

Abstract
This paper is the third in a series of reviews published in this issue resulting from the University of California Davis Cardiovascular Symposium 2014: Systems approach to understanding cardiac excitation–contraction coupling and arrhythmias: Na+ channel and Na+ transport. The goal of the symposium was to bring together experts in the field to discuss points of consensus and controversy on the topic of sodium in the heart. The present review focuses on cardiac Na+/Ca2+ exchange (NCX) and Na+/K+-ATPase (NKA). While the relevance of Ca2+ homeostasis in cardiac function has been extensively investigated, the role of Na+ regulation in shaping heart function is often overlooked. Small changes in the cytoplasmic Na+ content have multiple effects on the heart by influencing intracellular Ca2+ and pH levels thereby modulating heart contractility. Therefore it is essential for heart cells to maintain Na+ homeostasis. Among the proteins that accomplish this task are the Na+/Ca2+ exchanger (NCX) and the Na+/K+ pump (NKA). By transporting three Na+ ions into the cytoplasm in exchange for one Ca2+ moved out, NCX is one of the main Na+ influx mechanisms in cardiomyocytes. Acting in the opposite direction, NKA moves Na+ ions from the cytoplasm to the extracellular space against their gradient by utilizing the energy released from ATP hydrolysis. A fine balance between these two processes controls the net amount of intracellular Na+ and aberrations in either of these two systems can have a large impact on cardiac contractility. Due to the relevant role of these two proteins in Na+ homeostasis, the emphasis of this review is on recent developments regarding the cardiac Na+/Ca2+ exchanger (NCX1) and Na+/K+ pump and the controversies that still persist in the field.
The Na+/Ca2+ exchanger – structure, function and regulation
The session on ‘Na/Ca exchanger – structure, function and regulation’ included Michela Ottolia, Karin Sipido, John Bridge, Joshua Goldhaber, Andrew Edwards and Donald Bers as speakers, discussion leaders and panelists. Below is a summary of the current state of knowledge on the Na+/Ca2+ exchanger in cardiac ion homeostasis.
Molecular perspectives
The eukaryotic exchanger protein, as exemplified by the mammalian cardiac isoform NCX1.1 (indicated as NCX throughout the review), is organized into ten transmembrane segments (TMSs) (Liao et al. 2012; Ren & Philipson, 2013) with a large cytoplasmic loop between TMSs 5 and 6 that plays a regulatory role (Philipson et al. 2002). Ion transport is associated with two regions of intramolecular similarity named α repeats (Nicoll et al. 1996; Schwarz & Benzer, 1997; Shigekawa et al. 2002; Ottolia et al. 2005). They consist of TMSs 2–3 and TMSs 7–8 and their connecting links. The α repeats are highly conserved within the exchanger family to the extent that they are deemed the signature sequence of the Ca2+/cation exchanger class of proteins (Lytton, 2007). Their role in ion translocation has been confirmed by mutagenesis studies (Nicoll et al. 1996; Schwarz & Benzer, 1997; Shigekawa et al. 2002; Ottolia et al. 2005) and recently by the structure of the archaebacterial homologue NCX_Mj (Liao et al. 2012) (Fig. 1). In NCX_Mj the two α repeats are packaged in the core of the protein and form four ion binding sites (three for Na+ and one of Ca2+) which are reachable via two translocation pathways (Fig. 1). Essential amino acids responsible for Na+ and Ca2+ ion binding are conserved between the archaea and eukaryotic proteins and accessibility data indicate that the portions of α repeats are organized in a similar manner in the mammalian cardiac exchanger (John et al. 2013). These similarities are indicative of functional and mechanistic conservation between NCX_Mj and NCX. However, enthusiasm is tempered by the low sequence similarity between these two proteins outside the α repeats and the usefulness of NCX_Mj as a structural guide to investigate the properties of the mammalian homologue should be further investigated. For example, the links between TMSs 2 and 3 and TMSs 7 and 8 of the α-repeats appear to fold differently in the archaebacterial and mammalian exchangers (Fig. 1). Previous accessibility mapping studies using cysteine scanning mutagenesis and epitope tagging (Nicoll et al. 1996; Iwamoto et al. 1999, 2000; Shigekawa et al. 2002) indicate that these two portions of the cardiac NCX form re-entrant loops while the homologous regions in NCX_Mj (Liao et al. 2012) lack the re-entrant loop between TMSs 2 and 3 and have an additional transmembrane segment between TMSs 7 and 8 (Fig. 1). Many of these residues are intimately involved in ion transport as determined by mutagenesis studies.
Figure 1.
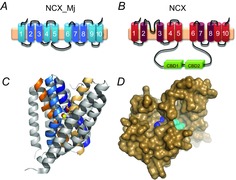
Molecular properties of the Na+/Ca2+ exchanger
The upper panels show the secondary topologies of the archaebacterial (A) and eukaryotic (B) exchangers. The conserved α-repeats (TMSs 2–3 and 7–8) are highlighted in darker shades. Note that the loop connecting TMS2 and TMS3 is modelled as a P loop in the eukaryotic NCX (B). The atomic structure of the archaebacterial NCX (NCX_Mj) (Liao et al. 2012) is shown in C. The transmembrane segments involved in ion transport are TMSs 2, 3, 7 and 8, coloured in cyan, dark blue, yellow and orange, respectively. The cytoplasmic ion translocation pathways of the cardiac exchanger have been investigated by cysteine scanning mutagenesis (John et al. 2013). A comparison between these data and the structure of NCX_Mj suggests that these two proteins have similar architecture. This is shown in D where the residues (shown in cyan and blue) of the cardiac exchanger found sensitive to thiol modification were mapped onto NCX_Mj inward structural model (shown as surface representation as seen from the cytoplasmic side). The accessibility data obtained from the eukaryotic exchanger correlates well with the NCX_Mj structure.
Regulation of the mammalian NCX has been elegantly demonstrated both at the functional and structural level. Allosteric regulation of NCX by cytoplasmic Na+ and Ca2+ ions occurs from within the large cytoplasmic loop that separates TMS 5 from TMS 6 (Philipson et al. 2002). This regulatory loop is absent from the NCX_Mj structure. However, the structures of two regulatory domains within this region of the eukaryotic exchanger are now available (Hilge et al. 2006; Nicoll et al. 2006; Besserer et al. 2007; Wu et al. 2009a,b, 2013). These two contiguous stretches of residues bind cytoplasmic Ca2+ and are identified as Ca2+ binding domains 1 and 2, respectively. They both adopt a β-sandwich fold organized in a head to tail fashion separated by only 5–7 amino acids. Binding of Ca2+ to these domains increases NCX activity (Hilgemann et al. 1992; Matsuoka et al. 1995; Ottolia et al. 2009) via molecular mechanisms not yet fully understood. The role of Ca2+ regulation in vivo is also uncertain as it is difficult to separate the effects of Ca2+ on the transport vs. regulatory sites. Recent evidence suggests that while Ca2+ ions bind rapidly to these sites at physiological levels (10–300 nm), its release is slow suggesting that in vivo NCX may be persistently activated during excitation–contraction (EC) coupling (Reeves & Condrescu, 2003; Bers & Ginsburg, 2007; Giladi et al. 2010; Ginsburg et al. 2013; John et al. 2013). Na+ ion regulation of NCX is less well studied. High cytoplasmic Na+ inactivates the Na+/Ca2+ exchanger (Hilgemann et al. 1992) consistent with a state-dependent model, but the mechanism is elusive. One hypothesis is that the binding of Na+ to its transport site drives the exchanger into an inactivated state (Hilgemann et al. 1992). However, mutagenesis studies have shown that a region within the large cytoplasmic loop is also involved in this process (Matsuoka et al. 1995). Whether NCX modulation by cytoplasmic Na+ is relevant to cardiac physiology remains to be established since relatively high intracellular Na+ concentrations (≥20 mm) are required to significantly inactivate NCX experimentally (Hilgemann et al. 1992; Matsuoka & Hilgemann, 1994; Matsuoka et al. 1995). Possibly, elevations in Na+ levels due to pathological conditions such as heart failure (Despa et al. 2002b; Despa & Bers, 2013) could influence NCX activity via Na+-dependent inactivation. Further investigations into NCX regulation by Na+ and Ca2+ are essential to determine the physiological impact of NCX on heart contractility.
