

SUMMARY
DNA damage associated with viral DNA synthesis can result in double strand breaks that threaten genome integrity and must be repaired. Here, we establish that the cellular Fanconi Anemia (FA) genomic stability pathway is exploited by HSV1 to promote viral DNA synthesis and enable its productive growth. Potent FA pathway activation in HSV1-infected cells resulted in monoubiquitination of FA effector proteins, FANCI and FANCD2 (FANCI-D2) and required the viral DNA polymerase. FANCD2 relocalized to viral replication compartments and FANCI-D2 interacted with a multi-subunit complex containing the virus-encoded single-stranded DNA-binding protein ICP8. Significantly, while HSV1 productive growth was impaired in monoubiquitination-defective FA patient cells, this restriction was partially surmounted by antagonizing the DNA-dependent protein kinase (DNA-PK), a critical enzyme required for non-homologous end-joining (NHEJ). This identifies the FA-pathway as a new cellular factor required for herpesvirus productive growth and suggests that FA-mediated suppression of NHEJ is a fundamental step in the viral lifecycle.
INTRODUCTION
DNA double strand breaks (DSBs) are potentially lethal lesions that can promote genome rearrangements. While exogenous agents like ionizing radiation produce DSBs, they also result from endogenous sources. Indeed, replication fork stalling at inter-strand DNA cross-links (ICLs) or collapsed forks at unrepaired, single-strand DNA nicks account for most endogenous DSBs (reviewed in Jackson & Bartek, 2009; Hartlerode & Scully, 2009; Ciccia & Elledge, 2010). To repair DSBs and preserve genome integrity, cells rely on either error-prone non-homologous end-joining (NHEJ) or the more accurate homologous recombination (HR) repair pathways (reviewed in Chapman et al., 2012). Although it is not well-understood how DNA repair choice between these distinct pathways is regulated in response to DSBs (Chapman et al., 2012), the cellular Fanconi Anemia (FA) pathway likely plays a critical role counteracting aspects of the NHEJ pathway to faithfully repair ICLs via HR-mediated events in mammalian cells (Adamo et al., 2010; Pace et al., 2010).
FA is a complex multigene disorder characterized by genome instability, congenital abnormalities, acute myeloid leukemia, and/or bone marrow failure and cancer predisposition (D’Andrea & Grompe, 2003; Kennedy & D’Andrea, 2005). The hallmark of FA cells is hypersensitivity to ICL-inducing agents, indicative of a repair defect in response to agents that block the replication fork. The FA pathway is composed of at least 15 known proteins corresponding to complementation groups mutated in FA patients: FANCA, B, C, D1, D2, E, F, G I, J, L M, N, O, and P (Moldovan & D’Andrea, 2009). In the presence of ICLs or stalled replication forks, the FA core E3 ligase complex (comprised of FANCA, B, C, E, F, G, L, and M) catalyzes the lysine site-specific monoubiquitination of two FA effector proteins, FANCI and FANCD2 (FANCI-D2), which promotes their recruitment to DNA damage repair sites in nuclear foci (Garcia-Higuera et al., 2001; Sims et al., 2007; Smogorzewska et al., 2007). Here, FANCI-D2 colocalizes with downstream FA proteins, FANCJ, FANCD1 (BRCA2), and FANCN (PALB2), to facilitate HR-mediated repair. While environmental toxins and endogenous metabolites are potential physiological sources of DNA damage that trigger FA pathway activation (Langevin et al., 2011; Rosado et al., 2011), the capacity of natural processes like infection to activate and possibly subvert FA pathway function remains relatively unexplored. To investigate the impact of virus infection on the FA pathway, we enlisted a powerful herpes simplex virus-1 (HSV1) model system.
Following delivery into the nucleus of the linear, dsDNA viral genome, which is GC rich and contains gaps and nicks (Wadsworth et al., 1976; Hyman et al., 1977; Jacob & Roizman, 1977; Roizman, 1979; Wilkinson & Weller, 2003), HSV1 gene expression proceeds as a temporally controlled cascade of differentially expressed genes (Roizman et al., 2006). The first viral transcripts encode key immediate-early (IE) regulatory proteins that in turn activate expression of HSV1 early genes, many of which participate in virus genome replication (Roizman et al., 2006). Prior to viral DNA synthesis, HSV1 replication proteins form pre-replication foci within the nucleus at locations that are likely sites of cellular DNA synthesis in uninfected cells (reviewed in Weller & Coen, 2012). Once viral DNA synthesis commences, pre-replication foci coalesce into larger replication compartments (RCs) that progressively occupy the nucleus while host chromatin becomes marginalized to the nuclear periphery (Weller & Coen, 2012). Viral DNA synthesis produces larger than unit length genome concatemers whose formation, while incompletely understood, likely involves recombination (Wilkinson & Weller, 2003). Furthermore, extensive genome isomerization dependent upon viral DNA replication results from intramolecular HR that is stimulated by DSBs (Sarisky & Weber, 1994; Wilkinson & Weller, 2003). Finally, late gene expression triggered by viral DNA synthesis signifies the final stage of the productive growth cycle and culminates in infectious virus production (Roizman et al., 2006).
As a DNA virus that replicates in the nucleus, HSV1 interacts with the cellular DNA damage response (DDR) network and triggers a complex response (reviewed in Weitzman et al., 2010; Weller, 2010). Whereas specific DDR pathways are activated by viral functions, others are disabled as HSV1 remodels the host response to prevent viral genome silencing and suppress intrinsic antiviral defenses. For example, recognition of linear viral genomes by cellular NHEJ proteins could elicit an antiviral response. In some cell types, the viral E3 ubiquitin ligase ICP0 counteracts this response by stimulating degradation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), a key NHEJ pathway component (Lees-Miller et al., 1996; Parkinson et al., 1999). In contrast, other cellular proteins involved in HR repair (Mre11, Rad50, Nbs1, Rad51) are recruited to viral pre-replicative sites and RCs, where they promote virus replication (Wilkinson & Weller, 2004; Lilley et al., 2005). Precisely how DNA repair choice is achieved upon HSV1 infection, the molecular basis underlying this switch between NHEJ and HR pathways, and its impact on productive viral replication remains poorly understood at best.
