

Abstract
In February 2014, a group of scientists convened as part of the University of California Davis Cardiovascular Symposium to bring together experimental and mathematical modelling perspectives and discuss points of consensus and controversy on the topic of sodium in the heart. This paper summarizes the topics of presentation and discussion from the symposium, with a focus on the role of aberrant sodium channels and abnormal sodium homeostasis in cardiac arrhythmias and pharmacotherapy from the subcellular scale to the whole heart. Two following papers focus on Na+ channel structure, function and regulation, and Na+/Ca2+ exchange and Na+/K+ ATPase. The UC Davis Cardiovascular Symposium is a biannual event that aims to bring together leading experts in subfields of cardiovascular biomedicine to focus on topics of importance to the field. The focus on Na+ in the 2014 symposium stemmed from the multitude of recent studies that point to the importance of maintaining Na+ homeostasis in the heart, as disruption of homeostatic processes are increasingly identified in cardiac disease states. Understanding how disruption in cardiac Na+-based processes leads to derangement in multiple cardiac components at the level of the cell and to then connect these perturbations to emergent behaviour in the heart to cause disease is a critical area of research. The ubiquity of disruption of Na+ channels and Na+ homeostasis in cardiac disorders of excitability and mechanics emphasizes the importance of a fundamental understanding of the associated mechanisms and disease processes to ultimately reveal new targets for human therapy.
Disruption of sodium homeostasis in cardiac disease
In the session on ‘Disruption of Na+ homeostasis, Sanda Despa, Brian O'Rourke, Donald Bers, Alicia Mattaizzi, William Louch, Christoph Maack and Sridharan Rajamani were presenters, discussion leaders and panelists. Below is a summary of the sessions and discussion.
Although it is well known that the normal cycling of intracellular Ca2+ in cardiac myocytes is critical for the normal electrical and mechanical functioning of the heart, the intracellular Na+ concentration ([Na+]i) is tightly coupled to Ca2+ homeostasis and is increasingly recognized as a modulating force of cellular excitability, frequency adaptation and cardiac contractility (Faber & Rudy, 2000; Grandi et al. 2010; Despa & Bers, 2013; Fig. 1). The direct coupling between intracellular Na+ and Ca2+ concentrations is mediated via the sodium–calcium exchanger (NCX), which exchanges 3 Na+ for each Ca2+, and comprises the primary cellular extrusion mechanism for Ca2+ (Blaustein & Lederer, 1999). The NCX can operate in both forward mode, during which it extrudes Ca2+, or can mediate Ca2+ influx when it operates in the ‘reverse mode’. The activity of NCX is sensitively tuned to changes in [Na+]i so that a millimolar increase in [Na+]i resulting from changes in the ion influx/efflux balance, can result in changes to NCX activity that alter Ca2+ homeostasis leading to intracellular Ca2+ loading in both cellular and sarcoplasmic reticulum (SR) compartments (Maack et al. 2006). A consequence of intracellular cardiac myocyte Ca2+ loading is stronger contraction (Pieske et al. 2002), but NCX-dependent Ca2+ loading may compromise the stability of the SR store, thus facilitating larger Ca2+ transients and/or spontaneous Ca2+ waves as in digitalis toxicity (Despa & Bers, 2013). If Ca2+ waves or transients are sufficiently large, the excess intracellular Ca2+ will be extruded via the NCX resulting in depolarizing current that may bring the membrane potential of the cell to the voltage threshold required for activation of the Na+ channel, causing delayed after-depolarizations (DADs) and arrhythmogenically triggered action potentials (Despa & Bers, 2013). For a more detailed description of structural and functional determinants of NCX, please refer to the white paper no. 3 (Shattock et al. 2015).
Figure 1.
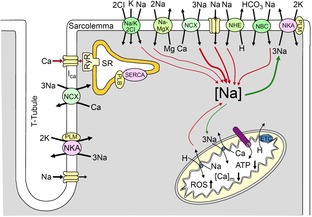
Schematic depiction of Na+ transport processes in the cardiac myocyte
The figure is reproduced from Despa & Bers (2013). PLM, phospholemman; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase. [Ca]m, mitochondrial Ca2+; ETC, electron transport chain; PLB, phospholamban; RyR, ryanodine receptor.
An additional important cellular mechanism for the maintenance of [Na+]i homeostasis is the sodium–potassium ATPase (NKA), which uses energy derived from hydrolysis of an ATP molecule, allowing extrusion of three Na+ ions in exchange for two K+ ions. The NKA is half-maximally activated between 10 and 22 mm [Na+]i and between 1 and 2 mm external K+ (Glitsch, 2001). Thus, at 4 mm normal extracellular K+, NKA is ∼70% saturated, with plenty of available ATP (5–10 mm) (half-maximal NKA activation is 80–150 μm) (Hilgemann et al. 1991). The regulation and function of NKA is covered in detail in white paper no. 3 (Shattock et al. 2015).
Cardiac myocytes also contain Na+ transport mechanisms that promote simultaneous maintenance of [Na+]i homeostasis and physiological pH including the sodium–hydrogen exchanger (NHE), which moves sodium into the cell in exchange for proton export (Leem et al. 1999). The sodium–bicarbonate symporter (NBC) is also present in myocytes and acts as an additional mechanism to couple [Na+]i homeostasis and pH (Leem et al. 1999).
In disease states, the importance of the proper coupling between multiple Na+ homeostatic mechanisms is evident. In ischaemia, failure of ion homeostasis starts with an influx of Na+ through the NHE (Eigel & Hadley, 1999) in an attempt to raise the acidified pH (through the extrusion of H+). In ischaemia/reperfusion injury, activation of the NHE and NBC, a pathological increase in the persistent/late Na+ current (INaL), Na+ entry through connexin hemichannels (at least in isolated myocytes) (Kondo et al. 2000), and NKA inhibition result in reverse mode NCX activity that leads to Ca2+ overload (Despa & Bers, 2013). During hypoxia, NHE in rabbit ventricular myocytes has been shown to dominate total Na+ influx (Bers et al. 2003). Inhibition of the NHE during ischaemic episodes attenuates the rise in intracellular [Na+]i (Avkiran, 2003; Baetz et al. 2003). Along with Na+ influx via the NHE, a parallel decrease in energy production due to mitochondrial dysfunction and loss of ATP results in reduced Na+ elimination through the Na+/K+ ATPase (Hale et al. 2008), which further augments [Na+]i. The above examples show how disruptions to [Na+]i homeostasis are evident in multiple disease states and occur via distinct mechanisms that can lead to a pathological cascade through coupled Na+ regulatory mechanisms that ultimately leads to disruptions in Ca2+ homeostasis.
Another key disease state marked by intracellular sodium dysregulation is heart failure (HF), where it has been shown that [Na+]i is elevated in humans and numerous animal models (Despa et al. 2002; Pieske et al. 2002; Baartscheer et al. 2003; Schillinger et al. 2006; Louch et al. 2010). Elevation of [Na+]i in HF may represent a compensatory adaptation that allows for an increase in Ca2+ influx via NCX, leading to improved contraction, as a type of physiological ‘digitalis’. However, increased [Na+]i has also been associated with diastolic dysfunction, as it impairs Ca2+ removal by NCX (Louch et al. 2010, 2012) and with energetic defects and oxidative stress (Kohlhaas et al. 2010; Bay et al. 2013; Nickel et al. 2014), of which the latter may contribute to the progression of the disease through inducing cell death and maladaptive remodelling (Nickel et al. 2014).
Question or controversy: sodium dysregulation
While there is little doubt that the sodium dysregulation is important in numerous cardiac pathologies, a detailed understanding of the precise mechanisms is missing.
Which are the critical pathways to Na+ overload in each disease?
How does disease-linked remodelling alter ion homeostatic mechanisms?
Which disease-linked changes in Na+ processes are compensatory?
While it is generally agreed upon that [Na+]i is increased in many forms of heart disease, the specific pathways responsible for the increase in intracellular Na+ in some diseases are still a matter of controversy. Increased Na+ entry through Na+ channels and NHE and decreased Na+ extrusion by reduced NKA activity have been reported for various animal models of disease. It could well be that the specific pathway is both species and model dependent. For example, NKA expression is reduced in failing human myocardium (Schwinger et al. 1999), although mRNA levels are unchanged (Allen et al. 1992). In the rat, mRNA and protein levels of the primary NKA α1 isoform are preserved in most HF models, whereas the protein levels of NKA α2 are apparently reduced, while NKA α3 is increased (Verdonck et al. 2003). In rabbit HF models all NKA isoforms have been shown to exhibit reduced protein expression in myocytes (Bossuyt et al. 2005). Clearly, a direct causative link between biochemical changes and function cannot be made because of potential confounding factors such as altered protein regulation, function or activity that are not measured with biochemical assays. An example here is the differential regulation of NKA by phospholemman (PLM) in HF, described in detail in white paper no. 3 (Shattock et al. 2015). Moreover, there are important changes in localization of NKA isoforms during heart failure. Swift et al. showed altered expression patterns of NKA α1 and NKA α2 isoforms, which were linked to altered Ca2+ extrusion from cardiomyocytes and consequent changes in contractile parameters (Swift et al. 2008).