Question or controversy: molecular perspectives
The crystal structure of the archaebacterial Na+/Ca2+ exchanger represents a milestone in the exchanger field and revealed, unexpectedly, the absence of re-entrant loops considered important for ion translocation in the eukaryotic homologues. Due to the low sequence similarity between the eukaryotic and archaebacterial exchanger, further studies are required to elucidate the properties of the mammalian NCX and to address the following questions:
Does the mammalian NCX differ structurally from the archaebacterial exchanger?
Evidence indicates that the mammalian NCX exists as a dimer in the membrane (Ren et al. 2008; John et al. 2011) while the archaebacterial exchanger crystalizes as a monomer. Does the oligomeric state of NCX affect its function?
NCX in excitation–contraction coupling
The Na+/Ca2+ exchanger catalyses the countertransport of three Na+ and one Ca2+ using the energy of the Na+ gradient as driving force. The cytoskeletal protein ankyrin B anchors NCX to the membrane and the NKA may be part of this complex suggesting the proximity of these two proteins (Mohler et al. 2005). In cardiomyocytes NCX appears widely distributed within the sarcolemma but there has been some controversy concerning the proportion of NCX molecules located in T-tubules (Frank et al. 1992; Despa et al. 2003), which would have considerable implications for EC coupling. For example, confocal and internal reflection imaging (Jayasinghe et al. 2009) show that 27% of NCX labelling in rat myocytes is within 150 nm of ryanodine receptor (RYR) puncta and that 45% of all NCX labelling is concentrated within puncta. As the authors explain these could be viewed as sites of concentrated Ca2+ entry or exit and is consistent with a fraction of NCX being located within couplons, i.e. junctions. This is consistent with earlier electrophysiological data suggesting that NCX can sense subsarcolemmal Ca2+ during sarcoplasmic reticulum (SR) release (Trafford et al. 1995; Weber et al. 2002); the dual time course of the NCX current during caffeine-induced SR Ca2+ release in rat myocytes is consistent with a fraction of NCX sensing early and high local Ca2+ (in the dyad) and another being activated with a delay (outside the dyad). Recent data indicate that L-type Ca2+ channels and NCX within the dyadic cleft report similar microdomain Ca2+ levels, with approximately 15% of all NCX located within close proximity to RyR in pig ventricular myocytes (Acsai et al. 2011). Besides results indicating that the triggering of SR Ca2+ release flux requires NCX which is activated by triggering Ca2+ current (Sobie et al. 2008), there is also evidence indicating that Na+ entry, via voltage-dependent Na+ channels, can drive NCX into the reverse mode thereby priming the dyadic cleft with a small amount of Ca2+, thus enhancing triggering by voltage-dependent Ca2+ channels (Fig. 2) (Larbig et al. 2010; Neco et al. 2010; Torres et al. 2010). Abrupt inhibition of NCX-mediated Ca2+ extrusion can instantaneously increase the frequency of Ca2+ sparks in resting cardiac myocytes (Goldhaber et al. 1999).
Figure 2.
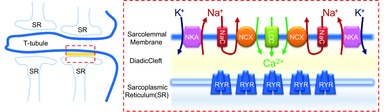
The Na+/Ca2+ exchanger primes the diadic cleft with Ca2+
The left panel shows the organization of the T-tubule and sarcoplasmic reticulum (SR) in an adult cardiac myocyte. The specialized junction between the T-tubule and the SR is highlighted in the red box. This region is further detailed on the right panel which depicts proteins important for excitation–contraction (EC) coupling (NKA, Na+/K+ pump; NCX, Na+/Ca2+ exchange; NaC, voltage-dependent Na+ channels; LCC, voltage-dependent Ca2+ channels; RYR, ryanodine receptor). Activation of the voltage-dependent Na+ channels increases the Na+ concentration in proximity of the Na+/Ca2+ exchanger, promoting its reverse mode. As a result, Ca2+ is transported into the diadic cleft, priming EC coupling by facilitating the opening of RYRs by Ca2+ entering through voltage-dependent Ca2+ channels.
Taken together, several lines of evidence suggest that a fraction of NCX is within the junctional space of the dyadic cleft. More studies to confirm the precise fraction and location of NCX within the junctional region would be of considerable value and will doubtless be forthcoming as the methods of high resolution confocal microscopy are refined. The data obtained in rodent models further need to be complemented with data from human and large mammal models which have a low T-tubular density and a large fraction of RyRs that are not within the dyadic cleft (Louch et al. 2004). Importantly, in disease, reorganization of the membrane structure and dyadic cleft occur and affect NCX interaction with RyR (Biesmans et al. 2011).
The Na+/Ca2+ exchanger is the major cardiac extrusion mechanism for the Ca2+ that enters via voltage-dependent Ca2+ channels with each beat. In conjunction with SR Ca2+ uptake, Ca2+ removal by NCX allows myocytes to relax. Concurrently, the coupled inward depolarizing Na+ current is a fundamental component of the action potential duration (Armoundas et al. 2003). Moreover, outward shifts in both NCX and NKA current at higher [Na+]i are important in determining action potential repolarization and duration (Grandi et al. 2010). In these roles, NCX is an important determinant of intracellular Ca2+ and Na+ and hence contractility. Direct evidence of the essential role of NCX in EC coupling comes from ventricular-specific conditional knockouts of NCX. Two main adaptive mechanisms appear relevant for the survival of these mice lacking an exchanger to provide Ca2+ efflux: (i) a reduction in Ca2+ influx via voltage-dependent Ca2+ channels, and (ii) an increase in the gain in the EC coupling; both of these are probably due to an increase in Ca2+ within the dyadic cleft (Pott et al. 2005).
In addition to maintaining both proper contraction and Na+ homeostasis, NCX has also been implicated in the generation of the pacemaker potential within the sinoatrial node (SA node). Rhythmic spontaneous release of Ca2+ from the SR via the RYR activates NCX resulting in a net inward current facilitating depolarization helping the pacemaker cells reach the threshold required for triggering an action potential. Several models of mice lacking NCX within the pacemaker cells have been generated with the goal of determining the role of NCX in the genesis of pacemaker activity (Gao et al. 2013; Groenke et al. 2013; Herrmann et al. 2013). Complete ablation of NCX from both the atria and SA node tissue abolishes atrial depolarization and sinus rhythm in an atrial-specific NCX knockout (KO) mouse (Groenke et al. 2013). Isolated SA node cells from these NCX KO mice are viable, but they have no spontaneous action potentials despite an intact funny current (If) through HCN4 channels. This clearly demonstrates the importance of NCX in SA node pacemaking.
Question or controversy: NCX in excitation–contraction coupling
The role of NCX in priming EC coupling is well documented but important questions remain:
Does NCX localize within the dyadic cleft? If so, what is the ratio of NCX in the cleft to those outside?
Does NCX interact with other proteins involved in EC coupling such as NKA or Na+ channels?
How does NCX prime the dyadic cleft to facilitate EC coupling?
NCX and heart pathologies
Under physiological conditions NCX ensures that intracellular Ca2+ is removed after each beat to allow for proper relaxation and cellular Ca2+ balance. However, common pathologies promote Ca2+ influx via NCX leading to Ca2+ accumulation which can accelerate myocardial necrosis, contractile failure and also trigger arrhythmias. This is seen for example during reperfusion after an ischaemic event when acidosis provokes Na+ overload by the Na+/H+ exchanger and slows or even reverses NCX. This example clearly demonstrates the tight coupling between Na+ and Ca2+ homeostasis.
