrast to the sparse distributions that typically attach to the outer surface of the epidermis in healthy individuals [93, 94]. Interestingly, within these follicles, multiple strains of P. acnes have been observed, but only certain strains, such as subtype IA, are associated with acne vulgaris [94–97]. A recent study utilizing 16S rRNA gene sequencing of P. acnes populations confirmed this finding by isolating certain subtypes of P. acnes from acne patients more frequently than others [98]. Interestingly, this group also reported a specific phylotype of P. acnes that associated more commonly with healthy subjects compared to acne patients, underscoring the importance of strain-specific profiles in P. acnes pathogenesis.
Overall, it appears that androgen-induced increases in sebum production during puberty may promote P. acnes colonization, but that this effect is not necessarily emblematic of disease. Rather, the growth of specific P. acnes strains may be required for acne lesions to develop into fully mature papules and pustules. Indeed, studies have reported a differential immune response in sebocytes and keratinocytes when exposed to alternative strains of P. acnes, a characteristic that could explain the ubiquity of P. acnes in both affected and unaffected individuals [82, 99].
Psoriasis
Psoriasis is a common inflammatory disease affecting approximately 2–3 % of the world’s population [100]. While multiple phenotypes exist, this condition is often characterized by well-demarcated erythematous plaques, resulting from chronic inflammation and the hyperproliferation of keratinocytes [101]. At onset, an initial inflammatory event is thought to precede plaque formation and induce the production of numerous proinflammatory cytokines. Further inflammation is then promoted by CD4+ Th1, Th17, and Th22 cells leading to distinct changes in skin architecture [102]. These include the thickening of epidermal cell layers, elongation of epidermal rete ridges, hypogranulosis, and parakeratosis [103].
Genome-wide association studies have largely supported these phenotypic observations with most identified defects belonging to the IL-23/Th17 axis, NF-κB pathway, and epidermal differentiation complex [104–106]. However, the major genetic determinant of psoriasis is found within the HLA-Cw0602* allele of the MHC class I molecule, HLA-C [107]. Mutations within this locus are thought to account for approximately 60 % of all psoriasis cases suggesting that CD8+ T cells may also play a major role in disease pathogenesis [108].
Although a number of pharmaceutical drugs are currently available to mediate the inflammatory nature of psoriasis, little is known with regard to the source of this inflammation. Physical trauma (Koebner’s phenomenon) and infection have both been associated with the induction of psoriatic flares [109, 110]. This is supported by the observation that surgical procedures and streptococcal throat infections often precede lesion formation [103, 111–113]. However, no study to date has identified an antigen capable of eliciting a complete psoriatic phenotype in healthy skin, despite links between superantigens and certain streptococcal surface proteins [114–116]. It is interesting to note that while infection of the throat with streptococcal species is the best-studied site of proclivity, Streptococcus is also a common resident of the skin [14–16]. As such, physical trauma and infection need not be mutually exclusive events if injury results in the presentation of streptococcal-associated (or alternative bacterial) antigens.
In this vein, a number of groups have attempted to characterize the microbiota of psoriasis plaques in search of inflammatory antigens and disease-associated microbial signatures. The first of these found an overabundance of Firmicutes in psoriasis skin compared to uninvolved skin, while Actinobacteria were significantly underrepresented at affected skin sites [117]. Psoriasis plaque communities were also more diverse than unaffected skin with elevated Streptococcus/Propionibacterium ratios. Unfortunately, this particular analysis employed an unmatched study design, raising the possibility that observed differences could also be due to variation between microbial communities at distinct topographical sites.
To address this concern, more recent studies have employed a matched control design that compares identical unaffected/affected skin sites. The first utilized skin biopsies to study microbial populations on the trunk, arms, and legs of affected individuals [118]. This group found no differences in alpha or beta diversity between psoriatic and normal skin. Moreover, when taking body site into account, no differences were observed between Firmicutes or Actinobacteria species at the trunk or limbs. Proteobacteria were found to be significantly greater in trunk psoriasis samples compared to the control group, however, this result was not significant when comparing the legs and arms of psoriasis subjects to controls. Similarly to above, the ratio of Streptococcus/Propionibacterium was elevated in the psoriasis group with respect to controls, but this result was largely due to the absence of Propionibacterium in a number of psoriasis samples, rather than significant fluctuations in Streptococcal species.
More recently, Alekseyenko et al. [119] compared swabs of psoriasis lesions to unaffected skin sites and demographically matched controls. While trending towards decreased alpha diversity, no significant differences in this metric were detected between lesions, unaffected sites, or control samples at the OTU level. There were also no differences in the relative abundances of Firmicutes or Actinobacteria. Notably, Proteobacteria were found at significantly higher levels in unaffected skin, in contrast to the above-mentioned study. Plaque specimens also displayed the greatest intragroup diversity while unaffected skin from psoriasis patients was more similar to control skin. This suggests that psoriasis plaques may be more permissive to alternative phylotypes, while unaffected skin may retain its ability to influence microbial populations.
In all, these studies indicate that skin bacterial communities from affected subjects may be shifted in a modest, but significant manner. Given the intrapersonal variability of the microbiota at sites with disease predilection, it is also possible that stochastic differences between subjects are masking additional, more subtle trends. For this reason, longitudinal comparisons of subjects may prove more valuable as a means to survey the skin over time and monitor each individual with respect to his/her unique microbial community. This is especially important when considering disorders such as psoriasis, in which alterations to the microbiota appear less pronounced.
Atopic dermatitis
Atopic dermatitis (AD) is a chronic inflammatory skin disease that affects 10–20 % of the childhood population [120]. This condition initially appears as an eczematous rash with pruritus and erythema, but during later stages of disease these lesions can mature into lichenified plaques [121]. AD also predisposes individuals to increased prevalence of asthma, allergic rhinitis, and food allergies—a condition known as the “atopic march” [122]. Unlike psoriasis, AD is a CD4+ Th2-mediated disorder with IL-4, IL-5, and IL-13 driving initial inflammatory events [123–126]. Upon sensitization, epidermal cells secrete proinflammatory cytokines such as thymic stromal lymphopoietin (TSLP), IL-25, and IL-33 [127–129]. This response then promotes a Th2-specific immune response which can lead to elevated infiltration by mast cells, eosinophils, and allergen-specific IgE [130–132].
