

Abstract
Filamin A (FlnA) has been associated with actin as cytoskeleton regulator. Recently its role in the cell has come under scrutiny for FlnA’s involvement in cancer development. FlnA was originally revealed as a cancer-promoting protein, involved in invasion and metastasis. However, recent studies have also found that under certain conditions, it prevented tumor formation or progression, confusing the precise function of FlnA in cancer development. Here, we try to decipher the role of FlnA in cancer and the implications for its dual role. We propose that differences in subcellular localization of FlnA dictate its role in cancer development. In the cytoplasm, FlnA functions in various growth signaling pathways, such as vascular endothelial growth factor, in addition to being involved in cell migration and adhesion pathways, such as R-Ras and integrin signaling. Involvement in these pathways and various others has shown a correlation between high cytoplasmic FlnA levels and invasive cancers. However, an active cleaved form of FlnA can localize to the nucleus rather than the cytoplasm and its interaction with transcription factors has been linked to a decrease in invasiveness of cancers. Therefore, overexpression of FlnA has a tumor-promoting effect, only when it is localized to the cytoplasm, whereas if FlnA undergoes proteolysis and the resulting C-terminal fragment localizes to the nucleus, it acts to suppress tumor growth and inhibit metastasis. Development of drugs to target FlnA and cause cleavage and subsequent localization to the nucleus could be a new and potent field of research in treating cancer.
Keywords: filamin A, metastasis, cancer, transcription, nucleus, cytoplasm, localization
Introduction
Filamin A (FlnA), also known as actin-binding protein 280 (ABP 280), was first identified as a non-muscle actin filament cross-linking protein (Stossel & Hartwig 1975). Subsequent reports revealed the importance of this protein in cytoplasmic gelation, cell contraction, and spreading (Hartwig & Stossel 1975). Since then, two other paralogous genes, filamin B (FlnB) and filamin C (FlnC), have been established as part of the filamin family. Filamins largely act as scaffolding molecules, facilitating protein–protein interactions and influencing protein cellular localization. Studies revealed the protein structures of the filamins and identified over 90 filamin-binding proteins involved in cell signaling, cell migration and adhesion, phosphorylation, proteolysis, ion channel regulation, transcription regulation, receptor activation, muscle development, and other important cell functions. Mutations in these molecules have can result in a wide range of disease phenotypes, including terminal osseous dysplasia, cardiovascular malformations, and neural defects (Nakamura et al. 2011).
The filamins, in particular, FlnA, are highly susceptible to proteolysis. Multiple studies showed that, while full-length FlnA is mainly localized to the cytoplasm, it can be cleaved to a 90-kDa fragment that can then localize to the nucleus (Ozanne et al. 2000, Loy et al. 2003, Wang et al. 2007, Bedolla et al. 2009). In the cytoplasm, full-length FlnA promotes the development of metastasis (recently reviewed in Yue et al. (2013)), whereas nuclear FlnA was shown to be necessary for inhibition of transcription and susceptibility to therapeutic interventions (Loy et al. 2003, Wang et al. 2007). In this review, we therefore focus on the opposing roles of FlnA in cancer progression depending on its localization. Reports in the literature have proposed blocking FlnA expression as a means of cancer therapy; however, we and others showed that completely eliminating FlnA will have serious deleterious consequences. Here, we propose, instead, to promote FlnA proteolysis – which was shown to have the required cancer-suppression effect.
FlnA is important for organogenesis during development
Early studies indicated that loss of FlnA was embryonically lethal (Feng et al. 2006). Absence of FlnA resulted in male lethality because of incomplete septation of the outflow tract of the heart, which produces common arterial trunk and midline fusion defects manifesting as sternum and palate abnormalities (Hart et al. 2006). In addition, carrier females exhibit misshapen pupils while a proportion of both male and female mutant mice have other cardiac defects including ventricular septal defect (Hart et al. 2006). However, mice that lack FlnA only in the megakaryocyte (MK) lineage were generated by pairing FlnAloxP mice with PF4-Cre mice and were shown to have severe macrothrombocytopenia because of the rapid clearance of FlnA-null platelets from circulation (Jurak Begonja et al. 2011). Further, FlnA was important for platelet formation because FlnAloxP PF4-Cre bone marrows and spleens had increased megakaryopoiesis and FlnA-null proplatelets released platelets more readily than controls in vitro (Jurak Begonja et al. 2011).
Mutations in the FlnA gene were linked to a large number of developmental diseases. Periventricular heterotopia is an X-linked dominant disorder, in which neurons fail to migrate into the cerebral cortex, but remain as nodules lining the ventricular surface. This disease has been linked to various mutations in FlnA, including a frameshift mutation (3045del5) in exon 21 (de Wit et al. 2009) and a cytosine-to-thymidine missense mutation (c. C1286T) in exon 9, resulting in a threonine-to-methionine amino acid substitution (Masruha et al. 2006). Various other mutations in other parts of the gene, resulting in protein misfolding, has been reported (Sheen et al. 2001, 2005, Parrini et al. 2006). Most hemizygous affected males die early during embryogenesis, whereas heterozygous females have normal intelligence but suffer from various neurological and vascular conditions. FlnA mutations have been linked to not only neurological disorders but cardiovascular problems as well (described in details in Zhou et al. (2007)), including familial cardiac valvular dystrophy (Kyndt et al. 2007).
The high rate of neurological and cardiovascular developmental disorders due to mutations in various parts of the FlnA gene has highlighted its importance in cell migration during development (Price et al. 1994, de Wit et al. 2009). Axonal pathfinding during embryogenesis is based on the migration of nerve growth cones, which relies strongly on f-actin, filamin, α-actinin, myosin, tropomyosin, talin, and vinculin (Letourneau & Shattuck 1989). Shortly thereafter, it was discovered that mutations in FlnA prevented migration of cerebral cortical neurons that led to incomplete neurological development (Fox et al. 1998). Because of this, FlnA mutations have been linked to a broad range of congenital malformations, affecting craniofacial structures, skeleton, brain, viscera, and urogenital tract, in four X-linked human disorders: otopalatodigital syndrome types 1 (OPD1) and 2 (OPD2), frontometaphyseal dysplasia, and Melnick–Needles syndrome (Robertson et al. 2003). Interestingly, the majority of the mutations are clustered into four regions of the gene: the actin-binding domain (ABD) and rod domain repeats 3, 10, and 14/15 (described later) (Robertson et al. 2003). Mutations in FlnA are also linked to an X-linked recessive form of chronic idiopathic intestinal pseudo-obstruction (CIIPX; Gargiulo et al. 2007) and to an X-inactivation mosaicism in the corneal epithelia (Douvaras et al. 2012).
The varieties of disorders that are associated with mutations in FlnA indicate its importance in cell and organ development. High filamin expression was reported in diverse organs including esophagus, stomach, intestine, aorta, lung, bladder, uterus, and ovary (Brown & Binder 1993). Filamin expression is somewhat tissue specific: FlnA and FlnB are ubiquitously expressed, while FlnC is limited to cardiac and skeletal cells (Maestrini et al. 1993, van der Flier & Sonnenberg 2001). Proper functioning of FlnA is ubiquitously required for maintenance and organ development (Zhou et al. 2010).
FlnA regulates organogenesis by its ability to induce cell migration through its actin-binding properties
FlnA’s role in organogenesis likely arises from its ability to regulate cell migration. Within the human genome, FlnA is located on chromosome Xq28 and consists of 48 exons spanning a little over 26 kb (Fig. 1A). The two other paralogous Fln genes in humans, FlnB and FlnC, are located on chromosome 3p14.3 and chromosome 7q32–35 respectively (Fig. 1A). The wide distribution of filamins throughout the human genome points to its high importance in development; it is likely that this arrangement prevents complete inactivation of all filamins due to malfunction of any one part. The FlnA gene has two known full-length mRNA variants. The two FlnA alternative splicing variants are very similar and only differ in the deletion of 24 nucleotides coding for amino acids 1649–1656, skipping an entire exon (Maestrini et al. 1993). The similarity between the two full-length FlnA transcripts has prevented studies to distinguish the expression patterns between the two variants (Maestrini et al. 1993, Uhlen et al. 2010). In addition, FlnA-spliced variants were identified, which could result in smaller C-terminal fragments of FlnA (predicted protein size 120, 106, or 85 kDa fragments) (Fig. 1B; Wiemann et al. 2001).
Figure 1.

(A) Within the human genome, FlnA is located on chromosome Xq28, while FlnB is located on chromosome 3p14.3, and FlnC is located on chromosome 7q32–35. (B) The FlnA gene has two known full-length mRNA variants, which only differ in the deletion of 24 nucleotides coding for amino acids 1649–1656 – skipping an entire exon. In addition, cDNA clones were identified that could result in smaller C-terminal fragments of FlnA (predicted 120, 106, or 85 kDa fragments).
The more common full-length transcript encodes a 280-kDa protein consisting of 2647 amino acids with several different domains. The full-length filamin contains an ABD at the N-terminal end followed by 24 immunoglobulin (Ig)-like repeat domains, which are folded into b-sheets and together form a rod structure (Ruskamo et al. 2012; Fig. 2A). The ABD of FlnA is similar to other ABDs and contains two calponin homology (CH) domains, with two actin-binding sites (ABS) in CH1 and one ABS in CH2 (Ruskamo & Ylanne 2009). These CH domains are conserved in FlnB and FlnC (Fig. 2A). In both FlnA and FlnC, there are also transmembrane domains in the ABD, not present in FlnB. Therefore, FlnA and FlnC, but not FlnB, are predicted to localize to the plasma membrane. On the other hand, FlnA and FlnB both contain nuclear localization signals (NLS) in the ABD, not present in FlnC, though all three isoforms have an NLS in repeat 14, and FlnA has a third NLS in repeat 20 (Fig. 2A).
Figure 2.
