

Abstract
Background
Genes that regulate stem cell function are suspected to exert adverse effects on prognosis in malignancy. However, diverse cancer stem cell signatures are difficult for physicians to interpret and apply clinically. To connect the transcriptome and stem cell biology, with potential clinical applications, we propose a novel computational “gene-to-function, snapshot-to-dynamics, and biology-to-clinic” framework to uncover core functional gene-sets signatures. This framework incorporates three function-centric gene-set analysis strategies: a meta-analysis of both microarray and RNA-seq data, novel dynamic network mechanism (DNM) identification, and a personalized prognostic indicator analysis. This work uses complex disease acute myeloid leukemia (AML) as a research platform.
Results
We introduced an adjustable “soft threshold” to a functional gene-set algorithm and found that two different analysis methods identified distinct gene-set signatures from the same samples. We identified a 30-gene cluster that characterizes leukemic stem cell (LSC)-depleted cells and a 25-gene cluster that characterizes LSC-enriched cells in parallel; both mark favorable-prognosis in AML. Genes within each signature significantly share common biological processes and/or molecular functions (empirical p = 6e-5 and 0.03 respectively). The 25-gene signature reflects the abnormal development of stem cells in AML, such as AURKA over-expression. We subsequently determined that the clinical relevance of both signatures is independent of known clinical risk classifications in 214 patients with cytogenetically normal AML. We successfully validated the prognosis of both signatures in two independent cohorts of 91 and 242 patients respectively (log-rank p < 0.0015 and 0.05; empirical p < 0.015 and 0.08).
Conclusion
The proposed algorithms and computational framework will harness systems biology research because they efficiently translate gene-sets (rather than single genes) into biological discoveries about AML and other complex diseases.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-015-0510-7) contains supplementary material, which is available to authorized users.
Keywords: Functional gene-set, Dynamic network biomarker, Relative effect analysis, Leukemic stem cell, AML outcome
Background
Acute myeloid leukemia (AML) has a high mortality rate. Leukemic stem cells (LSC) represent a rare self-renewing cellular subpopulation in each AML. Their chemo-resistant properties are associated with adverse outcomes [1,2]. However, the key events that confer stem cell-like characteristics to LSCs remain unclear. Still, gene expression markers for LSC among studies lack reproducibility, although the characterization of these genes is likely to reveal novel and tractable targets to improve treatment outcomes [3-5].
We hypothesize that one main limitation is that there is no definitive method to isolate LSC from bulk cell samples, and therefore, methods attempting to identify LSC signature are limited by cell heterogeneity. To overcome this problem, we have developed a novel computational systems biology analysis framework. This framework has a four-fold advantage: i) It enables multi-resource data integration and biologically functional interpretation by working on the scale of functional gene-sets; ii) It defines a cluster of functionally interpretable gene-sets shared among LSC populations generated by different labs; iii) It identifies mutual functional relationships from a network of spatiotemporally (disease developmentally) dynamic expression; iv) It provides a personalized leukemic prognostic indicator derived from the identified gene-set clusters. Here, we present the method and demonstrate its application to identify a common LSC signature. The identified LSC-associated biomarkers have a direct link to biological interpretation and clinical application.
First, to facilitate multi-resource data integration and biologically functional interpretation, knowledge-centric analysis (or the analysis of gene-sets) was developed by us and others, e.g., GSEA (Gene Set Enrichment Analysis) [6-8]. The analysis of gene-sets is superior to single-gene analysis in regard to noise and dimension reduction, as well as its desired biological interpretability [9]. However, most gene-set analysis methods impose inherent limits on low cross-dataset comparison or reproducibility from gene-by-sample measurements, as only the genes measured by all collected platforms can be interrogated together. Improved gene-set analyses condense transcriptomic data from gene-by-sample measurements (gene profiles) to gene-set-by-sample measurements (gene-set profiles), which are gene-coverage-difference tolerable and a breakthrough in genome analytics coordinates. Such gene-set-by-sample analyses facilitate the integration and analysis of multiple datasets, platforms, or layers of omics-data, by assigning them into a uniform gene-set scale [10-13]. Some transcription-focused methods, such as GSVA (Gene Set Variation Analysis), condense gene expression values into gene-set scores by evaluating sample-wise enrichments. Yet an open question is the sample-wised statistics before calculating gene-set scores, which limits a mechanistic representation of individual samples. To overcome these deficiencies, we have developed a gene-set-by-sample algorithm, FAIME (Functional Analysis of Individual Microarray (or RNA-seq) Expression) [7,8]. Note that for each sample, FAIME compares the cumulative effects of genes inside a gene-set with the effects of those outside. Additionally, FAIME employs an expression-based weight to rectify biases introduced by low-valued genes [14], and thus quantifies gene-sets primarily according to its highly expressed gene members. However, sensitivity remains a challenge as, at a significance level of false discovery rate (FDR) of 0.05, FAIME could identify hundreds of gene-sets, an impractical number for wet-lab validation. Therefore, we introduce in this study a new weighting parameter into the FAIME algorithm to better control the type-I error, especially for small gene-sets. Additionally, the integration of microarray and RNA-seq data is a new task that we have performed in this study, given the increase in their publication and availability.
Second, dynamic network biomarker (DNB) analysis has been developed on the gene level to address the challenge of temporal and spatial gene expression profiling. It identifies the disease biomarkers leading the whole system from the normal state to a disease state [15,16]. Here, we demonstrate the first dynamic network biomarker analysis on the gene-set level (termed dynamic network mechanism – DNM analysis). These gene-set-based network-relationship dynamics (rather than the static featured single gene-set up- or down-regulations) translate seemingly uninterpretable genomic data into distinct clinical prognoses. In particular, the identified gene-sets define a new biomarker for the characterized stem cell sub-population.
Finally, the goal of cancer treatment is to improve outcomes by earlier diagnosis and targeted therapy for each patient. We have developed the relative expression concept to build a personalized prognostic indicator on the gene level [17,18]. In this study, we expand the concept of relative expression to gene-set clusters and identify a prognostic indicator evaluated by three large cohorts. Meta-analysis is a powerful solution in identifying common LSC signatures shared among different LSC subpopulations with the challenge of small sample size and high cell heterogeneity. In this study, we pool sorted samples from eight labs, including microarray and RNA-seq data, into three groups: HSC+ (verified hematopoietic stem cells enriched, n = 23), LSC+ (LSC enriched, n = 77), and LSC- (LSC depleted, n = 59) cells. Enlarged sample size ensures reliable detection.
Molecular Signature Database (MSigDB) [19] is the most popular gene-set database that defines groups of genes associated with a common function, pathway or other characteristics using biological evidence. Built on MSigDB-defined gene-sets and the above four strategies (Figure 1), we generated gene-set profiles using FAIME (with a weighting parameter) and identified a cluster of three gene-sets with 30 genes representative for LSC-. We also generated gene-set profiles using the GSVA method and identified another cluster of three functional gene-sets with 25 genes in LSC+. The genes within each cluster of gene-sets display simultaneous co-variation with high mutual correlation exclusively within the corresponding cell subpopulations. Using available samples of primary AML patients, we further assessed the clinical relevance by prognosis and the biological relevance by ontology for both gene sets.
Figure 1.
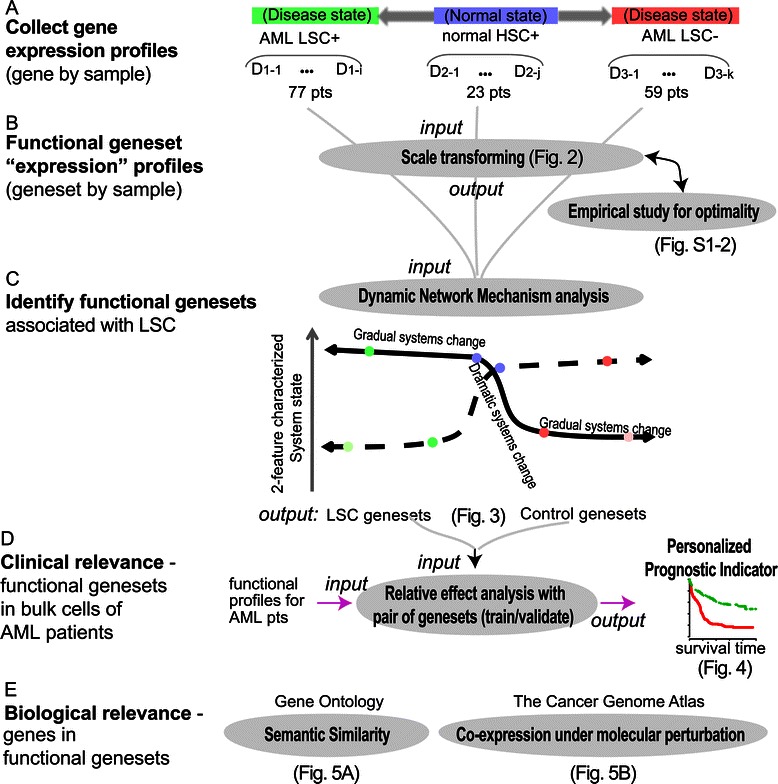
Flowchart of normalization and analysis of pooled molecular function profiles. Panel A) Collection of gene expression profiles. Datasets (D) of Acute Myeloid Leukemia (AML) positive or negative (1 and 2 respectively) for Leukemia Stem Cell (LSC), and of normal Hematopoietic Stem Cells (HSC) (3). Panel B) Calculation of gene-set profiles. Panel C) Identification of functional gene-sets associated with LSCs using the dynamic network mechanism analysis. A cartoon shows that different features characterize the dramatic systems stage changes in different ways. Panel D) Evaluation of clinical relevance in primary AML samples. Panel E) Evaluation of biological relevance using independent data resources.