Here, we show that HSV1 infection potently activates the host FA pathway by stimulating FANCI-D2 monoubiquitination. FA pathway activation required viral gene expression and was dependent upon the HSV1 DNA polymerase (Pol). Moreover, the viral single-strand DNA binding protein (SSB) ICP8 interacts with FANCD2 in infected cells. Significantly, while infection stimulated FANCD2 nuclear foci accumulation proximal to viral pre-replication centers and within viral RCs, FANCD2 redistribution was not dependent upon viral DNA synthesis. Finally, we show that HSV1 replication is FA pathway-dependent and can be partially restored in a monoubiquitination-defective FA mutant cell line by inhibiting DNA-PK to suppress NHEJ. Thus, restriction of HSV1 replication in FA-deficient cells reflects, in part, failure to suppress DNA-PK-mediated NHEJ. This work establishes the cellular FA pathway as a previously unrecognized host factor required for productive herpesvirus replication and suggests that FA-mediated suppression of NHEJ is a fundamental step in the virus lifecycle.
RESULTS
Activation of the cellular FA pathway by HSV1 infection
Cellular FA pathway activation can be monitored by the inducible monoubiquitination of FANCI-D2 proteins, which normally occurs after DNA damage in replicating cells. To investigate how HSV1-infection impacts the host FA pathway, cells were infected with either of two HSV1 strains (KOS or Patton) and FANCD2 monoubiquitination (Ub-FANCD2) assessed over time (Figs. 1A, S1A). Ub-FANCD2 accumulated by 9 hours post-infection (hpi) and persisted throughout the productive growth cycle (Fig. 1A). Monoubiquitinated FANCI also accumulated in HSV1-infected cells (Fig. S1B). FANCI-D2 deubiquitinase (DUB) USP1 abundance was reduced after infection and inversely correlated with Ub-FANCD2 and viral protein levels (Fig. 1A). While representative viral IE (ICP4, ICP0) and early (ICP8) proteins were observed in the absence of detectable Ub-FANCD2, late protein gC levels tracked most closely with Ub-FANCD2 accumulation (Fig. 1A). In FA patient-derived cells, HSV1 infection did not detectably induce Ub-FANCD2 without the essential FA core E3 ligase subunit, FANCA (FA-A + vector), ensuring that a viral E3 ligase was not required (Fig. 1B). FANCA-deficient cells complemented with wild-type FANCA (FA-A + FANCA WT) restored HSV1-dependent Ub-FANCD2 accumulation. Importantly, HSV1 gene expression was required to stimulate Ub-FANCD2 in FA-A corrected cells (FA-A + FANCA WT) since ultraviolet light (UV-C)-inactivated virus, which prevents transcription from the viral genome (see Fig. 2A schematic), did not activate the FA pathway (Fig. 1C).
Figure 1. Activation of the FA pathway in HSV1-infected cells.

A) Vero cells were mock-infected (M) or infected with WT HSV1 KOS (MOI=5). Total protein was harvested at different times post-infection (hPI), fractionated by SDS-PAGE and analyzed by immunoblotting using the indicated antibodies. Ub-FANCD2 denotes the slower- migrating, monoubiquitinated FANCD2. B) Patient-derived FA-A cells (FANCA-deficient) stably transduced with either empty expression vector only (FA-A vector) or functionally complemented with a WT FANCA cDNA (FA-A + FANCA WT) and treated as in A. C) FA-A cells complemented with FANCA WT were mock-infected (M) or infected with WT or UV-inactivated WT HSV1 KOS (MOI=1 or 5). Total protein harvested at 19 hpi was analyzed as in A.
Figure 2. FA pathway activation in HSV1-infected cells requires viral gene expression.
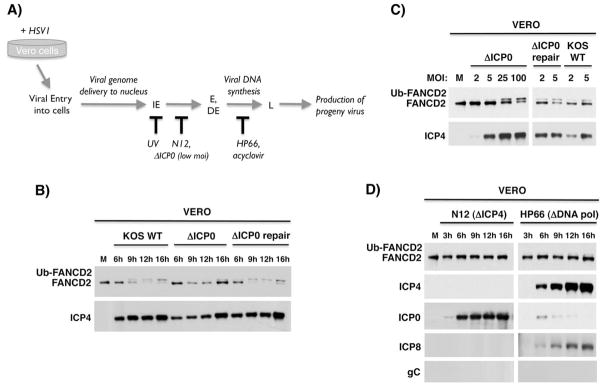
A) Temporal progression of the HSV1 productive replication cycle. Upon HSV1-infection of permissive Vero cells, the virus enters cells and the dsDNA viral genome is delivered to the nucleus. The viral gene expression program commences with transcription of immediate early (IE) genes, which in turn activate transcription of early (E) and delayed-early (DE) genes. This is followed by viral DNA synthesis, which activates transcription of late (L) viral genes, and culminates in infectious progeny virus production. The point at which different mutant viruses (ΔICP0 at low MOI, N12, HP66) or treatments (UV irradiated virus, acyclovir) arrest the viral lifecycle is shown below. B) Vero cells were mock-infected (M) or infected with WT KOS, ΔICP0 or ΔICP0-repair viruses (MOI = 5). Total protein was harvested at various times (hPI), fractionated by SDS-PAGE and analyzed by immunoblotting using the indicated antibodies. C) As in A except cells were mock-infected or infected at the indicated MOI (2, 5, 25,100). After 9 hpi, total protein was isolated and analyzed as in B. D) Vero cells were infected (MOI=5) with a virus deficient for ICP4 (N12) or the viral DNA Pol (HP66). Total protein isolated at the indicated time was analyzed as in B. In addition to FANCD2, accumulation of representative viral IE (ICP4, ICP0), early (ICP8) and late (gC) proteins were monitored. ICP0 is overexpressed in the absence of ICP4 (DeLuca et al., 1985), and preventing DNA synthesis limits ICP0 expression (Weinheimer & McKnight, 1987).
FA pathway activation by HSV1 is dependent upon the viral DNA polymerase
To more precisely delineate viral gene product(s) required for FA pathway activation, the capacity of different HSV1 mutants to stimulate Ub-FANCD2 was evaluated (Fig. 2A). First, the requirement for ICP0, the master regulatory viral gene required for growth at low MOI, was examined. In cells infected with an ICP0-deficient HSV1 (ΔICP0), Ub-FANCD2 induction was severely compromised at low, but not high MOI (Fig. 2B, C). Reintroducing the WT ICP0 gene into the ΔICP0 genome (ΔICP0 repair) restored its ability to stimulate Ub-FANCD2 in a manner indistinguishable from the WT parent strain (Fig. 2B, C). Thus, MOI-dependent induction of Ub-FANCD2 is a phenotype associated with ICP0-deficiency, but ICP0 is likely not directly required.