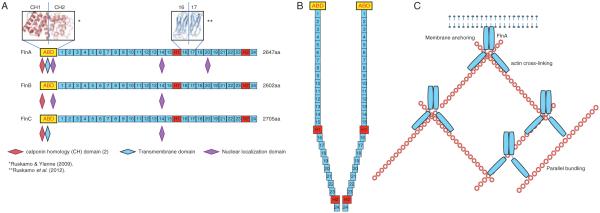
(A) FlnA is a 280 kDa protein consisting of 2647 amino acids with several different domains. There is an actin-binding domain (ABD) at the N-terminal end followed by 24 immunoglobulin (Ig)-like repeat domains, which are folded into β-sheets and together form a rod structure. The ABD of FlnA contains two calponin homology (CH) domains, with two actin-binding sites (ABS) in CH1 and one ABS in CH2. FlnA also contains a transmembrane domain in the ABD and three nuclear localization domains, located in repeats 1, 14, and 21. While the CH domains are conserved in FlnB and FlnC, FlnB does not contain the transmembrane domain and has only two nuclear localizations, located in repeats 1 and 14. FlnC does contain the transmembrane domain, but only has one nuclear localization in repeat 14. In all three, the 24 repeat domains following the ABD are interrupted by two hinge regions, the first (H1) is between repeats 15 and 16 and the second (H2) is between repeats 23 and 24. The repeat domains within FlnA have high sequence similarity and there is also high sequence similarity between the three filamin isoforms, with ~70% sequence similarity overall, and the lowest homology of only 45% at the hinge regions. (B) The molecules homodimerize by binding at repeat 24 and the hinge domains allow the protein to bend, so that when it dimerizes the structure is similar to a Y. (C) Dimerization allows FlnA to regulate the structure of the cell through actin binding in multiple ways. FlnA anchors the actin filamins at the membrane and can form them into perpendicular or parallel strands.
The 24 repeat domains at the C-terminal end of the filamins are interrupted by two hinge regions, the first (H1) between repeats 15 and 16 and the second (H2) between repeats 23 and 24 (Fig. 2A). The repeat domains within FlnA have high sequence similarity and there is also high homology between the three filamin isoforms, with ~70% of sequence similarity overall, and the lowest homology of only 45% at the hinge regions (van der Flier & Sonnenberg 2001). The molecules homodimerize through repeat 24 and the hinge domains allow the protein to bend, so that when it dimerizes, the structure is similar to a Y, with dimerization occurring at the 24th repeat (Robertson 2005; Fig. 2B).
Dimerization allows FlnA to regulate the structure of the cell through actin binding in multiple ways (Fig. 2C). The best known function of FlnA is to crosslink actin, providing organization and stability to actin that forms the cytoskeleton. This function of FlnA involves formation of homodimers at the C-terminal and binding to actin filamins with the N-terminal ABD (van der Flier & Sonnenberg 2001). As an integral part of the cytoskeleton, FlnA maintains cell rigidity and protein localization, and is of particular importance in cell adhesion and migration. In addition, FlnA also increases branching of actin proteins, which is required to organize the cytoskeleton and maintain the flexibility and rigidity of the cell (Stossel et al. 2001). FlnA is also crucial in cell migration – it is recruited to the sites of migration, such as filopodia and lamellipodia, and brings other proteins involved with this function to the cell surface to allow cell movement (Nakamura et al. 2011). These reports indicate that the major functions of FlnA in the cytoplasm or at the membrane appear to be the promotion of cell adhesion, migration, or rigidity. On the other hand, WT FlnA overexpression also inhibited neuronal migration, thereby indicating that an optimum levels of FlnA is required for proper development (Sarkisian et al. 2006).
FlnA acts as a scaffolding molecule to regulate various cell functions
FlnA has over 90 known protein-binding partners, many of which regulate the actin cytoskeleton (Fig. 3). Many of these proteins interact with FlnA as part of the signaling pathways to reorganize the actin cytoskeleton as a reaction to stress or other stimuli (Zhou et al. 2010). As FlnA is a large molecule, it is used as scaffolding for other proteins to bind and regulate the response to cell signaling. Most proteins interact with one of the Ig repeat domains of FlnA and because of the similarity between repeats, proteins can bind at multiple sites (Nakamura et al. 2011). In addition, most protein binding occurs at the C-terminal end, while the ABD is present at the N-terminal (van der Flier & Sonnenberg 2001, Stossel et al. 2001).
Figure 3.

FlnA has over 90 known protein-binding partners, many of which regulate the actin cytoskeleton. Actin binding occurs at the ABD and also between repeats 9 and 15. Most other proteins bind closer to the C-terminal domain, between repeats 16 and 24.
β1-integrin, which binds to repeat 21 of FlnA, is of critical importance in cell adhesion. FlnA binds to the cytoplasmic domain of β1-integrin and promotes cell adhesion and migration by facilitating ligand binding via inside-out signaling. FlnA and β1-integrin maintain a positively correlated relationship during cell spreading and migration, when one is decreased, so is the other (Kim et al. 2008). Disruption of this interaction by knockdown of FlnA decreases the amount of β1-integrin present at the cell membrane at the cell’s initial contact with the extracellular matrix, showing the importance of FlnA in cell adhesion (Kim & McCulloch 2011). Deregulation of this pathway is often found in various types of cancer and has been implicated for therapeutic targets (Barkan & Chambers 2011).
FlnA is often found co-localized with vimentin and both are found at sites of cell protrusions (Kim & McCulloch 2011). Overexpression of proteins such as RalA normally stimulates migration, but FlnA deficient cells were unable to migrate, indicating the crucial role of FlnA (Ohta et al. 1999). In addition, FlnA acts at these sites to bring multiple proteins including PtdIns(3,4,5)P(3), sphingosine kinase 1, and small GTPases to the sites of lamellipodia formation to promote cell migration (Maceyka et al. 2008, Takabayashi et al. 2010, Zhou et al. 2010). FlnA anchors the GTPases to the cell membrane, acting as a scaffolding protein for downstream targets to remodel the actin cytoskeleton and allow cell motility (Zhou et al. 2010).
Tissue factor (TF), the cellular receptor and cofactor for clotting factor VII/VIIa (FVII/VIIa), which plays a role in fibrin formation in embryogenesis, was found to co-localize with FlnA at the leading edge of spreading cells (Muller et al. 1999). FlnA is considered to be a ligand for the TF cytoplasmic domain and recruitment to TF adhesion contacts is associated with the reorganization of actin filaments, but these interactions are not associated with FlnA interactions with integrins (Ott et al. 1998). Lack of TF–FlnA interactions may result in incomplete implantation of the embryo into the endometrium and covering of the implantation location by a fibrin plug, which may also contribute to embryonic lethality in FlnA-null mice mentioned earlier (Feng et al. 2006). FlnA’s role in platelet adhesion also involves interaction with glycoprotein (GP) Ibα, which plays an important role in the regulation of the membrane skeleton and stability (Cranmer et al. 2011). The slit–diaphragm junctional adhesion protein, nephrin, is necessary for the development of podocyte morphology and transduces phosphorylation-dependent signals that regulate cytoskeletal dynamics (Venkatareddy et al. 2011). Upon activation, nephrin recruits and regulates a protein complex that includes SH2 domain containing 5′ inositol phosphatase (Ship2), filamin, and lamellipodin (Venkatareddy et al. 2011), resulting in lamellipodia formation. Finally, FlnA is also shown to regulate the cell cycle. Embryonic FlnA-null mice had reduced brain size and decreased neural progenitors, which were attributed to prolonged cell cycle (Lian et al. 2012). Suppression of FlnA led to prolongation of the cell cycle, principally in the M phase, whereas the cdk1 kinase Wee1 bound FlnA, demonstrated to show increased expression levels after loss of FlnA function, was associated with increased phosphorylation of cdk1 (Lian et al. 2012).
Regulation of FlnA expression and localization
A number of regulators of FlnA expression have been identified that determine which organs would express this molecule and at what phases of development. One protein that appears to regulate the functionality of FlnA is the FlnA-interacting protein (FILIP) that regulates cortical cell migration out of the ventricular zone (Nagano et al. 2002). FILIP interacts with FlnA and induces its degradation – therefore, most ventricular zone cells that overexpress FILIP fail to migrate in explants. FILIP siRNA results in FlnA overexpression and promotes the development and maintenance of a bipolar shape also in the subventricular and intermediate zones (Nagano et al. 2004). Thus FILIP regulates FlnA levels and determine the mode of migration of neurons entering the cortical plate (Sato & Nagano 2005).
Another regulator of FlnA expression is the vesicle transport ADP-ribosylation factor guanine exchange factor 2 gene (ARFGEF2). Transfection of a dominant-negative construct of ARFGEF2 in neuroblastoma cells partially blocked FlnA transport from the Golgi apparatus to the cell membrane, underscoring the importance of this protein in targeted transport of FlnA to the cell surface within neural progenitors (Lu et al. 2006). Refilins, a unique family of actin regulators, act as molecular switches to convert FlnA from an actin branching protein into one that bundles (Gay et al. 2011). The RefilinB/FlnA complex organizes the perinuclear actin filament bundles forming an actin cap. and the nuclear shape changes during epithelial–mesenchymal transition (EMT; Gay et al. 2011).
FlnA expression is also regulated by signal transduction pathways involving the MAP kinase pathway. MEKK4 (MAP3K4)(−/−) mice developed periventricular heterotopia associated with breaches in the neuroependymal lining, which largely comprised neurons that failed to reach the cortical plate. The expression of FlnA was elevated in MEKK4(−/−) forebrain, most notably near sites of failed neuronal migration while the recombinant MKK4 protein promoted the interaction between MEKK4 and Fln-A (Sarkisian et al. 2006). A novel group of evolutionarily conserved HECT ubiquitin ligases with an N-terminal filamin domain (HFNs) were deemed to regulate the filamin complex activity and cell type-specific motility through the breakdown of filamin complexes (Blagg et al. 2011). FlnA can also be regulated by proteasomal degradation, which is mediated by the E3 ubiquitin ligases ASB2α and ASB2β (Heuze et al. 2008). This regulation is of particular importance in the maturation of dendritic cells and inhibits their mobility by causing the degradation of FlnA (Lamsoul et al. 2013).
FlnA localization within the cell was found to be regulated by inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase complex-associated protein (IKBKAP), which encodes IKAP (ELP1), a protein associated with familial dysautonomia (FD), with defective neuronal development and maintenance (Johansen et al. 2008). Cytosolic IKAP that co-localized with FlnA along membrane ruffles, whereas downregulation of IKAP expression resulted in decreased FlnA membrane ruffles localization and defective actin cytoskeleton organization. Another mitochondria-anchored protein, protein kinase C, and casein kinase substrate in neurons protein 2 (PACSIN2) prevent filamin localization to membrane ruffles but not to stress fiber (Cousin et al. 2008). Thus, a family of proteins is involved in the regulation of FlnA expression in various parts of the cell.
Proteolysis of FlnA
One way by which FlnA can be quickly removed once it is no longer required is by proteolysis. The hinge regions are sites at which proteolysis occurs (van der Flier & Sonnenberg 2001). FlnA is susceptible to cleavage by calpain and caspase at the two hinge regions that can produce two proteins, a 170 kDa protein that includes the ABD and repeats 1–15 and a 110-kDa protein that is further cleaved to a 90-kDa protein that contains repeats 16–23 (Gorlin et al. 1990, Browne et al. 2000; Fig. 4). Susceptibility of FlnA to cleavage is regulated by its phosphorylation. Specific phosphorylation at S2152 in the 20th repeat is of particular importance because it is known to protect FlnA against cleavage (Garcia et al. 2006). FlnA can be phosphorylated at S2152 by several different kinases, including protein kinase A, protein kinase C, Ca2+/calmodulin-dependent protein kinase II, and p90 ribosomal S6 kinase (Wallach et al. 1978, Kawamoto & Hidaka 1984, Chen & Stracher 1989, Ohta & Hartwig 1996, Jay et al. 2000, Raynaud et al. 2006).