Results and discussion
Improved functional gene set profiles and the inter-dataset normalization allow cross-dataset comparisons
Some GSEA algorithms (e.g., the Bioconductor package PGSEA) suggest an applicable gene-set (GS) size ranging from 25 to 500 genes. However, half of the canonical pathways have only 10–30 gene members (MSigDB). To control the type-I error when testing on small GSs effectively, we improved the FAIME algorithm with a new adjusted parameter α (hereafter termed as FAIME.5 when α = 5). The larger α is, the higher the weight for the genes at the extreme top of expression ranks is, exponentially (Figure 2B). The parameter α thus acts as a “soft threshold” [20]. Previous studies have shown that both GSVA and FAIME exhibit better or comparable statistical power to that of the GSEA algorithm [7,21]. We compared FAIME.α with GSEA and GSVA in a simulation study. By empirically testing three integers (1,5, and 10) as candidate α values, we chose α = 5. FAIME.5 provided a better control of type-I errors than FAIME.1 but a higher accuracy than FAIME.10 for small GSs with less than 30 genes (Additional file 1: Figure S1, Additional file 2: Text S1: S Results 1.1). Even GSEA, FAIME and GSVA identified distinct GSs in an initial two-group comparison test, FAIME.1 and FAIME.5 shared over half of their identified GSs, and the FAIME shared more identified GSs with GSEA than GSVA (data not shown). Given that FAIME and GSVA have exhibited better or comparable statistical power to that of the GSEA algorithm in certain conditions of previous studies [7,21], we used FAIME.5 and GSVA in the subsequent LSC study. A vignette file with R source codes (Additional file 3: Text S2) shows how to run FAIME.α on a Bioconductor available gene expression dataset, using the MSigDB defined gene-sets of CGP (chemical and genetic perturbations) as a demo.
Figure 2.
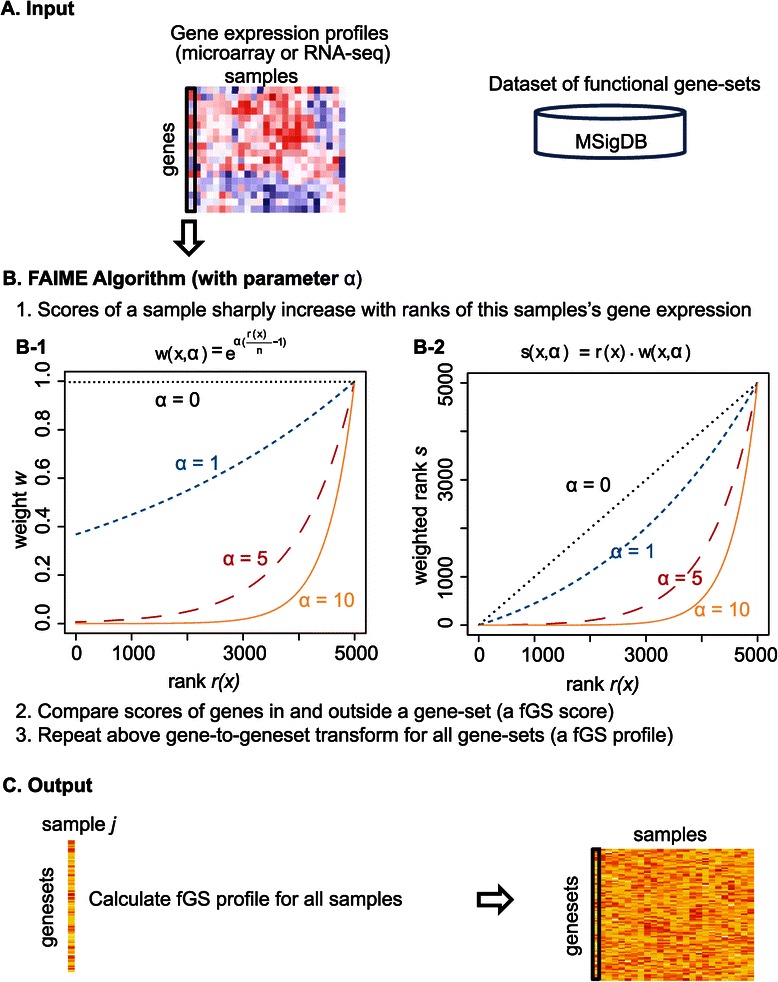
FAIME algorithm with a new parameter α outline. Panel A) The input for the FAIME algorithm is either a gene expression matrix in the form of log2 microarray expression values or RNA-seq counts, and a database of gene-sets. Panel B) Mechanism (or gene-set) score is defined as the difference between the scored expression of genes inside and outside a previously defined gene-set. B-1) Applying an increasingly larger α to the FAIME method. The weight (y-axis) is an exponential function of gene expression ranks (x-axis) adjusted by the parameter α. B-2) Weight-dependent qualitative scores sharply increase with gene rank. The score (y-axis) is the product of gene expression ranks (x-axis) and the rank’s weight adjusted by the parameter α. In each panel, the more highly expressed genes are ranked higher on the x-axis. The dashed line represents the score obtained with no weighting (i.e., ranking only). Panel C) Output of the algorithm as a matrix containing mechanism scores for each gene-set and sample.
The required sample size for functional gene set analysis was also discussed in the simulation study. With at least 20 samples per group, the three methods (FAIME, GSEA, and GSVA) could identify repeatable signatures in the scenarios in which half or more of genes in a GS exhibit strong signal-to-noise ratio (Additional file 1: Figure S1- C,D, dashed lines). Therefore, to ensure statistical power, we collected more than 20 samples per group (of microarray or RNA-seq data (Table 1 and Additional file 4: Table S1) - and performed meta-analysis on the GS level. Meta-analysis is one solution for the limited number of LSC+ sub-populations and their heterogeneity across studies. This study further meets an outstanding need to characterize LSC because, on average, only 16% (2%-28%, Additional file 1: Figure S2A) of reported LSC+ gene signatures were repeatable across five previous studies that attempted to distinguish malignant LSC in AML [1,22-24].
Table 1.
Summary of collected transcriptional and clinical data for AML LSC+, AML LSC-, and normal HSC+ samples
| GAL | CBX21 | GSE24006 | GSE30377 | GSE17054 | ETABM978 | GSM651554 | Group | |
|---|---|---|---|---|---|---|---|---|
| 1st author | Gal | Ishikawa F | Gentles AJ | Eppert K | Majeti R | Goardon N | Hu | |
| Journal | Leukemia | JAMA | JAMA leukemia | Nature Med | PNAS | Cancer Cell | Genome Res | |
| Year | 2006 | 2010 | 2011 | 2011 | 2009 | 2011 | 2011 | |
| PMID | 17039238 | 17952057 | 21177505 | 21873988 | 19218430 | 21251617 | 21795385 | |
| Engraftment verification for LSC+ | N/A | (NOD/SCID/IL)2r gamma (null) (NSG) | N/A | NOD/ShiLtSz-SCID (NODSCID) | N/A | NOD/SCID or NSG | N/A | |
| AML LCS+ (CD34 + CD38- | 8 | 13 | LSC+ (n = 77) | |||||
| AML LCS+ (CD34-CD38-) | 3 | |||||||
| AML LCS+ (CD34-CD38+) | 1 | |||||||
| AML LCS+ (CD34 + CD38+) | 8 | |||||||
| AML LCS+ (Lin-CD34 + CD38-CD90-) | ||||||||
| AML LCS+ (Lin-CD34 + CD38-CD90-CD45RA+) | 22 | |||||||
| GMP-like AML LSC+ (Lin-CD34 + CD38 + CD123+/loCD110-CD45RA + CD45RA+) | 22 | |||||||
| AML leukemia progenitor cell + (LPC+) (hCD34 + hCD38+) | 5 | 8 | 5 | LSC- (n = 59) | ||||
| AML CMP+ (Lin-CD34 + CD38+) | 7 | 1 | 1 | |||||
| AML MPP+ (Lin-CD34 + CD38-CD90-CD45RA-) | 2 | |||||||
| AML Blasts (Lin-CD34-) | 7 | 23 | ||||||
| normal HSC+ (CD34 + CD133+) | 1 | normal HSC+ (n = 23) | ||||||
| normal HSC+ (Lin-CD34 + CD38-) | ||||||||
| normal HSC+ (Lin-CD34 + CD38-) | ||||||||
| normal HSC+ (Lin-CD34 + CD38loCD36-) | ||||||||
| normal HSC+ (Lin-CD34 + CD38-CD90+) | 4 | |||||||
| normal HSC+ (Lin-CD34 + CD38-CD90 + CD45RA-) | 7 | 5 |
To make the samples per group from disparate resources comparable, we applied a straightforward inter-dataset normalization (z-transfer) on mechanism profiles that varied across observed datasets (Additional file 1: Figure S3). For both FAIME and GSVA methods, this inter-dataset normalization reduces the cross-sample variance regardless of gene-set sizes and re-scales gene-set scores to be symmetric and zero-centered (Additional file 1: Figure S3B). Subsequently, the majority of such normalized gene-set scores met the null hypothesis of conventional statistical models designed for gene expression, which is that few genes differ between phenotypes. Therefore, we can employ statistical models designed for gene expression profiles on these gene-set profiles, ensuring direct evaluation of gene-set significance with phenotypes. However, using the conventional hypergeometric test on 1320 canonical pathways each with five or more genes from the MSigDB (v4.0), we observed a large number exhibiting significant scores between two sub-populations (e.g., 210 FAIME-derived pathways meet the criteria of FDR < 0.05 and FC > 2, Bioconductor Limma package), suggesting the need for a proper model to concentrate on core pathways.
Dynamic network mechanism (DNM) analysis defines new LSC-representative signatures
To explore critical disease developmental signatures using the gene-set profiles, we developed a dynamic network mechanism (DNM) analysis. The hypothesis is that the deteriorations of patient’s condition in complex diseases such as AML are abrupt during the progression of disease and may be caused by a critical transition at a tipping point [15]. DNM is built on a method, called dynamical network biomarker (DNB), previously designed to capture early-warning signals before a critical transition from normal state to disease state [15,16]. The DNM analysis identified gene-sets are hereafter referred to as DNM gene-sets. Given gene-set profiles of populations with different disease states, the DNM analysis searches for the critical sub-network that exhibits a low variability and high dependence intra-population, but high variability and low dependence inter-population. Specifically, we evaluated the network nodes (the gene-sets) by the standard deviations for variability and pairwise Pearson correlation coefficients for dependence among three different cell sub-populations: HSC+, LSC+, and LSC- cells. We thereafter identified the critical transitional sub-population and, subsequently, the best cluster of gene-sets to distinguish this sub-population from the other two using a combined score of the variability and dependence of gene-sets. As a result, a new 30-gene signature best distinguishes the AML LSC- sub-population based on FAIME.5 profiles, and a new 25-gene signature best distinguishes the AML LSC+ population based on GSVA profiles (Table 2). Note that the DNM identified critical transitional sub-population from FAIME gene-set profiles or GSVA gene-set profiles is different, suggesting cell diversity. The number of involved genes from identified gene-sets is a practical number for wet-lab validation. Each signature is a cluster of 3 or 4 gene-sets showing simultaneous co-variation with mutual correlation exclusively within the corresponding cell subpopulations.