A large Na+ influx via other mechanisms has also been proposed to increase [Na+]i in HF. For example, a TTX-sensitive diastolic Na+ influx (implicating involvement of the voltage-gated Na+ channel, although the specific isoform is unclear) was observed to be upregulated in rabbits with pressure and volume overload-induced HF (Despa et al. 2002). NHE upregulation has also been documented in HF (Baartscheer et al. 2003). Acidosis was also shown to promote Na+ loading in the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) knockout mouse (Li et al. 2012). Na+ influx also occurs as a result of a gain of function of the Na+ channel in the form of the non-inactivating component INaL that will be detailed in the following sections.
Question or controversy: Na+ handling in disease
How do various isoforms of Na+ transport processes contribute to function or disease?
How does modification to individual Na+ transport isoforms alter function?
Do changes measured in animal models reflect human adaptive processes?
In summary, while dynamic beat-to-beat changes in [Na+]i allow physiological adaptation in non-diseased cardiac myocytes, perturbations to Na+ homeostasis occur through a variety of mechanisms in cardiac disease states. Disease-induced disruption to [Na+]i maintenance occurs because the pumps and exchangers described above are unable to adequately compensate for pathological Na+ loading without a consequent disruption in Ca2+ homeostasis. Not only does Ca2+ overload have the potential to disrupt electrical activity in the heart, it also has detrimental effects on cardiac energetics and metabolism.
Elevated [Na+]i is linked to disruptions in cardiac energetics and metabolism
Although increased [Na+]i may improve the contractile function of the diseased heart, elevated [Na+]i may have a pathological impact on cardiac metabolism and oxidative state. For example, the increase in [Na+]i and reverse mode NCX-mediated Ca2+ influx during the cardiac action potential is energetically less efficient than normal SR Ca2+ release and may contribute to a mismatch between energy supply and demand in the failing heart (Weisser-Thomas et al. 2003). Furthermore, when intracellular Ca2+ transients were triggered by NCX-mediated Ca2+ entry, the efficiency of mitochondrial Ca2+ uptake is substantially reduced, suggesting reduced efficiency in the transport mechanism necessary (of NCX vs. Ca2+ current and SR Ca2+ release) to drive Ca2+-induced stimulation of Krebs cycle processes (Kohlhaas & Maack, 2010). An interesting point to consider and that needs further clarification is what is the magnitude of mitochondrial Ca2+ uptake occurring on each heartbeat and how do kinetics of uptake, release and buffering impact cytoplasmic systolic and diastolic Ca2+ signals.
In failing cardiac myocytes, increased [Na+]i impairs energy supply-and-demand matching by promoting acceleration of mitochondrial Ca2+ efflux, via the mitochondrial Na+/Ca2+ exchanger (mNCE, or NCLX), which extrudes Ca2+ from the mitochondria in exchange for Na+ (Liu & O'Rourke, 2008; Palty et al. 2010). This effect, together with the decreased amplitude of cytosolic Ca2+ transients in HF, blunts mitochondrial Ca2+ signalling to the Krebs cycle during increased work, resulting in oxidation of the pyridine nucleotide pool (NAD(P)H), thus further compromising NADH supply for oxidative phosphorylation (Liu & O'Rourke, 2008). Additionally, since NADPH is required to maintain matrix antioxidant pathway flux, its oxidation causes cellular overload of reactive oxygen species (ROS) (Kohlhaas et al. 2010; Liu et al. 2010). ROS accumulation then contributes to oxidative modification of Ca2+ handling and ion channel targets to promote arrhythmias. This cascade of failures, stemming from [Na+]i overload, is thus hypothesized to provoke triggered arrhythmias (Liu et al. 2010), which, in the context of the altered electrophysiological substrate in HF, may induce sudden cardiac death (SCD). Interestingly, chronic inhibition of the mNCE during the induction of HF prevents these mitochondrial defects, and abrogates cardiac decompensation and sudden death, in a guinea pig model of HF/SCD (Liu et al. 2014).
Question or controversy: energetics and metabolism
How much mitochondrial Ca2+ uptake occurs on each heartbeat?
How do the kinetics of uptake, release and buffering influence cytoplasmic systolic and diastolic Ca2+ signals?
Abnormal sodium channel function in cardiac disease
In the session on ‘Na+-induced arrhythmias: from the cellular level to the whole heart’, Penelope James Weiss, Ole Sejersted, Jonathan Lederer, Zhilin Qu and Livia Hool comprised the group of presenters, discussion leaders and panelists. A summary of the session and discussion is below.
In addition to changes in [Na+]i homeostatic mechanisms in the heart, changes to the distribution and function of cardiac Na+ channels have been linked to disease manifestation and progression of both inherited and acquired cardiac arrhythmias. Either gain or loss of Na+ channel function can result, depending on the disease state, and both disruptions can lead to proarrhythmic consequences arising from alterations in cardiac conduction and repolarization.
Loss of Na+ channel function
In the case of loss of Na+ channel function, either as a result of disease-induced remodelling or as a result of drug application, reduced Na+ current (INa) can cause insufficient cellular excitability to allow propagation of electrical waves, leading to a well-known requirement for some types of re-entrant arrhythmias: conduction block.
One instance of remodelling of Na+ channels that plays a role in conduction block and arrhythmogenesis is in the infarct border zone where the electrical substrate is extensively remodelled compared to normal non-infarcted epicardium. The fact that progressive electrical remodelling that occurs in chronic disease states has been identified as a biomarker for sudden cardiac death indicates the critical importance of revealing its mechanisms (Gaudron et al. 2001; Goldberger et al. 2014). Na+ currents (as well as Ca2+ and K+ currents) in cells isolated from the epicardial border zone (EBZ) of 5-day- infarcted hearts have been shown to have both altered current amplitudes and changes in kinetics (Lue & Boyden, 1992; Aggarwal & Boyden, 1995; Pu & Boyden, 1997; Dun et al. 2004) consistent with reduced cellular action potential (AP) amplitudes and upstroke velocity and prolonged post-repolarization refractoriness. Within the re-entrant circuit, two distinct cell regions have been identified: cells from the central common pathway of the circuit (IZc) and cells from the outer pathway on the other side of the line of block (outer pathway, IZo; Baba et al. 2005). Cells from both of the infarct zone regions had reduced Na+ current density, but the cells from the IZo also exhibited slower Na+ channel kinetics for time to peak and current decay (Pu & Boyden, 1997). These changes in Na+ channel function along with observed changes to L-type Ca2+ currents give rise to electrical anisotropy that promotes stable lines of block within the zone (Cabo & Boyden, 2003). These stable lines of block then allow for the development of sustained re-entrant excitation and stable ventricular tachycardias in the EBZ (Janse & Wit, 1989).
If regional differences of ionic currents in cells of the EBZ are the mechanisms underlying the lines of block observed in the EBZ, then restoration of either the Na+ channel or the L-type Ca2+ channel availability, and thereby normal function, should be antiarrhythmic by disrupting the stability of the lines of block leading to termination of re-entry. Indeed, recent studies (Coronel et al. 2010) suggest that gene transfer-mediated overexpression of the skeletal muscle sodium channel SkM1 resulted in improved Na+ channel availability in cells of the EBZ because SkM1 channels have positively shifted kinetics of inactivation rendering them available to open at depolarized potentials at which cardiac Na+ channels are not available. SkM1 overexpression improved conduction and reduced the incidence of inducible ventricular tachycardia/ventricular fibrillation (VT/VF) post-myocardial infarction (Boink et al. 2012). Such approaches constitute the potential for new development of strategic interventions to restore electrical disruptions in the heart arising from electrically based remodelling.
Question or controversy: remodelling in the infarct border zone
How can stability of functional lines of block be disrupted to promote the termination of re-entry?
Gain of Na+ channel function
Recent work has focused on gain-of-function of the Na+ current because a range of cardiac diseases are marked by pathological increases in the persistent late Na+ current component (late Na+ current, INaL) that follows the rapid transient activation of INa. INaL is upregulated in many pathological conditions, such as in the failing and/or ischaemic heart, in the heart exposed to oxidative stress, and in hearts of patients with congenital long QT3 syndromes (Bennett et al. 1995; Wang et al. 1995; Maltsev et al. 1998a; Maltsev & Undrovinas, 2006; Song et al. 2006; Hund et al. 2008; Sossalla et al. 2010). Ca2+ dysregulation results in pathological effects to promote INaL (Mori et al. 2000; Wingo et al. 2004) via activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Wagner et al. 2006), increased mitochondrial oxidative phosphorylation (Belardinelli et al. 2006) and consequent increased ROS (Kohlhaas et al. 2010; Liu et al. 2010). Likewise increases in INaL, which may lead to an increase in intracellular Ca2+ can cause activation of CaMKII (Yao et al. 2011). Please see Chen-Izu et al. (2015) for detailed descriptions of Na+ channel regulation. Enhanced INaL leads to action potential prolongation, disruption of normal cellular repolarization, development of arrhythmia triggers, and propensity to ventricular arrhythmia. In heart failure, pharmacological targeting of INaL has been shown to result in: (1) stabilization of repolarization; (2) decrease in beat-to-beat action potential duration (APD) variability; and (3) improvement in Ca2+ handling and contractility (Maltsev et al. 1998b; Undrovinas et al. 1999, 2006; Undrovinas & Maltsev, 2008).