Figure 4.

FlnA is susceptible to proteolysis at the hinge regions. This cleavage can occur by calpain and caspase producing two proteins, a 170 kDa protein that includes the ABD and repeats 1–15 and a 110 kDa protein that is further cleaved to a 90 kDa protein that contains repeats 16–23. Susceptibility of FlnA to cleavage is regulated by its phosphorylation. Specific phosphorylation at S2152 in the 20th repeat is of particular importance because it is known to protect FlnA against cleavage. FlnA can be phosphorylated at S2152 by several different kinases including protein kinase A (cAMP-dependent protein kinase), protein kinase C, Ca2+/calmodulin-dependent protein kinase II, and p90 ribosomal S6 kinase. Other phosphorylation sites, including S2523, can influence location from the cytosol to the membrane and the interaction between FlnA surface receptors, but does not protect against cleavage.
Ca2+/calmodulin-dependent protein kinase II can phosphorylate S2523, which can result in a change of location from the cytosol to the membrane, regulating endothelial cell barrier function (Borbiev et al. 2001). p56lck can also cause phosphorylation of FlnA, but does not protect it against calpain cleavage, indicating that it phosphorylates a site outside of S2152. p56lck regulates the interaction between FlnA and surface receptors, including β2-integrins, and can also influence actin crosslinking (Goldmann 2002, Pal Sharma & Goldmann 2004). It is projected that following the completion of its function in the organ, FlnA undergoes proteolysis to prevent further activation of its downstream targets in order to prevent unwanted cell migration, cell adhesion, or cell cycle progression.
FlnA’s involvement in cancer
Cancer has been described as the product of developmental error leading to the acquisition of a unique cell character (de-differentiation) (da Costa 2001). Lineage tracing has shown that stem cells are mobilized to repair skin wounds and that this process may contribute to skin tumor development (Arwert et al. 2012). Tumorigenesis and wound repair, both depend on communication between epithelial cells, mesenchymal cells, and bone marrow-derived cells (Arwert et al. 2012). Similarly, exposure to certain chemicals and environmental factors, food, hormones, infectious agents, radiation, sunlight, and tobacco may trigger certain kinds of cancers. Genetics and propensity to obesity, diet, and physical activity determine the risks of developing cancer. Many of these factors may induce some type of injury to the affected organ resulting in de-differentiation (da Costa 2001).
Regeneration, or replacement, of lost or damaged cells involves the processes of de-differentiation, transdifferentiation, and reprograming (Jopling et al. 2011), and in many cases, cancer can be thought of as continued and unwanted regeneration that does not know how to stop, resulting in overexpression of certain key proteins. Whatever the cause, FlnA was seen to be overexpressed in multiple types of cancer, including prostate (Bedolla et al. 2009), breast (Alper et al. 2009, Tian et al. 2013), lung cancer (Uramoto et al. 2010), hemangiomas (Hosaka et al. 1985), colon cancer (Porter et al. 1993, Larriba et al. 2009), melanoma (Flanagan et al. 2001), neuroblastoma (Bachmann et al. 2006), squamous cell carcinoma (Kamochi et al. 2008), hepatic cholangiocarcinoma (Guedj et al. 2009), etc.
However, the puzzling factor regarding filamin overexpression in cancer is the outcome. On one hand, cyclin D1 interacted with FlnA to promote migration and invasive potential of breast cancer cells (Zhong et al. 2010). On the other hand, FlnA regulated focal adhesion disassembly and suppressed breast cancer cell migration and invasion (Xu et al. 2010). Similarly, in prostate cancer, FlnA binding promoted androgen receptor (AR) localization to the nucleus (Ozanne et al. 2000), which is thought to promote prostate cancer progression; however, once in the nucleus, it inhibited the transcriptional activity of the same transcription factor (Loy et al. 2003). Therefore, we argue that overexpression of FlnA has a tumor-promoting effect only when it is localized to the cytoplasm or plasma membrane, where it has the ability to promote tumor metastasis. On the other hand, despite overexpression, if FlnA undergoes proteolysis and the proteolysed fragments localize to the nucleus, where they regulate transcription, then they may act to suppress tumor growth and inhibit metastasis (Fig. 5). Ultimately, the function of FlnA in the cell, whether in cancer or in other diseases, depend on the binding partners available for its interaction – FlnA remains as a scaffolding protein that only act, whether in the cytoplasm or the nucleus, to promote the interaction of these proteins to either promote or prevent cancer.
Figure 5.
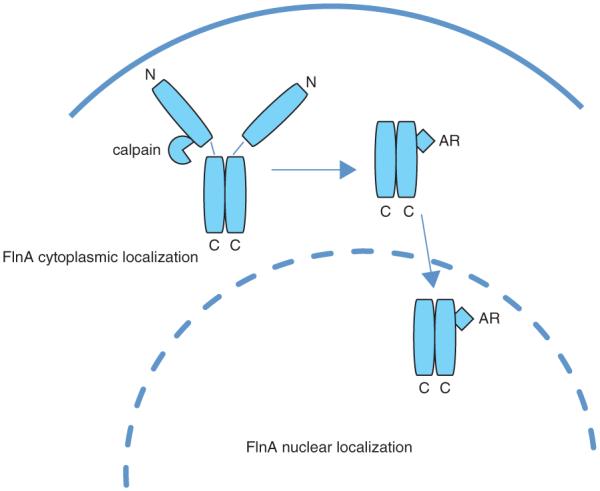
When FlnA is localized to the cytoplasm or plasma membrane, it has the ability to promote tumor metastasis through interaction with signaling molecules. If FlnA undergoes proteolysis and the proteolysed fragments localize to the nucleus, where they regulate transcription, then they may act to suppress tumor growth and inhibit metastasis.
FlnA promotes cancer growth and metastasis in the cytoplasm by interacting with signaling molecules
The development of cancer is a multistep process that transforms a normal cell to the one that evades apoptosis, grows uncontrollably, promotes angiogenesis to support the tumor, and finally invades surrounding tissue and metastasizes (Hanahan & Weinberg 2000). Through FlnA’s involvement in multiple cellular pathways, it can be linked as an important factor in all of these cancer-promoting steps. FlnA’s function in the cytoplasm as a scaffolding protein and its vital importance in cell adhesion and migration can transform it into an extremely potent cancer-promoting protein.
Targeting FlnA through knockdown experiments has given investigators an insight into FlnA’s role in cancer development and has shown its importance in cancer cell growth and metastasis. Crossing a conditionally overexpressed K-RAS mouse model (to induce lung tumor formations) with a conditional knockout FlnA mouse showed that in the absence of FlnA (albeit conditional, as an FlnA null mouse is embryonically lethal), K-RAS overexpression did not lead to lung tumor formation (Nallapalli et al. 2012). Isolated mouse fibroblasts from the K-RAS(+/+) FlnA(−/−) mouse lines showed reduced ability to proliferate and reduced motility as well as a 25% reduction of p-ERK and p-Akt in comparison with those expressing FlnA (Nallapalli et al. 2012). As K-RAS is a powerful oncogenic protein, it is very interesting to discover that without FlnA, K-RAS-driven lung cancer formation is halted.
Regulation of DNA damage
FlnA can be linked to an anti-apoptotic mechanism in cancer development through its involvement in double strand break repair (DSBR). Investigation of the role of FlnA in DSBR was initiated after an interaction between FlnA and BRCA2 was discovered by yeast two-hybrid screen and subsequent co-immunoprecipitation (Yuan & Shen 2001). A previous report showed that lack of FlnA led to susceptibility of DNA damage and G2 arrest (Meng et al. 2004). FlnA seems to act as a scaffolding protein for BRCA2 in the assembly of the repair complex. When cells were treated with radiation, lack of FlnA led to an increase of cell death, an increase in gH2AX nuclear foci (indicating double strand break), and a decrease in RAD51 formation on chromatin (Yue et al. 2009). Through this mechanism FlnA promotes cell survival and protects cancer cells against drug treatment.
Angiogenesis
Angiogenesis is another crucial part of cancer development. Tumors give rise to new blood vessels to continue their growth (Ricci-Vitiani et al. 2010, Wang et al. 2010). FlnA has been implicated in angiogenesis through links with vascular endothelial growth factor (VEGF; Uramoto et al. 2010). The expression of FlnA and VEGF was analyzed by immunohistochemistry in lung tumor samples. Positive expression of FlnA and VEGF was detected in the cytoplasm of tumor cells in 66/137 (48.2%) and 69/137 (50.4%) patients with lung cancer, respectively, while statistical analysis suggested that FlnA is mediating the angiogenesis pathway and is responsible for controlling the growth of lung tumors (Uramoto et al. 2010). In addition, studies on bone marrow angiogenesis found that four proteins: FlnA, vimentin, α-crystallin B, and 14-3-3ζ/δ protein, were overexpressed in endothelial cells present in multiple myeloma compared with normal endothelial cells (Berardi et al. 2011). The expression of FlnA in multiple myeloma was increased by VEGF, fibroblast growth factor 2, and hepatocyte growth factor, while using siRNA to knockdown their expression inhibited angiogenesis-related functions, such as spreading, migration, and tubular morphogenesis (Berardi et al. 2011).
Metastasis
Table 1 outlines a number of molecules that are shown to regulate FlnA-mediated cell migration and metastasis and only a few of these are outlined below. One mechanism by which FlnA is involved in promoting cancer metastasis is through activation of c-Met. c-Met is the only receptor for the hepatocyte growth factor and is extremely important in cell growth and motility and is a very potent oncogene. An association between FlnA and c-Met was first identified when irradiated fibroblasts expressed increased levels of both proteins (Kamochi et al. 2008). Even more importantly these cells promoted invasion and growth of human squamous cell carcinoma cells (Kamochi et al. 2008). Investigation of the mechanism by which these proteins interacted and promoted metastatic cancer cells showed that when tumors cells are deficient for FlnA, expression of c-Met was also significantly reduced and the cells exhibited poor migration and invasion ability when exposed to hepatocyte growth factor (HGF). In addition, AKT, a downstream signaling molecule, is also reduced in FlnA-deficient cells. FlnA was found to regulate c-Met expression through its interaction with Smad2 (Zhou et al. 2011).