Table 2.
Identified 30-gene and 25-gene signatures
| Symbol | Entrez gene name | Type(s)^ | Biomarker#^ | Symbol | Entrez gene name | Type(s)^ | Biomarker#^ |
|---|---|---|---|---|---|---|---|
| LSC- 30 genes | LSC+ 25 genes | ||||||
| ANLN | anillin, actin binding protein | other | APPBP2 | amyloid beta precursor protein (cytoplasmic tail) binding protein 2 | other | ||
| AURKA | aurora kinase A | kinase | E | ATXN3 | ataxin 3 | peptidase | |
| CCNA1 | cyclin A1 | other | CCND2 | cyclin D2 | other | D, E | |
| CCL5 | chemokine (C-C motif) ligand 5 | cytokine | D, E, U | DYNLL2 | dynein, light chain, LC8-type 2 | other | |
| CD38 | CD38 molecule | enzyme | E, P, U | ERC1 | ELKS/RAB6-interacting/CAST family member 1 | other | |
| CDC25B | cell division cycle 25B | phosphatase | ETV6 | ets variant 6 | transcription regulator | ||
| CDK1 | cyclin-dependent kinase 1 | kinase | GGNBP2 | gametogenetin binding protein 2 | other | ||
| CENPA | centromere protein A | other | GIMAP6 | GTPase, IMAP family member 6 | other | ||
| CLC | Charcot-Leyden crystal galectin | enzyme | KIF1B | kinesin family member 1B | transporter | ||
| CPA3 | carboxypeptidase A3 (mast cell) | peptidase | KIF1C | kinesin family member 1C | other | ||
| CSTA | cystatin A (stefin A) | other | KRAS | Kirsten rat sarcoma viral oncogene homolog | enzyme | D, E, P, R, U | |
| DDX53 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 53 | other | LOC728392 | uncharacterized LOC728392 | other | ||
| DLGAP5 | discs, large (Drosophila) homologassociated protein 5 | phosphatase | MAF | v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog | transcription regulator | ||
| HGF | hepatocyte growth factor (hepapoietin A; scatter factor) | growth factor | D, DP, E, P, U | MPO | myeloperoxidase | enzyme | D, E, U |
| IL36B | interleukin 36, beta | cytokine | MTERFD2 | MTERF domain containing 2 | other | ||
| KIAA0101 | KIAA0101 | other | NAV1 | neuron navigator 1 | enzyme | ||
| LATS2 | large tumor suppressor kinase 2 | kinase | PIAS1 | protein inhibitor of activated STAT, 1 | transcription regulator | U | |
| MBD3 | methyl-CpG binding domain protein 3 | other | PSMB6 | proteasome (prosome, macropain) subunit, beta type, 6 | peptidase | ||
| MND1 | meiotic nuclear divisions 1 homolog (S. cerevisiae) | other | SESN1 | sestrin 1 | other | ||
| MPO | myeloperoxidase | enzyme | D, E, U | SLC30A7 | solute carrier family 30 (zinc transporter), member 7 | transporter | |
| MS4A3 | membrane-spanning 4-domains, subfamily A, member 3 (hematopoietic cell-specific) | other | STK38 | serine/threonine kinase 38 | kinase | ||
| NDC80 | NDC80 kinetochore complex component | other | TMIE | transmembrane inner ear | other | ||
| OLFM4 | olfactomedin 4 | other | YARS2 | tyrosyl-tRNA synthetase 2, mitochondrial | enzyme | ||
| RNASE2 | ribonuclease, RNase A family, 2 (liver, eosinophil-derived neurotoxin) | enzyme | D | ZBTB10 | zinc finger and BTB domain containing 10 | other | |
| RNASE3 | ribonuclease, RNase A family, 3 | enzyme | E | ZNF384 | zinc finger protein 384 | transcription regulator | |
| SKA3 | spindle and kinetochore associated complex subunit 3 | other | |||||
| SPC25 | SPC25, NDC80 kinetochore complex component | other | |||||
| STAR | steroidogenic acute regulatory protein | transporter | |||||
| TOP2A | topoisomerase (DNA) II alpha 170 kDa | enzyme | D, E, P, RT | ||||
| ZWINT | ZW10 interacting kinetochore protein | other | |||||
#: D = diagnosis; DP = disease progression; P = prognosis; E = efficacy; RT = response to therapy; U = unspecified application.
^: Data resource: 2000–2014 Ingenuity Systems, Inc.
LSC- 30-gene signature
Based on the FAIME.5 profiles, DNM identified 3 out of 3403 gene-sets representing expression signatures of chemical and genetic perturbations (CGPs) that distinguish LSC- cells from the other two cell subpopulations. In the LSC- population, genes in those three gene-sets exhibited different mutual correlations but lost many of their original correlated partners from the normal HSC+ population (Figure 3A), defining a new 30 member gene-set to represent LSC- exclusively.
Figure 3.
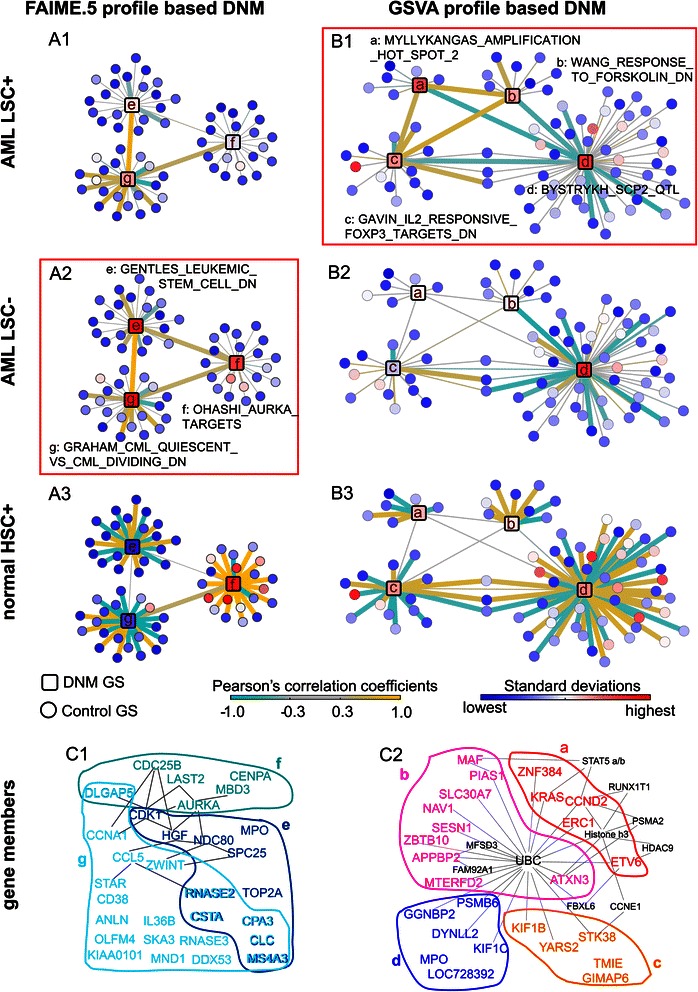
Dynamic Network Mechanism (DNM) analysis on functional gene-sets (gene-sets). Panel A) Network resulting from FAIME.5 profiles; Panel B) Network resulting from GSVA profiles. Each panel visually illustrates dynamics of the identified DNM gene-sets in each of the three sorted cell groups (1: AML LSC +, 2: AML LSC-, and 3: normal HSC+). Node color codes the standard deviation of a gene-set in the corresponding sample group, while line color codes the Pearson’s correlation coefficients between any two gene-sets. DNM gene-sets are represented as labeled squares and control gene-sets are represented as circles. The identified critical sample group for each analytical method is boxed in red (A1, B2). Line weight increases with correlation (>0.5 in Panel A and >0.4 in Panel B). Panel C) The colored groups of gene-members (a, b, c, d) or (e, f, g) corresponding to the above red-boxed DNM gene-sets respectively. The grey lines represent their pair-wise associations according to the Ingenuity knowledge database. Additional genes (black) that interacted with two or more identified genes in the Ingenuity database are also displayed.
Six (DLGAP5, RNASE2, CSTA, CPA3, CLC, MS4A3) out of the 30 genes shared significant overlap across the three gene-sets (p = 3.6e-15, Equation 7), indicating a functional cross-talk among the pre-defined gene-sets. Subsequent Ingenuity® connection analysis suggested that CDK1 and AURKA play the role of ‘hub’ in this 30-genes network. For example, the 30 genes included six candidate substrate proteins (MBD3, CDC25B, LATS2, DLGAP5, AURKA, CENPA) of AURKA [25] (Figure 3A, node f). AURKA is a mitotic kinase over-expressed in AML CD34 (+) /CD38 (−) cells relative to their CD34 (+)/CD38 (+) counterparts or CD34 (+) normal HSCs [26]. Given the clinical impact of AURKA [26,27], the 30-gene signature reveals promising molecular targets to eliminate chemotherapy-resistance in LSC.
Also identified were two known gene-sets derived from previous studies about LSC stemness (Additional file 4: Table S2). One gene-set represents 11 genes (SPC25, CPA3, NDC80, HGF, CSTA, CDK1, MS4A3, MPO, RNASE2, CLC, TOP2A) down-regulated in the quiescent CD34+ cells when compared to dividing CD34+ cells isolated from the peripheral blood of myeloid leukemia patients [28]. The other gene-set contains 19 LSC down-regulated genes (e.g., CD38, ZWINT, CCNA1) when compared to leukemia progenitor cells from AML tumor samples [22].
LSC+ 25-gene signature
The GSVA profiles identified four correlated gene-sets, consisting of 30 unique genes, that exclusively represent LSC+ cells (Figure 3B, Additional file 4: Table S3).