At least three distinct alterations in NaV1.5 (the cardiac isoform of the voltage gated Na channel) gating have been shown to increase INaL including the Na+ window currents, differential gating modalities, and non-equilibrium gating. These mechanisms were initially revealed via detailed electrophysiological study of mutations in SCN5A that resulted in long QT type 3 syndrome (LQT3) in patients. The window current describes the INa that is measurable in the voltage range where the steady-state inactivation curve and activation curve overlap (January & Riddle, 1989; Zeng & Rudy, 1995; Zaza et al. 2008). The current can be observed within the ‘window’ of voltage during cardiac repolarization or as a steady-state equilibrium current during voltage clamp. The window can be affected by changes to the activation and inactivation gating that result in widening of the voltage range. In addition to mutations and polymorphisms, there have been a number of physiological modulators identified including Ca2+, calmodulin and phosphorylation (discussed in Chen-Izu et al. 2015) that can increase the voltage range of the window (Zeng & Rudy, 1995).
The bursting of Na+ channels is a well-described gating mode where channels undergo a transient failure of channel inactivation. Maltsev and Undrovinas recorded INaL from heterologously expressed NaV1.5 in the absence of other isoforms (Maltsev & Undrovinas, 2006), showing that bursting channels were indeed of the same form as those underlying the transient inward Na+ current. Clancy et al. recorded and modelled the transitions from normally inactivating Na+ channels to bursting channels in heterologously expressed single NaV1.5 channels. A computational model based on these rates was then used to predict the magnitude and rate dependence of INaL expected from ensemble currents (Clancy et al. 2002).
Non-equilibrium gating describes another form of INaL that is not observed during typical voltage clamp depolarization protocols (Clancy et al. 2003). However, in response to a negative ramp current, a transient inward Na+ current is observed. The amplitude of the current is sensitively dependent on the rate of recovery from inactivation, where faster recovery or a shift in the voltage dependence of recovery from inactivation promotes the current. Just as for the window current, non-equilibrium INa is affected by a number of physiological modulators including calmodulin and phosphorylation (Herren et al. 2013; Chen-Izu et al. 2015).
Gating abnormalities of the cardiac Na+ channel were reported by Maltsev and Undrovinas in their description of a novel, ultraslow inactivating Na+ current, INaL, in both normal and failing human hearts (Maltsev et al. 1998a). The same group also showed an increased density and slower inactivation kinetics of INaL (Maltsev et al. 2007) in chronic heart failure as compared to normal hearts. In single channels, two modes of gating underlying late INa were observed: late scattered mode gating and burst mode of gating that had slower kinetics in failing human ventricular myocytes compared to non-failing ventricular myocytes (Maltsev & Undrovinas, 2006). Importantly, there were no differences in the unitary conductance of INaL current between normal and failing human hearts, suggesting a single population of channels in which this gating phenotype is upregulated in HF (Undrovinas et al. 2002).
Question or controversy: Na+ channel defects
What are the disease-specific mechanisms leading to pathological increases in INaL?
Are Na+ currents measured from human ‘non-failing’ myocytes the same as those from ‘normal’ myocytes?
Linking Na+ and Na+ channel abnormalities to arrhythmias
Disruptions in Na+-based processes in the heart foster arrhythmias by multiple mechanisms, depending on the specific perturbation to the Na+-linked process. Of major benefit to revealing and understanding the mechanisms of Na+-based arrhythmias is the development of numerous new experimental techniques including examples such as targeted subcellular imaging (Agarwal et al. 2011, 2014; Lederer et al. 2012; Despa et al. 2014), SR Ca2+ imaging (Wang et al. 2014), advances in electrophysiology (Horvath et al. 2013), ‘cell-in-gel’ and other techniques for mechanochemotransduction (Prosser et al. 2011; Jian et al. 2014), mitochondrial imaging (Zhou et al. 2014), and stem cell technologies (Matsa et al. 2014), just to name a few.
In addition to the advancement in experimental techniques, there have been significant gains in accessibility of modern computing power, computational speed and reduction in computing cost. Recent advances have also been made in numerical techniques and computing (Abramson et al. 2010; Rocha et al. 2011; Neic et al. 2012; Nivala et al. 2012), the implementation of customizable solvers such as Continuity, modelling platforms like CHASTE and OpenCMISS (Bernabeu et al. 2009; Bradley et al. 2011), and infrastructures aimed at facilitating standardization, interoperability and dissemination of models (e.g. CellML and FieldML; Bernabeu et al. 2009; Christie et al. 2009; Wimalaratne et al. 2009; Bradley et al. 2011; Quinn et al. 2011).
Mathematical models of cardiac physiology are widely used to complement experimental findings and clinical observations to improve understanding of cardiac electrical function in health and disease. Implementation of such models offers multiple advantages: In particular they enable exploration of high-dimensional models to determine how their range of dynamical behaviours corresponds to that of low-dimensional models. Emergent behaviours can be mapped back to underlying parameters through component dissection, to reveal mechanisms of emergent behaviours, a function for which there is no efficient comparable experimental counterpart.
Experimental approaches and computational modelling and simulation are complementary methods to determine how abnormalities in Na+ processes at the level of the cell can cause emergent arrhythmias in the whole heart. An example is a hallmark arrhythmia trigger in human heart failure resulting from Ca2+-induced DADs. When Na+ accumulation and Ca2+ overload occurs in cells, DADs arise because the cytosolic Ca2+ accumulation may ultimately exceed Ca2+ efflux via NCX and precipitate Ca2+ overload. A pathological version of the Ca2+-induced-Ca2+ release ensues, whereby there is spontaneous SR Ca2+ release leading to overloaded intracellular Ca2+ that is extruded by inward NCX current, which may depolarize the cell sufficiently to activate Na+ channels leading to the emergence of DADs and, if large enough, arrhythmogenically triggered action potentials. Because Na+-mediated Ca2+ overload does not occur uniformly in time or space, beat-to-beat variability in repolarization and emergent triggering of early after-depolarizations (EADs) and DADs occur unpredictably.
DADs occurring in a single myocyte are an insufficient source of current to trigger a premature beat in the whole heart because the current generated in a single cell is not enough to overcome the large electrotonically coupled downstream sink. Mathematical models and experiments in intact hearts have shown that DADs must occur simultaneously in many thousands of cells in order to generate an arrhythmia trigger (Hoeker et al. 2009; Xie et al. 2010; Myles et al. 2012). The precise mechanisms involved in the synchronization of DADs in many neighbouring cells remain to be resolved. However, there may be both local electrotonic (Sato et al. 2014) and Ca2+ handling effects, such as synchronous recovery of release channel function after the previous normal beat (Myles et al. 2012).
In the case of loss of Na+ channel function as described in the infarct border zone, a reduction in excitability at the cellular level emerges in coupled tissue as slowing of conduction velocity of the propagating depolarizing wave that drives cardiac excitation. Slow conduction can result in an increase in the ‘vulnerable window’ to result in unidirectional block and, if the conditions are favourable, retrograde conduction, promoting re-entrant arrhythmia in the organ (Mines, 1914; Allessie et al. 1973; Starmer et al. 1991, 1993; Starmer, 2002; Moreno et al. 2011). It is important to note that slow conduction, when promoted via use-dependent inhibition of INa by drugs for example, can dynamically prolong APD at the cellular level and QT interval at the organ level as a result of the intrinsic dynamical properties of Na+ channels and drug–channel interactions that give rise to the restitution relationship. The restitution relationship describes the correlation between APD and the preceding diastolic interval (DI). As the DI increases as a result of slow conduction, the subsequent AP will be relatively prolonged. If the DI is sufficiently long and other anomalies are present, reductions in repolarization reserve occur and even triggered arrhythmias such as early EADs may emerge (January & Riddle, 1989; Boutjdir & el-Sherif, 1991; Sicouri et al. 1997; Undrovinas & Maltsev, 2008; Zaza et al. 2008). Conversely, when the DI is very short, such as during rapid pacing or tachycardia and combined with other perturbations such as drugs or disease, the relationship between APD and DI may be very steep. In this situation, arrhythmogenic oscillation of the APD termed alternans can develop. All of these disruptions to normal cardiac electrical activity promote development of re-entrant arrhythmias and wavebreak causing fibrillation (Weiss et al. 2005).
The other effect of loss of Na+ channel function is slowed conduction that shortens the wavelength for re-entry. A slowing of the re-entrant wavefront acts to effectively increase the tissue size and may promote discordant alternans and allow induction and maintenance of arrhythmias (Weiss et al. 2011). Na+ channels that are altered by disease or drugs may exhibit slowed recovery from inactivation, which changes the conduction velocity (CV) restitution (the relationship describing the dependency of CV on the inter-beat interval) which combined with APD restitution and/or Ca2+ cycling instabilities promotes spatially discordant APD alternans to increase dispersion of refractoriness (Weiss et al. 2011; Sato & Clancy, 2013).