Table 1.
FlnA-interacting proteins that promote or inhibit metastasis
| Inhibits metastasis | Promotes metastasis |
|---|---|
| AR (Wang) | B1 integrin (Kim et al. 2007) |
| IGFBP5 (Abrass & Hansen 2010) | PtdIns(3,4,5)P(3) (Takabayashi et al. 2010) |
| RNA polymerase, POL III (Deng et al. 2012) | Sphingosine kinase 1 (Maceyka et al. 2008) |
| ASB2 (Heuze et al. 2008, Razinia et al. 2011) | BRCA2 (Yuan & Shen 2001) |
| PEBP2β (Yoshida et al. 2005) | VEGF (Uramoto et al. 2010) |
| FoxC1/PBX1 (Berry et al. 2005) | c-Met (Zhou) |
| Klotho (Camilli et al. 2011) | R-Ras (Gawecka et al. 2010) |
| Granzyme B (Browne et al. 2000) | RhoA |
| Cyclin B1 (Cukier et al. 2007) | |
| K-Ras (Nallapalli et al. 2012) | |
| cdc25C (Telles et al. 2011) | |
| BRCA1 (Velkova et al. 2010) | |
| Cyclin D1/cyclin-dependent kinase 4 (Zhong et al. 2010) | |
| Vimentin (MacPherson & Fagerholm 2010) | |
| MLL (De Braekeleer et al. 2009) | |
| Caveolin-1 (Ravid et al. 2008) | |
| TGF-β (Mishra & Marshall 2006) | |
| Arp2/3 (Flanagan et al. 2001) | |
| Pro-PrP (Li et al. 2010) | |
| EGFR (Sy) | |
| P73α (Ravid et al. 2008) |
A second mechanism by which FlnA is involved in promoting cell migration and metastasis is through its interaction with the small GTPases, R-Ras and RhoA. Similar to FlnA, R-Ras also regulates cell adhesion and migration. Using a yeast two-hybrid assay, investigators showed that R-Ras and FlnA interact, specifically at the third repeat of FlnA. Deletion of the third repeat of FlnA prevented R-Ras from binding to FlnA and decreased cell migration. Furthermore, knockdown of R-Ras inhibited FlnA from interacting with the fibronectin matrix assembly (Gawecka et al. 2010). In addition to R-Ras, FlnA also interacts with RhoA and promotes migration in head and neck squamous cell carcinoma cells. RhoA causes modification to the cytoskeleton by phosphorylation of FlnA (Bourguignon et al. 2006). R-Ras and RhoA are important cell signaling molecules that are often mutated or overexpressed in cancer (Mora et al. 2007, He et al. 2010). Through their interaction with FlnA they can easily promote cell migration and metastasis in cancer (Table 1).
FlnA can regulate RhoA through its interaction with Trio, a small guanine nucleotide-exchange factor (GEF) that activates a number of Rho GTPases by assisting the switch from GDP for GTP (Bellanger et al. 2000). Trio has two GEF domains (GEFD1 and GEFD2) that can activate RhoG, Rac, and RhoA and is also important for axon guidance and cell migration in the nervous system (Bellanger et al. 1998, Blangy et al. 2000). A yeast two-hybrid screen showed that the GEFD1 of Trio interacted with the C-terminal end of FlnA, specifically on repeat 23 and 24 (Bellanger et al. 2000). GEFD1 but not GEFD2 interacted with FlnA. A previous study on 3T3 cells showed that GEFD1 caused ruffle formation of actin fibers that contained high levels of FlnA (Bellanger et al. 1998). Using two human malignant melanoma cell lines, one lacking FlnA (M2) and a subline that stably expressed FlnA (M2A7), investigators showed that expression of GEFD1 in M2 caused no ruffle formation, while expression of GEFD1 in M2A7 caused dorsal ruffles. This FlnA-dependent formation was specific for Trio GEFD1, while overexpression of its target proteins, RhoG and Rac, caused ruffle formation regardless of the presence of FlnA. Furthermore, the ruffle formation was only seen when Trio GEFD1 and FlnA were physically linked; constructs of GEFD1 and FlnA lacking the necessary regions for binding prevented ruffle formation. These changes in the cytoskeletal formation by Trio indicate its importance in cancer development and metastasis through its link to FlnA (Bellanger et al. 2000).
An important step in cancer metastasis is the contraction of the cell and regulation of cell polarity. Cells undergoing locomotion must first expand by lamellae protrusions and then contract to induce the rest of the cell to move. This involved complex reorganization of the cytoskeleton is regulated by ROCK inactivation of Rac. Investigation has shown that this regulation of Rac and lamellae formation is mediated by the FlnA GTPase-activating protein (FilGAP). The yeast two-hybrid assay revealed this FlnA-binding protein, which had homologous domains to a family of RhoGAP proteins. To determine the function of FilGAP, the GAP domain was microinjected into Swiss 3T3 cells that where stimulated with either epidermal growth factor (EGF) or lysophosphatidic acid (LPA). FilGAP abolished filopodia formation in EGF-stimulated cells, which involved the activation of Cdc42 and Rac1, but not in LPA-stimulated cells which involved the activation of RhoA. The expression of an FlnA plasmid lacking repeat 23, where FilGAP binds, prevented the GAP activity of FilGAP, indicating the importance of FlnA in this pathway. SiRNA against FilGAP showed an increase in lamellae formation, demonstrating its importance in suppressing lamellae. ROCK was found to phosphorylate FilGAP, which inactivates Rac. This pathway shows both the importance of FlnA as a scaffolding molecule and as a remodeler of the cytoskeleton. Both of these have a large role in tumor development and spreading. The FilGAP pathway has been implicated in tumor cell migration and is largely dependent on FlnA to complete its activation (Saito et al. 2012).
Cancer is very rarely caused by the deregulation of one growth pathway alone. FlnA likely plays a role in cancer metastasis because of its involvement in multiple regulatory pathways (Gawecka et al. 2010, Zhou et al. 2011). As a result, overexpression of FlnA has been associated with highly metastatic cancers including prostate cancer, melanoma, and neuroblastoma (Bachmann et al. 2006, Coughlin et al. 2006, Zhu et al. 2007).
FlnA inhibits cancer growth and metastasis in the nucleus by interacting with transcription factors
FlnA’s role in cell biology is largely as a scaffolding molecule that acts to bring proteins together so that they can interact. In the cytoplasm often this results in cell migration and proliferation. Upon interaction with transcription factors, both in the cytoplasm and the nucleus, FlnA can regulate their transcriptional activity. FlnA was found to interact with PEBP2β in the cytoplasm and prevent its nuclear localization and interaction with Runx1 (Watanabe et al. 2005, Yoshida et al. 2005). Runx1 is an important transcription factor involved with hematopoietic cell differentiation (Okada et al. 1998). Furthermore Runx1 activity as a transcription factor is impaired without dimerization with PEBP2β (Niki et al. 1997). It was previously established that PEBP2β was present in the cytoplasm and brought into the nucleus by Runx1. FlnA was found to retain PEBP2β in the cytoplasm and prevent its interaction with Runx1 (Yoshida et al. 2005).
FlnA can also localize to the nucleus and this corresponds with a repression of some cell activities. FlnA has several nuclear localization sequences in repeats 1, 14, and 20. Ozanne et al. (2000) first showed that FlnA was crucial in the translocalization of the AR into the nucleus. During investigation of the mechanism by which the AR was transported to and from the nucleus, a yeast two-hybrid screen identified the C-terminal end of FlnA as an AR-interacting protein. Furthermore, in the absence of FlnA, AR was unable to enter the nucleus (Ozanne et al. 2000). FlnA’s role in the nucleus and specifically AR was further examined by a different group (Loy et al. 2003). They reported that the FlnA repeats 16–24 interacted with the hinge domain of AR. Assessment of FlnA’s role on AR transcriptional activity showed that increasing amounts of FlnA repressed AR induction of its transcriptional target prostate specific antigen (PSA) (Loy et al. 2003).
FlnA’s new role in the nucleus has led researchers to evaluate the differences that arise from the localization of the protein. Cytoplasmic FlnA is often highly overexpressed in metastatic cancers including breast and prostate. Specifically in metastatic prostate cancer, cytoplasmic FlnA is phosphorylated at S2152, which prevents its cleavage at the hinge region to the 90 kDa fragment, whereas in less aggressive or benign prostate tumors the 90 kDa FlnA was found in the nucleus (Bedolla et al. 2009). Inhibition of the AR by anti-androgen hormonal therapy is the prevalent treatment for metastatic prostate cancer; however, patients frequently become resistant to such treatment in advanced disease (hormone refractory prostate cancer, HRPC). Cytoplasmic full-length FlnA expression was increased in HRPC, indicating that lack of the nuclear 90 kDa FlnA could be a mechanism in which prostate cancer progresses (Bedolla et al. 2009). Transfection of a plasmid encoding the FlnA fragment that localizes to the nucleus caused a HRPC cell line, C4-2, to become sensitive to anti-androgen treatment (Wang et al. 2007). In addition to regulating AR in prostate cancer, FlnA has also been linked to other transcription factors and transcription machinery. Work with IGFBP5 showed that accumulation of the nuclear 90 kDa FlnA induced migration in glomerular mesangial cell by inducing transcription of laminin subunits (Berfield et al. 2006). Further research showed that IGFBP5 caused dephosphorylation of FlnA, leading to its cleavage by calpains. The C-terminal fragment of FlnA was attached to Smad3/4 and caused its nuclear translocation and binding to the promoter of the IGFBP5 target genes, which induced gene transcription (Abrass & Hansen 2010).
The 90 kDa FlnA fragment is not the only fragment that enters the nucleus. Full-length FlnA is also known to play a role in gene transcription in the nucleolus. It directly binds to RNA polymerase in the nucleolus to inhibit rRNA gene transcription (Deng et al. 2012). Knockdown of FlnA by siRNA caused increased rRNA expression, rDNA promoter activity, and cell proliferation. In vitro studies to immunodeplete FlnA from nuclear extracts caused a decrease in rDNA promoter-driven transcription. FlnA was also coimmunoprecipitated with several of the Pol I components; actin, TIF-IA, and RPA40, and in the absence of FlnA these proteins were found at a higher occupancy at the rDNA promoter (Deng et al. 2012). Full-length FlnA is also known to interact in the nucleus with the FOXC1 transcription factor (Berry et al. 2005). High levels of nuclear full-length FlnA in A7 melanoma cells inhibited FOXC1 from activating transcription. FOXC1 was further removed from transcription-rich areas in the nucleus and moved to a heterochromatin-rich region. FlnA is able to recruit PBX1 to the nucleus, which then interacted with FOXC1 and inhibited its transcriptional activity (Berry et al. 2005).