One DNM gene-set, stem cell proliferation-2 (Scp2) [29], included six genes (DYNLL2, GGNBP2, KIF1C, MPO, PSMB6, and LOC728392) that were physically mapped to the HSC proliferation quantitative trait locus (QTL) on chromosome 11 in mouse models (Figure 3B, node d). Scp2 is known to modulate the percentage of cells in S phase, and deletions of its corresponding region in human (maps to 5q31.1) have been associated with myelodysplastic syndrome and AML [29-31]. This gene-set is negatively correlated with the other three DNM gene-sets to represent LSC+ exclusively.
Another identified DNM gene-set contains five cancer genes (CCND2, ERC1, KRAS, ZNF384, ETV6) in the 12p13-p11.1 region with co-localized fragile sites (Figure 3B, node a). These amplified genes represent attractive targets for therapy, diagnostics, and prognostics [32]. This fact and the observation that 11 of these 25 genes are also significantly involved in cell death (ATXN3, CCND2, ERC1, ETV6, KIF1B, KIF1C, KRAS, MAF, MPO, PIAS1, STK38, p = 0.0068, Ingenuity pathway analysis - IPA, Ingenuity® Systems, www.ingenuity.com) indicate a clinical impact for prognosis.
Note that the DNM analysis on the gene-set level is designed to identify groups of genes with related functions and correlated expressions, rather than clustering samples on the gene-level. The expression changes of an identified gene might be subtle in one dataset or uncovered by its array. In fact, the joint gene-level expression pattern of the DNM identified gene-sets exhibits a correlation to data resources stronger than to cell types (Additional file 1: Figure S4, Additional file 5: Text S3). Regardless, the two identified gene clusters roughly clustered samples into two groups: LSC- samples or LSC+ samples. HSC+ samples are grouped together in each dataset. These results support the feasibility of using DNM analysis of an individual’s transcriptomic changes on a gene-set level to reveal functional biomarkers and biological underpinnings.
Clinical relevance - prognosis of DNM gene-sets in patients with primary AML
To test the clinical relevance of the dynamic network mechanism analysis identified LSC representative gene-sets, we first investigated the gene-set profiles derived from patients with all types of AML in three independent cohorts (GSE14468, n = 518 [33]; TCGA, n = 197 [34]; GSE12417, n = 242 [35]). Across the three cohorts, increased scores of the identified gene-sets are associated with favorable overall survival, as log-regression coefficients of both clusters of DNM gene-sets are averagely negative (−0.16 and −0.02 respectively). We therefore hypothesized that the patients’ survival can be stratified by the identified DNM gene-sets and their selected control gene-sets that stand for tumor suppressor effects and tumorigenesis effects respectively. The controls are gene-sets that are significantly correlated with DNM gene-sets in normal HSC+ but not in LSCs cells, the circled nodes in Figure 3A-B.
To validate that hypothesis, we designed a novel Relative Effect Analysis with Gene-set Pairs (RXA-GSP) model, built from a parameter-free model that we have designed for personalized prognosis [17]. RXA-GSP calculates a prognostic indicator comparing scores of the identified gene-sets with scores of selected control gene-sets (Equation 8). For each identified gene-set-cluster and using the training cohort (GSE14468 [33]), we selected from all the control gene-sets a prognostic subset (p < 0.01, cox-regression coefficient > 0, Additional file 4: Tables S2, and S3). We then validated the indicator in two independent validation cohorts (TCGA [34]; GSE12417 [35]). Interestingly, a negative indicator significantly predicted a shorter survival in both training and the two validation cohorts. Additional file 1: Figure S5A shows the Kaplan-Meier plots of the indicator built from the gene-sets derived from FAIME.5 profiles (log-rank p = 0.00043, 4.7e-6, and 0.00032 respectively; empirical p = 0.007, 0.0045, and 0.007 by permuting random gene-sets with the same number of genes respectively). Note that the empirical p-value is more severe than the log-rank tested p-value, as random signatures might also predict cancer outcome [36]. This LSC-representative indicator remained independent of all known prognostic indicators including cytogenetic risk groups or European LeukemiaNet (ELN) risk groups, using multivariate analyses on overall survival of primary AML patients in all three cohorts (Table 3).
Table 3.
Univariate and multivariate analyses of overall survival in patients with all types of AML, for the LSC- DNM gene-sets
| Dataset | Variate model | Variates | HR | 95% CI | p-value |
|---|---|---|---|---|---|
| GSE14468 (n = 518) | Univariate model | 3 DNM fGSs vs 4 control fGSs | 0.7 | 0.55-0.84 | 0.00043*** |
| ELN_RiskFavorable vs. Adverse | 0.3 | 0.20-0.40 | 2.15E-13*** | ||
| complex vs. others | 2.2 | 1.50-3.16 | 0.000024*** | ||
| 7q vs. others | 2.1 | 1.45-2.97 | 0.000051*** | ||
| ELN_RiskIntermediate-II vs. Adverse | 0.5 | 0.36-0.70 | 0.000053*** | ||
| Age group, years (≥60 vs. <60) | 1.7 | 1.29-2.28 | 0.00016*** | ||
| 3q vs. others | 2.1 | 1.32-3.42 | 0.0015** | ||
| ELN_RiskIntermediate-I vs. Adverse | 0.6 | 0.46-0.86 | 0.0035** | ||
| inv16 vs. others | 0.5 | 0.32-0.83 | 0.0057** | ||
| cebpa mutation vs. others | 0.6 | 0.67-0.95 | 0.028* | ||
| Multivariate model | 3 DNM fGSs vs 4 control fGSs | 0.7 | 0.60-0.91 | 0.0050** | |
| Age group, years (≥60 vs. <60) | 1.7 | 1.26-2.29 | 0.00046*** | ||
| ELN_RiskFavorable vs. Adverse | 0.4 | 0.26-0.76 | 0.0034** | ||
| complex vs. others | 1.6 | 0.96-2.58 | 0.07. | ||
| ELN_RiskIntermediate-II vs. Adverse | 0.7 | 0.43-1.23 | 0.23 | ||
| 7q vs. others | 1.3 | 0.82-2.09 | 0.26 | ||
| 3q vs. others | 1.3 | 0.71-2.27 | 0.42 | ||
| inv16 vs. others | 0.8 | 0.47-1.39 | 0.44 | ||
| cebpa mutation vs. others | 0.9 | 0.53-1.45 | 0.61 | ||
| ELN_RiskIntermediate-I vs. Adver | 0.9 | 0.53-1.48 | 0.65 | ||
| TCGA (n = 197) | Univariate model | 3 DNM fGSs vs 4 control fGSs | 0.46 | 0.32-0.64 | 4.66E-06*** |
| Age group, years (≥60 vs. <60) | 3.02 | 2.16-4.21 | 9.94E-12*** | ||
| gender | 0.88 | 0.64-1.22 | 0.44 | ||
| normal_karyotype vs. others | 1.12 | 0.81-1.55 | 0.50 | ||
| BM Blast(>50 vs. <=50) | 0.88 | 0.60-1.30 | 0.53 | ||
| Multivariate mode | 3 DNM fGSs vs 4 control fGSs | 0.60 | 0.42-0.86 | 0.0058** | |
| Age group, years (≥60 vs. <60) | 2.59 | 1.83-3.67 | 8.8E-08*** | ||
| GSE12417 (n = 242) | Univariate model | 3 DNM fGSs vs 4 control fGSs | 0.60 | 0.43-0.83 | 0.0021** |
| Age group, years (≥60 vs. <60) | 1.63 | 1.18-2.26 | 0.0029** | ||
| Multivariate model | 3 DNM fGSs vs 4 control fGSs | 0.60 | 0.43-0.83 | 0.0021** | |
| Age group, years (≥60 vs. <60) | 1.49 | 1.07-2.07 | 0.018* |
Significance code: ‘.’:p < .1; ‘*’: p < .05; ‘**’p < .01; ‘***’p < .001.
Significant univariate tested factors (p < .05) are used for multivariate test. Boldface highlights the results of DMN fGSs.
Next, we investigated the clinical relevance in patients with cytogenetically normal AML. A subclass of intermediate-risk AML, cytogenetically normal AML has a variety of outcomes: some affected individuals respond well to standard treatment while others may require more intensive therapy. We set the largest cytogenetically normal AML cohort for training (GSE12417, n = 242 cytogenetically normal AML [35]), and the other two datasets as validation (GSE14468, n = 214 cytogenetically normal AML [33]; TCGA, n = 91 cytogenetically normal AML [34]). For the LSC- gene-sets, significance of three out of four of the control gene-sets was repeatable, based on the FAIME.5 profiles (Additional file 4: Table S2). The observed prognosis (log-rank p = 0.0014, 0.0012, and 0.00082 respectively; empirical p = 0.014, 0.0045, and 0.013, respectively, Figure 4) remains significant in multivariate analysis, independent of age, KRAS mutation, and ELN risk classification (Table 4).
Figure 4.
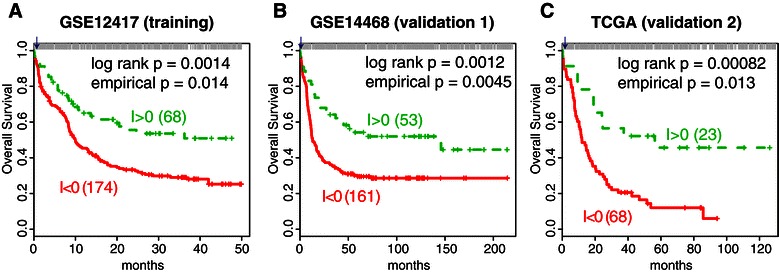
Prognostic analysis of patients with cytogenetically normal AML, based on DNM identified gene-set pairs. Kaplan–Meier plots of survival analysis on stratified samples with better outcome (green) or worse outcome (red). The Relative Effect Analysis with Gene-Set-Group Pairs (RXA-GSP) calculated a prognostic indicator by comparing three LSC- representative gene-sets (30 genes) with three normal control gene-sets (166 genes, Additional file 4: Table S2). The normalized FAIME.5 profiles are used. In each sub-panel, top bars mark the simulated p-values from which we estimated the empirical p-value for the actually observed log-rank p-value, the vertical line marked with an arrow. A RXA-GSP indicator I of less than 1 significantly indicates worse prognosis in the training cohort (GSE12417, Panel A) of cytogenetically normal AML patients and in two validation cohorts (GSE14468 and TCGA, Panels B and C) of cytogenetically normal AML patients.
Table 4.