Alterations in NCX expression are also associated with various cardiac pathologies including heart failure and post-ischaemic cardiac injury. Heart failure, a highly prevalent condition world-wide, is mostly due to ischaemic heart disease, valvular heart disease and hypertension. It is associated with high mortality as a result of contractile dysfunction and ventricular arrhythmia. While evidence indicates that NCX expression and function are significantly increased during the course of this pathology (Studer et al. 1994; Dipla et al. 1999; Pogwizd et al. 1999; Hobai & O'Rourke, 2000), this is not a characteristic of all stages of the disease nor in all types of heart failure (reviewed in Sipido et al. 2002; Antoons et al. 2012). The NCX function ultimately depends on prevailing Ca2+ and Na+ concentrations, as well as NCX distribution in the membrane and surface-to-volume changes. Increased NCX inward current in response to spontaneous SR Ca2+ release enhances afterdepolarizations, while the additional Ca2+ efflux reduces SR Ca2+ load. These events contribute to the arrhythmogenicity and depressed contractility associated with heart failure (Pogwizd et al. 1999; Pogwizd & Bers, 2002). Additionally, an increase in intracellular Na+ has been reported in cardiac myocytes during hypertrophy and heart failure with important implications for NCX activity and thereby on contractility. The rise in intracellular Na+ may also increase NCX reverse mode promoting Ca2+ influx (Despa et al. 2002a; Despa & Bers, 2013). Although moderate Ca2+ influx could potentially improve contractility linked to heart failure, Ca2+ overload may contribute to arrhythmias and cell death.
Because of these important roles of NCX, modulation of NCX function in heart failure has been suggested as a therapeutic target (Sipido et al. 2002; Pogwizd, 2003; Lee et al. 2005). For example, specific inhibitors for NCX could represent an alternative strategy in arrhythmia therapy (Antoons et al. 2012). However, pharmacological manipulation of NCX is problematic due to the lack of specific blockers. KB-R7943, long thought to be NCX specific, is in fact quite non-specific and seems to block other ionic currents (Reuter et al. 2002; Barrientos et al. 2009). Newer compounds such as SEA-0400 and SN-6 are more specific over other ion currents and more potent (Iwamoto et al. 2007; Niu et al. 2007). A unique and potentially desirable property of such blockers is their apparent preferential inhibition of NCX transport in the Ca2+ entry (reverse) mode, as initially proposed for KB-R7943 but later debated (Soma et al. 2006; Iwamoto et al. 2007). It is likely that this apparent selectivity is due to the asymmetric nature of the protein (i.e. different sensitivity of NCX for Na+ and Ca2+ at the opposite sides of the membrane) and the ionic conditions used to test these drugs. Under circumstances of high intracellular Na+, where reverse mode NCX activity dominates, NCX may reside in a state more sensitive to drug-mediated inhibition, thereby, leading to an apparently more efficient block. It should also be noted, however, that in more physiological conditions SEA-0400 will inhibit both forward and reverse mode to an equal extent (Ozdemir et al. 2008).
The development of a more selective inhibitor for NCX has strong clinical relevance for its potential therapeutic effects. However, the lack of structural information about the mammalian cardiac NCX is a major hindrance to achieving this goal and further studies are therefore essential to advance this field. Despite these limitations there have been a number of animal studies illustrating efficiency of NCX block in arrhythmias (Antoons et al. 2012). A certain lack of selectivity and reduction of L-type Ca2+ current (associated with NCX block) may be advantageous, as recently shown (Bourgonje et al. 2013).
Question or controversy: NCX and heart pathologies
At present there are no specific drugs for NCX. This prevents a full understanding of NCX's role in normal and pathophysiological conditions.
Additional structural information could help in developing new drugs. Can a selective drug block Ca2+ entry while maintaining Ca2+ extrusion?
What post-translational modifications does NCX undergo in pathological conditions that could alter its mode of operation?
Is activation or blockade of NCX likely to be of benefit in the treatment of heart pathologies?
The Na+/K+ pump – structure, function and regulation
In the session on ‘Na/K pump – structure, function and regulation’ Julie Bossuyt, Jerry Lingrel, Michael Shattock, Mordecai Blaustein, Jack Kaplan and Zi-Jian Xie were speakers, discussion leaders and panelists. Below is a summary of the materials presented and discussed in the session.
While the Na+/Ca2+ exchanger is quantitatively one of the main Na+ influx mechanisms in cardiac myocytes it is just one of a plethora of membrane transporters that utilize the energy in the trans-sarcolemmal Na+ gradient to move ions, substrates, amino acids and metabolites into or out of the cell. All of these transport mechanisms dissipate the Na+ gradient. Thus, the constant activity of the Na+/K+-ATPase (NKA, or Na+ pump) is essential for re-establishing and maintaining this gradient. In cardiac and vascular smooth muscle the principal isoforms of the NKA are α1 and α2 and their physiological role is controlled both by their unique and independent signalling pathways, and their discrete subcellular distribution.
In cardiac muscle, the α2 subunit has been suggested to play a specific subcellular role in preferentially regulating Na+ in the dyadic cleft while the α1 subunit plays more of a ‘housekeeping’ role regulating bulk cytoplasmic Na+ (Dostanic et al. 2005). In vascular smooth muscle and skeletal muscle evidence also implicates the α2 subunit in preferentially influencing contractile function (Juhaszova & Blaustein, 1997; Zhang et al. 2005; Radzyukevich et al. 2009). In the heart both isoforms (α1 and α2) associate with, and are regulated by, the FXYD protein phospholemman (PLM), with unphosphorylated PLM exerting a tonic inhibition on ion pumping (Pavlovic et al. 2007; Bossuyt et al. 2009). When PLM is phosphorylated (principally at Ser 63 and 68 by protein kinase C (PKC) or at Ser 68 by protein kinase A (PKA)) this inhibition is relieved and/or the pump is stimulated (Fuller et al. 2004; Despa et al. 2005). In this review we focus on recent advances in the regulation of NKA by PLM and studies elucidating the specific role of the α2 isoform in cardiac, vascular and skeletal muscle, and the role of the NKA in general in cardiovascular disease.
Molecular interactions between PLM and NKA
Stoichiometry considerations. Several NKA crystal structures have been resolved now, greatly advancing our understanding of pump function and molecular interactions of the pump complex (Morth et al. 2007; Ogawa et al. 2009; Shinoda et al. 2009; Toyoshima et al. 2011; Kanai et al. 2013). In all the crystal structures to date, the αβ complex, the minimal functional unit, was associated with a tissue-specific, regulatory FXYD protein (Kaplan, 2002; Jorgensen et al. 2003). Recently, a pool of ‘pump-free’ PLM multimers was identified in cardiac ventricle suggesting PLM expression is abundant enough to saturate αβ complexes (Wypijewski et al. 2013). PLM oligomerization, specifically tetramerization, is further supported by structural and fluorescence studies in recombinant systems (Beevers & Kukol, 2006; Bossuyt et al. 2006; Song et al. 2011). These PLM oligomers could represent an inactive storage pool of PLM or may have non-pump-related functions (e.g. form channels) but fluorescence resonance energy transfer (FRET) data suggest that, like for the phospholamban–sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) system, the PLM–PLM and PLM–NKA equilibria may be dynamically linked (Bossuyt et al. 2006; Song et al. 2011). That is, PLM phosphorylation shifts the PLM–NKA equilibrium towards oligomers thereby relieving inhibition of the pump and vice versa.
The Na+ pump as an αβFXYD unit (αβPLM for cardiac NKA) with a 1:1:1 stoichiometry appears to be the current consensus. Nonetheless, multiple studies have claimed that NKA can exist as an oligomer of multiple units ((αβFXYD)n) in native membranes (reviewed by Taniguchi et al. 2001), but this idea remains highly controversial and it is unknown whether NKA oligomerization occurs in cardiac myocytes. Also, the Kaplan group recently showed that multiple β subunits can associate with α in the pump complex, suggesting that NKA subunit composition may also not be so rigid (Clifford & Kaplan, 2008, 2009). Finally, it is not known whether other FXYD proteins are expressed in cardiac myocytes at baseline or under stress (e.g. FXYD5 was identified in cardiac homogenates; Lubarski et al. 2005).