Similarly to the aforementioned conditions, the underlying cause of AD pathology also remains unclear. Although both immune dysfunction and epidermal abnormalities have been implicated by GWAS analyses, loci associated with cutaneous barrier function have been associated most strongly with the disease, specifically mutations in the filament-aggregating protein, filaggrin [133]. Filaggrin is a major structural protein of the epidermis that aligns keratin filaments and contributes to the contractile strength of the stratum corneum [134]. Over time, filaggrin is also broken down into natural moisturizing factors and amino acid derivatives to assist in the hydration and acidification of the stratum corneum [135]. As such, this protein represents an essential member of the epidermal differentiation complex.
Because of the strong association between FLG mutations and AD, it is generally thought that disruptions to the epidermal barrier predispose the skin to allergen sensitization and immune dysfunction. However, this alteration in structure cannot fully explain the development of AD, as approximately 40 % of patients with FLG mutations often fail to develop the characteristic lesions seen in affected individuals [136]. FLG expression can also be downregulated in patients with wild type FLG alleles, suggesting that filaggrin levels and activity could be affected by peripheral means [137]. Indeed, exposure to the cytokines IL-4 and IL-13 can reduce expression of FLG, suggesting an alternative model in a subset of individuals whereby immune dysregulation could portend epidermal barrier abnormalities [138].
Interestingly, a number of studies also suggest that AD can promote colonization of the skin by Staphylococcus aureus. While S. aureus is a rather infrequent inhabitant of extranasal body sites in healthy individuals, it has been shown to colonize >80 % of patients with AD [139–141]. In support of this, a recent study utilizing 16S rRNA gene sequencing found that Staphylococcus species, specifically S. aureus and S. epidermidis, dominated atopic lesions, while the common skin residents Corynebacterium, Streptococcus, and Propionibacterium were all significantly reduced [142]. The relative abundances of S. aureus were also correlated with AD disease severity, similarly to previous reports, indicating an increased propensity for S. aureus to colonize AD lesions [140, 141, 143].
This increase in colonization has been hypothesized to occur for a number of reasons including a rise in the availability of S. aureus binding receptors, decreases in the expression of antimicrobial peptides (AMPs), and elevated levels of IL-4 expression. In this regard, the lack of an intact stratum corneum in AD skin could expose extracellular matrix proteins to the surface and promote S. aureus colonization. Indeed, S. aureus adherence to the skin is reduced following preincubation with fibrinogen or fibronectin, and S. aureus strains lacking fibrinogen- and fibronectin-binding proteins illustrate significantly impaired binding to AD skin [144, 145]. The cytokine IL-4 has also been shown to upregulate the production of fibronectin by dermal fibroblasts while binding of S. aureus to the skin is significantly impaired in IL-4 knockout mice [144, 146].
Unfortunately, the importance of antimicrobial peptides to S. aureus colonization remains unclear. It was initially thought that reduced expression of AMPs in atopic skin could eliminate a key barrier to S. aureus colonization. In support of this, numerous studies have reported decreased expression of AMPs in AD-affected skin compared to that of psoriatic lesions [147–149]. However, more recent data comparing the levels of antimicrobial peptides in AD skin to that of unaffected controls have shown increased expression of multiple AMPs including RNase 7, psoriasin, hBD-2, hBD-3, and LL-37 [150, 151]. Therefore, the previously ascribed reduction of AMPs in AD skin may be due more to the upregulation of these genes in psoriatic skin, rather than their decreased production in atopic individuals.
In all, it appears that both barrier disruptions and improper immune activation contribute to lesions in AD patients. While the underlying cause of inflammation remains unclear, it is likely that this determinant involves a combination of genetic and environmental factors. Notwithstanding, AD pathology consistently leads to shifts in skin microbial communities including an increase in staphylococcal species such as S. aureus. While this observation is a satisfying explanation for the increased prevalence of S. aureus infections in AD patients, it is perhaps more striking that this rate is not higher [152]. S. aureus levels have been found to reach 107 CFU/cm2 in uninfected individuals [139, 140], indicating that affected subjects may retain the ability to limit S. aureus pathogenesis despite a number of immune abnormalities. As such, a compartmentalized response in AD patients may exist, similarly to that seen in the gut, whereby atopic lesions can unintentionally promote the growth of S. aureus at the skin surface while simultaneously opposing infection of the underlying tissues.
Concluding remarks
Advances in sequencing technology have enhanced our ability to characterize cutaneous microbial communities in a more precise and accurate manner, and as a result, our knowledge regarding host–microbe interactions in skin health and disease is steadily increasing. As these insights are deepened and developed, a major challenge will be to translate this knowledge into strategies that improve skin health and cutaneous diagnostic techniques. Future analyses employing shotgun metagenomics and metabolomics are essential to this goal, as we work towards a better comprehension of skin microbial population dynamics. Indeed a recent study of the skin microbiome utilizing metagenomic approaches has contributed greatly to our understanding of skin bacterial communities [153]. Studies such as these are crucial to our perception of cutaneous microorganisms and can inform future experimental approaches. Only following these initial characterizations we can hope to truly appreciate the dysbiotic states associated with disease, and only then we can strive to successfully elucidate the importance of microbial inhabitants to hominal equilibria.
Acknowledgments
We thank Dr. John Seykora (University of Pennsylvania Department of Dermatology) for images in Fig. 1. AS is supported by the Department of Defense (DoD) through the National Defense Science and Engineering Graduate (NDSEG) Fellowship Program.