Univariate and multivariate analyses of overall survival in patients with cytogenetically normal AML, for the LSC- DNM gene-sets
| Dataset | Variate model | Variates | HR | 95% CI | p-value |
|---|---|---|---|---|---|
| GSE12417 (n = 242) | Univariate model | 3 DNM fGSs vs. 4 control fGSs | 0.53 | 0.35-0.79 | 0.0014** |
| Age group, years (≥60 vs. <60) | 1.63 | 1.18-2.26 | 0.0029** | ||
| Multivariate model | 3 DNM fGSs vs. 4 control fGSs | 0.6 | 0.37-0.83 | 0.0038** | |
| Age group, years (≥60 vs. <60) | 1.6 | 1.12-2.15 | 0.0083** | ||
| GSE14468 (n = 214) | Univariate model | 3 DNM fGSs v.s 4 control fGSs | 0.5 | 0.33-0.77 | 0.0012** |
| KRAS mutaion vs. others | 70.2 | 7.30-674.5 | 8.60E-13*** | ||
| ELN_risk (IntermediateI vs. Favorable) | 1.8 | 1.27-2.62 | 0.00095*** | ||
| Age group, years (≥60 vs. <60) | 1.4 | 0.89-2.18 | 0.15 | ||
| NPM1 mutation vs. others | 0.8 | 0.57-1.12 | 0.19 | ||
| CEBPA mutation vs. others | 0.7 | 0.42-1.22 | 0.22 | ||
| Gender | 0.9 | 0.62-1.21 | 0.39 | ||
| BM Blast (>50 vs. <=50) | 1.1 | 0.76-1.53 | 0.68 | ||
| NRAS mutation vs. others | 1.0 | 0.60-1.81 | 0.90 | ||
| Multivariate model | 3 DNM fGSs v.s 4 control fGSs | 0.5 | 0.35-0.83 | 0.0047** | |
| KRAS mutaion vs. others | 90.7 | 9.27-888.79 | 0.00011*** | ||
| ELN_risk (IntermediateI vs. Favorable) | 1.7 | 1.19-2.48 | 0.0039** | ||
| TCGA (n = 91) | Univariate model | 3 DNM fGSs vs. 4 control fGSs | 0.4 | 0.19-0.67 | 8.23E-04*** |
| Age group, years (≥60 vs. <60) | 2.5 | 1.58-4.09 | 6.53E-05*** | ||
| BM Blast (>50 vs. <=50) | 0.6 | 0.33-1.09 | 0.09. | ||
| Gender | 0.7 | 0.46-1.16 | 0.18 | ||
| Multivariate model | 3 DNM fGSs vs. 4 control fGSs | 0.4 | 0.23-0.84 | 0.012* | |
| Age group, years (≥60 vs. <60) | 2.1 | 1.32-3.47 | 0.0022** |
Significance code: ‘.’:p < .1; ‘*’: p < .05; ‘**’p < .01; ‘***’p < .001.
Significant univariate tested factors (p < .05) are used for multivariate test. Boldface highlights the results of DMN fGSs.
Finally, we tested the relative effect analysis with gene-set pairs on the four gene-sets that were derived from the GSVA profiles to represent LSC+ cells. We identified a significant prognostic indicator by training the control gene-sets using all AML patients (Additional file 1: Figure S5B, log-rank p = 5.4e-5, 2.5e-5, and 0.05 respectively; empirical p = 0.002, 0.0005, and 0.15 respectively). The indicator is independent of all known clinic risk groups with an exception that it is dependent on patient ages bipartitely at 60 years old in the TCGA cohort (Additional file 4: Table S5). Strikingly, this cytogenetically normal AML indicator showed significance in two larger-sized validation datasets (Additional file 1: Figure S5C, log-rank p = 1.3e-10, p = 0.0002, and 0.05 respectively; empirical p < 0.001, 0.001, and 0.08 respectively).
Additionally, as a side-by-side comparison with gene-level signature, we tested the prognostic power of the “LSC signature”, the weighted sum of 31 genes defined by Gentles et al. [22]. In patients from GSE14468 and GSE12417, high LSC scores were associated with worse overall and event-free survival among patients with either normal karyotypes or chromosomal abnormalities (log-rank p < 0.002, Additional file 1: Figure S6 A1-A2, B), which is in agreement with previous publications. However, the patients in the TCGA cohort exhibit an exception. Their survival could not be predicted using the LSC score (Additional file 1: Figure S6 A3, C). One possible explanation would be, at least partly, the over-representation of mutations in the TCGA cohort (99% compared to the expected 75% in AML patients [37]). In contrast, our geneset-level indicators showed unified prognoses in all three tested cohorts.
Biological relevance – Gene Ontology similarity, AML association, and correlation to RAS molecular activity
Gene Ontology (GO) provides curator-reviewed, standardized annotations for protein functions with a structured vocabulary. To evaluate co-functions of genes in the identified gene-set cluster, we employed semantic similarity of GO, a reliable computational method to exploit and classify coding gene functions (Additional file 2: Text S1: S Methods 2.4). For each DNM gene-set, two-thirds of the identified genes share molecular functional similarity or biological process similarity (semantic similarity score = 1, Figure 5A), explicating their shared dynamic phenotype in sorted cell populations and their consistent prognosis in primary AML samples. The 30 LSC- representative genes are intensively connected by 75 paired molecular functional similarity (empirical p = 6e-5, Figure 5A1), and the 25 LSC+ representative genes are intensively connected by 17 paired biological process similarity (empirical p = 0.03, Figure 5A2).
Figure 5.
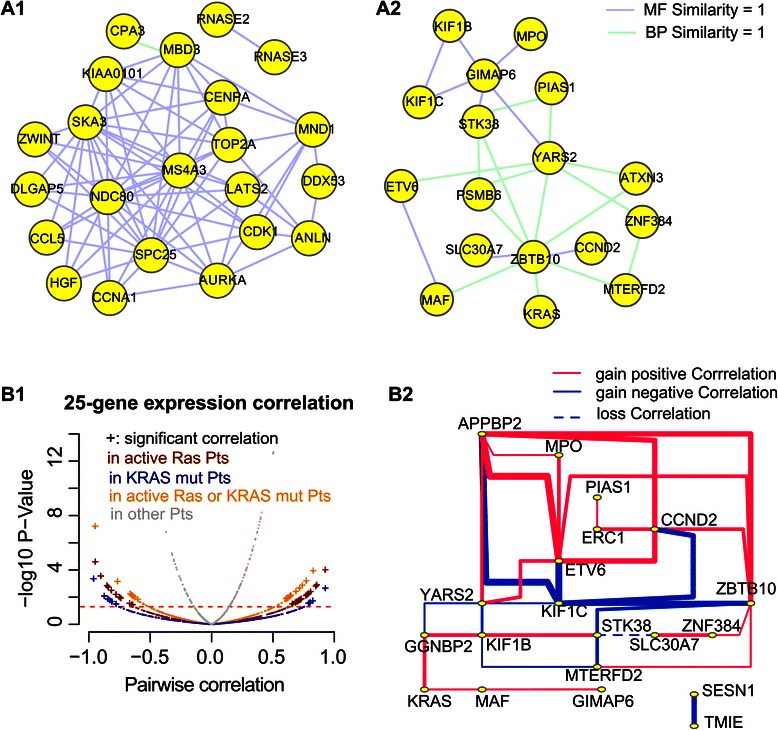
The computationally evaluated biological relevance of genes in the identified gene-sets. Panel A) Genes in the identified cluster of gene-sets share common functions. Sub-panel 1 represents the 30 LSC- representative genes and sub-panel 2 represents 25 LSC+ representative genes. Gene Ontology semantic similarity analysis reveals that 2/3 of the genes share the same function (the Lin distance =1). Line color codes the molecular function and biological process respectively. Panel B) Volcano plot (subpanel 1) of pairwise correlation tests between any two LSC+ 25-gene members (TCAG data). There are 31 significant co-expressions across 10 AML patients who show positive RAS activity (dark red). This co-expression disappears among the other 187 patients (grey), the 7 patients who carry somatic mutations of KRAS genes (blue), and the 16 patients carrying positive RAS activity or somatic RAS mutation (orange). Subpanel 2 illustrates the RAS activity-dependent co-expressed gene-pairs in a network. Comparing patients showing active molecular activity of RAS with patients showing normal RAS status, 31 pairs of genes gain significant co-expression, including 20 out of the 25 LSC+ representative genes (solid lines), whereas 1 gene-pair loses its co-expression in normal RAS status (the dashed line). Red lines represent positive correlations and blue lines represent negative correlations. Line widths correspond to Spearman correlation coefficients.
To further mine the biological and disease relevance of these gene-sets, we performed the Ingenuity pathway analysis (IPA) on the 30- and 25-gene sets respectively. Known cancer-related genes are over-represented in both: the LSC- 30-gene signature contains 22 cancer-associated genes (p = 0.0056), including three AML-associated genes (AURKA, MPO, TOP2A, p = 0.0048); the LSC+ 25-gene signature contains 10 cancer-associated genes, including another three AML-associated genes (ETV6, KRAS, MPO, p = 0.002). To interrogate novel AML-association in each signature, we re-ran IPA on the remaining LSC- representative (27-gene) subset and the remaining LSC+ representative (22-gene) subset excluding known AML genes. Interestingly, hematologic cancer genes (CCL5, CD38, CDK1, HGF, IL36B, NDC80, p = 0.0032) and genes linked with cell death of cancer cells (CD38, CDC25B, CDK1, p = 0.012) were overrepresented in the LSC- representative subset. Also, cancer cell transformation genes (CCND2, MAF, ZNF384, p = 0.018) and interphase of tumor cell lines (CCND2, PIAS1, STK38, ZBTB10, p = 0.000604) were enriched in the LSC+ representative subset. This result suggests a novel mechanism for AML tumorigenesis represented by these 30-gene and 25-gene signatures.
Finally, using available AML samples of the TCGA cohort [34], we investigated the relationship between the LSC+ 25-gene expression and the abnormal protein activity status of one gene member, RAS. The 25 genes gained co-expression (absolute Spearman coefficient >0.6 and p < 0.05) significantly among patients showing active RAS molecular activity (n = 10, empirical p = 0.048) but not among those carrying KRAS somatic mutation (n = 7, Additional file 4: Table S6) nor among those with normal RAS activity status (n = 187, Figure 5B).