NKA–PLM interactions. Physical interaction between PLM and the NKA α1 and α2 isoforms was shown via co-immunoprecipitation (Crambert et al. 2002; Fuller et al. 2004; Bossuyt et al. 2005; Silverman et al. 2005) and crosslinking (Lindzen et al. 2006) in heterologous expression systems and cardiac myocytes. Crosslinking (Lindzen et al. 2006) and SERCA homology predictions (Sweadner & Donnet, 2001; Li et al. 2004) suggest that the transmembrane domain (TM) of FXYDs could reside in a groove formed by TM2, TM6 and TM9 of the NKA α subunit. However, crystal structures show FXYD proteins to be in proximity to, but not inside, this groove. The latter may be only one of many physiological conformations but mutational analysis showed that the E960 residue on NKA TM9 and the F28 residue on PLM are critical for both the functional effects on the pump and NKA–PLM FRET (Khafaga et al. 2012). The co-immunoprecipitation was not completely abolished by these mutations indicating that additional PLM–NKA interaction sites contribute to the robust physical association of PLM with NKA. A similar approach showed that PLM phosphorylation dramatically decreased NKA–PLM FRET without eliminating co-immunoprecipitation (Bossuyt et al. 2006). This is consistent with NMR studies where PLM phosphorylation increases the mobility of the cytosolic tail which is normally tightly associated with the negatively charged plasmamembrane (Franzin et al. 2007a,b; Teriete et al. 2007). Thus, PLM phosphorylation alters the PLM–NKA interaction but does not result in a complete dissociation (as has been proposed for phospholamban Asahi et al. 2001; Karim et al. 2006).
Question or controversy: stoichiometry interactions
Recent findings have called into question the dogma of the Na+ pump complex as a functional heterodimer of α and β subunits that can associate with tissue-specific regulatory FXYD proteins.
Is the composition of the pump complex controlled via dynamic regulation of subunit expression or membrane abundance? Varying the composition would have functional consequences but to what extent does fine-tuning of the complex constituents occur? Or does this only become relevant in pathological situations such as heart failure?
If excess PLM is present in myocytes, possibly as PLM multimers, then what is their function?
Conversely, do ‘alternative’ complexes such as αβn or (αβ)n represent the pool of NKA with alternative functional roles (e.g. signalling, adhesion)?
Differential roles of α1 and α2 in cardiac EC coupling
Where are Na+ pump isoforms located? Of the four known NKA isoforms only α1–3 are expressed in cardiac muscle and their expression varies between species (Hensley et al. 1992; Sweadner et al. 1994; McDonough et al. 1996). The α1 isoform is both ubiquitous and quantitatively dominant, with the majority of species expressing significantly less, but functionally important, amounts of α2. On a subcellular level, the α2 isoform appears to be preferentially concentrated in T-tubular membranes while the α1 isoform is relatively uniformly distributed between T-tubular and external sarcolemmal membranes (Berry et al. 2007; Despa & Bers, 2007; Swift et al. 2007; Bossuyt et al. 2009). It is worth noting that the α1 subunit remains quantitatively comparable and even the dominant isoform within the T-tubular membrane (with α1:α2 density ratios reported as 1:1 (rat), Despa & Bers, 2007, 3:2 (rat), Swift et al. 2007, and 4:1 (mouse) (Berry et al. 2007); however, the disproportionate functional impact of the α2 isoform suggests α2 may be further concentrated in the junctional regions of the T-tubule – this has yet to be investigated (Fig. 3). This preferential localization of the α2 subunit close to the junctional SR has also been reported in both smooth (Juhaszova & Blaustein, 1997) and skeletal muscle (Lavoie et al. 1997).
Figure 3.
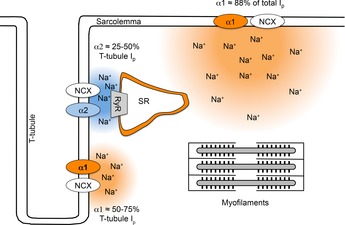
Diagram illustrating the relative distribution of Na+ pump α1 and α2 subunits in a cardiac myocyte
The NKA resides and regulates intracellular Na+ and Ca2+ (via NCX) in both external sarcolemma and T-tubular membranes. α1 is evenly distributed within the cells and is the dominant isoform providing around 88% of the total Na+ pump current (Ip). α2 is 4–5 times more concentrated in the T-tubules compared to external sarcolemma and provides only around 12–24% of the total Ip but up to 50% of the Ip in the T-tubules. Thus, it has been suggested that α1 subunits may principally be involved in controlling bulk cytoplasmic Na+. Functional evidence suggests that while both α1 and α2 subunits are expressed in T-tubular membranes, the possibility that the α2 subunit is relatively concentrated in the junctional areas (as shown above) remains to be structurally demonstrated. Estimates of pump current are based on the measurements of Berry et al. (2007).
Is there a preferential role for the α2 subunit in cardiac EC coupling? The relative concentration of α2 in both cardiac and smooth muscle in the specialized membranes close to other ion translocators and the EC coupling machinery, has led to the proposal that the α2 subunit preferentially regulates the Na+ (and hence Ca2+) concentration in a limited (junctional SR/T-tubular) subcellular compartment that then strongly influences contraction. In this model, the α1 subunit is proposed to play more of a ‘housekeeping’ role regulating Na+ in the bulk cytoplasmic compartment. Mice heterozygous for the α2 isoform (α2(+/–)) have a hyper-contractile phenotype (James et al. 1999), suggesting that Na+/K+ pumps containing the α2 subunit are functionally linked to the NCX. Further evidence for this functional compartmentation was provided by Dostanic et al. (2005) and Despa et al. (2012) who compared the effects of low-dose ouabain in wild-type hearts (expressing the ouabain-sensitive α2 isoform) with that in hearts expressing a genetically modified ouabain-resistant α2 isoform (‘SWAP’ mice). Dostanic et al., using SWAP mice, also demonstrated that the α1 isoform is physically and functionally associated with the Na+/Ca2+ exchanger and can also mediate inotropic responses to low-dose ouabain (Dostanic et al. 2004).
Question or controversy: EC coupling
While there is little doubt that the α2 isoform is important in the heart, a detailed understanding of its role is still missing.
Is there a preferential role for the NKA α2 subunit in EC coupling?
Within the T-tubule itself, are α2 isoforms further concentrated in regions close to the junctional SR?
Differential regulation of cardiac α1 and α2. The distinct distribution patterns of α1 and α2 suggest that the respective isoforms would be exposed to different signalling microdomains. Indeed, an early study in guinea pig ventricular myocytes found an exclusive link between PKA effects and NKA α1 isoforms, whereas PKC effects were targeted to α2 isoforms (Gao et al. 1999). Subsequent reports, however, found less divergent effects. Studies in both Xenopus oocytes and SWAP mice found that PKA activation increases the apparent Na+ affinity of both α1 and α2 (Bibert et al. 2008; Bossuyt et al. 2009), but in detubulated mouse myocytes β-adrenergic effects were limited to the α1 isoforms (Berry et al. 2007). FRET experiments in cells from the human embryonic kidney (HEK) cell line found that submaximal PKA activation had less effect on the PLM–NKA α2 isoform interaction than on the α1 isoform, indicating that the intrinsic sensitivity to PKA regulation is slightly different for the two isoforms (Bossuyt et al. 2009). The PKC effects on NKA are more complex. Even at the level of the overall effect there are some discrepancies: most agree that PKC stimulates NKA (Wang et al. 1998; Gao et al. 1999; Han et al. 2006; Bibert et al. 2008; Fuller et al. 2009); however, inhibition was also reported (Lundmark et al. 1999; Buhagiar et al. 2001; White et al. 2009). Some of these differences could be due to which PKC isoform is engaged or the intracellular Ca2+ level used in the experiment (since Ca2+ can modulate PKC effects on NKA). The mechanism of the increased pump activity also remains unclear as changes in Vmax alone (Han et al. 2006; Bibert et al. 2008) or both Vmax and Na+ affinity (Bossuyt et al. 2009) were reported. These mechanistic differences could be due to differential phosphorylation of PLM by PKA and PKC (Han et al. 2006; Fuller et al. 2009). In oocytes, PKC also affected only the turnover rate of the α2 isoforms (Bibert et al. 2008). In SWAP mice on the other hand, PKC increased the Na+ affinities (via phospholemman) of both isoforms while only increasing the Vmax for the α2 isoform (Bossuyt et al. 2009). Interestingly, in contrast to PKA, PKC similarly affected PLM–NKA FRET for both NKA isoforms. Thus, despite recent advances, the differential regulation of NKA isoforms by protein kinases has not been clearly established.