Abbreviations
- OTU
Operational taxonomic unit
- PCR
Polymerase chain reaction
- qPCR
Quantitative polymerase chain reaction
- LC
Langerhans cells
- CLA
Cutaneous leukocyte antigen
- HPV
Human papillomavirus
- PID
Primary immunodeficiency
- GF
Germ free
- SPF
Specific pathogen free
- AD
Atopic dermatitis
References
- 1.Telofski LS, Morello AP, 3rd, Mack Correa MC, Stamatas GN. The infant skin barrier: can we preserve, protect, and enhance the barrier? Dermatol Res Pract. 2012;2012:198789. doi: 10.1155/2012/198789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9(4):244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simpson CL, Patel DM, Green KJ. Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat Rev Mol Cell Biol. 2011;12(9):565–580. doi: 10.1038/nrm3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchs E, Raghavan S. Getting under the skin of epidermal morphogenesis. Nat Rev Genet. 2002;3(3):199–209. doi: 10.1038/nrg758. [DOI] [PubMed] [Google Scholar]
- 5.Chu DC. Development and structure of skin. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, editors. Fitzpatrick’s dermatology in general medicine. 8. New York: Mcgraw-Hill Medical; 2012. [Google Scholar]
- 6.Kirschner N, Brandner JM. Barriers and more: functions of tight junction proteins in the skin. Ann NY Acad Sci. 2012;1257:158–166. doi: 10.1111/j.1749-6632.2012.06554.x. [DOI] [PubMed] [Google Scholar]
- 7.Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6(4):328–340. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- 8.Zouboulis CC, Boschnakow A. Chronological ageing and photoageing of the human sebaceous gland. Clin Exp Dermatol. 2001;26(7):600–607. doi: 10.1046/j.1365-2230.2001.00894.x. [DOI] [PubMed] [Google Scholar]
- 9.Roth RR, James WD. Microbial ecology of the skin. Annu Rev Microbiol. 1988;42:441–464. doi: 10.1146/annurev.mi.42.100188.002301. [DOI] [PubMed] [Google Scholar]
- 10.Puhvel SM, Reisner RM, Sakamoto M. Analysis of lipid composition of isolated human sebaceous gland homogenates after incubation with cutaneous bacteria. Thin-layer chromatography. J Invest Dermatol. 1975;64(6):406–411. doi: 10.1111/1523-1747.ep12512337. [DOI] [PubMed] [Google Scholar]
- 11.Leyden JJ, McGinley KJ, Mills OH, Kligman AM. Age-related changes in the resident bacterial flora of the human face. J Invest Dermatol. 1975;65(4):379–381. doi: 10.1111/1523-1747.ep12607630. [DOI] [PubMed] [Google Scholar]
- 12.Lu C, Fuchs E. Sweat gland progenitors in development, homeostasis, and wound repair. Cold Spring Harbor Perspect Med. 2014 doi: 10.1101/cshperspect.a015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giacomoni PU, Mammone T, Teri M. Gender-linked differences in human skin. J Dermatol Sci. 2009;55(3):144–149. doi: 10.1016/j.jdermsci.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324(5931):1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao Z, Tseng CH, Pei Z, Blaser MJ. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci USA. 2007;104(8):2927–2932. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staudinger T, Pipal A, Redl B. Molecular analysis of the prevalent microbiota of human male and female forehead skin compared to forearm skin and the influence of make-up. J Appl Microbiol. 2011;110(6):1381–1389. doi: 10.1111/j.1365-2672.2011.04991.x. [DOI] [PubMed] [Google Scholar]
- 18.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA. 2008;105(46):17994–17999. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ling Z, Liu X, Luo Y, Yuan L, Nelson KE, Wang Y, Xiang C, Li L. Pyrosequencing analysis of the human microbiota of healthy Chinese undergraduates. BMC Genom. 2013;14:390. doi: 10.1186/1471-2164-14-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dekio I, Hayashi H, Sakamoto M, Kitahara M, Nishikawa T, Suematsu M, Benno Y. Detection of potentially novel bacterial components of the human skin microbiota using culture-independent molecular profiling. J Med Microbiol. 2005;54(Pt 12):1231–1238. doi: 10.1099/jmm.0.46075-0. [DOI] [PubMed] [Google Scholar]
- 21.Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D, Nomicos E, Park M, Kong HH, Segre JA. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498(7454):367–370. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandby-Moller J, Poulsen T, Wulf HC. Epidermal thickness at different body sites: relationship to age, gender, pigmentation, blood content, skin type and smoking habits. Acta Dermato-venereol. 2003;83(6):410–413. doi: 10.1080/00015550310015419. [DOI] [PubMed] [Google Scholar]
- 23.Shuster S, Black MM, McVitie E. The influence of age and sex on skin thickness, skin collagen and density. Br J Dermatol. 1975;93(6):639–643. doi: 10.1111/j.1365-2133.1975.tb05113.x. [DOI] [PubMed] [Google Scholar]
- 24.Pochi PE, Strauss JS. Endocrinologic control of the development and activity of the human sebaceous gland. J Invest Dermatol. 1974;62(3):191–201. doi: 10.1111/1523-1747.ep12676783. [DOI] [PubMed] [Google Scholar]
- 25.Green JM, Bishop PA, Muir IH, Lomax RG. Gender differences in sweat lactate. Eur J Appl Physiol. 2000;82(3):230–235. doi: 10.1007/s004210050676. [DOI] [PubMed] [Google Scholar]
- 26.Kim MK, Patel RA, Shinn AH, Choi SY, Byun HJ, Huh CH, Park KC, Youn SW. Evaluation of gender difference in skin type and pH. J Dermatol Sci. 2006;41(2):153–156. doi: 10.1016/j.jdermsci.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Ehlers C, Ivens UI, Moller ML, Senderovitz T, Serup J. Females have lower skin surface pH than men. A study on the surface of gender, forearm site variation, right/left difference and time of the day on the skin surface pH. Skin Res Technol. 2001;7(2):90–94. doi: 10.1034/j.1600-0846.2001.70206.x. [DOI] [PubMed] [Google Scholar]
- 28.Jacobi U, Gautier J, Sterry W, Lademann J. Gender-related differences in the physiology of the stratum corneum. Dermatology. 2005;211(4):312–317. doi: 10.1159/000088499. [DOI] [PubMed] [Google Scholar]