In contrast, when cells were infected with an ICP4-deficient virus (ΔICP4), Ub-FANCD2 was no longer detected (Fig. 2D). Importantly, USP1 levels were still reduced in cells infected with ΔICP4 even though Ub-FANCD2 was not detected (Fig S2). Thus, the virus-induced reduction in USP1 abundance can be genetically separated from FA pathway activation as assessed by Ub-FANCD2 accumulation. In the absence of ICP4, the viral genome is delivered into the nucleus and transcription of IE genes occurs (Fig. 2A); however, the productive replication cycle arrests and only a restricted set of proteins accumulate (ICP0, 6, 27, 22, 47; DeLuca et al., 1985). Thus, events requiring progression of the viral lifecycle beyond the IE stage were likely required to activate the host FA pathway (Fig. 2A). To address this possibility, cells were infected with the HP66 mutant virus, which is deficient in the catalytic subunit of the HSV1 DNA Pol. (UL30; Marcy et al., 1990). Unlike cells infected with ΔICP4, the viral lifecycle proceeds into the early phase in HP66-infected cells and viral proteins required for DNA replication, including the SSB protein ICP8, are produced (Fig. 2A, D). However, without the viral DNA Pol, DNA synthesis cannot initiate and the infection is blocked at the boundary between early and late stages of the productive growth cycle preventing expression of late genes like gC (Fig. 2A, D; Marcy et al., 1990). Interestingly, Ub-FANCD2 induction was severely inhibited in HP66-infected cells, establishing that HSV1-induced FA pathway activation was dependent upon the viral DNA Pol (Fig. 2D). Blocking viral DNA synthesis with the antiviral drug acyclovir similarly prevented FA pathway activation (Fig. S3). This suggests that FA pathway activation in response to HSV1 is activated by viral DNA synthesis or a closely linked event like late gene expression, and agrees with recent studies demonstrating that FA proteins sense DNA crosslink damage at active replication forks (Knipscheer et al., 2009; Shen et al., 2009; Nakanashi et al., 2011).
Association of cellular FA pathway components with viral replication proteins in HSV1-infected cells
To determine if any HSV1 polypeptides physically-associated with host FA proteins, infected cell proteins metabolically-labeled with 35S-containing amino acids were immunoprecipitated (IP’ed) using antibodies specific for FANCD2 or FANCI (Fig. 3A). As HSV1 infection impairs ongoing host protein synthesis (reviewed in Walsh & Mohr, 2011; Mohr & Sonenberg, 2012), radiolabeled proteins overwhelmingly, but not exclusively, represent newly synthesized, viral polypeptides. Analysis of labeled proteins retained in immune complexes by SDS-PAGE followed by autoradiography revealed associated polypeptides of apparent molecular weights 130–150 kDa with FANCI, and 50–80 kDa with FANCD2 (Fig. 3B). As their association with FANCD2 and FANCI was also observed in HP66-infected cells (Fig. 3B), virus-induced FA pathway activation and viral DNA synthesis were not required. Finally, these proteins were specifically associated with host FA components as they were not detected using antisera against an unrelated protein, the cellular 4E-BP1 translation repressor (Fig. 3B).
Figure 3. Association of FANCD2-FANCI with specific virus-encoded proteins in HSV1-infected cells.

A) Experimental plan to identify HSV1 polypeptides associated with cellular FA proteins. Vero cells were infected (MOI=10) with either WT HSV1 or a mutant virus that arrests its growth cycle prior to initiating DNA synthesis (HP66). After 7h, cultures were metabolically labeled with 35S met / cys for 3h. Under these conditions, host protein synthesis is impaired and the vast majority of newly synthesized, radiolabeled polypeptides are HSV1-encoded. A cell-free lysate prepared using non-ionic detergent was nuclease-treated to degrade single and ds nucleic acid, and subsequently IP’ed using anti-FANCD2, anti-FANCI, or a control affinity-purified rabbit antisera that recognizes an unrelated protein (the translational repressor 4E-BP1). B) Immune complexes isolated from infected, metabolically-labeled lysates described and treated as in A were fractionated by SDS-PAGE and analyzed by autoradiography. A sample of input lysate is shown (right panel). Radiolabeled proteins with molecular weights of 130–150 kDA (+), 80 kDA (o), and 50 kDA (*) are indicated on the autoradiogram. Molecular weight standards with their indicated relative molecular weight (kDa) appear to the left. C) Anti-FANCI and anti-FANCD2 immune complexes isolated from HSV1-infected (WT vs. HP66) cells were analyzed by tandem MS. HSV1-encoded DNA replication and repair proteins (gene products) associated with FANCI-D2, the number of spectral counts (Spec#), the number of peptides identified (Pep#), % amino acid coverage (%AA), and the antibody used for IP are shown. D) Non-ionic detergent lysates prepared from Vero cells mock-infected (M) or infected as in A were nuclease-treated and IP’ed using the specified antisera. Immune complexes were fractionated by SDS-PAGE and analyzed by immunoblotting (IB) using the indicated antisera. A sample of input lysate is shown (right panel). * indicates IgG heavy chain. E) As in D except samples were IP’ed using anti-ICP8 or an isotype matched control antisera (anti-raptor).
To determine the identity of viral proteins associated with FANCI-D2, endogenous FANCD2 and FANCI were IP’ed from non-ionic detergent lysates prepared from virus-infected cells and proteins eluted from immune complexes analyzed by shotgun tandem mass spectrometry (Washburn et al., 2001). Peptides derived from HSV1 proteins involved in virus genome localization (ICP4), DNA synthesis (DNA Pol, UL42 DNA Pol processivity factor, UL5 helicase, ICP8 SSB), DNA repair (dUTPase) and recombination (UL12 alkaline endonuclease) were detected in FANCD2 and FANCI immune complexes (Fig. 3C, tables S1, S2). These interactions were specific as only a limited number of HSV1 proteins were found in FANCI-D2 immune complexes analyzed by SDS-PAGE and mass spectrometry, while numerous more abundant viral proteins were in fact not detected (Fig. 3 and table S1, S2).
Among HSV1 proteins involved in DNA replication, repair, and recombination, peptides from the SSB ICP8 were the most abundant and consistently detected in immune complexes isolated from WT or HP66-infected cells. As ICP8 is a component of nuclear RCs in infected cells, this suggested that ICP8 might physically interact with FA effector proteins. Indeed, IP experiments demonstrated that FANCI associated with ICP8 in HSV1-infected cells using antisera specific for either FANCI (Figs. 3C, D) or ICP8 (Fig. 3E). Likewise, the viral DNA pol processivity factor UL42, UL12 alkaline nuclease and ICP4, a viral transcription factor that marks sites of incoming viral genomes (Everett & Murray, 2005), were also validated as FANCI or D2-associated proteins (Fig. 3D). UL12 was also detected in anti-ICP8 immune complexes (Fig. 3E), consistent with earlier reports (Thomas et al., 1992).