A gain of function of the Na+ channel during disease that results in an increased INaL has also been linked to arrhythmias associated with acquired diseases such as heart failure and post-myocardial infarction remodelling, due to their impact on action potential duration and repolarization abnormalities. Approximately 40% of chronic heart failure patients die due to sudden cardiac death, with ventricular tachycardia and fibrillation documented in 80% of patients (Nikolic et al. 1982; Bayes de Luna et al. 1989; Undrovinas & Maltsev, 2008). Conditions and diseases that lead to an increased INaL exhibit electrical instability (due to after-depolarizations, beat-to-beat variability in repolarization, ventricular arrhythmias), mechanical instability (impaired diastolic relaxation and ventricular wall tension, increased diastolic and decreased systolic force generation), as well as mitochondrial dysfunction. This sets up a cascade leading to further ischaemia and abnormal contraction in a pathological feedback loop. Failing canine ventricular myocytes with prolonged APs, Ca2+ transients and substantial diastolic Ca2+ accumulation leading to spontaneous Ca2+ release were shown to improve with TTX and ranolazine (a selective INaL inhibitor; Wasserstrom et al. 2009; Undrovinas et al. 2010; Antzelevitch et al. 2011). These results are additional strong indications of the link between pathological INaL to the induction of deranged Ca2+ homeostasis at the cellular level. A subsequent study using human ventricular myocytes (Maltsev et al. 1998a) similarly found improvement with TTX.
New therapeutic approaches for Na+-linked arrhythmias
In the session on ‘Therapeutics’, Robert Kass, Colleen Clancy, Luiz Belardinelli, András Varró, Lazlso Csernoch, Antonio Zaza, Bernard Fermini and Crystal Ripplinger presented and discussed current issues in the field. The section below summarizes these interactions.
As described above, both gain- and loss-of-function in the cardiac Na+ channel can result in dangerous proarrhythmic consequences by altering cardiac repolarization and conduction. Thus, the prospect of targeted pharmacological treatment to modify Na+-based arrhythmias has fuelled historical and recent pursuit of new drugs. However, the history of antiarrhythmic drug failures makes careful and reliable assessment of drug effects on cardiac rhythms a preclinical necessity to ensure safety and efficacy.
The difficulty in predicting drug effects on the electrical activity of the heart is clear from both the failure of large clinical trials to demonstrate drug safety for multiple antiarrhythmic drug classes (for example, the CAST (1989) and SWORD (Waldo et al. 1996) clinical trials), and from the market withdrawal of otherwise promising drugs for treating cardiac dysrhythm, psychiatric disorders, gastrointestinal symptoms and infection following unexpected sudden cardiac death (Drici & Barhanin, 2000). These events have resulted in a burdensome regulatory process for preclinical drugs that have discouraged the development and emergence of potentially therapeutic agents for clinical use.
The reasons that it has been so difficult to predict ion channel-targeting drug effects on cardiac electrical activity are that most antiarrhythmic drugs have complex interactions with multiple channels, conformational state specificity, bioactive metabolites and neutral and charged drug fractions. Drugs alter the action potential waveform, which in turn affects drug potency. Thus, it is extremely difficult to know how intended antiarrhythmic drugs that primarily target ion channels will alter emergent electrical activity in the whole heart.
In the context of pharmacology that is now being developed to target pathologically enhanced INaL, it must be emphasized that selective INaL inhibition needs to be carefully determined (Szel et al. 2011; Belardinelli et al. 2013). In order to assess the specificity of INaL inhibition, the effect of a candidate drug needs to be thoroughly screened for its effects on both peak and late INa. Because peak INa may be differentially affected by drugs at various heart rates or diastolic voltages, the screening tests must include an array of assessments to ensure that selectivity of INaL versus peak INa persists under a variety of physiological conditions. For example, drugs that have relatively rapid unbinding kinetics may not exhibit obvious effects on peak INa unless the stimulation rate is sufficiently high. Moreover, new drugs aimed at selective inhibition of INaL must be effective in cardiac disease states where hyperkalaemia, hypoxia, acidosis and ischaemia are likely.
Question or controversy: predicting therapy
How can critical determinants of drug effects in the whole heart be predicted from drug targets, kinetics and active metabolites?
Selective INaL inhibition needs to be clearly defined and careful determination of drug effects on peak INa needs to be considered at multiple stimulation frequencies.
Recently, the US Food and Drug Administration (FDA) and other stakeholders have suggested the potential implementation of a new pro-arrhythmia safety protocol that includes testing for the effects of drugs on multiple cardiac ion channels and integration of this information using computational modelling and simulation approaches, and stem cell technologies (Chi, 2013).
Modelling and simulations for predictive pharmacology
Cardiac modelling and simulation has recently been utilized to investigate mechanisms of Na+ channel blocking drugs that both reduce peak Na+ current and that specifically target INaL. A recent study investigated lidocaine (lignocaine) effects in a multiscale computational model (Cardona et al. 2010). The authors demonstrated both anti-fibrillatory effects in normal tissue and predicted the potential for pro-arrhythmia with lidocaine during pathologies including acidosis and ischaemia, as has also been demonstrated experimentally due to differential effects of drugs on channels from the normal and infarct border zones (Pu et al. 1998).
Moreno et al. (2011) also implemented modelling and simulation approaches to investigate the mechanisms of failure of the once promising antiarrhythmic drug flecainide, the subject of the cardiac arrhythmia suppression trial (CAST), which, in the clinical trial showed increased mortality with flecainide over placebo (CAST, 1989). In the computational modelling and simulation study, the dynamical complexity of the drug kinetics was modelled for both charged and neutral drug fractions. After developing the drug–channel model, a simulation in cells first confirmed experimental findings: no overt pro-arrhythmic potential was ever observed at the cellular level (Moreno et al. 2011). In tissue level simulations, the outcome was very different. Substantial use-dependent block by flecainide (an intrinsic dynamical property of channel block) was predicted in the model to result in failed impulse conduction, a higher dimensional phenomenon that emerged as a result of increased electrotonic load in coupled tissue. Pro-arrhythmic conduction block led to development of tachycardia indicated by spiral wave re-entry, which was verified experimentally (Moreno et al. 2011). This emergent phenomenon was linked back to the fundamental mechanism: the drug kinetics of unblock, identified as the basic mechanism of failure. Moreover, the study indicated that the kinetics of drug interactions for lidocaine promoted safety in higher dimensions as indicated by no re-entrant arrhythmias in the presence of lidocaine in normal tissue.
Question or controversy: modelling and simulation for predictive therapy
It is still not possible to predictively link structural perturbations to pharmacological effects in the whole heart.
How to make the model, ‘as simple as possible, but not simpler’?
Disease-induced enhancement of INaL promotes the development of arrhythmogenic after-depolarizations, triggered arrhythmic activity, and torsades de pointes in cardiac ventricular myocytes, cardiac tissue and intact hearts (Boutjdir & el-Sherif, 1991; Sicouri et al. 1997; Clancy & Rudy, 1999; Song et al. 2004; Wu et al. 2006). Pharmacological targeting of INaL has been shown to improve cardiac electrical function in myocytes challenged by cardiac glycosides, hydrogen peroxide, pharmacological enhancement of INaL, and even with drugs that block hERG channels (IKr; the rapidly activating component of the delayed rectifier potassium current) and reduce repolarization reserve (Ver Donck et al. 1993; Haigney et al. 1994; Le Grand et al. 1995; Sicouri et al. 1997; Song et al. 2004, 2006; Wu et al. 2006; Undrovinas & Maltsev, 2008; Sossalla et al. 2010; Wu et al. 2011).
Recently, modelling and simulation have been used to probe and predict effects of the selective INaL inhibitor ranolazine in pathological situations (Moreno et al. 2013). Simulations of clinically relevant concentrations of drug were used to predict the cellular level effects of Na+ channel blockade using both ranolazine and its active metabolites on hERG, which have potent blocking effects in the therapeutically relevant range. The model was used to predict if therapeutic effects of targeted pharmacological treatment by ranolazine prevailed over the unintended pathological block of hERG for normalizing arrhythmia triggers (EADs) in bradycardia-dependent arrhythmias in LQT3, as well as tachyarrhythmogenic triggers arising from heart failure-induced remodelling (e.g. DADs). Model predictions suggested that acute targeting of INaL with ranolazine is an effective therapeutic strategy in diverse arrhythmia-provoking situations that arise from a common pathway of increased pathological INaL.
Trenor et al. developed a tool for in silico preclinical anti-arrhythmic drug safety assessment that predicted the impact of the IKr/INaL ratio of steady-state block of drug candidates on ‘torsadogenic’ biomarkers that they defined as AP duration, triangulation, reverse rate dependence, transmural dispersion of repolarization and electrocardiogram QT interval (Trenor et al. 2013).
Although the studies described above included detailed descriptions of the kinetics of drug interactions with ion channels, it is important to note that even detailed kinetic models are phenomenological, for example a Markov model of ion channel dynamics or drug channel interactions is a phenomenological representation that greatly simplifies the underlying molecular mechanics. In the studies described above, it is difficult, if not impossible, to predictively link atomic scale anomalies to higher order phenomena, or to predict how structural perturbations might affect pharmacological effects in the whole heart. An important aspect of modelling and simulation as it relates to the prediction of disease processes and pharmacology is choosing the level of detail in the model. There must be a match between the required complexity of the model and its predictive capacity, so as not to introduce unnecessary degrees of freedom that result in vastly over-determined models. In other words, the modeller must be concerned with the issue of determining how to, as Einstein suggested, make the model ‘as simple as possible, but not simpler.’