Conclusion
The examination of FlnA in cancer development is still underway, but already it is considered to have an important function in cancer development and the progression to metastasis. In the cytoplasm, FlnA binds to actin to give structure to and remodel the cytoskeleton and, as importantly, acts as a scaffolding molecule, facilitating protein interaction (Zhou et al. 2010). FlnA has been reported to interact with over 90 proteins, which indicates the numerous pathways that FlnA can affect and its importance in the cell (Stossel et al. 2001). FlnA is of particular importance in facilitating cell adhesion and migration by directing β1 integrin to the site of cell attachment and through its interaction with vimentin (Kim et al. 2008, Kim & McCulloch 2011). In addition, FlnA has been widely implicated in having an important role in cancer development and being a possible target for future therapies.
In regard to FlnA’s involvement in cancer development, FlnA can cause two opposite outcomes of cancer depending on its subcellular localization. By being present in the cytoplasm and interacting with cell signaling molecules FlnA can promote cell growth and metastasis; on the other hand, when acting in the nucleus and with transcription factors, FlnA causes inhibition of cell growth and prevents metastasis. In the cytoplasm FlnA acts in various signaling pathways and interacts with several significant oncogenic proteins including K-RAS, R-Ras, VEGF, and c-Met (Gawecka et al. 2010, Uramoto et al. 2010, Zhou et al. 2011, Nallapalli et al. 2012). Through these interactions FlnA aids their functions and enhances cell growth. In the nucleus, FlnA interacts with several transcription factors including AR, Pol III, and FOXC1 (Ozanne et al. 2000, Berry et al. 2005, Deng et al. 2012). FlnA causes a general inhibition of their activities at the promoter regions on the DNA, causing growth inhibition or cell death. Targeting FlnA in the cytoplasm through the inhibition of the phosphorylation of S2152 would result in cleavage of the full-length protein into the 90 kDa form. Subsequent localization to the nucleus would then inhibit various transcription factors and result in the inhibition of cancer growth (Wang et al. 2007). Work toward this end is already in progress in prostate cancer. Genistein-combined polysaccharide is a soy derivative that can reduce phosphorylation of FlnA S2152 in prostate cancer cells (Mooso et al. 2012). This results in translocalization of the 90 kDa FlnA and modification of AR transcription. Using this therapy or others to induce cleavage of FlnA, in other cancers, would cause subsequent localization to the nucleus and repress various transcription factors and induce cell quiescence or death.
Acknowledgments
Funding
During the composition of this manuscript, salary support for one or more authors was provided partly by a Biomedical Laboratory Research and Development (BLRD) service Merit Award (I01BX000400) from the Department of Veterans Affairs and by Award # CA133209 from the National Cancer Institute. The work reported here does not represent the views or opinions of the Department of Veteran Affairs or the United States Government.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the review.
References
- Abrass CK, Hansen KM. Insulin-like growth factor-binding protein-5-induced laminin gamma1 transcription requires filamin A. Journal of Biological Chemistry. 2010;285:12925–12934. doi: 10.1074/jbc.M109.061754. doi:10.1074/jbc.M109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper O, Stetler-Stevenson WG, Harris LN, Leitner WW, Ozdemirli M, Hartmann D, Raffeld M, Abu-Asab M, Byers S, Zhuang Z, et al. Novel anti-filamin-A antibody detects a secreted variant of filamin-A in plasma from patients with breast carcinoma and high-grade astrocytoma. Cancer Science. 2009;100:1748–1756. doi: 10.1111/j.1349-7006.2009.01244.x. doi:10.1111/j.1349-7006.2009.01244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nature Reviews. Cancer. 2012;12:170–180. doi: 10.1038/nrc3217. doi:10.1038/nrc3217. [DOI] [PubMed] [Google Scholar]
- Bachmann AS, Howard JP, Vogel CW. Actin-binding protein filamin A is displayed on the surface of human neuroblastoma cells. Cancer Science. 2006;97:1359–1365. doi: 10.1111/j.1349-7006.2006.00327.x. doi:10.1111/j.1349-7006.2006.00327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan D, Chambers AF. β1-integrin: a potential therapeutic target in the battle against cancer recurrence. Clinical Cancer Research. 2011;17:7219–7223. doi: 10.1158/1078-0432.CCR-11-0642. doi:10.1158/1078-0432.CCR-11-0642. [DOI] [PubMed] [Google Scholar]
- Bedolla RG, Wang Y, Asuncion A, Chamie K, Siddiqui S, Mudryj MM, Prihoda TJ, Siddiqui J, Chinnaiyan AM, Mehra R, et al. Nuclear versus cytoplasmic localization of filamin A in prostate cancer: immunohistochemical correlation with metastases. Clinical Cancer Research. 2009;15:788–796. doi: 10.1158/1078-0432.CCR-08-1402. doi:10.1158/1078-0432.CCR-08-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanger JM, Lazaro JB, Diriong S, Fernandez A, Lamb N, Debant A. The two guanine nucleotide exchange factor domains of Trio link the Rac1 and the RhoA pathways in vivo. Oncogene. 1998;16:147–152. doi: 10.1038/sj.onc.1201532. doi:10.1038/sj.onc.1201532. [DOI] [PubMed] [Google Scholar]
- Bellanger JM, Astier C, Sardet C, Ohta Y, Stossel TP, Debant A. The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nature Cell Biology. 2000;2:888–892. doi: 10.1038/35046533. doi:10.1038/35046533. [DOI] [PubMed] [Google Scholar]
- Berardi S, Caivano A, Ria R, Nico B, Savino R, Terracciano R, De Tullio G, Ferrucci A, De Luisi A, Moschetta M, et al. Four proteins governing overangiogenic endothelial cell phenotype in patients with multiple myeloma are plausible therapeutic targets. Oncogene. 2011;31:2258–2269. doi: 10.1038/onc.2011.412. doi:10.1038/onc.2011.412. [DOI] [PubMed] [Google Scholar]
- Berfield AK, Hansen KM, Abrass CK. Rat glomerular mesangial cells require laminin-9 to migrate in response to insulin-like growth factor binding protein-5. American Journal of Physiology. Cell Physiology. 2006;291:C589–C599. doi: 10.1152/ajpcell.00623.2005. doi:10.1152/ajpcell.00623.2005. [DOI] [PubMed] [Google Scholar]
- Berry FB, O’Neill MA, Coca-Prados M, Walter MA. FOXC1 transcriptional regulatory activity is impaired by PBX1 in a filamin A-mediated manner. Molecular and Cellular Biology. 2005;25:1415–1424. doi: 10.1128/MCB.25.4.1415-1424.2005. doi:10.1128/MCB.25.4.1415-1424.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagg SL, Battom SE, Annesley SJ, Keller T, Parkinson K, Wu JM, Fisher PR, Thompson CR. Cell type-specific filamin complex regulation by a novel class of HECT ubiquitin ligase is required for normal cell motility and patterning. Development. 2011;138:1583–1593. doi: 10.1242/dev.063800. doi:10.1242/dev.063800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangy A, Vignal E, Schmidt S, Debant A, Gauthier-Rouviere C, Fort P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. Journal of Cell Science. 2000;113:729–739. doi: 10.1242/jcs.113.4.729. [DOI] [PubMed] [Google Scholar]
- Borbiev T, Verin AD, Shi S, Liu F, Garcia JG. Regulation of endothelial cell barrier function by calcium/calmodulin-dependent protein kinase II. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2001;280:L983–L990. doi: 10.1152/ajplung.2001.280.5.L983. [DOI] [PubMed] [Google Scholar]
- Bourguignon LY, Gilad E, Brightman A, Diedrich F, Singleton P. Hyaluronan–CD44 interaction with leukemia-associated RhoGEF and epidermal growth factor receptor promotes Rho/Ras co-activation, phospholipase C epsilon-Ca2+ signaling, and cytoskeleton modification in head and neck squamous cell carcinoma cells. Journal of Biological Chemistry. 2006;281:14026–14040. doi: 10.1074/jbc.M507734200. doi:10.1074/jbc.M507734200. [DOI] [PubMed] [Google Scholar]
- Brown KD, Binder LI. Expression of the cytoskeletal-associated protein filamin in adult rat organs. Experimental Cell Research. 1993;209:325–332. doi: 10.1006/excr.1993.1317. doi:10.1006/excr.1993.1317. [DOI] [PubMed] [Google Scholar]
- Browne KA, Johnstone RW, Jans DA, Trapani JA. Filamin (280-kDa actin-binding protein) is a caspase substrate and is also cleaved directly by the cytotoxic T lymphocyte protease granzyme B during apoptosis. Journal of Biological Chemistry. 2000;275:39262–39266. doi: 10.1074/jbc.C000622200. doi:10.1074/jbc.C000622200. [DOI] [PubMed] [Google Scholar]
- Camilli TC, Xu M, O’Connell MP, Chien B, Frank BP, Subaran S, Indig FE, Morin PJ, Hewitt SM, Weeraratna AT. Loss of Klotho during melanoma progression leads to increased filamin cleavage, increased Wnt5A expression, and enhanced melanoma cell motility. Pigment Cell & Melanoma Research. 2011;24:175–186. doi: 10.1111/j.1755-148X.2010.00792.x. doi:10.1111/j.1755-148X.2010.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Stracher A. In situ phosphorylation of platelet actin-binding protein by cAMP-dependent protein kinase stabilizes it against proteolysis by calpain. Journal of Biological Chemistry. 1989;264:14282–14289. [PubMed] [Google Scholar]
- da Costa LF. Return of de-differentiation: why cancer is a developmental disease. Current Opinion in Oncology. 2001;13:58–62. doi: 10.1097/00001622-200101000-00012. doi:10.1097/00001622-200101000-00012. [DOI] [PubMed] [Google Scholar]