- 29.Ohman H, Vahlquist A. In vivo studies concerning a pH gradient in human stratum corneum and upper epidermis. Acta Dermato-venereol. 1994;74(5):375–379. doi: 10.2340/0001555574375379. [DOI] [PubMed] [Google Scholar]
- 30.Blaser MJ, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Estrada I, Gao Z, Clemente JC, Costello EK, Knight R. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. ISME J. 2013;7(1):85–95. doi: 10.1038/ismej.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hospodsky D, Pickering AJ, Julian TR, Miller D, Gorthala S, Boehm AB, Peccia J. Hand bacterial communities vary across two different human populations. Microbiology. 2014;160(Pt 6):1144–1152. doi: 10.1099/mic.0.075390-0. [DOI] [PubMed] [Google Scholar]
- 32.Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 2012;4(10):77. doi: 10.1186/gm378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akaza N, Akamatsu H, Sasaki Y, Takeoka S, Kishi M, Mizutani H, Sano A, Hirokawa K, Nakata S, Matsunaga K. Cutaneous Malassezia microbiota of healthy subjects differ by sex, body part and season. J Dermatol. 2010;37(9):786–792. doi: 10.1111/j.1346-8138.2010.00913.x. [DOI] [PubMed] [Google Scholar]
- 34.Callewaert C, Kerckhof FM, Granitsiotis MS, Van Gele M, Van de Wiele T, Boon N. Characterization of Staphylococcus and Corynebacterium clusters in the human axillary region. PLoS ONE. 2013;8(8):e70538. doi: 10.1371/journal.pone.0070538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeeuwen PL, Boekhorst J, van den Bogaard EH, de Koning HD, van de Kerkhof PM, Saulnier DM, van Swam II, van Hijum SA, Kleerebezem M, Schalkwijk J, Timmerman HM. Microbiome dynamics of human epidermis following skin barrier disruption. Genome Biol. 2012;13(11):R101. doi: 10.1186/gb-2012-13-11-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans NJ, Rutter N. Development of the epidermis in the newborn. Biol Neonate. 1986;49(2):74–80. doi: 10.1159/000242513. [DOI] [PubMed] [Google Scholar]
- 37.Cartlidge P. The epidermal barrier. Semin Neonatol. 2000;5(4):273–280. doi: 10.1053/siny.2000.0013. [DOI] [PubMed] [Google Scholar]
- 38.Kalia YN, Nonato LB, Lund CH, Guy RH. Development of skin barrier function in premature infants. J Invest Dermatol. 1998;111(2):320–326. doi: 10.1046/j.1523-1747.1998.00289.x. [DOI] [PubMed] [Google Scholar]
- 39.Nikolovski J, Stamatas GN, Kollias N, Wiegand BC. Barrier function and water-holding and transport properties of infant stratum corneum are different from adult and continue to develop through the first year of life. J Invest Dermatol. 2008;128(7):1728–1736. doi: 10.1038/sj.jid.5701239. [DOI] [PubMed] [Google Scholar]
- 40.Stamatas GN, Nikolovski J, Luedtke MA, Kollias N, Wiegand BC. Infant skin microstructure assessed in vivo differs from adult skin in organization and at the cellular level. Pediatr Dermatol. 2010;27(2):125–131. doi: 10.1111/j.1525-1470.2009.00973.x. [DOI] [PubMed] [Google Scholar]
- 41.Stamatas GN, Nikolovski J, Mack MC, Kollias N. Infant skin physiology and development during the first years of life: a review of recent findings based on in vivo studies. Int J Cosmet Sci. 2011;33(1):17–24. doi: 10.1111/j.1468-2494.2010.00611.x. [DOI] [PubMed] [Google Scholar]
- 42.Giusti F, Martella A, Bertoni L, Seidenari S. Skin barrier, hydration, and pH of the skin of infants under 2 years of age. Pediatr Dermatol. 2001;18(2):93–96. doi: 10.1046/j.1525-1470.2001.018002093.x. [DOI] [PubMed] [Google Scholar]
- 43.Capone KA, Dowd SE, Stamatas GN, Nikolovski J. Diversity of the human skin microbiome early in life. J Invest Dermatol. 2011;131(10):2026–2032. doi: 10.1038/jid.2011.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA. 2010;107(26):11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cotterill JA, Cunliffe WJ, Williamson B, Bulusu L. Age and sex variation in skin surface lipid composition and sebum excretion rate. Br J Dermatol. 1972;87(4):333–340. doi: 10.1111/j.1365-2133.1972.tb07419.x. [DOI] [PubMed] [Google Scholar]
- 46.Nagata R, Nagano H, Ogishima D, Nakamura Y, Hiruma M, Sugita T. Transmission of the major skin microbiota, Malassezia, from mother to neonate. Pediatr Int. 2012;54(3):350–355. doi: 10.1111/j.1442-200X.2012.03563.x. [DOI] [PubMed] [Google Scholar]
- 47.Jimenez E, Fernandez L, Marin ML, Martin R, Odriozola JM, Nueno-Palop C, Narbad A, Olivares M, Xaus J, Rodriguez JM. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr Microbiol. 2005;51(4):270–274. doi: 10.1007/s00284-005-0020-3. [DOI] [PubMed] [Google Scholar]
- 48.Jimenez E, Marin ML, Martin R, Odriozola JM, Olivares M, Xaus J, Fernandez L, Rodriguez JM. Is meconium from healthy newborns actually sterile? Res Microbiol. 2008;159(3):187–193. doi: 10.1016/j.resmic.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 49.Steel JH, Malatos S, Kennea N, Edwards AD, Miles L, Duggan P, Reynolds PR, Feldman RG, Sullivan MHF. Bacteria and inflammatory cells in fetal membranes do not always cause preterm labor. Pediatr Res. 2005;57(3):404–411. doi: 10.1203/01.PDR.0000153869.96337.90. [DOI] [PubMed] [Google Scholar]
- 50.Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6(237):237ra265. doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stout MJ, Conlon B, Landeau M, Lee I, Bower C, Zhao Q, Roehl KA, Nelson DM, Macones GA, Mysorekar IU. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am J Obstet Gynecol. 2013;208(3):226, e221–227. doi: 10.1016/j.ajog.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grice EA, Kong HH, Renaud G, Young AC, Bouffard GG, Blakesley RW, Wolfsberg TG, Turner ML, Segre JA. A diversity profile of the human skin microbiota. Genome Res. 2008;18(7):1043–1050. doi: 10.1101/gr.075549.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013;4:1431. doi: 10.1038/ncomms2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heath WR, Carbone FR. The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol. 2013;14(10):978–985. doi: 10.1038/ni.2680. [DOI] [PubMed] [Google Scholar]