We conclude that the LSC- signature of 30-gene or the LSC+ signature of 25-gene significantly share biological processes and molecular functions. Although mutations of RAS genes usually cause an intrinsic activation of RAS pathway in AML, it was RAS molecular activity, not genetic mutation, that perturbed the LSC+ signature of 25-gene from that of the control. The literature suggests in parallel that it was RAS molecular activity rather than its somatic mutations that exhibited a prognostic quality [38].
Conclusion
Computational strategies
Diverse signatures derived from the analysis of LSC gene expression profiles at the gene level confirm the heterogeneity of AML [5]. However, analyzing functional gene-sets can reveal common networks that are important for regulating LSC functions [39]. By meta-analysis and inter-dataset normalization, we have improved the reproducibility of characterizing clinically relevant LSC-signatures on the gene-set level. The other advantage of gene-set based algorithms is their ability to build functional profiles, facilitating computational identification and subsequent biological interpretation [7,21]. Building on gene-set-by-sample profiles, we successfully integrated microarray and RNA-seq data and performed two novel gene-set-analysis methods to reveal critical gene-sets for disease diagnosis and prognosis.
A precise gene-set-by-sample profile is a necessary prerequisite for functional class scoring approaches (reviewed by Khatri et al. [40]). It is the pan-genome weighting approach that more heavily weights highly-expressed genes and thus distinguishes FAIME.α from other gene-set analysis methods. Specifically, it biases of not only extremely high-valued genes by ranking but also the noise of low-valued genes by weighting, per sample. GSVA, on the other hand, heavily weights the two-tails of statistics and is sample-wise dependent [21]. FAIME.5 is an improved gene-set algorithm characterized by individualized, non-parametric, and un-supervised statistic (i.e., without the use of sample-wise estimation of differential expression).
We introduced novel dynamic network biomarker analysis on the gene-set level (DNM analysis) to represent LSC sub-populations. The identified gene-sets define new biomarkers for the regulation of stem cell function in AML, by characterizing dynamic features rather than the static differences. DNM has the ability to discovery significant regulatory changes of functional gene-sets across disease progression stages. By taking into account of a critical transition from normal state to disease state during cell development, DNM outperforms other gene-set analytic strategies in capturing critical signatures. The proposed RXA-GSP (Relative Effect Analysis with Functional gene-set-Group Pairs algorithm) is a parameter-free model with respect to gene-sets and is designed to bridge cancer biology from the lab to the clinic [17,18]. It can be extended to other applications when analyzing biological imbalance within a patient. RXA-GSP naturally fits the “two-hit hypothesis” for malignancy: one leads to uncontrolled cellular proliferation and evasion of apoptosis and the other adjusts inhibition of differentiation [41]. It is particularly useful for efficiently translating microarray or RNA-seq data to clinical discoveries.
Altogether, researchers can apply these proposed computational strategies to study other diseases in a systematic “gene-to-function, snapshot-to-dynamics, and biology-to-clinic” framework. Previously, we have successfully demonstrated the application of gene-to-function (FAIME) on head and neck cancer, snapshot-to-dynamics (DNB) on diabetes, and biology-to-clinic (RXA-GSP) on breast cancer [7,16,17]. Increasing evidence suggests the feasibility of analyzing an individual’s transcriptome on a pathway-level for clinical decision-making and precise mechanism comprehension. Additionally, different methods perform differently under different conditions or parameter settings in their ability to detect the complex abnormality in cancer. Researchers need to make ad hoc modifications and select among analysis algorithms to make biological discoveries.
Biological discoveries
This work sheds promising insight into gene-set profiles by defining the LSC- signature of 30-gene and the LSC+ signature of 25-gene for prognosis in AML. These signatures suggest potential therapeutic regimens to eradicate quiescent, chemotherapy-resistant LSCs, because they exhibit two key characterizes - independence of cell cycle status and not substrates for drug efflux pump proteins [39]. The LSC- signature of 30-gene exposed a promising molecular target, AURKA [26,27], to potentially eliminate chemotherapy-resistance in LSC. The LSC+ signature of 25-gene included known cancer genes in the 12p13-p11.1 region [32] as well as genes in the 5q31.1 region [29] that are known to be associated with myelodysplastic syndrome and AML. The only common gene from both signatures, myeloperoxidase (MPO), is implicated as a biomarker for favorable prognosis in AML and its transcription levels reflect epigenetic modification [42]. This is not surprising because MPO is known to be expressed by the myeloid lineage in AML but not in HSC [43]. Given the complex system changes in tumor progression, we expect that new gene-set clusters could be prioritized using alternative GSEA methods.
The two identified clusters of gene-sets significantly predicted overall survival for 1478 primary AML patients, regardless of the inter-patient and intra-patient variability of AML phenotypes. Their prognostic independence is consistent with the published LSC-derived prognostic signature. However, previously identified genes are associated with adverse prognosis in AML [22]. Our results suggest that the regulation of stem cell function in AML also contains a favorable prognostic component. Notably, the ability of LSC- representative gene-sets to significantly stratify 548 patients with cytogenetically normal AML is intriguing for precision medicine, as cytogenetically normal AML is difficult to stratify for patient outcomes. Evidence from the literature further supports clinical relevance of the identified DNM gene-sets and the selected control gene-sets. These LSC- control gene-sets included published LSC highly-expressed genes that were previously associated with unfavorable event-free survival in patients with AML [22]. The LSC+ control gene-sets included fenretinide-down-regulated genes that were significantly correlated with poor-prognosis in AML patients [44]. In contrast, the LSC- DNM gene-sets are down-expressed in LSC and consist of genes indicating good outcome (and/or) genes being repressed by forskolin or IL2, two AML drugs that stop blood cell differentiation or induce prolonged remissions in advanced AML [45]. Genes associated with the Scp2 GWAS study [29] are potential new drug targets according to our results.
Genes from the identified functional gene-sets significantly share biological processes and molecular functions, suggesting a new aspect of stem cell biology. The questions of how these genes coordinate in the regulation of stem cell function or determine “stemness” warrant future investigation. We confirmed the biological co-expression of the LSC+ signature of 25-gene in AML patients carrying positive RAS molecular activity. As previous publications attest, it was RAS molecular activity rather than gene mutation that disturbed the identified LSC+ signaling and showed a prognostic factor.
In conclusion, LSC, or leukemia-initiating cell is a rare cellular subpopulation that must be eradicated to cure a patient of leukemia. However, their underlying mechanisms remain a biological conundrum, partly due to limited sample size and inter-patient and intra-patient variability. This study proposes a comprehensive knowledge-driven systematic analysis to functionally characterize LSC collected by different laboratories, followed by a novel dynamic network analysis. Two identified LSC- subpopulation-specific gene-set clusters, showing significant biological and clinical relevance, have been validated independently. The proposed framework extends our ability to re-use multiple layers of “omics”-data, to derive a new gene-set from coordinated gene-sets, and to discover new prognostic indicators, thus bridging cancer biology in the lab and the clinic.
Methods
Data
Patient samples
All datasets were previously published (Additional file 4: Table S4) and are publicly available. We have additionally received written authorization from the authors of GSE14468 to re-use the survival time of their samples. All samples of patients with AML were obtained at the time of diagnosis and with informed consent at corresponding hospitals, and study protocols were approved by the institutional review boards of corresponding institutes and hospitals [34,35,46,47].
Gene expression
We performed a literature review from PubMed, GEO, and ArrayExpress for three keywords (“stem cell”, “AML”, and “prognosis”) in October 2012. We collected nine LSC studies (Table 1 and Additional file 4: Table S1), two large primary AML datasets [35,46] (n > 200 each) and samples from TCGA [34] (Additional file 4: Table S4). We pooled sorted cell samples into three groups defined by cell surface markers: AML LSC-enriched cells (LSC+), AML LSC-depleted cells (LSC-), and normal samples of sorted HSC-enriched cells (HSC+). Only functionally defined LSC+ samples (n = 77) were investigated, showing in vivo validated leukemia stemness in xenograft models. The HSC+ samples (n = 23) included diverse microarrays and RNA-seq measurements (Table 1).
Functional gene-set
We studied three categories of previously defined functional gene-sets from MSigDB (version 4.0) [19], with variable numbers of member genes (Additional file 2: Text S1: S Methods 2.1).
Functional gene-set profiles
Collapse multiple measurements per gene
To convert the gene-by-sample values into the gene-set-by-sample scores, we first collapsed the gene expression profiles to one value per sample for each unique gene, using a selective collapsing strategy with the highest average expression. This strategy has previously led to the best inter-study consistency [48]. This step is necessary to ensure equal gene representation as some genes have multiple measurements on a microarray.
Calculate gene-set profiles
We calculated the FAIME-scores [7], the GSVA-scores [21], and the new FAIME.α-scores using Equations 1, 2, 3, from all genes for each sample (j) (hereafter referred to as the “gene-set profile”).
The null statistical hypothesis of a FAIME score is that the weighted ranks of genes inside a gene-set and outside the gene-set are drawn from the same distribution. In our calculations, we first converted the original gene expression values to ranks (r (x) j) to reduce the influence of potential outliers. Then we converted the ranks per sample into a new scale (s (x) j) of continuous values to softly award highly-expressed genes [20]. FAIME.5 employs qualitative scores sharply decreasing along gene ranks by assigning the parameter α = 5 (Equation 1). This strategy filters out low expressed genes and maintains cohort-independence.
Let i denote the gene-set index, by j the sample index, x the gene index, and n the total number of measured genes. Then
| 1 |
where the parameter α controls the rate at which the weights rise when moving the list from the lowest expression ranks to the highest. A sequence of rapidly increased weights (large α) is sensitive to the genes at the extreme top of expression ranks (Figure 2B). Our previous research has shown that the ability to discriminate between signal and noise critically depends on the original dataset [14], and a value of α = 1 was used in previous data [7].