Sub-nuclear localization of FANCD2 in response to HSV1-infection
A hallmark of FA pathway activation in response to DNA ICLs and replication stress is FANCD2 (and FANCI) accumulation into discrete nuclear foci (Garcia-Higuera et al., 2001; Taniguchi et al., 2002; Smogorzewska et al., 2007). Similarly, herpesvirus DNA synthesis occurs in defined, nuclear RCs (Fig 4A). However, prior to initiating viral DNA synthesis, pre-replication foci form composed of several HSV1 replication proteins, one of which is ICP8 (Figs 4A, S4A; Weller & Coen, 2012). Given FANCI and ICP8 physically associate (Fig. 3), and ICP8 marks specific subnuclear sites destined to form RCs, we investigated whether viral DNA synthesis and/or FANCA (FA core E3 ligase component) was required for FANCD2 nuclear foci formation. To compare between active viral RCs (WT) or pre-replicative foci (HP66), Vero cells were infected with either WT or the HP66 DNA Pol mutant virus and FANCD2 foci formation assessed by indirect immunofluorescence. Intriguingly, FANCD2 foci formation was enhanced in both WT and HP66-infected cells (Fig. 4B, right panel and Fig. S4C). As a positive control, uninfected cells were treated with the DNA replication inhibitor hydroxyurea (HU). As shown in Figure 2, Ub-FANCD2 was at best barely detectable in HP66-infected cells, suggesting Ub-FANCD2 accumulation may not be required for HSV1-induced FANCD2 foci formation. However, it remained possible that even the exceptionally low levels of Ub-FANCD2 in HP66-infected cells may be sufficient to stimulate FANCD2 foci formation near pre-replication sites. To address this, we determined whether FANCA was required for HSV1-induced FANCD2 nuclear foci formation. Importantly, FANCD2 nuclear foci formation was largely inhibited in FANCA-deficient cells, but not in the corrected cells (Fig. 4B, middle and left panels; Fig. S4B). While many FANCD2 foci were proximal to ICP8-containing structures, they did not appear to completely colocalize irrespective of infection with a replication proficient (WT) or deficient (HP66) virus (Fig. 4B). Other host DNA repair proteins (53BP1, BRCA1, Mdc1, γH2aX) also accumulate at nuclear foci distinct from viral genomes (Lilley et al., 2011). Thus, HSV1 infection stimulated redistribution of the cellular FANCD2 protein into nuclear foci in a manner i) dependent on FANCA; and ii) independent of viral DNA synthesis. Notably, in cells infected with a WT virus capable of HSV1 DNA synthesis where FA pathway activation was readily observed (see Fig 1), FANCD2 foci accumulated within viral RCs in a FA-A-dependent manner in all of the ICP8-positive cells (Fig 4, KOS WT panels).
Figure 4. Redistribution of FANCD2 in response to HSV1-infection and accumulation proximal to nuclear viral replication compartments (RCs).

A) Maturation of HSV1 pre-replication foci into nuclear RCs is dependent upon viral DNA synthesis. The HSV1-encoded SSB ICP8 is one of several viral replication proteins that accumulate at discrete foci (depicted as red dots) distributed within host nuclei (blue). Viral DNA synthesis triggers rearrangement of replication foci into an organized RC within nuclei. Since the HP66 mutant lacks a functional viral DNA pol and cannot replicate viral DNA, ICP8 only accumulates in pre-replication foci and the viral lifecycle arrests at this point. Pre-replication foci mature into RCs in cells infected with WT virus. B) Vero cells or FA-A cells stably transduced with either empty expression vector only (FA-A + vector) or a WT FANCA cDNA (FA-A + FANCA WT) were mock-infected or infected (MOI=5) with HP66 or WT KOS (10 h for FA-A cells; 8 h for Vero cells) and subsequently processed for indirect immunofluorescence. As a positive control to detect FANCD2 nuclear foci after DNA damage, cells were treated with 2 mM hydroxyurea (HU) for 8 or 10 h. Cells were co-stained to detect either ICP8 (red), FANCD2 (green), or DAPI (blue) and a representative image for each condition is shown.
Productive herpesvirus replication is dependent upon the host FA pathway
While HSV1 infection potently stimulated FANCD2 relocalization and monoubiquitination, it was unclear whether the FA pathway was required for productive herpesvirus replication. To evaluate the role of FA in productive viral growth, FANCA-deficient and corrected cells were infected with a WT HSV1 GFP-reporter strain in which EGFP-coding sequences were fused to the late viral Us11 gene. Viral replication and spread were monitored in live cells by fluorescent and phase contrast microscopy. Remarkably, the number of EGFP-positive cells and cytopathic effect (cpe) were dramatically reduced in FANCA-deficient cells compared to the WT corrected cells over a wide range of MOIs (Fig. 5A). Notably, this MOI-dependent growth phenotype was most penetrant at lower MOI (Fig. 5A). To measure the extent to which viral replication was impaired in FANCA-deficient cells, infectious virus produced after 3 days was quantified by plaque assay in permissive Vero cells. In the absence of FANCA, infectious virus production was reduced by nearly 1,000-fold at low MOI and more than 100-fold at higher MOI (Fig. 5B). This growth defect was not limited to patient-derived FA-deficient cells, but was partially recapitulated in primary human fibroblasts where FANCD2 was depleted using siRNA (Fig. 5C; Fig. S5).
Figure 5. The cellular FA pathway is required for productive HSV1 replication.