- 55.Yu N, Zhang S, Zuo F, Kang K, Guan M, Xiang L. Cultured human melanocytes express functional toll-like receptors 2–4, 7 and 9. J Dermatol Sci. 2009;56(2):113–120. doi: 10.1016/j.jdermsci.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 56.Schuler G, Steinman RM. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J Exp Med. 1985;161(3):526–546. doi: 10.1084/jem.161.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kubo A, Nagao K, Yokouchi M, Sasaki H, Amagai M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J Exp Med. 2009;206(13):2937–2946. doi: 10.1084/jem.20091527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, de Bovis B, Devilard E, Viret C, Azukizawa H, Kissenpfennig A, Malissen B. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med. 2010;207(1):189–206. doi: 10.1084/jem.20091964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, Brooks AG, Heath WR. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. 2009;10(5):488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- 60.Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, Butcher EC. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol. 2007;8(3):285–293. doi: 10.1038/ni1433. [DOI] [PubMed] [Google Scholar]
- 61.Fuhlbrigge RC, Kieffer JD, Armerding D, Kupper TS. Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature. 1997;389(6654):978–981. doi: 10.1038/40166. [DOI] [PubMed] [Google Scholar]
- 62.Campbell JJ, Haraldsen G, Pan J, Rottman J, Qin S, Ponath P, Andrew DP, Warnke R, Ruffing N, Kassam N, Wu L, Butcher EC. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature. 1999;400(6746):776–780. doi: 10.1038/23495. [DOI] [PubMed] [Google Scholar]
- 63.Schaerli P, Ebert L, Willimann K, Blaser A, Roos RS, Loetscher P, Moser B. A skin-selective homing mechanism for human immune surveillance T cells. J Exp Med. 2004;199(9):1265–1275. doi: 10.1084/jem.20032177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10(5):524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 65.Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, Carbone FR, Mueller SN. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477(7363):216–219. doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- 66.Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36(5):873–884. doi: 10.1016/j.immuni.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, Ryan AF, Di Nardo A, Gallo RL. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol. 2010;130(9):2211–2221. doi: 10.1038/jid.2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wanke I, Steffen H, Christ C, Krismer B, Gotz F, Peschel A, Schaller M, Schittek B. Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J Invest Dermatol. 2011;131(2):382–390. doi: 10.1038/jid.2010.328. [DOI] [PubMed] [Google Scholar]
- 69.Percoco G, Merle C, Jaouen T, Ramdani Y, Benard M, Hillion M, Mijouin L, Lati E, Feuilloley M, Lefeuvre L, Driouich A, Follet-Gueye ML. Antimicrobial peptides and pro-inflammatory cytokines are differentially regulated across epidermal layers following bacterial stimuli. Exp Dermatol. 2013;22(12):800–806. doi: 10.1111/exd.12259. [DOI] [PubMed] [Google Scholar]
- 70.Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR, Hooper LV, Schmidt RR, von Aulock S, Radek KA, Huang CM, Ryan AF, Gallo RL. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15(12):1377–1382. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Z, MacLeod DT, Di Nardo A. Commensal bacteria lipoteichoic acid increases skin mast cell antimicrobial activity against vaccinia viruses. J Immunol. 2012;189(4):1551–1558. doi: 10.4049/jimmunol.1200471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuo IH, Yoshida T, De Benedetto A, Beck LA. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol. 2013;131(2):266–278. doi: 10.1016/j.jaci.2012.12.1563. [DOI] [PubMed] [Google Scholar]
- 73.Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci USA. 2013;110(37):15061–15066. doi: 10.1073/pnas.1307855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garcia-Garcera M, Coscolla M, Garcia-Etxebarria K, Martin-Caballero J, Gonzalez-Candelas F, Latorre A, Calafell F. Staphylococcus prevails in the skin microbiota of long-term immunodeficient mice. Environ Microbiol. 2012;14(8):2087–2098. doi: 10.1111/j.1462-2920.2012.02756.x. [DOI] [PubMed] [Google Scholar]
- 75.Oh J, Freeman AF, Park M, Sokolic R, Candotti F, Holland SM, Segre JA, Kong HH. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013;23(12):2103–2114. doi: 10.1101/gr.159467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, Spencer S, Hall JA, Dzutsev A, Kong H, Campbell DJ, Trinchieri G, Segre JA, Belkaid Y. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337(6098):1115–1119. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shen W, Li W, Hixon JA, Bouladoux N, Belkaid Y, Dzutzev A, Durum SK. Adaptive immunity to murine skin commensals. Proc Natl Acad Sci USA. 2014;111(29):E2977–E2986. doi: 10.1073/pnas.1401820111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scholz F, Badgley BD, Sadowsky MJ, Kaplan DH. Immune mediated shaping of microflora community composition depends on barrier site. PLoS ONE. 2014;9(1):e84019. doi: 10.1371/journal.pone.0084019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bickers DR, Lim HW, Margolis D, Weinstock MA, Goodman C, Faulkner E, Gould C, Gemmen E, Dall T. The burden of skin diseases: 2004 a joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol. 2006;55(3):490–500. doi: 10.1016/j.jaad.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 80.James WD. Clinical practice. Acne. N Engl J Med. 2005;352(14):1463–1472. doi: 10.1056/NEJMcp033487. [DOI] [PubMed] [Google Scholar]
- 81.Kurokawa I, Danby FW, Ju Q, Wang X, Xiang LF, Xia L, Chen W, Nagy I, Picardo M, Suh DH, Ganceviciene R, Schagen S, Tsatsou F, Zouboulis CC. New developments in our understanding of acne pathogenesis and treatment. Exp Dermatol. 2009;18(10):821–832. doi: 10.1111/j.1600-0625.2009.00890.x. [DOI] [PubMed] [Google Scholar]