Given a set of genes {x 1 ,…,x m}∈ gene-set i of size |gene-set i |, we scored its activity function A() using Equation 2 and that for its complementary set .
| 2 |
Equation 3 defines a gene-set score which is the difference between the activity function of genes inside and outside a previously defined gene-set.
| 3 |
Inter-dataset normalization
The goal of normalization was to compensate for technical differences and thus to make gene-set profiles from different resources comparable [49,50]. Inter-dataset normalization is implemented using the z-transform on the gene-set profile f. j = {f 1 ,f 2 ,…f N} per sample j by dividing by the standard deviation of the centered values, which is denoted y. j in Equation 4. Y is a combined matrix where rows correspond to pathways and columns correspond to samples. This type of standardization has been applied to two types of analyses: integrating gene-set scores of differentially expressed genes and analyzing trait-associated genetic markers [13].
| 4 |
Unbiased simulation
To evaluate statistical power (sensitivity) and type-I error, we carried out a simulation study as previously described (Additional file 1: Figure S1) [21]. In short, normalized and log-transformed gene expression values were simulated for each gene a = {1,2,…p} and sample-group b = {1,2} using the linear additive model in Equation 5,
| 5 |
where gene specific effect is α α ~ N(0,1), sample specific effect is β b ~ N(μ j ,σ j ), and random noise is e iab ~ N(0,1).
In the simulation study, we modeled p = 5000 genes and two groups of samples each with different sample size n = {10, 20, 40, 60, 80, 100}. We randomly built a differentially expressed (DE) gene-set of 30 differentially-expressed genes and a non-DE gene-set of 30 no-changed genes, considering the following two facts for the DE gene-set. 1) The fraction of differentially expressed genes in the DE set varied, 50% and 80% respectively. 2) The expected signal-to-noise-ratio varied between weak and strong, meaning that the magnitude of DE set between two sample groups was 0.5 and 1. The simulation was done by setting μ 1 = μ 2 = 0 with σ 1 = σ 2 = 1 for all genes in the non-DE set but μ 1 = m, μ2 = 0 with σ 1 = σ 2 = 0.5 for a certain fraction of genes in the DE set. Then we applied GSEA, GSVA, and FAIME modulating the parameter α = {1, 5, 10} (Equations 1, 2, 3) on the simulated gene expression data to generate the simulated gene-set scores respectively. We repeated the above simulations assigning the DE and non-DE gene-sets with x = {10, 20, 80, and 100}, i.e., 0.2%, 0.4%, 1.6%, and 2% of the modeled 5000 genes. The statistical power was a fraction of significance for the DE set (true positive) whereas the empirical type-I error was a fraction of the non-adjusted significance (p < 0.05, two-sample t-test) of the non-DE set. We adjusted the p-values for multiple-testing across 1000 simulations [51] and set a significance level of FDR = 0.05.
Dynamic Network Mechanism (DNM) analysis
DNM is built on dynamic network biomarker analysis (DNB) [15], a model-free method to detect the dynamics of disease developmental stage changes using gene expression profiles. Developed for gene-set profiling, DNM identifies a cluster of gene-sets that highly co-fluctuate in a “critical stage” (or disease transition stage). It explores the dynamic character of network nodes (gene-sets) and links (alteration between gene-set pairs) across sample groups. Given a sample group t = {LSC+, LSC-, and normal HSC+} and the assigned control group (e.g., the normal HSC+), we performed the following five-step calculation in our DNM analysis Figure 3.
To acquire candidate gene-sets with the highest intra-group variation, we calculated the standard deviation (SD) of every gene-set across samples within this sample group. Then, we picked gene-set t, the top 5% of all gene-sets having the highest SDs.
To cluster the gene-set t into highly co-variable modules in a sample group t of interest, termed C t I, we calculated the ‘intra-module’ correlation of SDs (gene-sets were compared in pairs). We ran an unsupervised hierarchical clustering on overall pairwise Pearson correlation coefficients (PCC). This agglomerative hierarchical clustering was performed in R using complete linkage on the pairwise PCC distance (1-PCC) in gene-set t. A module of gene-set t consists of gene-sets that satisfy a significant level (p-value ≤ 0.01). Applying a threshold of p-value ≤ 0.001 or p-value ≤ 0.05 also leads to comparable DNMs.
To estimate ‘inter-module’ co-variation within this sample group t (C t I), we calculated the correlation of SDs between C t I members and other control gene-sets. We also estimated ‘cross-module’ co-variation between this sample group t and control group (C t O). The control gene-sets preserved the top n highest correlations with any C t I members in the control group but outside the C t I set. We applied n = 5 to small- or middle-sized gene-sets (pathways and motifs) and n = 20 to the larger-sized gene-sets (the CGPs).
To determine the critical sample group, we calculated a combination score S t I in Equation 6 [15] for each module C t I in a given sample group t.
| 6 |
where SD tI is the average SD value of gene-sets in the C t I, PCC I represents the average PCC value of gene-set-pairs in the C t I, and PCC o the average PCC value of gene-sets between this C t I and C t O. The C t I with the largest S t I score was then determined to comprise the DNM markers and the corresponding sample group t to be the critical sample group (disease stage).
-
5)
Additionally, to assess the significance of gene overlap among DNM gene-sets for a critical sample group, we calculated the hypergeometric probability using Equation 7 (Additional file 2: Text S1: S Methods for inference). Given A, B, and C to be the number of gene-set size for three DNM gene-sets respectively, N = 22000 the background number of human genes, n the observed number of shared genes among DNM gene-sets, we calculated:
| 7 |
Prognosis of DNM gene-sets on primary AML samples
Previously, we proposed the concept of relative-expression of gene-set pairs [17] which calculates a prognostic indicator (Equation 8) for each patient. An indicator of Relative Effect Analysis with Gene-set Pairs (RXA-GSP) compares effects of DNM gene-sets with selected control gene-sets, instead of genes, for each patient.
| 8 |
Based on the DNM analysis, we selected control gene-sets {C} meeting the following two criteria: 1) significant univariate analyses on overall survival of AML patients (log-rank p < 0.01), and 2) a positive cox-regression coefficient (i.e., increased scores of selected control gene-sets are associated with poor outcome). We then derived a sum-value S j in Equation 9 for the DNM gene-set group {S} and the control gene-set group {C}, respectively. The sum-value is a linear combination of the gene-set scores (indexed by i) weighted by the respective gene-set’ cox-regression coefficient in the training set [47].
| 9 |
Gene-set scores of all identified DNM gene-sets and their corresponding control gene-sets were calculated for primary AML patients in three independent cohorts to identify an indicator (Additional file 4: Table S4). We set the largest cohort as the training set and used the other two cohorts for validation. The treatment-related AML patients and patients with prior malignancy or leukemogenic agent exposure were excluded.
The Kaplan-Meier and the univariate Cox regression analyses on both the training set and validation set demonstrated the prognosticative power of the indicator. We then asked whether the novel indicator remained prognostic after adjusting by all other significant variables that had a univariate p < 0.05, using multivariate testing.
Additionally, an empirical evaluation was performed as suggested [52], by replacing the FAIME scores in Equation 8 with randomly picked |S| and |C| gene-sets from 3420 CGPs in the MSigDB database and repeating the above two calculations X = 2000 times, where the cardinality of a set S is denoted |S|. Then we estimated the chance of observing the observed log-rank p-values using Equation 10, the fraction of simulated p-values that are less than or equal to the observed p-values.
| 10 |
Acknowledgements
We thank Dr. Gangqing Hu from National Heart, Lung and Blood Institute, NIH, for providing the RPKM values for one CD34+ RNA-seq data, Peter J.M. Valk, Ruud Delwel, and Bob Lowenberg from Department of Hematology, Erasmus University Medical Center, Rotterdam, the Netherlands, for providing the detailed survival information of their patients in the dataset GSE14468 to support our secondary data analyses. We acknowledge The Cancer Genome Atlas Consortium for providing detailed raw data. We thank Joshua Stevens_Stein for his assistance editing this manuscript.
This work was supported by the National Institutes of Health [R21 CA167305-01A1 to Y.X., B.W., C.J, C.M.J]; 973 Project of China [2014CB910504 to C.L.], National Natural Science Foundation of China [61134013, 91029301, and 11326035 to C.L.], the Knowledge Innovation Program of CAS [KSCX2-EW-R-01 to C.L.], and 863 Project [2012AA020406 to C.L.]. Funding for open access charge: National Institutes of Health [R21 CA167305-01A1].
Abbreviations
- AML
Acute myeloid leukemia
- CGP
Chemical and genetic perturbations
- CtI
‘Inter-module’ co-variation within this sample group t
- CtO
‘Cross-module’ co-variation between this sample group t and control group
- DNB
Dynamic network biomarker
- DNM
Dynamic network mechanism
- SD
Standard deviation
- ELN
European LeukemiaNet
- FAIME
Functional analysis of individual microarray (or RNA-seq) expression
- FDR
False discovery rate
- IPA
Ingenuity pathway analysis
- GO
Gene ontology
- GS
Gene set
- GSEA
Gene set enrichment analysis
- GSVA
Gene set variation analysis
- HSC+
Verified hematopoietic stem cells enriched
- LSC
Leukemic stem cell
- LSC+
LSC enriched
- LSC-
LSC depleted
- MSigDB
Molecular signature database
- PCC
Pearson correlation coefficients
- RXA-GSP
Relative effect analysis with gene-set pairs
Additional files
Statistical power and type-I error rate in unbiased simulations. Figure S2. Published LSC associated gene signatures and their significantly enriched canonical pathways. Figure S3. Distribution of pathway profiles for collected samples. Figure S4. Heatmap of genes in the two DNM identified gene clusters from different datasets of three cell subpopulations. Figure S5. Kaplan-Meier plots of patients with primary AML, based on DNM identified gene-set pairs. Figure S6. Kaplan-Meier plots of patients with primary AML, based on Gentles “LSC signature” [22].
Text S1. Supplementary results, methods, and the limitation.
Text S2. R Code and demonstration for the function rungene2pathway.
Nine studies and eight published gene lists pertaining to AML LSC. Table S2. LSC- representative DNM gene-sets (30-gene) and their control pairs in the normal state, derived from gene-set profiles using the FAIME.5 algorithm. Table S3. LSC+ representative DNM gene-sets (25-gene) and their control pairs in the normal states, derived from gene-set profiles using the GSVA algorithm. Table S4. Three independent primary AML studies. Table S5. Univariate and multivariate analyses of overall survival in patients with all types of AML, for the LSC+ representative DNM gene-sets. Table S6. The somatic mutations (of genes in the identified DNM gene-sets) and their patient information in the TCGA database.