A) FA-A cells stably transduced with either empty expression vector only (FA-A + vector, right panel) or complemented with a WT FANCA cDNA (FA-A + FANCA WT, left panel) were infected at the indicated MOI with the WT EGFP-HSV1 reporter strain (GFP-HSV1). This strain contains EGFP-fused to the virus Us11 late gene and is indistinguishable from WT virus in its replicative capacity (Benboudjema et al., 2003). After 3 d, live cells were evaluated by phase contrast and epifluorescence microscopy. Images of representative fields are shown. B) Infectious virus produced in the experiment shown in panel A was quantified by plaque assay in Vero cells. * p=0.0098; ** p=0.00001. C) Primary human fibroblasts transfected with non-silencing, control siRNA or FANCD2 siRNA (Fig. S5) were infected with GFP-HSV1 (MOI = 10−3) and evaluated as in A after 2 d post-infection (Left panel). Infectious virus produced was quantified as in B (Right panel). * p=0.0282. D) FA-A + vector or FA-A + FANCA WT cells were infected (MOI=5) with HP66 or WT KOS, and total genomic DNA was isolated at the indicated times. Relative viral genomic DNA levels were determined by qPCR using primers complementary to the HSV1 ICP27 gene. Input DNA was normalized by amplifying the RPL19 gene. E) FA-G cells stably transduced with either empty expression vector only (FA-G + vector) or functionally complemented with a WT FANCG cDNA (FA-G + FANCG WT) were infected (MOI= 0.5 × 10−3) with the WT EGFP-HSV1 reporter strain (GFP-HSV1). After 2 d, live cells were evaluated by microscopy as in A (Left panel). Infectious virus produced was quantified as in B (Right panel). * p=0.001. F) BRCA2-deficient cells stably transfected with empty expression vector (BRCA2 vector) or functionally reconstituted with chromosome 13, which contains a WT BRCA2 gene (BRCA2 WT), were infected (MOI= 5 × 10−3) and analyzed as in E. * p=0.009
Having shown FANCA-deficient cells expressed viral IE proteins (Fig. 1B) but did not accumulate FANCD2 nuclear foci in viral RCs following HSV1-infection (Fig. 4), the impact of FA-pathway function on viral DNA synthesis was investigated by qPCR. Unlike cells expressing WT FANCA, FANCA-deficient cells were markedly impaired in their ability to accumulate viral DNA. Notably, FANCA-deficient cells contained nearly 16-fold less viral DNA by 10 hpi than an isogenic cell line expressing WT FANCA (Fig. 5D). Furthermore, viral DNA levels in FANCA-deficient cells at 10 hpi was still less than the amount measured at 5.5 hpi WT FANCA-expressing cells (Fig. 5D). This demonstrates that normal HSV DNA synthesis is dependent upon the cellular FA genomic stability pathway, and suggests impaired growth of HSV1 in FANCA-deficient cells results, in part, from insufficient nascent viral DNA accumulation.
To determine if HSV1 replication was specifically dependent upon FANCA alone, or indicative of a general FA pathway requirement, viral growth in FA patient-derived cells harboring mutations in different FA DNA repair pathway complementation groups was evaluated. In particular, we focused on cells deficient for FANCG, which together with FANCA is one of eight FA core complex components that comprise a multisubunit ubiquitin E3 ligase, and FANCD1 (alternatively known as BRCA2), one of several FA proteins acting downstream of FANCA and FANCG involved in strand invasion and resolution of recombinant intermediates (Kennedy & D’Andrea, 2005; Moldovan & D’Andrea, 2011). In both cases, HSV1 replication and spread was reduced approximately 75-fold in these FA-deficient cell lines and restored by introduction of the corresponding wild-type FANCG or FANCD1 allele (Fig. 5 E, F). This establishes the cellular FA pathway as a critical host factor important for herpesvirus replication. While other viruses interfere with host DDR pathway function to foster their replication, this is the first example of a virus that requires cellular FA genomic stability pathway activity to promote its productive growth.
Suppressing NHEJ in FA-deficient cells enhances HSV1 replication
HSV1-induced FA pathway activation was dependent upon the viral DNA Pol and likely required viral DNA synthesis, suggesting that the host FA pathway might play a critical role preventing and/or repairing ds breaks associated with complications arising during viral DNA replication. While NHEJ or HR can repair these lesions, the FA pathway counteracts some NHEJ activities and thereby fosters repair via HR-related pathways (Pace et al., 2010; Adamo et al., 2010). Indeed, in FA-deficient cells, repair of DNA damage through NHEJ could potentially restrict HSV1 replication. To determine if reduced HSV1 replication in FA-deficient cells results, in part, from aberrant NHEJ, viral replication was measured in FANCA-deficient cells in the presence or absence of NU7441, a selective, small-molecule DNAPKcs inhibitor (Leahy et al., 2004). DNAPKcs is the catalytic subunit of DNA-PK, a key early component involved in NHEJ (reviewed in Jackson & Bartek, 2009; Lieber, 2010). Strikingly, FANCA-deficient cells exhibited enhanced EGFP fluorescence and cpe (phase) after 3 days in independently-infected, NU7441-treated vs DMSO-treated control cultures (Fig. 6A). Quantification of infectious virus production revealed that NU7441-treatment enhanced virus replication by nearly 25-fold (Fig. 6A). This demonstrates that DNAPKcs activity restricts HSV1 replication in FA-deficient cells, and that a specific DNAPKcs chemical inhibitor partially overcomes this block and allows viral replication to proceed in FA-deficient cells.
Figure 6. Inhibiting DNA-PKcs partially restores HSV-1 replication in FA-deficient cells.

A) FA-A cells stably transduced with empty expression vector only (FA-A + vector) were infected with EGFP-HSV-1 (MOI = 0.05) in the presence of the DNA-PKcs inhibitor NU7441 (1μM) or vehicle control (DMSO). After 3 d, live cells were evaluated by phase contrast and epifluorescence microscopy. Images of representative fields are shown for three separate experiments (left panel). Infectious virus produced was quantified by plaque assay in Vero cells (right panel). * p=0.0101. B) FA-A cells as in A were transduced with a lentivirus expressing control, non-silencing shRNA (ctr), or one of two shRNAs targeting Ku80 (Ku80(1), Ku80(2)). After 4 d, total protein was harvested and the abundance of Ku80 vs tubulin (loading control) evaluated by immunoblotting. C). As in B except cells transduced with the indicated shRNA-expressing lentivirus were infected with GFP-HSV1 (MOI= 5 × 10−2). After 6 d, live cells were evaluated by microscopy (left panel) and infectious virus quantified (right panel) as in A. * p= 0.0003. D) Model depicting the proposed role of the FA pathway in counteracting aberrant NHEJ activity during HSV1 genome replication. Viral infection co-opts the cellular FA pathway by stimulating redistribution and monoubiquitination of FA effector proteins, FANCI and FANCD2, near viral replication sites. The precise role of monoubiquitinated FANCI-D2 at these sites remains unclear but may involve counteracting illegitimate DNA transactions by NHEJ during viral replication. Thus, the cellular FA pathway is subverted by HSV1 to enable efficient viral genome replication and promote productive viral growth.