- 82.Nagy I, Pivarcsi A, Kis K, Koreck A, Bodai L, McDowell A, Seltmann H, Patrick S, Zouboulis CC, Kemeny L. Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infect Inst Pasteur. 2006;8(8):2195–2205. doi: 10.1016/j.micinf.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 83.Kim J, Ochoa MT, Krutzik SR, Takeuchi O, Uematsu S, Legaspi AJ, Brightbill HD, Holland D, Cunliffe WJ, Akira S, Sieling PA, Godowski PJ, Modlin RL. Activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. J Immunol. 2002;169(3):1535–1541. doi: 10.4049/jimmunol.169.3.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vowels BR, Yang S, Leyden JJ. Induction of proinflammatory cytokines by a soluble factor of Propionibacterium acnes: implications for chronic inflammatory acne. Infect Immun. 1995;63(8):3158–3165. doi: 10.1128/iai.63.8.3158-3165.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Norris JF, Cunliffe WJ. A histological and immunocytochemical study of early acne lesions. Br J Dermatol. 1988;118(5):651–659. doi: 10.1111/j.1365-2133.1988.tb02566.x. [DOI] [PubMed] [Google Scholar]
- 86.Jeremy AH, Holland DB, Roberts SG, Thomson KF, Cunliffe WJ. Inflammatory events are involved in acne lesion initiation. J Invest Dermatol. 2003;121(1):20–27. doi: 10.1046/j.1523-1747.2003.12321.x. [DOI] [PubMed] [Google Scholar]
- 87.Layton AM, Morris C, Cunliffe WJ, Ingham E. Immunohistochemical investigation of evolving inflammation in lesions of acne vulgaris. Exp Dermatol. 1998;7(4):191–197. doi: 10.1111/j.1600-0625.1998.tb00323.x. [DOI] [PubMed] [Google Scholar]
- 88.Qin M, Pirouz A, Kim MH, Krutzik SR, Garban HJ, Kim J. Propionibacterium acnes induces IL-1beta secretion via the NLRP3 inflammasome in human monocytes. J Invest Dermatol. 2014;134(2):381–388. doi: 10.1038/jid.2013.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kistowska M, Gehrke S, Jankovic D, Kerl K, Fettelschoss A, Feldmeyer L, Fenini G, Kolios A, Navarini A, Ganceviciene R, Schauber J, Contassot E, French LE. IL-1beta drives inflammatory responses to Propionibacterium acnes in vitro and in vivo. J Invest Dermatol. 2014;134(3):677–685. doi: 10.1038/jid.2013.438. [DOI] [PubMed] [Google Scholar]
- 90.Li ZJ, Choi DK, Sohn KC, Seo MS, Lee HE, Lee Y, Seo YJ, Lee YH, Shi G, Zouboulis CC, Kim CD, Lee JH, Im M. Propionibacterium acnes Activates the NLRP3 inflammasome in human sebocytes. J Invest Dermatol. 2014 doi: 10.1038/jid.2014.221. [DOI] [PubMed] [Google Scholar]
- 91.Agak GW, Qin M, Nobe J, Kim MH, Krutzik SR, Tristan GR, Elashoff D, Garban HJ, Kim J. Propionibacterium acnes induces an IL-17 response in acne vulgaris that is regulated by vitamin A and vitamin D. J Invest Dermatol. 2014;134(2):366–373. doi: 10.1038/jid.2013.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kistowska M, Meier B, Proust T, Feldmeyer L, Cozzio A, Kuendig T, Contassot E, French LE. Propionibacterium acnes promotes Th17 and Th17/Th1 responses in acne patients. J Invest Dermatol. 2014 doi: 10.1038/jid.2014.290. [DOI] [PubMed] [Google Scholar]
- 93.Alexeyev OA, Lundskog B, Ganceviciene R, Palmer RH, McDowell A, Patrick S, Zouboulis C, Golovleva I. Pattern of tissue invasion by Propionibacterium acnes in acne vulgaris. J Dermatol Sci. 2012;67(1):63–66. doi: 10.1016/j.jdermsci.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 94.Jahns AC, Lundskog B, Ganceviciene R, Palmer RH, Golovleva I, Zouboulis CC, McDowell A, Patrick S, Alexeyev OA. An increased incidence of Propionibacterium acnes biofilms in acne vulgaris: a case-control study. Br J Dermatol. 2012;167(1):50–58. doi: 10.1111/j.1365-2133.2012.10897.x. [DOI] [PubMed] [Google Scholar]
- 95.Lomholt HB, Kilian M. Population genetic analysis of Propionibacterium acnes identifies a subpopulation and epidemic clones associated with acne. PLoS ONE. 2010;5(8):e12277. doi: 10.1371/journal.pone.0012277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McDowell A, Barnard E, Nagy I, Gao A, Tomida S, Li H, Eady A, Cove J, Nord CE, Patrick S. An expanded multilocus sequence typing scheme for Propionibacterium acnes: investigation of ‘pathogenic’, ‘commensal’ and antibiotic resistant strains. PLoS ONE. 2012;7(7):e41480. doi: 10.1371/journal.pone.0041480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kwon HH, Yoon JY, Park SY, Suh DH. Analysis of distribution patterns of Propionibacterium acnes phylotypes and Peptostreptococcus species from acne lesions. Br J Dermatol. 2013;169(5):1152–1155. doi: 10.1111/bjd.12486. [DOI] [PubMed] [Google Scholar]
- 98.Fitz-Gibbon S, Tomida S, Chiu BH, Nguyen L, Du C, Liu M, Elashoff D, Erfe MC, Loncaric A, Kim J, Modlin RL, Miller JF, Sodergren E, Craft N, Weinstock GM, Li H. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J Invest Dermatol. 2013;133(9):2152–2160. doi: 10.1038/jid.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nagy I, Pivarcsi A, Koreck A, Szell M, Urban E, Kemeny L. Distinct strains of Propionibacterium acnes induce selective human beta-defensin-2 and interleukin-8 expression in human keratinocytes through toll-like receptors. J Invest Dermatol. 2005;124(5):931–938. doi: 10.1111/j.0022-202X.2005.23705.x. [DOI] [PubMed] [Google Scholar]
- 100.Crow JM. Psoriasis uncovered. Nature. 2012;492(7429):S50–S`51. doi: 10.1038/492S50a. [DOI] [PubMed] [Google Scholar]
- 101.Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445(7130):866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 102.Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385–422. doi: 10.1146/annurev-pathol-011811-132448. [DOI] [PubMed] [Google Scholar]
- 103.Schon MP, Boehncke WH. Psoriasis. N Engl J Med. 2005;352(18):1899–1912. doi: 10.1056/NEJMra041320. [DOI] [PubMed] [Google Scholar]