In addition to phosphorylation, other post-translational modifications, such as glutathionylation and palmitoylation of cysteine residues within the pump complex, can regulate NKA function (Figtree et al. 2009; Howie et al. 2013). Oxidant stress and modification of NKA thiols has long been known to inhibit the NKA activity (Shattock & Matsuura, 1993; Haddock et al. 1995) and, more recently, White et al. have shown that angiotensin II-mediated oxidant stress can inhibit Na+/K+ pump activity via a PKC and nitric oxide synthase-dependent mechanism (White et al. 2009). While the α, β and PLM subunits of NKA have all been reported to be palmitoylated (Howie et al. 2013), and the β subunit preferentially glutathionylated (Figtree et al. 2009), at present whether regulation via such thiol modification differs between α subunit isoforms is unknown. Interestingly, a recent report from Gao et al. shows that transmural gradients of angiotensin II may differentially regulate Na+/K+ pump function in response to mechanical load (Gao et al. 2014).
The Na+ pump in hypertrophy and heart failure
Evidence and significance of Na+ dysregulation. In most mammalian hearts with a long action potential, intracellular Na+ ([Na+]i)is maintained at around 4–8 mm (Harrison et al. 1992; Despa et al. 2002a). In mice and rats, the combination of a high heart rate and short action potential is associated with a higher Na+ of around 10–20 mm (Shattock & Bers, 1989; Despa et al. 2002a). As discussed earlier, the maintenance of this large trans-sarcolemmal inward gradient is essential for numerous transport and electrogenic processes and its dissipation in various pathologies such as ischaemia/reperfusion (Tani & Neely, 1989; Neubauer et al. 1992), hypertrophy or heart failure (Pogwizd et al. 2003; Verdonck, 2003) is clearly detrimental. In hypertrophy and heart failure many aspects of EC coupling are altered; however, in combination with these the elevation in intracellular Na+ may contribute to the negative force–frequency relationship, slowed relaxation and arrhythmias (Pieske, 2002). Recently, O'Rourke and colleagues have also shown that elevation of cytoplasmic Na+ can impair mitochondrial energetics (Liu & O'Rourke, 2008; Kohlhaas et al. 2010; Liu et al. 2010). Mitochondrial Ca2+ ([Ca2+]m) plays a key role in linking ATP production to ATP demand (i.e. mechanical activity) and as Ca2+ rises in the cell, so does [Ca2+]m; this activates mitochondrial enzymes to step-up ATP production (Liu & O'Rourke, 2008; Kohlhaas et al. 2010). This relationship, which crucially matches ATP supply to demand, is blocked when cytosolic Na+ is elevated (Liu et al. 2010). The rise in Na+ activates Na+/Ca2+ exchange in the inner mitochondrial membrane and this keeps [Ca2+]m low preventing ATP supply meeting demand, leaving the heart metabolically compromised. Not only might this contribute to the known metabolic insufficiency in failing hearts but Kohlhaas et al. have shown that this mechanism increases mitochondrial free radical formation in failing hearts, further exacerbating injury (Kohlhaas et al. 2010).
While a component of the elevation of [Na+]i may reflect an increase in Na+ influx (Despa et al. 2002b), there is a large body of evidence showing that Na+/K+ pump function may also be compromised (Pogwizd et al. 2003; Verdonck et al. 2003a,b; Boguslavskyi et al. 2014b). Specifically in cardiac hypertrophy many studies have shown that Na+/K+ pump function, and/or expression, is reduced (Pogwizd et al. 2003; Verdonck et al. 2003a,b; Bossuyt et al. 2005; Boguslavskyi et al. 2014b).
Role of PLM in hypertrophy and failure. Despite the importance of PLM in regulating intracellular Na+, and the considerable evidence suggesting that intracellular Na+ is elevated in hypertrophied and failing myocardium, and contributes to diastolic dysfunction, the role of PLM and its phosphorylation in the overloaded and failing heart has not been systematically characterized. Bossuyt et al. have reported hyper-phosphorylation of PLM in a rabbit model of volume overload-induced dilated failure (Bossuyt et al. 2005) while El-Armouche et al. (2011) have reported hypo-phosphorylation of PLM in failing human hearts, attributable to down-regulation of Inhibitor-1 and increased PP-1 phosphatase activity. Recently, Boguslavskyi et al. (2014b) and Correll et al. (2014) have reported a reduction in both PLM expression and phosphorylation in mouse models of hypertrophy that, in the Boguslavskyi study, has been directly related to declining Na+/K+ pump function and intracellular Na+ elevation.
The lack of consensus regarding the PLM phosphorylation status may reflect different stages of the disease process or different animal models. Simply reporting hypo- or hyper-phosphorylation of PLM also does not distinguish between an effect that causally influences the progression of the disease from one that is simply a bystander – reporting dynamic disease-induced changes in PKA and PKC signalling pathways. In order to test this, Boguslavskyi et al. have used a mutant PLM3SA knock-in mouse (in which PLM has been rendered unphosphorylatable) (Boguslavskyi et al. 2014b). They showed that preventing PLM phosphorylation in mice subjected to aortic constriction further inhibits NKA function, increases cellular Na+ overload and exacerbates adverse hypertrophic remodelling. This suggests that in heart failure, PLM hypo-phosphorylation in mice is not simply a bystander effect (reporting dynamic disease-induced changes in PKA and PKC signalling) but is causally involved in disease progression.
Role of the α2 subunit in hypertrophy and failure. The α2 subunit has been suggested to not only play a preferential role in normal EC coupling but also in the pathological response to cardiac hypertrophy and heart failure. In a recent study, Correll et al. have reported that the over-expression of α2 (but not α1) preferentially protects against adverse remodelling in the hypertrophic heart and improves Ca2+ and Na+ handling. In this study, the ‘forced’ over-expression of α2 leads to a reduction in expression and phosphorylation of PLM and an overall increase in the Na+ sensitivity of the NKA. They suggest that the α2 subunit is ‘less regulated by PLM’, thus providing a mechanism by which α2 can efficiently couple to NCX to maintain ion (and particularly Ca2+) regulation. Adaptive increases in α2 expression are also seen in mice expressing an unphosphorylatable form of PLM (PLM3SA), perhaps allowing the normalization of EC coupling despite an increase in the bulk cytoplasmic Na+ concentration (Boguslavskyi et al. 2014b).
Since cardiac remodelling and dysfunction in heart failure are exacerbated by preventing PLM phosphorylation (Boguslavskyi et al. 2014b), and α2 overexpression is protective (Correll et al. 2014), then this raises the interesting possibility that Na+/K+ pump stimulation may be therapeutically useful in heart failure (Rasmussen & Figtree, 2007; Shattock, 2009) and, in particular, isoform-selective α2 stimulation. Superficially, this seems to fly in the face of 200 or more years of digitalis use to treat heart failure. However, it would not be the first time that counter-intuitive therapeutics prove beneficial as demonstrated by the historic aversion to negative inotropes in heart failure and the remarkable recent success of β blockers. In fact, digitalis seems to only provide symptomatic relief and, in the largest trial of its kind, the Digitalis Investigation Group (DIG) concluded that digoxin reduced hospitalization due to worsening heart failure symptoms but had no long-term effect on mortality (Hobbs, 1997). This issue is, however, undoubtedly more complex, not least because of the presence of endogenous ouabains in heart failure patients and the known ouabain–digoxin antagonism (see Blaustein, 2014 for review).
Question or controversy: heart failure
What do we need for the treatment of heart failure – Na+/K+ pump inhibitors or stimulators?
The preferential role of the α2 subunit in EC coupling may mean that an isoform-specific inhibitor might be able to induce an ionotropic benefit WITHOUT triggering Ca2+ overload and arrhythmias. Is it possible to achieve an increase in SR Ca2+ load and positive inotropy WITHOUT risking Ca2+ overload?
Conversely, it could be argued that the protective role of the α2 subunit in heart failure suggests that an isoform-specific (α2) stimulator might prevent Na+ and Ca2+ overload and limit adverse remodelling.
Isoform-specific roles in control of blood pressure
The control of blood pressure, and specifically the aetiology of primary hypertension is now recognized to have both peripheral and central components. There is mounting evidence that the misregulation of the NKA may contribute to both.