The restriction of HSV1 growth in FANCA-deficient cells by DNA-PK was further investigated using RNAi to deplete Ku80, a critical DNA-PK regulatory subunit (Lieber, 2010). Upon Ku80-depletion in FANCA-deficient cells using two different shRNAs, replication of the HSV1-EGFP reporter strain and virus-induced cpe markedly increased compared to cultures expressing control, non-silencing shRNA (Fig. 6B, C). Quantification of this data by plaque assay revealed that Ku80-depletion enhanced productive viral growth by approximately 20–30-fold (Fig. 6C). Thus, antagonizing DNA-PK via two independent methodologies (Ku80 depletion vs chemical kinase inhibitor) partially restored productive HSV1 growth in FANCA-deficient cells. Taken together, this suggests that HSV1 activates the host FA pathway as a means to suppress NHEJ and foster HR-related DNA recombination and repair pathways during viral DNA synthesis in infected cells (Fig. 6D).
DISCUSSION
With linear dsDNA genomes that contain nicks and gaps (Wadsworth et al., 1976; Hyman et al., 1977; Jacob & Roizman, 1977; Roizman, 1979; Wilkinson & Weller, 2003), herpesviruses trigger an intricate DNA damage response upon their delivery into the host nucleus. While select DDR signals are disabled by the virus to remodel the host response, prevent viral genome silencing and suppress intrinsic antiviral defenses (reviewed in Weller, 2010; Weitzman et al., 2010), others are activated (Tarakanova et al., 2007). Here, we show that the cellular FA pathway is stimulated upon HSV1-infection of normal human cells. This involves monoubiquitination of the essential effector proteins FANCI and FANCD2, and a reduction in levels of the FA antagonist USP1. FANCD2 physically associated with the HSV1 SSB ICP8, accumulated near intranuclear viral pre-replication sites in a FA core complex-dependent manner prior to viral DNA synthesis, and was found within nuclear virus RCs. Significantly, productive viral replication is markedly inhibited in FA-deficient cells and this growth defect can be rescued, in part, by interfering with NHEJ. This establishes that the cellular FA pathway is activated by a human herpesvirus and enlisted to stimulate its lytic replication. Furthermore, it suggests that the FA pathway functions in part to suppress NHEJ during the productive HSV1 growth cycle.
The cellular FA pathway participates in repair of DNA ICLs and maintains genomic stability during DNA replication. Consequently, the FA pathway may be poised to play an important role in the lifecycle of many DNA viruses that replicate in the nucleus. While we establish that the FA pathway is critical for HSV1 replication, other DNA viruses may not be as reliant. For instance, while adenovirus and SV40 reportedly activate the FA pathway, viral replication was at best only modestly reduced when the FA pathway was impaired (Biochuk et al., 2010; Cherubini et al., 2011). In contrast, loss of FANCD2 stimulates papillomavirus genome amplification and productive replication, indicating that the intact FA pathway likely suppresses viral growth (Hoskins et al., 2012; Gulbahce et al., 2012). Thus, the much greater dependence of herpesviruses on the cellular FA pathway for their replication is striking compared to other DNA viruses, representing the first example of a virus that has functionally integrated host FA pathway activation into its productive growth cycle. This may reflect underlying differences in virus genome structure and biology, and key similarities with the types of damage repaired by the FA pathway. Herpesvirus genomes are significantly more GC rich than other DNA viruses and their hosts, rendering them more susceptible to endogenous ICLs (Roizman, 1979).
Despite recruiting ATR to HSV1 replication centers, the viral SSB ICP8 and helicaseprimase restrict recruitment of essential downstream ATR pathway proteins, preventing ATR signaling to RPA and phosphorylation of the ATR substrate Chk1 (Mohni et al., 2010, 2013b). This could potentially promote fork collapse, which might benefit HSV1 (Mohni et al., 2013b). Notably, fork collapse may be capitalized upon to create a critical role for the FA pathway to assist herpesvirus genome replication. The resulting FANCI-D2 monoubiquitination, perhaps triggered by ATR phosphorylation of FANCI or a different mechanism, would promote recruitment and/or formation of specialized nucleoprotein complexes, including endonucleases, translesion synthesis (TLS) polymerases and HR proteins, required to repair damaged viral DNA or resolve problematic replication structures (Ishiai et al., 2008; Kottemann et al., 2013).
A distinctive aspect of HSV replication involves generating longer than unit length concatemers that are processed into genome monomers upon encapsidation (Weller & Coen, 2012). While the incoming viral genome has been proposed to circularize upon infection and concatemer formation initiated by a rolling circle replication mechanism, this idea has been challenged by evidence supporting a role for recombination-dependent replication similar to that observed in bacteriophage λ or T4 (Lo Piano et al., 2011). Rad51, a key host protein required for HR, accumulates in HSV1 RCs consistent with a role for HR in viral replication (Wilkinson & Weller, 2004). In addition, NHEJ-mediated circularization of input HSV1 genomes may be intermediates on the path to stable repression of lytic genes or a state resembling latency in neurons, while linear genomes are in fact templates for lytic gene expression and subsequent productive replication (Jackson & DeLuca, 2003). While latency in neurons and genome repression would be favored by NHEJ-mediated genome circularization, FA activation would counteract NHEJ, antagonize genome circularization, and support productive viral growth. Notably, NHEJ is thought to be the predominate pathway for repairing dsDNA breaks in non-dividing, differentiated cells including mature neurons (Lee & McKinnon, 2007). Thus, the method involved in repairing viral genome damage could conceivably play an important role in the lytic-latent decision fundamental to herpesvirus biology (Jackson & DeLuca, 2003; Lilley et al., 2010).
A key enzyme involved in NHEJ is DNA-PK (Jackson & Bartek, 2009). Consistent with models proposing NHEJ restricts productive HSV1 growth, DNA-PKcs degradation has been reported in a limited number of, but not all, cell lines that support lytic viral replication (Lees-Miller et al., 1996; Parkinson et al, 1999). In addition, DNA-PK-deficient murine cells support greater levels of HSV1 replication (Parkinson et al, 1999; Taylor & Knipe, 2004). In cells where DNA-PK is present and not degraded, recruitment of FA proteins along with other host DSB repair factors to virus RCs may represent a more physiological means to suppress NHEJ and perhaps favor HR. Using FA to inhibit NHEJ via DNA-PK has the added benefit of disabling a DNA sensing innate immune component (Ferguson et al., 2012).
In contrast to FA pathway activation in uninfected cells, the FA effector proteins FANCI-D2 concentrate near sites of HSV1 replication prior to the initiation of DNA synthesis. This likely requires physical interaction with viral replication proteins like ICP8. However, potent FA pathway activation, as shown by Ub-FANCD2, is dependent upon the viral DNA pol, suggesting that HSV1 DNA synthesis is involved. The HSV1-induced reduction in USP1 levels, which possibly involves the inhibition of host protein synthesis in infected cells, may also contribute to the sustained monoubiquitination of FANCI-D2.