- 104.Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129(6):1339–1350. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- 105.Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, Barton A, Band G, Bellenguez C, Bergboer JG, Blackwell JM, Bramon E, Bumpstead SJ, Casas JP, Cork MJ, Corvin A, Deloukas P, Dilthey A, Duncanson A, Edkins S, Estivill X, Fitzgerald O, Freeman C, Giardina E, Gray E, Hofer A, Huffmeier U, Hunt SE, Irvine AD, Jankowski J, Kirby B, Langford C, Lascorz J, Leman J, Leslie S, Mallbris L, Markus HS, Mathew CG, McLean WH, McManus R, Mossner R, Moutsianas L, Naluai AT, Nestle FO, Novelli G, Onoufriadis A, Palmer CN, Perricone C, Pirinen M, Plomin R, Potter SC, Pujol RM, Rautanen A, Riveira-Munoz E, Ryan AW, Salmhofer W, Samuelsson L, Sawcer SJ, Schalkwijk J, Smith CH, Stahle M, Su Z, Tazi-Ahnini R, Traupe H, Viswanathan AC, Warren RB, Weger W, Wolk K, Wood N, Worthington J, Young HS, Zeeuwen PL, Hayday A, Burden AD, Griffiths CE, Kere J, Reis A, McVean G, Evans DM, Brown MA, Barker JN, Peltonen L, Donnelly P, Trembath RC. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42(11):985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang XJ, Huang W, Yang S, Sun LD, Zhang FY, Zhu QX, Zhang FR, Zhang C, Du WH, Pu XM, Li H, Xiao FL, Wang ZX, Cui Y, Hao F, Zheng J, Yang XQ, Cheng H, He CD, Liu XM, Xu LM, Zheng HF, Zhang SM, Zhang JZ, Wang HY, Cheng YL, Ji BH, Fang QY, Li YZ, Zhou FS, Han JW, Quan C, Chen B, Liu JL, Lin D, Fan L, Zhang AP, Liu SX, Yang CJ, Wang PG, Zhou WM, Lin GS, Wu WD, Fan X, Gao M, Yang BQ, Lu WS, Zhang Z, Zhu KJ, Shen SK, Li M, Zhang XY, Cao TT, Ren W, Zhang X, He J, Tang XF, Lu S, Yang JQ, Zhang L, Wang DN, Yuan F, Yin XY, Huang HJ, Wang HF, Lin XY, Liu JJ. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat Genet. 2009;41(2):205–210. doi: 10.1038/ng.310. [DOI] [PubMed] [Google Scholar]
- 107.Trembath RC, Clough RL, Rosbotham JL, Jones AB, Camp RD, Frodsham A, Browne J, Barber R, Terwilliger J, Lathrop GM, Barker JN. Identification of a major susceptibility locus on chromosome 6p and evidence for further disease loci revealed by a two stage genome-wide search in psoriasis. Hum Mol Genet. 1997;6(5):813–820. doi: 10.1093/hmg/6.5.813. [DOI] [PubMed] [Google Scholar]
- 108.Mallon E, Newson R, Bunker CB. HLA-Cw6 and the genetic predisposition to psoriasis: a meta-analysis of published serologic studies. J Invest Dermatol. 1999;113(4):693–695. doi: 10.1046/j.1523-1747.1999.00724.x. [DOI] [PubMed] [Google Scholar]
- 109.Norrlind R. Significance of infections in origin of psoriasis. Acta Rhematol Scand. 1955;1:135–144. [PubMed] [Google Scholar]
- 110.Miller RA. The Koebner phenomenon. Int J Dermatol. 1982;21(4):192–197. doi: 10.1111/j.1365-4362.1982.tb02070.x. [DOI] [PubMed] [Google Scholar]
- 111.Whyte HJ, Baughman RD. Acute guttate psoriasis and streptococcal infection. Arch Dermatol. 1964;89:350–356. doi: 10.1001/archderm.1964.01590270036008. [DOI] [PubMed] [Google Scholar]
- 112.Telfer NR, Chalmers RJ, Whale K, Colman G. The role of streptococcal infection in the initiation of guttate psoriasis. Arch Dermatol. 1992;128(1):39–42. [PubMed] [Google Scholar]
- 113.Ladizinski B, Lee KC, Wilmer E, Alavi A, Mistry N, Sibbald RG. A review of the clinical variants and the management of psoriasis. Adv Skin Wound Care. 2013;26(6):271–284. doi: 10.1097/01.ASW.0000429778.10020.67. [DOI] [PubMed] [Google Scholar]
- 114.Boehncke WH. Psoriasis and bacterial superantigens—formal or causal correlation? Trends Microbiol. 1996;4(12):485–489. doi: 10.1016/s0966-842x(97)82910-1. [DOI] [PubMed] [Google Scholar]
- 115.Sigmundsdottir H, Sigurgeirsson B, Troye-Blomberg M, Good MF, Valdimarsson H, Jonsdottir I. Circulating T cells of patients with active psoriasis respond to streptococcal M-peptides sharing sequences with human epidermal keratins. Scand J Immunol. 1997;45(6):688–697. doi: 10.1046/j.1365-3083.1997.d01-438.x. [DOI] [PubMed] [Google Scholar]
- 116.Gudmundsdottir AS, Sigmundsdottir H, Sigurgeirsson B, Good MF, Valdimarsson H, Jonsdottir I. Is an epitope on keratin 17 a major target for autoreactive T lymphocytes in psoriasis? Clin Exp Immunol. 1999;117(3):580–586. doi: 10.1046/j.1365-2249.1999.01013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE. 2008;3(7):e2719. doi: 10.1371/journal.pone.0002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fahlen A, Engstrand L, Baker BS, Powles A, Fry L. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch Dermatol Res. 2012;304(1):15–22. doi: 10.1007/s00403-011-1189-x. [DOI] [PubMed] [Google Scholar]
- 119.Alekseyenko AV, Perez-Perez GI, De Souza A, Strober B, Gao Z, Bihan M, Li K, Methe BA, Blaser MJ. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome. 2013;1(1):31. doi: 10.1186/2049-2618-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shaw TE, Currie GP, Koudelka CW, Simpson EL. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131(1):67–73. doi: 10.1038/jid.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358(14):1483–1494. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 122.Spergel JM. From atopic dermatitis to asthma: the atopic march. Ann Allergy Asthma Immunol. 2010;105(2):99–106. doi: 10.1016/j.anai.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 123.Furue M, Ogata F, Ootsuki M, Ishibashi Y. Hyperresponsibility to exogeneous interleukin 4 in atopic dermatitis. J Dermatol. 1989;16(3):247–250. doi: 10.1111/j.1346-8138.1989.tb01258.x. [DOI] [PubMed] [Google Scholar]
- 124.Jeong CW, Ahn KS, Rho NK, Park YD, Lee DY, Lee JH, Lee ES, Yang JM. Differential in vivo cytokine mRNA expression in lesional skin of intrinsic vs. extrinsic atopic dermatitis patients using semiquantitative RT-PCR. Clin Exp Allergy. 2003;33(12):1717–1724. doi: 10.1111/j.1365-2222.2003.01782.x. [DOI] [PubMed] [Google Scholar]
- 125.Hamid Q, Naseer T, Minshall EM, Song YL, Boguniewicz M, Leung DY. In vivo expression of IL-12 and IL-13 in atopic dermatitis. J Allergy Clin Immunol. 1996;98(1):225–231. doi: 10.1016/s0091-6749(96)70246-4. [DOI] [PubMed] [Google Scholar]
- 126.Lee GR, Flavell RA. Transgenic mice which overproduce Th2 cytokines develop spontaneous atopic dermatitis and asthma. Int Immunol. 2004;16(8):1155–1160. doi: 10.1093/intimm/dxh117. [DOI] [PubMed] [Google Scholar]