Central role of α2 in regulation of blood pressure. While primary hypertension involves the interplay of genetic and environmental factors, such as high-salt intake, obesity, insulin resistance, low physical activity levels and stress, there is now increasing evidence that elements of such hypertension may be neurogenic in origin (Huang et al. 2006; Esler et al. 2010). A number of studies have shown that the level of Na+ in the cerebrospinal fluid (CSF) is a critical factor contributing to the pressor response induced by elevated dietary Na+ (Huang et al. 1998, 2001, 2004; Van Huysse et al. 2011). Van Huysse et al. (2011) have shown that the rise in blood pressure in response to CSF Na+ elevation is mediated by the central release of an endogenous ouabain-like substance that specifically targets the ouabain-sensitive α2 subunit of the NKA. The cell types and CNS pathways involved in this signalling pathway have yet to be identified. However, in the brain the α2 NKA isoform is concentrated in the glia where, coincidentally, 90% of brain angiotensinogen is also found (Schinke et al. 1999). Since, upregulation of the glial renin–angiotensin system specifically raises blood pressure (Morimoto et al. 2001, 2002), this cell type may be central in the pressor response to Na+ load.
Question or controversy: blood pressure control – central role of NKA
What is/are the cellular sources and identities of the endogenous ouabain-like substances that mediate the pressor response to Na+ elevation in CSF?
In human hypertension, does this pathway preferentially target the α2 NKA isoform in the CNS as it does in mice?
Does this offer a novel therapeutic target for the treatment of primary hypertension?
Peripheral role of α2 in the regulation of blood pressure. In addition to a central role for the NKA in blood pressure control, the activity of the Na+/K+ pump can also locally regulate both endothelial and vascular smooth muscle (VSM) function. The NKA in VSM, as in myocardial cells, sets and maintains the Na+ and K+ electrochemical gradient and, in this way, modulates the activity of other membrane ion channels and signalling pathways (Shelly et al. 2004; Zhang et al. 2005). The relatively high input impedance of VSM cells also means that the electrogenic pump can theoretically directly and substantially modulate membrane potential (and hence Ca2+ influx) with activation of the pump causing hyperpolarization and relaxation, and inhibition causing depolarization and vasoconstriction (Casteels et al. 1977; Quinn et al. 2000; Burns et al. 2004; Molin et al. 2005). However, the extent to which electrogenic NKA activity in vascular smooth muscle contributes to membrane potential varies between experimental preparation and vascular beds. Selective α2 inhibition by low-dose ouabain also can mediate vasoconstriction in the absence of changes in membrane potential. Thus, while NKA activity can undoubtedly modulate vascular tone, in many studies it is difficult to dissociate effects arising from NKA-induced changes in membrane potential from those induced by changes in ion channels or secondary to changes in local ion gradients, NCX activity etc. The situation is further complicated by studies showing that the effects of NKA inhibition or stimulation differ between intact vessels and those in which the endothelium has been denuded – suggesting that both smooth muscle and endothelial NKA activity can modulate vascular tone in intact tissues (see below).
As in other tissues the vascular NKA is expressed as a heterodimer composed of a catalytic α subunit and a glycosylated β subunit (Lingrel & Kuntzweiler, 1994). Of the four known α isoforms (α1–4) only α1 and α2 were found in VSM cells (Shelly et al. 2004; Pritchard et al. 2010). These isoforms have different cellular distribution, expression and functional significance. In VSM cells the α1 isoform is by far the most abundant and is thought to play a ‘house-keeping’ role controlling bulk cytoplasmic Na+ (Juhaszova & Blaustein, 1997; Weston et al. 2002). In rodents, this α1 isoform is relatively insensitive to ouabain. The α2 ouabain-sensitive isoform, while much less abundant, has a highly localized cellular distribution and is suggested to play a role in regulating Na+ in microdomains with preferential access to other membrane transporters (Juhaszova & Blaustein, 1997; Shelly et al. 2004; Pritchard et al. 2010). The close association between the α2 subunit, NCX and the intracellular SR membrane has been suggested to give the α2 subunit a preferential role in regulating Ca2+ (and hence constriction) in VSM cells (Juhaszova & Blaustein, 1997; Arnon et al. 2000; Lee et al. 2006; Linde et al. 2012). Recently it has been shown that NKA and Ca2+-handling proteins form a functionally coupled unit where the concentration of Na+ in a subcellular microdomain modulates submembrane Ca2+ and hence SR Ca2+ load (Matchkov et al. 2007; Pritchard et al. 2010). Reduction of NKA activity (specifically of α2) by pharmacological inhibition or transgenesis leads to enhanced agonist-induced vascular contractility and myogenic tone (Zhang et al. 2005). Matchkov et al. showed that α2, through modulation of local Ca2+, is very important in gap junction coupling between VSM cells and hence affects vasomotion and the spread of vasoconstriction and relaxation (Matchkov et al. 2007, 2012). There is also a large body of evidence showing that NKA is important for NO- and endothelium-derived hyperpolarizing factor (EDHF)-dependent responses (Gupta et al. 1994; Sathishkumar et al. 2005; Leung et al. 2006; Dora et al. 2008; Matchkov et al. 2012). Furthermore, the α2 subunit is present at myo-endothelial junctions where local increases of K+ concentration (due to the opening of endothelial Ca2+-activated K+ channels) can activate the Na+/K+ pump and induce VSM cell hyperpolarization and relaxation (Dora et al. 2008).
Given the involvement of NKA (in particular α2) in the regulation of myogenic tone, vasomotion, cell-to-cell coupling, and NO- and EDHF-dependent relaxation it is perhaps not surprising that NKA activity may be an important modulator of blood pressure (BP) in vivo. Circulating endogenous ouabain, for example, has been shown to be involved in hypertension in both animals and humans (see below) (Manunta et al. 1999; Schoner, 2002). Furthermore, in mice expressing ouabain-resistant α2 NKA, Dostanic et al. showed that ouabain-induced hypertension is mediated by the α2 subunit (Dostanic et al. 2005). Further experiments with these mice revealed the importance of ouabain-binding sites on NKA in ACTH-induced hypertension (Dostanic-Larson et al. 2005; Lorenz et al. 2008). Results obtained from other genetic models with global down-regulation or over-expression of NKA demonstrate that the activity of NKA has a major impact on BP (Zhang et al. 2005; Pritchard et al. 2007). Interestingly, in mice with cardiovascular-specific knockout of α2, basal BP was not affected, but the hypertensive response to ACTH treatment was blunted (Rindler et al. 2011).
In the vasculature, as in cardiac and skeletal muscle, NKA is associated with FXYD1 (phospholemman) (Palmer et al. 1991; Rembold et al. 2005). Dey et al. demonstrated that PLM phosphorylation by PKC activates the α2 isoform pulmonary artery smooth muscle cells (Dey et al. 2012). Recently, Boguslavskyi et al. have shown that PLM phosphorylation is critically important in modulating the constrictor response of aortic smooth muscle to phenylephrine and the relaxing effects of an NO donor (Boguslavskyi et al. 2014a). In the heart, Pavlovic et al. have shown that NO stimulates NKA via PKC-mediated PLM phosphorylation (Pavlovic et al. 2013). Whether similar pathways exist in the vasculature is yet to be determined.
In summary, direct (endogenous cardiotonic steroids, local K+) or indirect (PLM dependent) regulation of vascular NKA activity affects myogenic tone through altering Na+ and Ca2+ homeostasis and/or stimulating Src-dependent signalling (see below) which significantly influences blood pressure in vivo.
Question or controversy: blood pressure control – vascular smooth muscle
Does the NKA (and specifically the α2 subunit) play a role in regulating vascular tone and if so, are these effects mediated by electrogenic NKA transport and changes in membrane potential or by changes in transmembrane ionic concentrations?
What are the relative roles of smooth muscle vs. endothelial NKA in regulating tone?
Is vascular smooth muscle NKA regulated by phospholemman and, if so, via which signalling pathways?
Is this signalling–PLM–NKA–tone pathway defective in hypertension and/or does it provide a therapeutic target for the treatment of hypertension?