- Coughlin MF, Puig-de-Morales M, Bursac P, Mellema M, Millet E, Fredberg JJ. Filamin-A and rheological properties of cultured melanoma cells. Biophysical Journal. 2006;90:2199–2205. doi: 10.1529/biophysj.105.061267. doi:10.1529/biophysj.105.061267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin H, Desimone DW, Alfandari D. PACSIN2 regulates cell adhesion during gastrulation in Xenopus laevis. Developmental Biology. 2008;319:86–99. doi: 10.1016/j.ydbio.2008.04.007. doi:10.1016/j.ydbio.REF4=10.1111/j.1349-7006.2006.00327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranmer SL, Ashworth KJ, Yao Y, Berndt MC, Ruggeri ZM, Andrews RK, Jackson SP. High shear-dependent loss of membrane integrity and defective platelet adhesion following disruption of the GPIbα–filamin interaction. Blood. 2011;117:2718–2727. doi: 10.1182/blood-2010-07-296194. doi:10.1182/blood-2010-07-296194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukier IH, Li Y, Lee JM. Cyclin B1/Cdk1 binds and phosphorylates Filamin A and regulates its ability to cross-link actin. FEBS Letters. 2007;581:1661–1672. doi: 10.1016/j.febslet.2007.03.041. doi:10.1016/j.febslet.2007.REF8=10.1038/35046533. [DOI] [PubMed] [Google Scholar]
- De Braekeleer E, Douet-Guilbert N, Morel F, Le Bris MJ, Meyer C, Marschalek R, Ferec C, De Braekeleer M. FLNA, a new partner gene fused to MLL in a patient with acute myelomonoblastic leukaemia. British Journal of Haematology. 2009;146:693–695. doi: 10.1111/j.1365-2141.2009.07824.x. doi:10.1111/j.1365-2141.2009.07824.x. [DOI] [PubMed] [Google Scholar]
- Deng W, Lopez-Camacho C, Tang JY, Mendoza-Villanueva D, Maya-Mendoza A, Jackson DA, Shore P. Cytoskeletal protein filamin A is a nucleolar protein that suppresses ribosomal RNA gene transcription. PNAS. 2012;109:1524–1529. doi: 10.1073/pnas.1107879109. doi:10.1073/pnas.1107879109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douvaras P, Liu W, Mort RL, McKie L, West KM, Cross SH, Morley SD, West JD. Normal X-inactivation mosaicism in corneas of heterozygous FlnaDilp2/+ female mice – a model of human filamin A (FLNA) diseases. BMC Research Notes. 2012;5:122. doi: 10.1186/1756-0500-5-122. doi:10.1186/1756-0500-5-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Chen MH, Moskowitz IP, Mendonza AM, Vidali L, Nakamura F, Kwiatkowski DJ, Walsh CA. Filamin A (FLNA) is required for cell–cell contact in vascular development and cardiac morphogenesis. PNAS. 2006;103:19836–19841. doi: 10.1073/pnas.0609628104. doi:10.1073/pnas.0609628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori JL, Zhu TN, O’Connell MP, Hoek KS, Indig FE, Frank BP, Morris C, Kole S, Hasskamp J, Elias G, et al. Filamin A modulates kinase activation and intracellular trafficking of epidermal growth factor receptors in human melanoma cells. Endocrinology. 2009;150:2551–2560. doi: 10.1210/en.2008-1344. doi:10.1210/en.2008-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan LA, Chou J, Falet H, Neujahr R, Hartwig JH, Stossel TP. Filamin A, the Arp2/3 complex, and the morphology and function of cortical actin filaments in human melanoma cells. Journal of Cell Biology. 2001;155:511–517. doi: 10.1083/jcb.200105148. doi:10.1083/jcb.200105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier A, Sonnenberg A. Structural and functional aspects of filamins. Biochimica et Biophysica Acta. 2001;1538:99–117. doi: 10.1016/s0167-4889(01)00072-6. doi:10.1016/S0167-4889(01)00072-6. [DOI] [PubMed] [Google Scholar]
- Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, Scheffer IE, Dobyns WB, Hirsch BA, Radtke RA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. doi: 10.1016/s0896-6273(00)80651-0. doi:10.1016/S0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- Garcia E, Stracher A, Jay D. Calcineurin dephosphorylates the C-terminal region of filamin in an important regulatory site: a possible mechanism for filamin mobilization and cell signaling. Archives of Biochemistry and Biophysics. 2006;446:140–150. doi: 10.1016/j.abb.2005.12.006. doi:10.1016/j.abb.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Gargiulo A, Auricchio R, Barone MV, Cotugno G, Reardon W, Milla PJ, Ballabio A, Ciccodicola A, Auricchio A. Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. American Journal of Human Genetics. 2007;80:751–758. doi: 10.1086/513321. doi:10.1086/513321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawecka JE, Griffiths GS, Ek-Rylander B, Ramos JW, Matter ML. R-Ras regulates migration through an interaction with filamin A in melanoma cells. PLoS ONE. 2010;5:e11269. doi: 10.1371/journal.pone.0011269. doi:10.1371/journal.pone.0011269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay O, Gilquin B, Nakamura F, Jenkins ZA, McCartney R, Krakow D, Deshiere A, Assard N, Hartwig JH, Robertson SP, et al. RefilinB (FAM101B) targets filamin A to organize perinuclear actin networks and regulates nuclear shape. PNAS. 2011;108:11464–11469. doi: 10.1073/pnas.1104211108. doi:10.1073/pnas.1104211108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann WH. p56(lck) Controls phosphorylation of filamin (ABP-280) and regulates focal adhesion kinase (pp125(FAK)) Cell Biology International. 2002;26:567–571. doi: 10.1006/cbir.2002.0900. doi:10.1006/cbir.2002.0900. [DOI] [PubMed] [Google Scholar]
- Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. Journal of Cell Biology. 1990;111:1089–1105. doi: 10.1083/jcb.111.3.1089. doi:10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj N, Zhan Q, Perigny M, Rautou PE, Degos F, Belghiti J, Farges O, Bedossa P, Paradis V. Comparative protein expression profiles of hilar and peripheral hepatic cholangiocarcinomas. Journal of Hepatology. 2009;51:93–101. doi: 10.1016/j.jhep.2009.03.017. doi:10.1016/j.jhep.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. doi:10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hart AW, Morgan JE, Schneider J, West K, McKie L, Bhattacharya S, Jackson IJ, Cross SH. Cardiac malformations and midline skeletal defects in mice lacking filamin A. Human Molecular Genetics. 2006;15:2457–2467. doi: 10.1093/hmg/ddl168. doi:10.1093/hmg/ddl168. [DOI] [PubMed] [Google Scholar]
- Hartwig JH, Stossel TP. Isolation and properties of actin, myosin, and a new actinbinding protein in rabbit alveolar macrophages. Journal of Biological Chemistry. 1975;250:5696–5705. [PubMed] [Google Scholar]
- He M, Cheng Y, Li W, Liu Q, Liu J, Huang J, Fu X. Vascular endothelial growth factor C promotes cervical cancer metastasis via up-regulation and activation of RhoA/ROCK-2/moesin cascade. BMC Cancer. 2010;10:170. doi: 10.1186/1471-2407-10-170. doi:10.1186/1471-2407-10-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuze ML, Lamsoul I, Baldassarre M, Lad Y, Leveque S, Razinia Z, Moog-Lutz C, Calderwood DA, Lutz PG. ASB2 targets filamins A and B to proteasomal degradation. Blood. 2008;112:5130–5140. doi: 10.1182/blood-2007-12-128744. doi:10.1182/blood-2007-12-128744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M, Murase N, Orito T, Mori M. Immunohistochemical evaluation of factor VIII related antigen, filament proteins and lectin binding in haemangiomas. Virchows Archiv. A, Pathological Anatomy and Histopathology. 1985;407:237–247. doi: 10.1007/BF00710649. doi:10.1007/BF00710649. [DOI] [PubMed] [Google Scholar]
- Jay D, Garcia EJ, Lara JE, Medina MA, de la Luz Ibαrra M. Determination of a cAMP-dependent protein kinase phosphorylation site in the C-terminal region of human endothelial actin-binding protein. Archives of Biochemistry and Biophysics. 2000;377:80–84. doi: 10.1006/abbi.2000.1762. doi:10.1006/abbi.2000.1762. [DOI] [PubMed] [Google Scholar]
- Johansen LD, Naumanen T, Knudsen A, Westerlund N, Gromova I, Junttila M, Nielsen C, Bottzauw T, Tolkovsky A, Westermarck J, et al. IKAP localizes to membrane ruffles with filamin A and regulates actin cytoskeleton organization and cell migration. Journal of Cell Science. 2008;121:854–864. doi: 10.1242/jcs.013722. doi:10.1242/jcs.013722. [DOI] [PubMed] [Google Scholar]
- Jopling C, Boue S, Izpisua Belmonte JC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nature Reviews. Molecular Cell Biology. 2011;12:79–89. doi: 10.1038/nrm3043. doi:10.1038/nrm3043. [DOI] [PubMed] [Google Scholar]
- Jurak Begonja A, Hoffmeister KM, Hartwig JH, Falet H. FlnA-null megakaryocytes prematurely release large and fragile platelets that circulate poorly. Blood. 2011;118:2285–2295. doi: 10.1182/blood-2011-04-348482. doi:10.1182/blood-2011-04-348482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamochi N, Nakashima M, Aoki S, Uchihashi K, Sugihara H, Toda S, Kudo S. Irradiated fibroblast-induced bystander effects on invasive growth of squamous cell carcinoma under cancer–stromal cell interaction. Cancer Science. 2008;99:2417–2427. doi: 10.1111/j.1349-7006.2008.00978.x. doi:10.1111/j.1349-7006.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S, Hidaka H. Ca2+-activated, phospholipid-dependent protein kinase catalyzes the phosphorylation of actin-binding proteins. Biochemical and Biophysical Research Communications. 1984;118:736–742. doi: 10.1016/0006-291x(84)91456-6. doi:10.1016/0006-291X(84)91456-6. [DOI] [PubMed] [Google Scholar]
- Kim H, McCulloch CA. Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Letters. 2011;585:18–22. doi: 10.1016/j.febslet.2010.11.033. doi:10.1016/j.febslet.2010.11.033. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Park JS, Um SJ. Filamin A negatively regulates the transcriptional activity of p73α in the cytoplasm. Biochemical and Biophysical Research Communications. 2007;362:1101–1106. doi: 10.1016/j.bbrc.2007.08.148. doi:10.1016/j.bbrc.2007.08.148. [DOI] [PubMed] [Google Scholar]
- Kim H, Sengupta A, Glogauer M, McCulloch CA. Filamin A regulates cell spreading and survival via β1 integrins. Experimental Cell Research. 2008;314:834–846. doi: 10.1016/j.yexcr.2007.11.022. doi:10.1016/j.yexcr.2007.11.022. [DOI] [PubMed] [Google Scholar]
- Kyndt F, Gueffet JP, Probst V, Jaafar P, Legendre A, Le Bouffant F, Toquet C, Roy E, McGregor L, Lynch SA, et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115:40–49. doi: 10.1161/CIRCULATIONAHA.106.622621. doi:10.1161/CIRCULATIONAHA.106.622621. [DOI] [PubMed] [Google Scholar]