While the HSV1 replication defect in FA-deficient cells is partially suppressed by inhibiting NHEJ, precisely how the virus utilizes the FA pathway can now be investigated. Using chromosomally-integrated reporters to distinguish between cellular DSB repair pathways utilized in HSV1-infected cells, single-strand annealing (SSA) activity was found to increase compared to HR, NHEJ, and Alternative-NHEJ (Schumacher et al, 2012). Stimulation of SSA was dependent upon the viral alkaline endonuclease, UL12, emphasizing similarities with recombination-dependent replication in λ phage. Besides interacting with the dsDNA break-sensing complex MRN (Balasubramanian et al., 2010), we found UL12 associated with FANCI in HSV1-infected cells (Fig. 3C, D, Table S1). Intriguingly, FANCD2 (and likely FANCI) monoubiquitination is required for SSA, suggesting an early role for monoubiquitinated FA effector proteins in homologous DSB repair pathway choice (Nakanishi et al., 2005). In the future, the role of SSA in herpesvirus genome replication and how the cellular FA pathway is critical for this process needs to be explored. By exploiting the herpesvirus lifecycle, a powerful new physiological model system can be harnessed to investigate how the FA pathway maintains genomic stability during DNA replication.
EXPERIMENTAL PROCEDURES
Immunoprecipitation
For metabolic labeling, Vero cells (1 × 106) were mock-infected or infected with WT HSV1 KOS or HP66 (MOI=10). At 7 hpi, cells were incubated for 3 h in 3mL of met-free DMEM containing 1 mCi 35S-met/cys mixture (Amersham, NEG072). For mass spectrophotometric analysis, Vero cells seeded into 100 mm plates (2 × 106 cells/dish) and infected as described above. To IP FANCD2, approx.. 9 × 106 cells were used for each condition (mock, WT, HP66), whereas 8 × 107 cells were used for FANCI IPs. At 9 hpi, cells were washed with PBS and lysed in NP-40 lysis buffer (50 mM HEPES-KOH pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40 including Complete Mini protease inhibitor cocktail (Roche) and/or phosSTOP (Roche) phosphatase inhibitor. To digest nucleic acids, Benzonase Nuclease (Novagen) was included in the lysis buffer and samples rocked for 1 h at 4°C. Extracts were clarified by centrifugation at 10,000 × g (10 min, 4°C). After pre-clearing supernatants using normal rabbit serum to remove non-specific binding proteins as described (Walsh & Mohr, 2006), 2–6 μg affinity purified rabbit anti-FANCD2, FANCI, or 4E-BP1 were added for 2.5 h at 4°C. Protein A Sepharose (20–50 μL settled bed volume) was subsequently added, and the incubation continued for 1 h at 4°C. After washing the collected beads 4 times in 0.5 mL NP-40 lysis buffer, bound proteins were eluted in sample buffer, boiled and analyzed by SDS-PAGE using a Nupage 4–12% MES gel (Invitrogen). Labeled proteins were visualized by exposing the fixed, dried gel to X-ray film. Unlabeled proteins were transferred to PVDF and analyzed by immunoblotting.
To prepare samples for multidimensional protein identification Technology (MudPIT) and LTQ Orbitrap mass spectrophotometric analysis, bound proteins were eluted from the beads by boiling in sample buffer lacking bromophenol blue, precipitated with 20% TCA, resuspended in 8 M urea and processed with ProteasMAX (Promega, Madison, WI) per the manufacturer’s instructions. Samples were reduced with 5 mM TCEP (tris(2-carboxyethyl)-phosphine) for 20 min at RT, alkylated in the dark with 10 mM Iodoacetamide for 20 min. and quenched with 25 mM TCEP. Proteins were digested over-night at 37°C with Sequencing Grade Modified Trypsin (Promega, Madison, WI) and the reaction stopped by acidification with formic acid at 5% final.
Supplementary Material
HIGHLIGHTS.
The Fanconi Anemia (FA) genomic stability pathway is activated by HSV1
FA effector proteins are monoubiquitinated and recruited to viral replication centers
Virus replication is impaired in FA-deficient cells and restored by inhibiting DNA-PK
FA pathway is a new cellular factor required for productive herpesvirus replication
Acknowledgments
We thank members of the Mohr lab and A. Wilson for many discussions, Y. Deng at the NYU SOM Microscopy core for assistance, and are most grateful to D. Coen, A. D’Andrea, T. Taniguchi, M. Grompe, S. Weller, and N. DeLuca for generously providing virus strains, FA patient-derived cells, antisera, and shRNA vectors. This work was supported by NIH Grants AI073898 and GM056927 (to I.M.), GM084244 (to T.T.H.), P41 GM103533 (to J.R.Y.), and American Cancer Society grant RSG-12-158-01-DMC (to T.T.H). HK was supported in part by an Academy of Finland grant (123356, 218425), JNS was supported by NIA Fellowship F32 AG039127, and CM was supported in part by NIH training Grant T32 AI007180.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. Preventing non-homologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;39:25–35. doi: 10.1016/j.molcel.2010.06.026. [DOI] [PubMed] [Google Scholar]
- Balasubramanian N, Bai P, Buchek G, Korza G, Weller SK. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J Virol. 2010;84:12504–12514. doi: 10.1128/JVI.01506-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benboudjema L, Mulvey M, Gao Y, Pimplikar SW, Mohr I. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J Virol. 2003;77:9192–9203. doi: 10.1128/JVI.77.17.9192-9203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boichuk S, Hu L, Hein J, Gjoerup OV. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol. 2010;84:007–8020. doi: 10.1128/JVI.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Cherubini G, Naim V, Caruso P, Burla R, Bogliolo M, Cundari E, Benihoud K, Saggio I, Rosselli F. The FANC pathway is activated by adenovirus infection and promotes viral replication-dependent recombination. Nuc Acids Res. 2011;39:5459–5473. doi: 10.1093/nar/gkr084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- DeLuca N, McCarthy AM, Schaffer P. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J Virol. 1985;56:558–570. doi: 10.1128/jvi.56.2.558-570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Murray J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol. 2005;79:5078–5089. doi: 10.1128/JVI.79.8.5078-5089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife. 2012;1:e00047. doi: 10.7554/eLife.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gulbahce N, Yan H, Dricot A, Padi M, Byrdsong D, Franchi R, Lee DS, Rozenblatt-Rosen O, Mar JC, Calderwood MA, et al. Viral perturbations of host networks reflect disease etiology. PLoS Comput Biol. 2012;8(6):e1002531. doi: 10.1371/journal.pcbi.1002531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins EE, Morreale RJ, Werner SP, Higginbotham JM, Laimins LA, Lambert PF, Brown DR, Gillison ML, Nuovo GJ, Witte DP, et al. The fanconi anemia pathway limits human papillomavirus replication. J Virol. 2012;86:8131–8138. doi: 10.1128/JVI.00408-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman RW, Oakes JE, Kudler L. In vitro repair of the preexisting nicks and gaps in herpes simplex virus DNA. Virology. 1977;76:286–294. doi: 10.1016/0042-6822(77)90303-8. [DOI] [PubMed] [Google Scholar]
- Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol. 2008;15:1138–11346. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SA, DeLuca NA. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc Natl Acad Sci USA. 2003;100:7871–7876. doi: 10.1073/pnas.1230643100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob RJ, Roizman B. Anatomy of herpes simplex virus DNA VIII. Properties of the replicating DNA. J Virol. 1977;23:394–411. doi: 10.1128/jvi.23.2.394-411.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19:2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, Smith GC. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett. 2004;14:6083–6087. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- Lee Y, McKinnon PJ. Responding to DNA double strand breaks in the nervous system. Neuroscience. 2007;145:1365–1374. doi: 10.1016/j.neuroscience.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol. 1996;70:7471–7477. doi: 10.1128/jvi.70.11.7471-7477.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Ann Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci USA. 2005;102:5844–5849. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011;7(6):e1002084. doi: 10.1371/journal.ppat.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010;29:943–955. doi: 10.1038/emboj.2009.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Piano A, Martinez-Jimenez MI, Zecchi L, Ayora S. Recombination-dependent concatemeric viral DNA replication. Virus Res. 2011;160:1–14. doi: 10.1016/j.virusres.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Marcy AI, Yager DR, Coen DM. Isolation and characterization of herpes simplex virus mutants containing engineered mutations at the DNA polymerase locus. J Virol. 1990;64:2208–2216. doi: 10.1128/jvi.64.5.2208-2216.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Dee AR, Smith S, Schumacher AJ, Weller SK. Efficient Herpes Simplex Virus 1 Replication Requires Cellular ATR Pathway Proteins. J Virol. 2013;87:531–542. doi: 10.1128/JVI.02504-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Livingston CM, Cortez D, Weller SK. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J Virol. 2010;84:12152–12164. doi: 10.1128/JVI.01643-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Smith S, Dee AR, Schumacher AJ, Weller SK. Herpes simplex virus type 1 single strand DNA binding protein and helicase / primase complex disable cellular ATR signaling. PLoS Path. 2013;9(10):e1003652. doi: 10.1371/journal.ppat.1003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr I, Sonenberg N. Host translation at the nexus of infection and immunity. Cell Host Microbe. 2012;12:470–483. doi: 10.1016/j.chom.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Ann Rev Genet. 2011;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Cavallo F, Perrouault L, Giovannangeli C, Moynahan ME, Barchi M, Brunet E, Jasin M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat Struct Mol Biol. 2011;18:500–503. doi: 10.1038/nsmb.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia mono-ubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci USA. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 2010;329:219–223. doi: 10.1126/science.1192277. [DOI] [PubMed] [Google Scholar]
- Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol. 1999;73:650–657. doi: 10.1128/jvi.73.1.650-657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B. The structure and isomerization of herpes simplex virus genomes. Cell. 1979;16:481–494. doi: 10.1016/0092-8674(79)90023-0. [DOI] [PubMed] [Google Scholar]
- Roizman B, Knipe D, Whitley R. Herpes simplex virus. In: Knipe D, Howley P, Griffin D, Lamb R, Martin M, Straus S, editors. Fields Virology. 5. Philadelphia: Lippincott-Williams & Wilkins; 2006. pp. 2501–2601. [Google Scholar]
- Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat Struct Mol Biol. 2011;18:1432–1434. doi: 10.1038/nsmb.2173. [DOI] [PubMed] [Google Scholar]
- Sarisky RT, Weber PC. Requirement for double strand breaks but not for specific DNA sequences in herpes simplex virus type 1 genome isomerization events. J Virol. 1994;68:34–47. doi: 10.1128/jvi.68.1.34-47.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, Weller SK. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog. 2012;8(8):e1002862. doi: 10.1371/journal.ppat.1002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Do H, Li Y, Chung WH, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, Li L. Recruitment of fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell. 2009;35:716–723. doi: 10.1016/j.molcel.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims AE, Spiteri E, Sims RJ, 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, et al. FANCI is a second mono-ubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002;100:2414–2420. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- Tarakanova VL, Leung-Pineda V, Hwang S, Yang C-W, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HW., 4th γ-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe. 2007;1:275–286. doi: 10.1016/j.chom.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TJ, Knipe DM. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol. 2004;78:5856–5866. doi: 10.1128/JVI.78.11.5856-5866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MS, Gao M, Knipe DM, Powell KL. Association between the herpes simplex virus major DNA-binding protein and alkaline nuclease. J Virol. 1992;66:1152–1161. doi: 10.1128/jvi.66.2.1152-1161.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth S, Hayward GS, Roizman B. Anatomy of herpes simplex virus DNA. V Terminally repetitive sequences. J Virol. 1976;16:503–512. doi: 10.1128/jvi.17.2.503-512.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Mohr I. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 2004;18:660–672. doi: 10.1101/gad.1185304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Mohr I. Assembly of an active translation initiation factor complex by a viral protein. Genes Dev. 2006;20:461–472. doi: 10.1101/gad.1375006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Mohr I. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol. 2011;9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- Weinheimer SP, McKnight SL. Transcriptional and post-transcriptional controls establish the cascade of herpes simplex virus protein synthesis. J Mol Biol. 1987;195:819–833. doi: 10.1016/0022-2836(87)90487-6. [DOI] [PubMed] [Google Scholar]
- Weitzman MD, Lilley CE, Chaurushiya MS. Genomes in conflict: maintaining genome integrity during virus infection. Ann Rev Microbiol. 2010;64:61–81. doi: 10.1146/annurev.micro.112408.134016. [DOI] [PubMed] [Google Scholar]
- Weller SK. Herpes simplex virus reorganizes the cellular DNA repair and protein quality control machinery. PLoS Pathog. 2010;6:e1001105. doi: 10.1371/journal.ppat.1001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller SK, Coen DM. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol. 2012;4(9):a013011. doi: 10.1101/cshperspect.a013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life. 2003;55:451–458. doi: 10.1080/15216540310001612237. [DOI] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol. 2004;78:4783–4796. doi: 10.1128/JVI.78.9.4783-4796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.