Na+/K+ pump function and skeletal muscle
Skeletal muscle fatigue. While many other mechanisms (such as Pi accumulation, acidosis, ATP depletion, generation of reactive oxygen species etc.) have been implicated in fatigue (see Allen et al. 2008 for review), there is substantial evidence that extracellular K+ accumulation (and the associated disruption of intracellular Na+ and K+ concentrations) is central to this process (Medbo & Sejersted, 1990; Juel et al. 2000; Clausen, 2003; Allen et al. 2008). The NKA plays a central role in limiting extracellular K+ accumulation during exercise (Clausen, 2003). Interventions that stimulate the pump (such as the β2-adrenoceptor agonist salbutamol) can decrease K+ accumulation and limit fatigue (Clausen, 2003), while interventions that inhibit the pump (such as ouabain) increase K+ accumulation and exacerbate fatigue (Nielsen & Clausen, 1996). It is now widely accepted that the increased activity of the NKA during exercise ‘protects’ skeletal muscle against fatigue during exercise (Juel et al. 2000; Clausen, 2003). One of the strongest drivers for activation of the Na+/K+ pump is the elevation of intracellular Na+. However, it is also clear that sympathetic stimulation (which inevitably accompanies exercise in vivo) also directly activates the pump and can increase its basal turn-over by up to 20-fold (Allen et al. 2008). This appears to be particularly important when extracellular K+ is elevated (Nielsen & de Paoli, 2007). An increasing number of studies have implicated PLM as the link between cell signalling and exercise-induced NKA stimulation (Thomassen et al. 2010, 2011, 2013; Benziane et al. 2011). PLM phosphorylation is increased by exercise via a PKCα-dependent mechanism (Thomassen et al. 2011) possibly in response to intracellular Ca2+ elevation, although the exact signalling pathways involved have yet to be determined.
Role of the α2 subunit. While quantitatively the α1 subunit dominates cardiac muscle, in skeletal muscle up to 87% of total NKA expression is estimated to be α2 (He et al. 2001). As in the heart, α2 in skeletal muscle appears to preferentially play a key role in regulation of contraction and protection against fatigue. Radzyukevich et al. (2013) used a skeletal muscle-specific α2 knockout to demonstrate that α2, but not α1, is responsible for acute upregulation of NKA activity and protects skeletal muscle against fatigue (Radzyukevich et al. 2013). This and other studies also show that the functional α2 subunit is preferentially located in the T-tubules and it is potassium regulation within the T-tubular lumen that may be critical in determining fatigue (Lavoie et al. 1997; Williams et al. 2001; Heiny et al. 2010). A large fraction of skeletal muscle α2 may also be resident in submembrane vesicles readily available for trafficking to and from the plasma membrane in response to receptor stimulation or demand (Hundal et al. 1992; Al-Khalili et al. 2003).
Question or controversy: skeletal muscle
Does the phosphorylation of phospholemman regulate skeletal muscle NKA and, if so, via which signalling pathways?
Does dysregulation of these pathways contribute to muscle fatigue?
Endogenous cardiotonic steroids
Realization that cardiotonic steroids (CTS) are present in nanomolar concentrations in the serum of experimental animals and humans has intensified research into their physiological and pathophysiological roles over the last 20 years (Hamlyn et al. 1982; Bagrov et al. 1998; Gallice et al. 1998; Gonick et al. 1998; Manunta et al. 1999; Harwood et al. 2001; Periyasamy et al. 2001; Komiyama et al. 2005; Kennedy et al. 2006; Fedorova et al. 2009; Li et al. 2010). Interestingly, a recent study using a highly sensitive UPLC–MS–MS technique failed to detect ouabain per se in human plasma suggesting that the CTS detected in other studies using radioimmunoassays represent compounds that are structurally similar, but not identical, to ouabain (Baecher et al. 2014). CTS are divided into two structurally distinct groups, cardenolides (ouabain-like compounds and digoxin) and bufadienolides (marinobufagenin, bufalin and telocinobufagin). Evidence is accumulating that most CTS are synthesized from cholesterol in the adrenal glands and possibly hypothalamus (Laredo et al. 1995; Dmitrieva et al. 2000; Murrell et al. 2005). The trigger for biosynthesis initiation is complex, with serum concentrations of CTS increasing in response to kidney dysfunction (Komiyama et al. 2005; Stella et al. 2008; Tian et al. 2010), volume expansion (Manunta et al. 2006), salt accumulation in the brain (Huang et al. 2004), adrenocorticotropic hormone, angiotensin II, vasopressin, and phenylephrine (Laredo et al. 1995; Shah et al. 1999). Growing evidence suggests that CTS secretion is involved in the pathogenesis of a number of diseases such as hypertension, pre-eclampsia and uraemic cardiomyopathy.
Cardiotonic steroids in blood pressure control. The mechanisms that govern CTS-mediated hypertension development have not yet been fully elucidated; however, evidence is accumulating that in response to high salt intake, a ouabain-like compound is secreted by the hypothalamus (Murrell et al. 2005), whereas both ouabain and marinobufagenin are secreted by the adrenal glands (Laredo et al. 1995; Dmitrieva et al. 2000) and that this leads to both central and peripheral blood pressure elevation (Fig. 4). Specifically, high salt intake elevates both plasma and cerebrospinal fluid Na+ and this induces the secretion of CTS by the adrenals and the hypothalamus (Huang et al. 2004). Intracerebroventricular infusion of Na+-rich cerebrospinal fluid (Van Huysse et al. 2011) or ouabain (Huang et al. 2004) increased sympathetic nerve activity, heart rate and blood pressure in rodents and these effects were partially reversed by Digibind (anti-digoxin Fab® fragments). On the other hand, chronic intravenous infusion of ouabain or marinobufagenin at concentrations comparable with in vivo plasma levels lead to an increase in arterial pressure and cardiac hypertrophy (Pamnani et al. 1994; Ferrandi et al. 2004; Kennedy et al. 2006). Furthermore, hypertension induced either by salt loading, ouabain infusion or pre-eclampsia is reduced by immunoneutralization with anti-CTS antibodies (Fedorova et al. 2002, 2007) or CTS antagonists rostafuroxin (Ferrari et al. 2006) and resibufogenin (Horvat et al. 2010). It is important to emphasize that ouabain binds to the Digibind with high affinity, whereas marinobufagenin binding affinity is lower (Pullen et al. 2004) suggesting that a ouabain-like substance is the dominant CTS in blood pressure regulation. Whereas the effects of brain-derived endogenous ouabain require further characterization, plasma ouabain produced by the adrenal glands can contribute to acute vasoconstriction via inhibition of the α2 sodium pumps in the smooth muscle, as discussed in the previous section ‘Isoform-specific roles in control of blood pressure’. Interestingly, 3 days of salt loading in healthy individuals did not result in a change in blood pressure (Manunta et al. 2006) suggesting that acute salt-loading in a healthy individual has no pathological consequences. However, it is clear that during chronic salt loading or chronic ouabain infusion, hypertension persists. It is likely that this is mediated via a combination of chronic elevation of brain Na+, leading to increased sympathetic nerve activity, and the direct effects of endogenous ouabain on the smooth and cardiac muscle, potentially resulting in increased cardiac output, increased total peripheral resistance and thus elevation in blood pressure. Furthermore, reported CTS-mediated activation and internalization of the Na+/K+–Src–epidermal growth factor receptor (EGFR) complex (Liu et al. 2002, 2004, 2005) as well as up-regulated arterial expression of the NCX1, SERCA2 and TRPC6 (Blaustein et al. 2012; Pulina et al. 2013) could contribute to arterial remodelling of the affected tissue and thus leading to established hypertension. Whereas both ouabain and marinobufagenin have been implicated in the regulation of blood pressure and hypertension development, most of the currently available data imply that ouabain is the dominant CTS. Interestingly, digoxin has been shown to oppose ouabain-induced hypertension in rats (Kimura et al. 2000; Manunta et al. 2000) and possibly even in humans (Abarquez, 1967). Song et al. (2014) report that low doses of digoxin can antagonize the effects of ouabain, possibly explaining the recently reported beneficial effects of low doses of digoxin therapy in heart failure patients (Ahmed et al. 2006).
Figure 4.
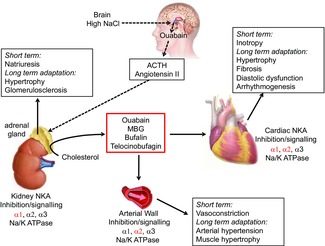
Proposed biosynthesis and function of endogenous cardiotonic steroids
Synthesis of endogenous cardiotonic steroids occurs in the adrenal cortex from cholesterol and is proposed to be under the control of adrenocorticotropic hormone (ACTH) and angiotensin II. These are produced in response to high salt and the resultant ouabain synthesis in the hypothalamus. Acute effects of endogenous cardiotonic steroids are mediated via inhibition of the NKA and lead to increased cardiac output, Na+ excretion and vasoconstriction (NKA isoform-specific effects in each tissue are shown in red). Chronic exposure to cardiotonic steroids leads to adaptational remodelling changes of the affected tissue, possibly resulting in hypertrophy, fibrosis, hypertension and arrhythmogenesis. MBG, marinobufagenin. Adapted from Pavlovic (2014).