- Lamsoul I, Metais A, Gouot E, Heuze ML, Lennon-Dumenil AM, Moog-Lutz C, Lutz PG. ASB2α regulates migration of immature dendritic cells. Blood. 2013;122:533–541. doi: 10.1182/blood-2012-11-466649. doi:10.1182/blood-2012-11-466649. [DOI] [PubMed] [Google Scholar]
- Larriba MJ, Martin-Villar E, Garcia JM, Pereira F, Pena C, de Herreros AG, Bonilla F, Munoz A. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis. 2009;30:1459–1468. doi: 10.1093/carcin/bgp140. doi:10.1093/carcin/bgp140. [DOI] [PubMed] [Google Scholar]
- Letourneau PC, Shattuck TA. Distribution and possible interactions of actin-associated proteins and cell adhesion molecules of nerve growth cones. Development. 1989;105:505–519. doi: 10.1242/dev.105.3.505. [DOI] [PubMed] [Google Scholar]
- Li C, Yu S, Nakamura F, Pentikainen OT, Singh N, Yin S, Xin W, Sy MS. Pro-prion binds filamin A, facilitating its interaction with integrin β1, and contributes to melanomagenesis. Journal of Biological Chemistry. 2010;285:30328–30339. doi: 10.1074/jbc.M110.147413. doi:10.1074/jbc.M110.147413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian G, Lu J, Hu J, Zhang J, Cross SH, Ferland RJ, Sheen VL. Filamin A regulates neural progenitor proliferation and cortical size through Wee1-dependent Cdk1 phosphorylation. Journal of Neuroscience. 2012;32:7672–7684. doi: 10.1523/JNEUROSCI.0894-12.2012. doi:10.1523/JNEUROSCI.0894-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy CJ, Sim KS, Yong EL. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. PNAS. 2003;100:4562–4567. doi: 10.1073/pnas.0736237100. doi:10.1073/pnas.0736237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Tiao G, Folkerth R, Hecht J, Walsh C, Sheen V. Overlapping expression of ARFGEF2 and Filamin A in the neuroependymal lining of the lateral ventricles: insights into the cause of periventricular heterotopia. Journal of Comparative Neurology. 2006;494:476–484. doi: 10.1002/cne.20806. doi:10.1002/cne.20806. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Alvarez SE, Milstien S, Spiegel S. Filamin A links sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 at lamellipodia to orchestrate cell migration. Molecular and Cellular Biology. 2008;28:5687–5697. doi: 10.1128/MCB.00465-08. doi:10.1128/MCB.00465-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson M, Fagerholm SC. Filamin and filamin-binding proteins in integrin-regulation and adhesion. Focus on: "FilaminA is required for vimentin-mediated cell adhesion and spreading". American Journal of Physiology. Cell Physiology. 2010;298:C206–C208. doi: 10.1152/ajpcell.00505.2009. doi:10.1152/ajpcell.00505.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestrini E, Patrosso C, Mancini M, Rivella S, Rocchi M, Repetto M, Villa A, Frattini A, Zoppe M, Vezzoni P, et al. Mapping of two genes encoding isoforms of the actin binding protein ABP-280, a dystrophin like protein, to Xq28 and to chromosome 7. Human Molecular Genetics. 1993;2:761–766. doi: 10.1093/hmg/2.6.761. doi:10.1093/hmg/2.6.761. [DOI] [PubMed] [Google Scholar]
- Masruha MR, Caboclo LO, Carrete H, Jr, Cendes IL, Rodrigues MG, Garzon E, Yacubian EM, Sakamoto AC, Sheen V, Harney M, et al. Mutation in filamin A causes periventricular heterotopia, developmental regression, and West syndrome in males. Epilepsia. 2006;47:211–214. doi: 10.1111/j.1528-1167.2006.00390.x. doi:10.1111/j.1528-1167.2006.00390.x. [DOI] [PubMed] [Google Scholar]
- Meng X, Yuan Y, Maestas A, Shen Z. Recovery from DNA damage-induced G2 arrest requires actin-binding protein filamin-A/actin-binding protein 280. Journal of Biological Chemistry. 2004;279:6098–6105. doi: 10.1074/jbc.M306794200. doi:10.1074/jbc.M306794200. [DOI] [PubMed] [Google Scholar]
- Mishra L, Marshall B. Adaptor proteins and ubiquinators in TGF-β signaling. Cytokine & Growth Factor Reviews. 2006;17:75–87. doi: 10.1016/j.cytogfr.2005.09.001. doi:10.1016/j.cytogfr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Mooso BA, Vinall RL, Tepper CG, Savoy RM, Cheung JP, Singh S, Siddiqui S, Wang Y, Bedolla RG, Martinez A, et al. Enhancing the effectiveness of androgen deprivation in prostate cancer by inducing Filamin A nuclear localization. Endocrine-Related Cancer. 2012;19:759–777. doi: 10.1530/ERC-12-0171. doi:10.1530/ERC-12-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora N, Rosales R, Rosales C. R-Ras promotes metastasis of cervical cancer epithelial cells. Cancer Immunology and Immunotherapy. 2007;56:535–544. doi: 10.1007/s00262-006-0205-z. doi:10.1007/s00262-006-0205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Albrecht S, Golfert F, Hofer A, Funk RH, Magdolen V, Flossel C, Luther T. Localization of tissue factor in actin-filament-rich membrane areas of epithelial cells. Experimental Cell Research. 1999;248:136–147. doi: 10.1006/excr.1999.4395. doi:10.1006/excr.1999.4395. [DOI] [PubMed] [Google Scholar]
- Nagano T, Yoneda T, Hatanaka Y, Kubota C, Murakami F, Sato M. Filamin A-interacting protein (FILIP) regulates cortical cell migration out of the ventricular zone. Nature Cell Biology. 2002;4:495–501. doi: 10.1038/ncb808. doi:10.1038/ncb808. [DOI] [PubMed] [Google Scholar]
- Nagano T, Morikubo S, Sato M. Filamin A and FILIP (Filamin A-Interacting Protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. Journal of Neuroscience. 2004;24:9648–9657. doi: 10.1523/JNEUROSCI.2363-04.2004. doi:10.1523/JNEUROSCI.2363-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adhesion & Migration. 2011;5:160–169. doi: 10.4161/cam.5.2.14401. doi:10.4161/cam.5.2.14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallapalli RK, Ibrahim MX, Zhou AX, Bandaru S, Naresh S, Redfors B, Pazooki D, Zhang Y, Boren J, Cao Y, et al. Targeting filamin A reduces K-RAS-induced lung adenocarcinomas and endothelial response to tumor growth in mice. Molecular Cancer. 2012;11:50. doi: 10.1186/1476-4598-11-50. doi:10.1186/1476-4598-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki M, Okada H, Takano H, Kuno J, Tani K, Hibino H, Asano S, Ito Y, Satake M, Noda T. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. PNAS. 1997;94:5697–5702. doi: 10.1073/pnas.94.11.5697. doi:10.1073/pnas.94.11.5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Hartwig JH. Phosphorylation of actin-binding protein 280 by growth factors is mediated by p90 ribosomal protein S6 kinase. Journal of Biological Chemistry. 1996;271:11858–11864. doi: 10.1074/jbc.271.20.11858. doi:10.1074/jbc.271.27.16294. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. The small GTPase RalA targets filamin to induce filopodia. PNAS. 1999;96:2122–2128. doi: 10.1073/pnas.96.5.2122. (doi:10.1073/pnas.96.5.2122) (doi:10.1073/pnas.96.5.2122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Watanabe T, Niki M, Takano H, Chiba N, Yanai N, Tani K, Hibino H, Asano S, Mucenski ML, et al. AML1(−/−) embryos do not express certain hematopoiesis-related gene transcripts including those of the PU.1 gene. Oncogene. 1998;17:2287–2293. doi: 10.1038/sj.onc.1202151. doi:10.1038/sj.onc.1202151. [DOI] [PubMed] [Google Scholar]
- Ott I, Fischer EG, Miyagi Y, Mueller BM, Ruf W. A role for tissue factor in cell adhesion and migration mediated by interaction with actin-binding protein 280. Journal of Cell Biology. 1998;140:1241–1253. doi: 10.1083/jcb.140.5.1241. doi:10.1083/jcb.140.5.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozanne DM, Brady ME, Cook S, Gaughan L, Neal DE, Robson CN. Androgen receptor nuclear translocation is facilitated by the f-actin cross-linking protein filamin. Molecular Endocrinology. 2000;14:1618–1626. doi: 10.1210/mend.14.10.0541. doi:10.1210/me.14.10.1618. [DOI] [PubMed] [Google Scholar]
- Pal Sharma C, Goldmann WH. Phosphorylation of actin-binding protein (ABP-280; filamin) by tyrosine kinase p56lck modulates actin filament cross-linking. Cell Biology International. 2004;28:935–941. doi: 10.1016/j.cellbi.2004.09.006. doi:10.1016/j.cellbi.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Parrini E, Ramazzotti A, Dobyns WB, Mei D, Moro F, Veggiotti P, Marini C, Brilstra EH, Dalla Bernardina B, Goodwin L, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain. 2006;129:1892–1906. doi: 10.1093/brain/awl125. doi:10.1093/brain/awl125. [DOI] [PubMed] [Google Scholar]
- Porter RM, Holme TC, Newman EL, Hopwood D, Wilkinson JM, Cuschieri A. Monoclonal antibodies to cytoskeletal proteins: an immunohistochemical investigation of human colon cancer. Journal of Pathology. 1993;170:435–440. doi: 10.1002/path.1711700406. doi:10.1002/path.1711700406. [DOI] [PubMed] [Google Scholar]
- Price MG, Caprette DR, Gomer RH. Different temporal patterns of expression result in the same type, amount, and distribution of filamin (ABP) in cardiac and skeletal myofibrils. Cell Motility and the Cytoskeleton. 1994;27:248–261. doi: 10.1002/cm.970270306. doi:10.1002/cm.970270306. [DOI] [PubMed] [Google Scholar]
- Ravid D, Chuderland D, Landsman L, Lavie Y, Reich R, Liscovitch M. Filamin A is a novel caveolin-1-dependent target in IGF-I-stimulated cancer cell migration. Experimental Cell Research. 2008;314:2762–2773. doi: 10.1016/j.yexcr.2008.06.004. doi:10.1016/j.yexcr.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Raynaud F, Jond-Necand C, Marcilhac A, Furst D, Benyamin Y. Calpain 1-gamma filamin interaction in muscle cells: a possible in situ regulation by PKC-α. International Journal of Biochemistry & Cell Biology. 2006;38:404–413. doi: 10.1016/j.biocel.2005.09.020. doi:10.1016/j.biocel.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Razinia Z, Baldassarre M, Bouaouina M, Lamsoul I, Lutz PG, Calderwood DA. The E3 ubiquitin ligase specificity subunit ASB2α targets filamins for proteasomal degradation by interacting with the filamin actin-binding domain. Journal of Cell Science. 2011;124:2631–2641. doi: 10.1242/jcs.084343. doi:10.1242/jcs.084343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. doi:10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- Robertson SP. Filamin A: phenotypic diversity. Current Opinion in Genetics & Development. 2005;15:301–307. doi: 10.1016/j.gde.2005.04.001. doi:10.1016/j.gde.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, Kenwrick SJ, Kim CA, Morava E, Newbury-Ecob R, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nature Genetics. 2003;33:487–491. doi: 10.1038/ng1119. doi:10.1038/ng1119. [DOI] [PubMed] [Google Scholar]