Stem cells for predictive pharmacology
The potential for personalized medicine via drug screening in patients’ own induced pluripotent stem cell (hiPSC)-derived cardiomyocytes (hiPSC-CMs) (Braam et al. 2010) is another developing and exciting application at the interface of molecular and clinical information (Liang et al. 2013; Sallam et al. 2014). Patient-specific hiPSC-CMs containing unknown genotype profiles, or with known polymorphisms and/or mutations in cardiac ion channels can be used to qualitatively and quantitatively assess variability in drug responses (Matsa et al. 2014). Thus far, iPSC-CMs have been used to successfully model arrhythmic disorders, with excellent agreement between altered cardiac channel function and emergent electrophysiological phenotypes in the inherited long QT syndromes and catecholaminergic polymorphic ventricular tachycardia (Sallam et al. 2014). Indeed, the ion channel, Ca2+ handling and signalling in hiPSC-CMs still differ substantially from those in adult cardiac myocytes.
Question or controversy: patient-derived induced pluripotent stem cell (hiPSC)-derived cardiomyocytes
Ca2+ handling and signalling in hiPSC-CMs still differ substantially from those in adult cardiac myocytes.
Many challenges need to be overcome before stem cell technologies enter the mainstream for drug screening and therapy.
Terrenoire et al. recently demonstrated the usefulness of such an approach in a study where they derived iPSCs from a long QT syndrome patient with complex genetics (Terrenoire et al. 2013). They identified a de novo mutation in the SCN5A (F1473C) gene encoding the NaV1.5, and a polymorphism (K897T) in KCNH2, the gene encoding hERG. Biophysical and pharmacological analysis of ion channels expressed in iPSC-CMs demonstrated that the disease was a primary consequence of the NaV1.5 defect and was not influenced by the KCNH2 polymorphism. The mutation resulted in a gain-of-function in INaL, which resulted in delayed repolarization, a prolonged QT interval, and increased risk of arrhythmia. They also found a uniquely steep fast-rate-dependent reduction in INaL that especially facilitated pharmacological inhibition by the Na+ channel inhibitor mexiletine. Of critical importance, the experiments revealed rate-dependent properties of ion currents and drug interactions that were unique to the patient's iPSC-CMs, and that were corroborated in a successful patient treatment regimen. This study is an example of the potential of iPSC-CM approaches in developing patient-specific clinical regimens (Terrenoire et al. 2013).
While the study described above focused on a gain of function perturbation in the Na+ channel, and its cellular level effects, iPSC-CMs can also be cultured in monolayer or grown on scaffolds to investigate patient-specific metrics related to loss of Na+ channel function, especially conduction velocity. For example, measurements of voltage wavefronts in monolayers of iPSC-CMs via optical mapping has recently been demonstrated (Chen et al. 2011).
The potential for expansion of stem cell technologies in the cardiac therapeutic arena is vast (Liang et al. 2013; Mordwinkin et al. 2013). For example, these cells may prove extremely useful to reveal some of the most basic variations in drug responses that might include the influence of sex, polytherapy, hormones and drug effects in the context of genetically based diseases. Examples include the apparent differential effects of hERG blockade in males and females, oral contraceptive effects on cardiac risk, or to determine the electrophysiological effects of β-blockers in LQT3 patients (Kurokawa et al. 2008; Ahrens-Nicklas et al. 2009; Abu-Zeitone et al. 2014). However, many challenges still need to be overcome in order for stem cell technologies to enter the mainstream for screening and therapy. Toward this goal, best practices are in development to improve the maturity and homogeneity of electrical activity in iPSC-derived myocytes (Bett et al. 2013; Hulot et al. 2014).
Summary
In order to begin to translate the new findings on Na+ derangement in the heart and its link to disease processes to effective patient interventions, there must be a specific and concerted collaboration between the many scientific investigators at the bench, the computer terminal and the bedside. Such an effort would allow more immediate application of critical research findings and predictions to more rapidly improve outcomes for patients with cardiac disease states that are marked by disruptions in ion homeostasis. Consideration of patient-specific disease and therapeutics should be part of the consortium effort. A critical requirement for developing interdisciplinary efforts to promote understanding and knowledge among stakeholders in the field is to ‘get them together in a room’ for discussion and debate regarding the most critical aspects that need to be addressed. Indeed, the UC Davis Cardiovascular Symposium in 2014 on Na+ in the heart aimed to do exactly this: bring together the world's topical experts to engage in scientific dialogue and debate to move the field forward.
Acknowledgments
We thank the meeting organizers, Advisory Board and all conference participants for their contributions to the scientific exchange and constructive discussions. For conference information see https://basicscience.ucdmc.ucdavis.edu/ucd-cvs-2014/index.html.
Glossary
- AP
action potential
- APD
action potential duration
- DAD
delayed after-depolarization
- EAD
early after-depolarization
- EBZ
epicardial border zone
- HF
heart failure
- INa
Na+ current
- INaL
late Na+ current
- iPSC-CMs
induced pluripotent stem cell-derived cardiomyocytes
- LQT3
long QT type 3 syndrome
- NBC
sodium–bicarbonate symporter
- NCX
sodium–calcium exchanger
- NHE
sodium–hydrogen exchanger
- NKA
sodium–potassium ATPase
- ROS
reactive oxygen species
- SR
sarcoplasmic reticulum
Additional information
Competing interests
None declared.
Funding
The authors acknowledge grant support from the National Institutes of Health (P01-HL080101 to D.M.B.) and additional grants to other authors that allowed them to participate. The authors would also like to acknowledge the many participating sponsors that support the UC Davis Cardiovascular Symposium. Detailed sponsor information can be found on the conference website.
References
- Abramson D, Bernabeu MO, Bethwaite B, Burrage K, Corrias A, Enticott C, Garic S, Gavaghan D, Peachey T, Pitt-Francis J, Pueyo E, Rodriguez B, Sher A. Tan J. High-throughput cardiac science on the Grid. Philos Trans A Math Phys Eng Sci. 2010;368:3907–3923. doi: 10.1098/rsta.2010.0170. [DOI] [PubMed] [Google Scholar]
- Abu-Zeitone A, Peterson DR, Polonsky B, McNitt S. Moss AJ. Oral contraceptive use and the risk of cardiac events in patients with long QT syndrome. Heart Rhythm. 2014;11:1170–1175. doi: 10.1016/j.hrthm.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, MacDougall DA, Tyser R, Pugh SD, Calaghan SC. Harvey RD. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J Mol Cell Cardiol. 2011;50:500–509. doi: 10.1016/j.yjmcc.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Yang PC, Rice M, Singer CA, Nikolaev VO, Lohse MJ, Clancy CE. Harvey RD. Role of membrane microdomains in compartmentation of cAMP signaling. PLoS One. 2014;9:e95835. doi: 10.1371/journal.pone.0095835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal R. Boyden PA. Diminished Ca2+ and Ba2+ currents in myocytes surviving in the epicardial border zone of the 5-day infarcted canine heart. Circ Res. 1995;77:1180–1191. doi: 10.1161/01.res.77.6.1180. [DOI] [PubMed] [Google Scholar]
- Ahrens-Nicklas RC, Clancy CE. Christini DJ. Re-evaluating the efficacy of β-adrenergic agonists and antagonists in long QT-3 syndrome through computational modelling. Cardiovasc Res. 2009;82:439–447. doi: 10.1093/cvr/cvp083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen PD, Schmidt TA, Marsh JD. Kjeldsen K. Na,K-ATPase expression in normal and failing human left ventricle. Basic Res Cardiol. 1992;87(Suppl. 1):87–94. doi: 10.1007/978-3-642-72474-9_7. [DOI] [PubMed] [Google Scholar]
- Allessie MA, Bonke FI. Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of trachycardia. Circ Res. 1973;33:54–62. [PubMed] [Google Scholar]
- Antzelevitch C, Burashnikov A, Sicouri S. Belardinelli L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011;8:1281–1290. doi: 10.1016/j.hrthm.2011.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avkiran M. Basic biology and pharmacology of the cardiac sarcolemmal sodium/hydrogen exchanger. J Card Surg. 2003;18(Suppl. 1):3–12. doi: 10.1046/j.1540-8191.18.s1.2.x. [DOI] [PubMed] [Google Scholar]
- Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R. Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–1024. doi: 10.1016/s0008-6363(02)00809-x. [DOI] [PubMed] [Google Scholar]
- Baba S, Dun W, Cabo C. Boyden PA. Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation. 2005;112:2386–2396. doi: 10.1161/CIRCULATIONAHA.105.534784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetz D, Bernard M, Pinet C, Tamareille S, Chattou S, El Banani H, Coulombe A. Feuvray D. Different pathways for sodium entry in cardiac cells during ischemia and early reperfusion. Mol Cell Biochem. 2003;242:115–120. [PubMed] [Google Scholar]