- 127.Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, de Waal-Malefyt Rd R, Bazan F, Kastelein RA, Liu YJ. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 128.Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, Hippe A, Corrigan CJ, Dong C, Homey B, Yao Z, Ying S, Huston DP, Liu YJ. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204(8):1837–1847. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Savinko T, Matikainen S, Saarialho-Kere U, Lehto M, Wang G, Lehtimaki S, Karisola P, Reunala T, Wolff H, Lauerma A, Alenius H. IL-33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors. J Invest Dermatol. 2012;132(5):1392–1400. doi: 10.1038/jid.2011.446. [DOI] [PubMed] [Google Scholar]
- 130.Soter NA. Morphology of atopic eczema. Allergy. 1989;44(Suppl 9):16–19. doi: 10.1111/j.1398-9995.1989.tb04310.x. [DOI] [PubMed] [Google Scholar]
- 131.Kagi MK, Joller-Jemelka H, Wuthrich B. Correlation of eosinophils, eosinophil cationic protein and soluble interleukin-2 receptor with the clinical activity of atopic dermatitis. Dermatology. 1992;185(2):88–92. doi: 10.1159/000247419. [DOI] [PubMed] [Google Scholar]
- 132.Liu FT, Goodarzi H, Chen HY. IgE, mast cells, and eosinophils in atopic dermatitis. Clin Rev Allergy Immunol. 2011;41(3):298–310. doi: 10.1007/s12016-011-8252-4. [DOI] [PubMed] [Google Scholar]
- 133.van den Oord RA, Sheikh A. Filaggrin gene defects and risk of developing allergic sensitisation and allergic disorders: systematic review and meta-analysis. BMJ. 2009;339:b2433. doi: 10.1136/bmj.b2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol. 2013;131(2):280–291. doi: 10.1016/j.jaci.2012.12.668. [DOI] [PubMed] [Google Scholar]
- 135.Rawlings AV, Harding CR. Moisturization and skin barrier function. Dermatol Ther. 2004;17(Suppl 1):43–48. doi: 10.1111/j.1396-0296.2004.04s1005.x. [DOI] [PubMed] [Google Scholar]
- 136.Henderson J, Northstone K, Lee SP, Liao H, Zhao Y, Pembrey M, Mukhopadhyay S, Smith GD, Palmer CN, McLean WH, Irvine AD. The burden of disease associated with filaggrin mutations: a population-based, longitudinal birth cohort study. J Allergy Clin Immunol. 2008;121(4):872–877, e879. doi: 10.1016/j.jaci.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 137.Kezic S, O’Regan GM, Yau N, Sandilands A, Chen H, Campbell LE, Kroboth K, Watson R, Rowland M, McLean WH, Irvine AD. Levels of filaggrin degradation products are influenced by both filaggrin genotype and atopic dermatitis severity. Allergy. 2011;66(7):934–940. doi: 10.1111/j.1398-9995.2010.02540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, DeBenedetto A, Schneider L, Beck LA, Barnes KC, Leung DY. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2009;124(3 Suppl 2):R7–R12. doi: 10.1016/j.jaci.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 139.Leyden JJ, Marples RR, Kligman AM. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol. 1974;90(5):525–530. doi: 10.1111/j.1365-2133.1974.tb06447.x. [DOI] [PubMed] [Google Scholar]
- 140.Nilsson EJ, Henning CG, Magnusson J. Topical corticosteroids and Staphylococcus aureus in atopic dermatitis. J Am Acad Dermatol. 1992;27(1):29–34. doi: 10.1016/0190-9622(92)70151-5. [DOI] [PubMed] [Google Scholar]
- 141.Higaki S, Morohashi M, Yamagishi T, Hasegawa Y. Comparative study of staphylococci from the skin of atopic dermatitis patients and from healthy subjects. Int J Dermatol. 1999;38(4):265–269. doi: 10.1046/j.1365-4362.1999.00686.x. [DOI] [PubMed] [Google Scholar]
- 142.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, Turner ML, Segre JA. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22(5):850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gilani SJ, Gonzalez M, Hussain I, Finlay AY, Patel GK. Staphylococcus aureus re-colonization in atopic dermatitis: beyond the skin. Clin Exp Dermatol. 2005;30(1):10–13. doi: 10.1111/j.1365-2230.2004.01679.x. [DOI] [PubMed] [Google Scholar]
- 144.Cho SH, Strickland I, Tomkinson A, Fehringer AP, Gelfand EW, Leung DY. Preferential binding of Staphylococcus aureus to skin sites of Th2-mediated inflammation in a murine model. J Invest Dermatol. 2001;116(5):658–663. doi: 10.1046/j.0022-202x.2001.01331.x. [DOI] [PubMed] [Google Scholar]
- 145.Cho SH, Strickland I, Boguniewicz M, Leung DY. Fibronectin and fibrinogen contribute to the enhanced binding of Staphylococcus aureus to atopic skin. J Allergy Clin Immunol. 2001;108(2):269–274. doi: 10.1067/mai.2001.117455. [DOI] [PubMed] [Google Scholar]
- 146.Postlethwaite AE, Holness MA, Katai H, Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J Clin Investig. 1992;90(4):1479–1485. doi: 10.1172/JCI116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347(15):1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 148.Nomura I, Goleva E, Howell MD, Hamid QA, Ong PY, Hall CF, Darst MA, Gao B, Boguniewicz M, Travers JB, Leung DY. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol. 2003;171(6):3262–3269. doi: 10.4049/jimmunol.171.6.3262. [DOI] [PubMed] [Google Scholar]
- 149.de Jongh GJ, Zeeuwen PL, Kucharekova M, Pfundt R, van der Valk PG, Blokx W, Dogan A, Hiemstra PS, van de Kerkhof PC, Schalkwijk J. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol. 2005;125(6):1163–1173. doi: 10.1111/j.0022-202X.2005.23935.x. [DOI] [PubMed] [Google Scholar]
- 150.Ballardini N, Johansson C, Lilja G, Lindh M, Linde Y, Scheynius A, Agerberth B. Enhanced expression of the antimicrobial peptide LL-37 in lesional skin of adults with atopic eczema. Br J Dermatol. 2009;161(1):40–47. doi: 10.1111/j.1365-2133.2009.09095.x. [DOI] [PubMed] [Google Scholar]
- 151.Harder J, Dressel S, Wittersheim M, Cordes J, Meyer-Hoffert U, Mrowietz U, Folster-Holst R, Proksch E, Schroder JM, Schwarz T, Glaser R. Enhanced expression and secretion of antimicrobial peptides in atopic dermatitis and after superficial skin injury. J Invest Dermatol. 2010;130(5):1355–1364. doi: 10.1038/jid.2009.432. [DOI] [PubMed] [Google Scholar]
- 152.G R (1975) Atopic Dermatits. WB Saunders Co:19
- 153.Oh J, Byrd AL, Deming C, Conlan S, NISC Comparative Sequencing Program. Kong HH, Segre JA. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514(7520):59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]