- Ruskamo S, Ylanne J. Structure of the human filamin A actin-binding domain. Acta Crystallographica. Section D. Biological Crystallography. 2009;65:1217–1221. doi: 10.1107/S0907444909037330. doi:10.1107/S0907444909037330. [DOI] [PubMed] [Google Scholar]
- Ruskamo S, Gilbert R, Hofmann G, Jiang P, Campbell ID, Ylanne J, Pentikainen U. The C-terminal rod 2 fragment of filamin A forms a compact structure that can be extended. Biochemical Journal. 2012;446:261–269. doi: 10.1042/BJ20120361. doi:10.1042/BJ20120361. [DOI] [PubMed] [Google Scholar]
- Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto-Torii K, Torii M, Flavell RA, Rakic P. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron. 2006;52:789–801. doi: 10.1016/j.neuron.2006.10.024. doi:10.1016/j.neuron.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Nagano T. Involvement of filamin A and filamin A-interacting protein (FILIP) in controlling the start and cell shape of radially migrating cortical neurons. Anatomical Science International. 2005;80:19–29. doi: 10.1111/j.1447-073x.2005.00101.x. doi:10.1111/j.1447-073x.2005.00101.x. [DOI] [PubMed] [Google Scholar]
- Saito K, Ozawa Y, Hibino K, Ohta Y. FilGAP, a Rho/Rho-associated protein kinase-regulated GTPase-activating protein for Rac, controls tumor cell migration. Molecular Biology of the Cell. 2012;23:4739–4750. doi: 10.1091/mbc.E12-04-0310. doi:10.1091/mbc.E12-04-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen VL, Dixon PH, Fox JW, Hong SE, Kinton L, Sisodiya SM, Duncan JS, Dubeau F, Scheffer IE, Schachter SC, et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Human Molecular Genetics. 2001;10:1775–1783. doi: 10.1093/hmg/10.17.1775. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Jansen A, Chen MH, Parrini E, Morgan T, Ravenscroft R, Ganesh V, Underwood T, Wiley J, Leventer R, et al. Filamin A mutations cause periventricular heterotopia with Ehlers–Danlos syndrome. Neurology. 2005;64:254–262. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- Stossel TP, Hartwig JH. Interactions between actin, myosin, and an actin-binding protein from rabbit alveolar macrophages. Alveolar macrophage myosin Mg-2+-adenosine triphosphatase requires a cofactor for activation by actin. Journal of Biological Chemistry. 1975;250:5706–5712. [PubMed] [Google Scholar]
- Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nature Reviews. Molecular Cell Biology. 2001;2:138–145. doi: 10.1038/35052082. doi:10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- Takabayashi T, Xie MJ, Takeuchi S, Kawasaki M, Yagi H, Okamoto M, Tariqur RM, Malik F, Kuroda K, Kubota C, et al. LL5β directs the translocation of filamin A and SHIP2 to sites of phosphatidylinositol 3,4,5-triphosphate (PtdIns(3,4,5)P3) accumulation, and PtdIns(3,4,5)P3 localization is mutually modified by co-recruited SHIP2. Journal of Biological Chemistry. 2010;285:16155–16165. doi: 10.1074/jbc.M109.081901. doi:10.1074/jbc.M109.081901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telles E, Gurjar M, Ganti K, Gupta D, Dalal SN. Filamin A stimulates cdc25C function and promotes entry into mitosis. Cell Cycle. 2011;10:776–782. doi: 10.4161/cc.10.5.14954. doi:10.4161/cc.10.5.14954. [DOI] [PubMed] [Google Scholar]
- Tian H-M, Liu X-H, Han W, Zhao L-L, Yuan B, Yuan C-J. Differential expression of filamin A and its clinical significance in breast cancer. Oncology Letters. 2013;6:681–686. doi: 10.3892/ol.2013.1454. doi:10.3892/ol.2013.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, et al. Towards a knowledge-based Human Protein Atlas. Nature Biotechnology. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. doi:10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- Uramoto H, Akyurek LM, Hanagiri T. A positive relationship between filamin and VEGF in patients with lung cancer. Anticancer Research. 2010;30:3939–3944. [PubMed] [Google Scholar]
- Velkova A, Carvalho MA, Johnson JO, Tavtigian SV, Monteiro AN. Identification of Filamin A as a BRCA1-interacting protein required for efficient DNA repair. Cell Cycle. 2010;9:1421–1433. doi: 10.4161/cc.9.7.11256. doi:10.4161/cc.9.7.11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatareddy M, Cook L, Abuarquob K, Verma R, Garg P. Nephrin regulates lamellipodia formation by assembling a protein complex that includes Ship2, filamin and lamellipodin. PLoS ONE. 2011;6:e28710. doi: 10.1371/journal.pone.0028710. doi:10.1371/journal.pone.0028710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Davies PJ, Pastan I. Cyclic AMP-dependent phosphorylation of filamin in mammalian smooth muscle. Journal of Biological Chemistry. 1978;253:4739–4745. [PubMed] [Google Scholar]
- Wang Y, Kreisberg JI, Bedolla RG, Mikhailova M, deVere White RW, Ghosh PM. A 90 kDa fragment of filamin A promotes Casodex-induced growth inhibition in Casodex-resistant androgen receptor positive C4-2 prostate cancer cells. Oncogene. 2007;26:6061–6070. doi: 10.1038/sj.onc.1210435. doi:10.1038/sj.onc.1210435. [DOI] [PubMed] [Google Scholar]
- Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. doi:10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Yoshida N, Satake M. Biological implications of filamin A-bound PEBP2β/CBFβ retention in the cytoplasm. Critical Reviews in Eukaryotic Gene Expression. 2005;15:197–206. doi: 10.1615/critreveukargeneexpr.v15.i3.20. doi:10.1615/CritRevEukarGen-eExpr.v15.i3.20. [DOI] [PubMed] [Google Scholar]
- Wiemann S, Weil B, Wellenreuther R, Gassenhuber J, Glassl S, Ansorge W, Bocher M, Blocker H, Bauersachs S, Blum H, et al. Toward a catalog of human genes and proteins: sequencing and analysis of 500 novel complete protein coding human cDNAs. Genome Research. 2001;11:422–435. doi: 10.1101/gr.154701. doi:10.1101/gr.GR1547R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit MC, Kros JM, Halley DJ, de Coo IF, Verdijk R, Jacobs BC, Mancini GM. Filamin A mutation, a common cause for periventricular heterotopia, aneurysms and cardiac defects. Journal of Neurology, Neurosurgery, and Psychiatry. 2009;80:426–428. doi: 10.1136/jnnp.2008.149419. doi:10.1136/jnnp.2008.149419. [DOI] [PubMed] [Google Scholar]
- Xu Y, Bismar TA, Su J, Xu B, Kristiansen G, Varga Z, Teng L, Ingber DE, Mammoto A, Kumar R, et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. Journal of Experimental Medicine. 2010;207:2421–2437. doi: 10.1084/jem.20100433. doi:10.1084/jem.20100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Ogata T, Tanabe K, Li S, Nakazato M, Kohu K, Takafuta T, Shapiro S, Ohta Y, Satake M, et al. Filamin A-bound PEBP2β/CBFb is retained in the cytoplasm and prevented from functioning as a partner of the Runx1 transcription factor. Molecular and Cellular Biology. 2005;25:1003–1012. doi: 10.1128/MCB.25.3.1003-1012.2005. doi:10.1128/MCB.25.3.1003-1012.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Shen Z. Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response. Journal of Biological Chemistry. 2001;276:48318–48324. doi: 10.1074/jbc.M102557200. doi:10.1074/jbc.M102557200. [DOI] [PubMed] [Google Scholar]
- Yue J, Wang Q, Lu H, Brenneman M, Fan F, Shen Z. The cytoskeleton protein filamin-A is required for an efficient recombinational DNA double strand break repair. Cancer Research. 2009;69:7978–7985. doi: 10.1158/0008-5472.CAN-09-2177. doi:10.1158/0008-5472.CAN-09-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Huhn S, Shen Z. Complex roles of filamin-A mediated cytoskeleton network in cancer progression. Cell & Bioscience. 2013;3:7. doi: 10.1186/2045-3701-3-7. doi:10.1186/2045-3701-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Yeow WS, Zou C, Wassell R, Wang C, Pestell RG, Quong JN, Quong AA. Cyclin D1/cyclin-dependent kinase 4 interacts with filamin A and affects the migration and invasion potential of breast cancer cells. Cancer Research. 2010;70:2105–2114. doi: 10.1158/0008-5472.CAN-08-1108. doi:10.1158/0008-5472.CAN-08-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Boren J, Akyurek LM. Filamins in cardiovascular development. Trends in Cardiovascular Medicine. 2007;17:222–229. doi: 10.1016/j.tcm.2007.08.001. doi:10.1016/j.tcm.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Zhou AX, Hartwig JH, Akyurek LM. Filamins in cell signaling, transcription and organ development. Trends in Cell Biology. 2010;20:113–123. doi: 10.1016/j.tcb.2009.12.001. doi:10.1016/j.tcb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Zhou AX, Toylu A, Nallapalli RK, Nilsson G, Atabey N, Heldin CH, Boren J, Bergo MO, Akyurek LM. Filamin A mediates HGF/c-MET signaling in tumor cell migration. International Journal of Cancer. 2011;128:839–846. doi: 10.1002/ijc.25417. doi:10.1002/ijc.25417. [DOI] [PubMed] [Google Scholar]
- Zhu TN, He HJ, Kole S, D’Souza T, Agarwal R, Morin PJ, Bernier M. Filamin A-mediated down-regulation of the exchange factor Ras-GRF1 correlates with decreased matrix metalloproteinase-9 expression in human melanoma cells. Journal of Biological Chemistry. 2007;282:14816–14826. doi: 10.1074/jbc.M611430200. doi:10.1074/jbc.M61143020REF110=10.1615/CritRevEukarGeneExpr.v15.i3.20. [DOI] [PubMed] [Google Scholar]