- Bay J, Kohlhaas M. Maack C. Intracellular Na+ and cardiac metabolism. J Mol Cell Cardiol. 2013;61:20–27. doi: 10.1016/j.yjmcc.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Bayes de Luna A, Coumel P. Leclercq JF. Ambulatory sudden cardiac death: mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am Heart J. 1989;117:151–159. doi: 10.1016/0002-8703(89)90670-4. [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Liu G, Smith-Maxwell C, Wang WQ, El-Bizri N, Hirakawa R, Karpinski S, Li CH, Hu L, Li XJ, Crumb W, Wu L, Koltun D, Zablocki J, Yao L, Dhalla AK, Rajamani S. Shryock JC. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J Pharmacol Exp Ther. 2013;344:23–32. doi: 10.1124/jpet.112.198887. [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC. Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl. 4):iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N. George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Bernabeu MO, Bordas R, Pathmanathan P, Pitt-Francis J, Cooper J, Garny A, Gavaghan DJ, Rodriguez B, Southern JA. Whiteley JP. CHASTE: incorporating a novel multi-scale spatial and temporal algorithm into a large-scale open source library. Philos Trans A Math Phys Eng Sci. 2009;367:1907–1930. doi: 10.1098/rsta.2008.0309. [DOI] [PubMed] [Google Scholar]
- Bers DM, Barry WH. Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- Bett GC, Kaplan AD, Lis A, Cimato TR, Tzanakakis ES, Zhou Q, Morales MJ. Rasmusson RL. Electronic ‘expression’ of the inward rectifier in cardiocytes derived from human-induced pluripotent stem cells. Heart Rhythm. 2013;10:1903–1910. doi: 10.1016/j.hrthm.2013.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP. Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Boink GJ, Lau DH, Shlapakova IN, Sosunov EA, Anyukhovsky EP, Driessen HE, Dun W, Chen M, Danilo P, Jr, Rosen TS, Ozgen N, Duffy HS, Kryukova Y, Boyden PA, Robinson RB, Brink PR, Cohen IS. Rosen MR. SkM1 and Cx32 improve conduction in canine myocardial infarcts yet only SkM1 is antiarrhythmic. Cardiovasc Res. 2012;94:450–459. doi: 10.1093/cvr/cvs107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt J, Ai X, Moorman JR, Pogwizd SM. Bers DM. Expression and phosphorylation of the Na-pump regulatory subunit phospholemman in heart failure. Circ Res. 2005;97:558–565. doi: 10.1161/01.RES.0000181172.27931.c3. [DOI] [PubMed] [Google Scholar]
- Boutjdir M. el-Sherif N( Pharmacological evaluation of early afterdepolarisations induced by sea anemone toxin (ATXII) in dog heart. Cardiovasc Res. 1991;25:815–819. doi: 10.1093/cvr/25.10.815. [DOI] [PubMed] [Google Scholar]
- Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R. Mummery CL. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010;4:107–116. doi: 10.1016/j.scr.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Bradley C, Bowery A, Britten R, Budelmann V, Camara O, Christie R, Cookson A, Frangi AF, Gamage TB, Heidlauf T, Krittian S, Ladd D, Little C, Mithraratne K, Nash M, Nickerson D, Nielsen P, Nordbo O, Omholt S, Pashaei A, Paterson D, Rajagopal V, Reeve A, Rohrle O, Safaei S, Sebastian R, Steghofer M, Wu T, Yu T, Zhang HY. Hunter P. OpenCMISS: A multi-physics & multi-scale computational infrastructure for the VPH/Physiome project. Prog Biophys Mol Biol. 2011;107:32–47. doi: 10.1016/j.pbiomolbio.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Cabo C. Boyden PA. Electrical remodeling of the epicardial border zone in the canine infarcted heart: a computational analysis. Am J Physiol Heart Circ Physiol. 2003;284:H372–H384. doi: 10.1152/ajpheart.00512.2002. [DOI] [PubMed] [Google Scholar]
- Cardona K, Trenor B, Molto G, Martinez M, Ferrero JM, Jr, Starmer F. Saiz J. Exploring the role of pH in modulating the effects of lidocaine in virtual ischemic tissue. Am J Physiol Heart Circ Physiol. 2010;299:H1615–H1624. doi: 10.1152/ajpheart.00425.2010. [DOI] [PubMed] [Google Scholar]
- CAST. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N Engl J Med. 1989;321:406–412. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- Chen A, Lieu DK, Freschauf L, Lew V, Sharma H, Wang J, Nguyen D, Karakikes I, Hajjar RJ, Gopinathan A, Botvinick E, Fowlkes CC, Li RA. Khine M. Shrink-film configurable multiscale wrinkles for functional alignment of human embryonic stem cells and their cardiac derivatives. Adv Mater. 2011;23:5785–5791. doi: 10.1002/adma.201103463. [DOI] [PubMed] [Google Scholar]
- Chen-Izu, et al. 2015.
- Chi KR. Revolution dawning in cardiotoxicity testing. Nat Rev Drug Discov. 2013;12:565–567. doi: 10.1038/nrd4083. [DOI] [PubMed] [Google Scholar]
- Christie GR, Nielsen PMF, Blackett SA, Bradley CP. Hunter PJ. FieldML: concepts and implementation. Philos Trans A Math Phys Eng Sci. 2009;367:1869–1884. doi: 10.1098/rsta.2009.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE. Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999;400:566–569. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]
- Clancy CE, Tateyama M. Kass RS. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J Clin Invest. 2002;110:1251–1262. doi: 10.1172/JCI15928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE, Tateyama M, Liu H, Wehrens XH. Kass RS. Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation. 2003;107:2233–2237. doi: 10.1161/01.CIR.0000069273.51375.BD. [DOI] [PubMed] [Google Scholar]
- Coronel R, Lau DH, Sosunov EA, Janse MJ, Danilo P, Jr, Anyukhovsky EP, Wilms-Schopman FJ, Opthof T, Shlapakova IN, Ozgen N, Prestia K, Kryukova Y, Cohen IS, Robinson RB. Rosen MR. Cardiac expression of skeletal muscle sodium channels increases longitudinal conduction velocity in the canine 1-week myocardial infarction. Heart Rhythm. 2010;7:1104–1110. doi: 10.1016/j.hrthm.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S. Bers DM. Na+ transport in the normal and failing heart – remember the balance. J Mol Cell Cardiol. 2013;61:2–10. doi: 10.1016/j.yjmcc.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, Pogwizd SM. Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Despa S, Shui B, Bossuyt J, Lang D, Kotlikoff MI. Bers DM. Junctional cleft [Ca2+]i measurements using novel cleft-targeted Ca2+ sensors. Circ Res. 2014;115:339–347. doi: 10.1161/CIRCRESAHA.115.303582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drici MD. Barhanin J. Cardiac K+ channels and drug-acquired long QT syndrome. Therapie. 2000;55:185–193. [PubMed] [Google Scholar]
- Dun W, Baba S, Yagi T. Boyden PA. Dynamic remodeling of K+ and Ca2+ currents in cells that survived in the epicardial border zone of canine healed infarcted heart. Am J Physiol Heart Circ Physiol. 2004;287:H1046–H1054. doi: 10.1152/ajpheart.00082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigel BN. Hadley RW. Contribution of the Na+ channel and Na+/H+ exchanger to the anoxic rise of [Na+] in ventricular myocytes. Am J Physiol Heart Circ Physiol. 1999;277:H1817–H1822. doi: 10.1152/ajpheart.1999.277.5.H1817. [DOI] [PubMed] [Google Scholar]
- Faber GM. Rudy Y. Action potential and contractility changes in [Na+]i overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudron P, Kugler I, Hu K, Bauer W, Eilles C. Ertl G. Time course of cardiac structural, functional and electrical changes in asymptomatic patients after myocardial infarction: their inter-relation and prognostic impact. J Am Coll Cardiol. 2001;38:33–40. doi: 10.1016/s0735-1097(01)01319-5. [DOI] [PubMed] [Google Scholar]
- Glitsch HG( Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol Rev. 2001;81:1791–1826. doi: 10.1152/physrev.2001.81.4.1791. [DOI] [PubMed] [Google Scholar]
- Goldberger JJ, Basu A, Boineau R, Buxton AE, Cain ME, Canty JM, Jr, Chen PS, Chugh SS, Costantini O, Exner DV, Kadish AH, Lee B, Lloyd-Jones D, Moss AJ, Myerburg RJ, Olgin JE, Passman R, Stevenson WG, Tomaselli GF, Zareba W, Zipes DP. Zoloth L. Risk stratification for sudden cardiac death: a plan for the future. Circulation. 2014;129:516–526. doi: 10.1161/CIRCULATIONAHA.113.007149. [DOI] [PubMed] [Google Scholar]
- Grandi E, Pasqualini FS. Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48:112–121. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigney MC, Lakatta EG, Stern MD. Silverman HS. Sodium channel blockade reduces hypoxic sodium loading and sodium-dependent calcium loading. Circulation. 1994;90:391–399. doi: 10.1161/01.cir.90.1.391. [DOI] [PubMed] [Google Scholar]
- Hale SL, Shryock JC, Belardinelli L, Sweeney M. Kloner RA. Late sodium current inhibition as a new cardioprotective approach. J Mol Cell Cardiol. 2008;44:954–967. doi: 10.1016/j.yjmcc.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Herren AW, Bers DM. Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol. 2013;305:H431–H445. doi: 10.1152/ajpheart.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D, Nagel G. Gadsby DC. De Weer P. Kaplan JH. The Sodium Pump: Recent Developments. New York: Rockefeller University Press; 1991. Na/K pump currents in giant membrane patches excised from ventricular myocytes; pp. 543–547. [Google Scholar]
- Hoeker GS, Katra RP, Wilson LD, Plummer BN. Laurita KR. Spontaneous calcium release in tissue from the failing canine heart. Am J Physiol Heart Circ Physiol. 2009;297:H1235–H1242. doi: 10.1152/ajpheart.01320.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath B, Banyasz T, Jian Z, Hegyi B, Kistamas K, Nanasi PP, Izu LT. Chen-Izu Y. Dynamics of the late Na+ current during cardiac action potential and its contribution to afterdepolarizations. J Mol Cell Cardiol. 2013;64:59–68. doi: 10.1016/j.yjmcc.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulot JS, Stillitano F, Salem JE, Kovacic JC, Fuster V. Hajjar RJ. Considerations for pre-clinical models and clinical trials of pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther. 2014;5:1. doi: 10.1186/scrt390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hund TJ, Decker KF, Kanter E, Mohler PJ, Boyden PA, Schuessler RB, Yamada KA. Rudy Y. Role of activated CaMKII in abnormal calcium homeostasis and INa remodeling after myocardial infarction: insights from mathematical modeling. J Mol Cell Cardiol. 2008;45:420–428. doi: 10.1016/j.yjmcc.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse MJ. Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049–1169. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- January C. Riddle J. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- Jian Z, Han H, Zhang T, Puglisi J, Izu LT, Shaw JA, Onofiok E, Erickson JR, Chen YJ, Horvath B, Shimkunas R, Xiao W, Li Y, Pan T, Chan J, Banyasz T, Tardiff JC, Chiamvimonvat N, Bers DM, Lam KS. Chen-Izu Y. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal. 2014;7:ra27. doi: 10.1126/scisignal.2005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O'Rourke B. Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhaas M. Maack C. Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation. 2010;122:2273–2280. doi: 10.1161/CIRCULATIONAHA.110.968057. [DOI] [PubMed] [Google Scholar]