Text S3. A tab-delimited text file recording the normalized expression of the 54 identified genes.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XY, LC, and JMC designed the projects; JC supervised the prognostic analysis; XY, LM, and BW analyzed data, WZ modeled the hypergeometric probability; AD, JJ, KO, and XY interpreted the biological meaning; XY, LM, WZ, and AD wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Xinan Holly Yang, Email: xyang2@uchicago.edu.
Meiyi Li, Email: limeiyi@sibs.ac.cn.
Bin Wang, Email: bwang@peds.bsd.uchicago.edu.
Wanqi Zhu, Email: 1213.ghs@gmail.com.
Aurelie Desgardin, Email: adesgard@bsd.uchicago.edu.
Kenan Onel, Email: konel@peds.bsd.uchicago.edu.
Jill de Jong, Email: jdejong@peds.bsd.uchicago.edu.
Jianjun Chen, Email: jchen@medicine.bsd.uchicago.edu.
Luonan Chen, Email: lnchen@sibs.ac.cn.
John M Cunningham, Email: jmcunningham@uchicago.edu.
References
- 1.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25(11):1315–21. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 2.Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105(11):4163–9. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horton SJ, Huntly BJ. Recent advances in acute myeloid leukemia stem cell biology. Haematologica. 2012;97(7):966–74. doi: 10.3324/haematol.2011.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 5.Wiseman DH, Greystoke BF, Somervaille TC. The variety of leukemic stem cells in myeloid malignancy. Oncogene. 2014;33(24):3091–8. doi: 10.1038/onc.2013.269. [DOI] [PubMed] [Google Scholar]
- 6.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X, Regan K, Huang Y, Zhang Q, Li J, Seiwert TY, et al. Single sample expression-anchored mechanisms predict survival in head and neck cancer. PLoS Comput Biol. 2012;8(1):e1002350. doi: 10.1371/journal.pcbi.1002350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang X, Li H, Regan K, Li J, Huang Y, Lussier YA. Towards mechanism classifiers: expression-anchored gene ontology signature predicts clinical outcome in lung adenocarcinoma patients. AMIA Annu Symp Proc. 2012;2012:1040–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Tarca AL, Bhatti G, Romero R. A comparison of gene set analysis methods in terms of sensitivity, prioritization and specificity. PLoS One. 2013;8(11):e79217. doi: 10.1371/journal.pone.0079217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26(12):i237–45. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;36(10):1090–8. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 12.Drier Y, Sheffer M, Domany E. Pathway-based personalized analysis of cancer. Proc Natl Acad Sci U S A. 2013;110(16):6388–93. doi: 10.1073/pnas.1219651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong Q, Ancona N, Hauser ER, Mukherjee S, Furey TS. Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets. Genome Res. 2012;22(2):386–97. doi: 10.1101/gr.124370.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X, Bentink S, Scheid S, Spang R. Similarities of ordered gene lists. J Bioinform Comput Biol. 2006;4(3):693–708. doi: 10.1142/S0219720006002120. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Liu R, Liu ZP, Li M, Aihara K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci Rep. 2012;2:342. doi: 10.1038/srep00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Zeng T, Liu R, Chen L. Detecting tissue-specific early warning signals for complex diseases based on dynamical network biomarkers: study of type 2 diabetes by cross-tissue analysis. Brief Bioinform. 2014;15(2):229–43. doi: 10.1093/bib/bbt027. [DOI] [PubMed] [Google Scholar]
- 17.Yang X, Vasudevan P, Parekh V, Penev A, Cunningham JM. Bridging cancer biology with the clinic: relative expression of a GRHL2-mediated gene-set pair predicts breast cancer metastasis. PLoS One. 2013;8(2):e56195. doi: 10.1371/journal.pone.0056195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X, Ai X, Cunningham JM. Computational prognostic indicators for breast cancer. Cancer Manage Res. 2014;6:301–12. doi: 10.2147/CMAR.S46483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lottaz C, Yang X, Scheid S, Spang R. OrderedList–a bioconductor package for detecting similarity in ordered gene lists. Bioinformatics. 2006;22(18):2315–6. doi: 10.1093/bioinformatics/btl385. [DOI] [PubMed] [Google Scholar]
- 21.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304(24):2706–15. doi: 10.1001/jama.2010.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Jonge HJ, Woolthuis CM, Vos AZ, Mulder A, van den Berg E, Kluin PM, et al. Gene expression profiling in the leukemic stem cell-enriched CD34+ fraction identifies target genes that predict prognosis in normal karyotype AML. Leukemia. 2011;25(12):1825–33. doi: 10.1038/leu.2011.172. [DOI] [PubMed] [Google Scholar]
- 24.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–93. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 25.Ohashi S, Sakashita G, Ban R, Nagasawa M, Matsuzaki H, Murata Y, et al. Phospho-regulation of human protein kinase Aurora-A: analysis using anti-phospho-Thr288 monoclonal antibodies. Oncogene. 2006;25(59):7691–702. doi: 10.1038/sj.onc.1209754. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Ikezoe T, Nishioka C, Nobumoto A, Udaka K, Yokoyama A. CD34(+)/CD38(−) acute myelogenous leukemia cells aberrantly express Aurora kinase A. Int J Cancer. 2013;133(11):2706–19. doi: 10.1002/ijc.28277. [DOI] [PubMed] [Google Scholar]
- 27.Kojima K, Shimanuki M, Shikami M, Andreeff M, Nakakuma H. Cyclin-dependent kinase 1 inhibitor RO-3306 enhances p53-mediated Bax activation and mitochondrial apoptosis in AML. Cancer Sci. 2009;100(6):1128–36. doi: 10.1111/j.1349-7006.2009.01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham SM, Vass JK, Holyoake TL, Graham GJ. Transcriptional analysis of quiescent and proliferating CD34+ human hemopoietic cells from normal and chronic myeloid leukemia sources. Stem Cells. 2007;25(12):3111–20. doi: 10.1634/stemcells.2007-0250. [DOI] [PubMed] [Google Scholar]
- 29.Bystrykh L, Weersing E, Dontje B, Sutton S, Pletcher MT, Wiltshire T, et al. Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nat Genet. 2005;37(3):225–32. doi: 10.1038/ng1497. [DOI] [PubMed] [Google Scholar]
- 30.Boultwood J, Lewis S, Wainscoat JS. The 5q-syndrome. Blood. 1994;84(10):3253–60. [PubMed] [Google Scholar]
- 31.Lai F, Godley LA, Joslin J, Fernald AA, Liu J, Espinosa R, 3rd, et al. Transcript map and comparative analysis of the 1.5-Mb commonly deleted segment of human 5q31 in malignant myeloid diseases with a del(5q) Genomics. 2001;71(2):235–45. doi: 10.1006/geno.2000.6414. [DOI] [PubMed] [Google Scholar]
- 32.Myllykangas S, Himberg J, Bohling T, Nagy B, Hollmen J, Knuutila S. DNA copy number amplification profiling of human neoplasms. Oncogene. 2006;25(55):7324–32. doi: 10.1038/sj.onc.1209717. [DOI] [PubMed] [Google Scholar]
- 33.Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113(13):3088–91. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research N Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112(10):4193–201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol. 2011;7(10):e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118(20):5593–603. doi: 10.1182/blood-2011-03-343988. [DOI] [PubMed] [Google Scholar]
- 38.Illmer T, Thiede C, Fredersdorf A, Stadler S, Neubauer A, Ehninger G, et al. Activation of the RAS pathway is predictive for a chemosensitive phenotype of acute myelogenous leukemia blasts. Clin Cancer Res. 2005;11(9):3217–24. doi: 10.1158/1078-0432.CCR-04-2232. [DOI] [PubMed] [Google Scholar]
- 39.Becker MW, Jordan CT. Leukemia stemness signatures step toward the clinic. Cell Stem Cell. 2011;9(3):185–6. doi: 10.1016/j.stem.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 40.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;8(2):e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 42.Itonaga H, Imanishi D, Wong YF, Sato S, Ando K, Sawayama Y, et al. Expression of myeloperoxidase in acute myeloid leukemia blasts mirrors the distinct DNA methylation pattern involving the downregulation of DNA methyltransferase DNMT3B. Leukemia. 2014;28(7):1459–66. doi: 10.1038/leu.2014.15. [DOI] [PubMed] [Google Scholar]
- 43.Leong AS-Y, Cooper K, Leong FJW-M. Manual of Diagnostic Antibodies for Immunohistology Entnode. London: Greenwich Medical Media Ltd; 1999. [Google Scholar]
- 44.Zhang H, Mi JQ, Fang H, Wang Z, Wang C, Wu L, et al. Preferential eradication of acute myelogenous leukemia stem cells by fenretinide. Proc Natl Acad Sci U S A. 2013;110(14):5606–11. doi: 10.1073/pnas.1302352110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meloni G, Foa R, Vignetti M, Guarini A, Fenu S, Tosti S, et al. Interleukin-2 may induce prolonged remissions in advanced acute myelogenous leukemia. Blood. 1994;84(7):2158–63. [PubMed] [Google Scholar]
- 46.Noordermeer SM, Sanders MA, Gilissen C, Tonnissen E, van der Heijden A, Dohner K, et al. High BRE expression predicts favorable outcome in adult acute myeloid leukemia, in particular among MLL-AF9-positive patients. Blood. 2011;118(20):5613–21. doi: 10.1182/blood-2011-06-359182. [DOI] [PubMed] [Google Scholar]
- 47.Li Z, Herold T, He C, Valk PJ, Chen P, Jurinovic V, et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol. 2013;31(9):1172–81. doi: 10.1200/JCO.2012.44.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller JA, Cai C, Langfelder P, Geschwind DH, Kurian SM, Salomon DR, et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics. 2011;12:322. doi: 10.1186/1471-2105-12-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics. 2012;13(2):204–16. doi: 10.1093/biostatistics/kxr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim WK, Wang K, Lefebvre C, Califano A. Comparative analysis of microarray normalization procedures: effects on reverse engineering gene networks. Bioinformatics. 2007;23(13):i282–8. doi: 10.1093/bioinformatics/btm201. [DOI] [PubMed] [Google Scholar]
- 51.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc B (Methodological) 1995;57(1):289–300. [Google Scholar]
- 52.Li J, Lenferink AE, Deng Y, Collins C, Cui Q, Purisima EO, et al. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat Commun. 2010;1:34. doi: 10.1038/ncomms1033. [DOI] [PMC free article] [PubMed] [Google Scholar]