- Kondo RP, Wang SY, John SA, Weiss JN. Goldhaber JI. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J Mol Cell Cardiol. 2000;32:1859–1872. doi: 10.1006/jmcc.2000.1220. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Tamagawa M, Harada N, Honda S, Bai CX, Nakaya H. Furukawa T. Acute effects of oestrogen on the guinea pig and human IKr channels and drug-induced prolongation of cardiac repolarization. J Physiol. 2008;586:2961–2973. doi: 10.1113/jphysiol.2007.150367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer WJ, Hagen BM. Zhao G. Cell biology. Superresolution subspace signaling. Science. 2012;336:546–547. doi: 10.1126/science.1222540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem CH, Lagadic-Gossmann D. Vaughan-Jones RD. Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J Physiol. 1999;517:159–180. doi: 10.1111/j.1469-7793.1999.0159z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand B, Vie B, Talmant JM, Coraboeuf E. John GW. Alleviation of contractile dysfunction in ischemic hearts by slowly inactivating Na+ current blockers. Am J Physiol Heart Circ Physiol. 1995;269:H533–H540. doi: 10.1152/ajpheart.1995.269.2.H533. [DOI] [PubMed] [Google Scholar]
- Li L, Louch WE, Niederer SA, Aronsen JM, Christensen G, Sejersted OM. Smith NP. Sodium accumulation in SERCA knockout-induced heart failure. Biophys J. 2012;102:2039–2048. doi: 10.1016/j.bpj.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, Nguyen PK, Bers DM, Robbins RC. Wu JC. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677–1691. doi: 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Brown DA. O'Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol. 2010;49:728–736. doi: 10.1016/j.yjmcc.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. O'Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Takimoto E, Dimaano VL, Demazumder D, Kettlewell S, Smith GL, Sidor A, Abraham T. O'Rourke B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a guinea pig model of heart failure. Circ Res. 2014;115:44–54. doi: 10.1161/CIRCRESAHA.115.303062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Hougen K, Mørk HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G. Sejersted OM. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J Physiol. 2010;588:465–478. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Stokke MK, Sjaastad I, Christensen G. Sejersted OM. No rest for the weary: diastolic calcium homeostasis in the normal and failing myocardium. Physiology (Bethesda) 2012;27:308–323. doi: 10.1152/physiol.00021.2012. [DOI] [PubMed] [Google Scholar]
-
Lue WM. Boyden PA. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in
 and the transient outward current. Circulation. 1992;85:1175–1188. doi: 10.1161/01.cir.85.3.1175. [DOI] [PubMed] [Google Scholar]
and the transient outward current. Circulation. 1992;85:1175–1188. doi: 10.1161/01.cir.85.3.1175. [DOI] [PubMed] [Google Scholar] - Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T. O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M. Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998a;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Tanimura M, Lesch M, Goldstein S. Undrovinas AI. Relationship between action potential, contraction-relaxation pattern, and intracellular Ca2+ transient in cardiomyocytes of dogs with chronic heart failure. Cell Mol Life Sci. 1998b;54:597–605. doi: 10.1007/s000180050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Silverman N, Sabbah HN. Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA. Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116–127. doi: 10.1016/j.cardiores.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsa E, Sallam K. Wu JC. Cardiac stem cell biology: glimpse of the past, present, and future. Circ Res. 2014;114:21–27. doi: 10.1161/CIRCRESAHA.113.302895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mines G. On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. Trans R Soc Can. 1914;8:43–53. [Google Scholar]
- Mordwinkin NM, Lee AS. Wu JC. Patient-specific stem cells and cardiovascular drug discovery. JAMA. 2013;310:2039–2040. doi: 10.1001/jama.2013.282409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JD, Yang PC, Bankston JR, Grandi E, Bers DM, Kass RS. Clancy CE. Ranolazine for congenital and acquired late INa-linked arrhythmias: in silico pharmacological screening. Circ Res. 2013;113:e50–61. doi: 10.1161/CIRCRESAHA.113.301971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JD, Zhu ZI, Yang PC, Bankston JR, Jeng MT, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, Ripplinger CM, Kass RS. Clancy CE. A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci Transl Med. 2011;3:98ra83. doi: 10.1126/scitranslmed.3002588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Konno T, Ozawa T, Murata M, Imoto K. Nagayama K. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-sensitivity. Biochemistry. 2000;39:1316–1323. doi: 10.1021/bi9912600. [DOI] [PubMed] [Google Scholar]
- Myles RC, Wang L, Kang C, Bers DM. Ripplinger CM. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. 2012;110:1454–1464. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neic A, Liebmann M, Hoetzl E, Mitchell L, Vigmond EJ, Haase G. Plank G. Accelerating cardiac bidomain simulations using graphics processing units. IEEE Trans Biomed Eng. 2012;59:2281–2290. doi: 10.1109/TBME.2012.2202661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel A, Kohlhaas M. Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. 2014;73:26–33. doi: 10.1016/j.yjmcc.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Nikolic G, Bishop RL. Singh JB. Sudden death recorded during Holter monitoring. Circulation. 1982;66:218–225. doi: 10.1161/01.cir.66.1.218. [DOI] [PubMed] [Google Scholar]
- Nivala M, de Lange E, Rovetti R. Qu Z. Computational modeling and numerical methods for spatiotemporal calcium cycling in ventricular myocytes. Front Physiol. 2012;3:114. doi: 10.3389/fphys.2012.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D. Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieske B, Maier LS, Piacentino V, 3rd, Weisser J, Hasenfuss G. Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- Prosser BL, Ward CW. Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- Pu J, Balser JR. Boyden PA. Lidocaine action on Na+ currents in ventricular myocytes from the epicardial border zone of the infarcted heart. Circ Res. 1998;83:431–440. doi: 10.1161/01.res.83.4.431. [DOI] [PubMed] [Google Scholar]
- Pu J. Boyden PA. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res. 1997;81:110–119. doi: 10.1161/01.res.81.1.110. [DOI] [PubMed] [Google Scholar]
- Quinn TA, Granite S, Allessie MA, Antzelevitch C, Bollensdorff C, Bub G, Burton RAB, Cerbai E, Chen PS, Delmar M, DiFrancesco D, Earm YE, Efimov IR, Egger M, Entcheva E, Fink M, Fischmeister R, Franz MR, Garny A, Giles WR, Hannes T, Harding SE, Hunter PJ, Iribe G, Jalife J, Johnson CR, Kass RS, Kodama I, Koren G, Lord P, Markhasin VS, Matsuoka S, McCulloch AD, Mirams GR, Morley GE, Nattel S, Noble D, Olesen SP, Panfilov AV, Trayanova NA, Ravens U, Richard S, Rosenbaum DS, Rudy Y, Sachs F, Sachse FB, Saint DA, Schotten U, Solovyova O, Taggart P, Tung L, Varro A, Volders PG, Wang K, Weiss JN, Wettwer E, White E, Wilders R, Winslow RL. Kohl P. Minimum Information about a Cardiac Electrophysiology Experiment (MICEE): Standardised reporting for model reproducibility, interoperability, and data sharing. Prog Biophys Mol Biol. 2011;107:4–10. doi: 10.1016/j.pbiomolbio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha BM, Campos FO, Amorim RM, Plank G, dos Santos RW, Liebmann M. Haase G. Accelerating cardiac excitation spread simulations using graphics processing units. Concurr Comp-Pract E. 2011;23:708–720. [Google Scholar]
- Sallam K, Kodo K. Wu JC. Modeling inherited cardiac disorders. Circ J. 2014;78:784–794. doi: 10.1253/circj.cj-14-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D, Bartos DC, Ginsburg KS. Bers DM. Depolarization of cardiac membrane potential synchronizes calcium sparks and waves in tissue. Biophys J. 2014;107:1313–1317. doi: 10.1016/j.bpj.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D. Clancy CE. Cardiac electrophysiological dynamics from the cellular level to the organ level. Biomed Eng Comput Biol. 2013;5:69–75. doi: 10.4137/BECB.S10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillinger W, Teucher N, Christians C, Kohlhaas M, Sossalla S, Van Nguyen P, Schmidt AG, Schunck O, Nebendahl K, Maier LS, Zeitz O. Hasenfuss G. High intracellular Na+ preserves myocardial function at low heart rates in isolated myocardium from failing hearts. Eur J Heart Fail. 2006;8:673–680. doi: 10.1016/j.ejheart.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Schwinger RH, Wang J, Frank K, Müller-Ehmsen J, Brixius K, McDonough AA. Erdmann E. Reduced sodium pump α1, α3, and β1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation. 1999;99:2105–2112. doi: 10.1161/01.cir.99.16.2105. [DOI] [PubMed] [Google Scholar]
- Shattock, et al. 2015.
- Sicouri S, Antzelevitch D, Heilmann C. Antzelevitch C. Effects of sodium channel block with mexiletine to reverse action potential prolongation in in vitro models of the long term QT syndrome. J Cardiovasc Electrophysiol. 1997;8:1280–1290. doi: 10.1111/j.1540-8167.1997.tb01019.x. [DOI] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Wagner S, Maier LS. Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Wu L. Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, Schmitto JD, Seipelt R, Schondube FA, Hasenfuss G, Belardinelli L. Maier LS. Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–2342. doi: 10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- Starmer CF. How antiarrhythmic drugs increase the rate of sudden cardiac death. Int J Bifurcat Chaos. 2002;12:1953–1968. [Google Scholar]
- Starmer CF, Biktashev VN, Romashko DN, Stepanov MR, Makarova ON. Krinsky VI. Vulnerability in an excitable medium: analytical and numerical studies of initiating unidirectional propagation. Biophys J. 1993;65:1775–1787. doi: 10.1016/S0006-3495(93)81233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starmer CF, Lastra AA, Nesterenko VV. Grant AO. Proarrhythmic response to sodium-channel blockade. Theoretical model and numerical experiments. Circulation. 1991;84:1364–1377. doi: 10.1161/01.cir.84.3.1364. [DOI] [PubMed] [Google Scholar]
- Swift F, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, Louch WE, Sjaastad I. Sejersted OM. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase α2-isoform in heart failure. Cardiovasc Res. 2008;78:71–78. doi: 10.1093/cvr/cvn013. [DOI] [PubMed] [Google Scholar]
- Szel T, Koncz I, Jost N, Baczko I, Husti Z, Virag L, Bussek A, Wettwer E, Ravens U, Papp JG. Varro A. Class I/B antiarrhythmic property of ranolazine, a novel antianginal agent, in dog and human cardiac preparations. Eur J Pharmacol. 2011;662:31–39. doi: 10.1016/j.ejphar.2011.04.042. [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Wang K, Tung KW, Chung WK, Pass RH, Lu JT, Jean JC, Omari A, Sampson KJ, Kotton DN, Keller G. Kass RS. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141:61–72. doi: 10.1085/jgp.201210899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenor B, Gomis-Tena J, Cardona K, Romero L, Rajamani S, Belardinelli L, Giles WR. Saiz J. In silico assessment of drug safety in human heart applied to late sodium current blockers. Channels (Austin) 2013;7:249–262. doi: 10.4161/chan.24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas A. Maltsev VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem. 2008;6:348–359. doi: 10.2174/187152508785909447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas AI, Belardinelli L, Undrovinas NA. Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17(Suppl. 1):S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas AI, Maltsev VA, Kyle JW, Silverman N. Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Maltsev VA. Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas NA, Maltsev VA, Belardinelli L, Sabbah HN. Undrovinas A. Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci. 2010;60:245–257. doi: 10.1007/s12576-010-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonck F, Volders PG, Vos MA. Sipido KR. Intracellular Na+ and altered Na+ transport mechanisms in cardiac hypertrophy and failure. J Mol Cell Cardiol. 2003;35:5–25. doi: 10.1016/s0022-2828(02)00280-8. [DOI] [PubMed] [Google Scholar]
- Ver Donck L, Borgers M. Verdonck F. Inhibition of sodium and calcium overload pathology in the myocardium: a new cytoprotective principle. Cardiovasc Res. 1993;27:349–357. doi: 10.1093/cvr/27.3.349. [DOI] [PubMed] [Google Scholar]
- Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM. Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo AL, Camm AJ, deRuyter H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ. Veltri EP. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol. Lancet. 1996;348:7–12. doi: 10.1016/s0140-6736(96)02149-6. [DOI] [PubMed] [Google Scholar]
- Wang L, Myles RC, De Jesus NM, Ohlendorf AK, Bers DM. Ripplinger CM. Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: ryanodine receptor refractoriness during alternans and fibrillation. Circ Res. 2014;114:1410–1421. doi: 10.1161/CIRCRESAHA.114.302505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA. Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- Wasserstrom JA, Sharma R, O'Toole MJ, Zheng J, Kelly JE, Shryock J, Belardinelli L. Aistrup GL. Ranolazine antagonizes the effects of increased late sodium current on intracellular calcium cycling in rat isolated intact heart. J Pharmacol Exp Ther. 2009;331:382–391. doi: 10.1124/jpet.109.156471. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Nivala M, Garfinkel A. Qu Z. Alternans and arrhythmias: from cell to heart. Circ Res. 2011;108:98–112. doi: 10.1161/CIRCRESAHA.110.223586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Qu Z, Chen P-S, Lin S-F, Karagueuzian HS, Hayashi H, Garfinkel A. Karma A. The dynamics of cardiac fibrillation. Circulation. 2005;112:1232–1240. doi: 10.1161/CIRCULATIONAHA.104.529545. [DOI] [PubMed] [Google Scholar]
- Weisser-Thomas J, Piacentino V, 3rd, Gaughan JP, Margulies K. Houser SR. Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc Res. 2003;57:974–985. doi: 10.1016/s0008-6363(02)00732-0. [DOI] [PubMed] [Google Scholar]
- Wimalaratne SM, Halstead MDB, Lloyd CM, Crampin EJ. Nielsen PF. Biophysical annotation and representation of CellML models. Bioinformatics. 2009;25:2263–2270. doi: 10.1093/bioinformatics/btp391. [DOI] [PubMed] [Google Scholar]
- Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ. Balser JR. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol. 2004;11:219–225. doi: 10.1038/nsmb737. [DOI] [PubMed] [Google Scholar]
- Wu L, Ma J, Li H, Wang C, Grandi E, Zhang P, Luo A, Bers DM, Shryock JC. Belardinelli L. Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation. 2011;123:1713–1720. doi: 10.1161/CIRCULATIONAHA.110.000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Shryock JC, Song Y. Belardinelli L. An increase in late sodium current potentiates the proarrhythmic activities of low-risk QT-prolonging drugs in female rabbit hearts. J Pharmacol Exp Ther. 2006;316:718–726. doi: 10.1124/jpet.105.094862. [DOI] [PubMed] [Google Scholar]
- Xie Y, Sato D, Garfinkel A, Qu Z. Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Fan P, Jiang Z, Viatchenko-Karpinski S, Wu Y, Kornyeyev D, Hirakawa R, Budas GR, Rajamani S, Shryock JC. Belardinelli L. Nav1.5-dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. Am J Physiol Cell Physiol. 2011;301:C577–C586. doi: 10.1152/ajpcell.00125.2011. [DOI] [PubMed] [Google Scholar]
- Zaza A, Belardinelli L. Shryock JC. Pathophysiology and pharmacology of the cardiac ‘late sodium current.’. Pharmacol Ther. 2008;119:326–339. doi: 10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Zeng J. Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Solhjoo S, Millare B, Plank G, Abraham MR, Cortassa S, Trayanova N. O'Rourke B. Effects of regional mitochondrial depolarization on electrical propagation: implications for arrhythmogenesis. Circ Arrhythm Electrophysiol. 2014;7:143–151. doi: 10.1161/CIRCEP.113.000600. [DOI] [PMC free article] [PubMed] [Google Scholar]