Question or controversy: endogenous cardiotonic steroids
What is the biological significance of the diversity of endogenous CTS?
Do individual CTS play specific roles in the development of cardiovascular dysfunction?
Can we exploit knowledge of the antagonism between CTS molecules to therapeutic advantage?
Cardiotonic steroids in chronic kidney disease. It has been evident for many years that patients with chronic kidney disease (CKD) and end-stage renal disease (ESRD) are at increased risk of developing cardiovascular disease (Tyralla & Amann, 2002; Best et al. 2004). Renal failure results in a cardiomyopathy characterized by left ventricular hypertrophy, reduced ejection fraction, myocardial fibrosis and arrhythmias (Tyralla & Amann, 2002; Best et al. 2004). Although the cause of cardiomyopathy development is likely to be multifactorial, McMahon and colleagues have observed elevated diastolic intracellular Ca2+ (due to compromised NCX activity) in the hearts of rats with impaired renal function (McMahon et al. 1996, 2002, 2006; Donohoe et al. 2000).
Considering that serum levels of ouabain, telocinobufagin and marinobufagenin are substantially elevated in patients with ESRD (Komiyama et al. 2005; Stella et al. 2008) and animals with CKD (Kennedy et al. 2006, 2008), it is reasonable to propose that CTS-mediated Na+/K+ inhibition and subsequent increase in intracellular Na+ could be the mechanism responsible for the observed myocardial Ca2+ overload and could explain contractile dysfunction, negative force–frequency relationship, increased susceptibility to arrhythmias and left ventricular hypertrophy. Ca2+ overload in the smooth muscle could also lead to the development of hypertension as discussed in the previous section. In a rat model of CKD, Pavlovic et al. have shown that cardiac Na+/K+ pump is inhibited and that this inhibition is attenuated by the removal of CTS using DigiFAB® (Pavlovic, 2010). On the other hand, Liu and colleagues have proposed an alternative model of CTS-mediated cardiomyopathy development. This model proposes that a fraction of Na+/K+ pump subunits are localized in the caveolae and are not involved in transport of Na+ and K+ ions but instead act as receptors for CTS (Pierre & Xie, 2006). These ‘inactive’ pumps are physically associated with other key signalling proteins such as EGFR and Src (Wang et al. 2004; Liang et al. 2007). Binding of the CTS leads to activation of hypertrophic and fibrotic signalling cascades via Na+/K+–Src extracellular signal-regulated kinase (Li et al. 2010), independently of changes in intracellular Na+ and Ca2+ (Liu et al. 2000). This alternative signalling pathway does not account for the reported Na+/K+ pump inhibition that can be reversed by anti-CTS antibodies in patients with diabetes (Bagrov et al. 2005) or ESRD (Periyasamy et al. 2001), nor the accompanying increases in intracellular Na+ and Ca2+ reported by other groups (Peng et al. 1996; Dong et al. 2004; Andrikopoulos et al. 2011). Na+/K+ pump inhibition via CTS alone can explain the cardiovascular dysfunction observed in CKD patients, whereas the signalling model can only account for the hypertrophy and hypertension development. Whether CTS induce hypertrophic growth in the heart via the ionic or signalling pathway remains to be resolved, however, the weight of evidence in support of both pathways makes it likely that they act in conjunction with each other.
Question or controversy: CKD-induced cardiomyopathy
Although the CTS binding site is highly conserved across the evolutionary spectrum (Lingrel, 2010), rats and mice possess an Asn122His substitution in their NKA α1 isoform, making it less sensitive to CTS (Price & Lingrel, 1988). Considering that most studies are conducted in rat and mouse, a question of relevance of some of the data to human physiology requires investigation.
The relative contribution of the CTS signalling vs. effects on ion transport of the NKA in CKD and hypertension have yet to be determined.
Acknowledgments
We thank the meeting organizers, Advisory Board and all conference participants for their contributions to the scientific exchange and constructive discussions. For conference information see https://basicscience.ucdmc.ucdavis.edu/ucd-cvs-2014/index.html.
Glossary
- CKD
chronic kidney disease
- CTS
cardiotonic steroids
- EC
excitation–contraction
- EGFR
epidermal growth factor receptor
- ESRD
end-stage renal disease
- FRET
fluorescence resonance energy transfer
- NCX
Na+/Ca2+ exchanger
- NKA
Na+/K+-ATPase (Na+/K+ pump)
- PKA
protein kinase A
- PKC
protein kinase C
- PLM
phospholemman
- RYR
ryanodine receptor
- SERCA
sarco(endo)plasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- TMS
transmembrane segment
- VSM
vascular smooth muscle
Additional information
Competing interests
None declared.
Funding
The authors acknowledge grant support from the British Heart Foundation (RG/12/4/29426; to M.J.S.) and National Institutes of Health (P01-HL080101 to D.M.B.) and additional grants to other authors that allowed them to participate. Sponsors who subsidized conference costs can be found at the website below.
References
- Abarquez RF., Jr Digitalis in the treatment of hypertension. A preliminary report. Acta Med Philipp. 1967;3:161–170. [PubMed] [Google Scholar]
- Acsai K, Antoons G, Livshitz L, Rudy Y. Sipido KR. Microdomain [Ca2+] near ryanodine receptors as reported by L-type Ca2+ and Na+/Ca2+ exchange currents. J Physiol. 2011;589:2569–2583. doi: 10.1113/jphysiol.2010.202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed A, Rich MW, Fleg JL, Zile MR, Young JB, Kitzman DW, Love TE, Aronow WS, Adams KF., Jr Gheorghiade M. Effects of digoxin on morbidity and mortality in diastolic heart failure: the ancillary digitalis investigation group trial. Circulation. 2006;114:397–403. doi: 10.1161/CIRCULATIONAHA.106.628347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khalili L, Yu M. Chibalin AV. Na+,K+-ATPase trafficking in skeletal muscle: insulin stimulates translocation of both α1- and α2-subunit isoforms. FEBS Lett. 2003;536:198–202. doi: 10.1016/s0014-5793(03)00047-4. [DOI] [PubMed] [Google Scholar]
- Allen DG, Lamb GD. Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- Andrikopoulos P, Baba A, Matsuda T, Djamgoz MB, Yaqoob MM. Eccles SA. Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J Biol Chem. 2011;286:37919–37931. doi: 10.1074/jbc.M111.251777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoons G, Willems R. Sipido KR. Alternative strategies in arrhythmia therapy: evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol Ther. 2012;134:26–42. doi: 10.1016/j.pharmthera.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL. O'Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. doi: 10.1161/01.RES.0000080932.98903.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon A, Hamlyn JM. Blaustein MP. Ouabain augments Ca2+ transients in arterial smooth muscle without raising cytosolic Na+ Am J Physiol Heart Circ Physiol. 2000;279:H679–H691. doi: 10.1152/ajpheart.2000.279.2.H679. [DOI] [PubMed] [Google Scholar]
- Asahi M, Green NM, Kurzydlowski K, Tada M. MacLennan DH. Phospholamban domain IB forms an interaction site with the loop between transmembrane helices M6 and M7 of sarco(endo)plasmic reticulum Ca2+ ATPases. Proc Natl Acad Sci USA. 2001;98:10061–10066. doi: 10.1073/pnas.181348298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baecher S, Kroiss M, Fassnacht M. Vogeser M. No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS. Clin Chim Acta. 2014;431:87–92. doi: 10.1016/j.cca.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Bagrov AY, Fedorova OV, Dmitrieva RI, Howald WN, Hunter AP, Kuznetsova EA. Shpen VM. Characterization of a urinary bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension. 1998;31:1097–1103. doi: 10.1161/01.hyp.31.5.1097. [DOI] [PubMed] [Google Scholar]
- Bagrov YY, Manusova NB, Egorova IA, Fedorova OV. Bagrov AY. Endogenous digitalis-like ligands and Na/K-ATPase inhibition in experimental diabetes mellitus. Front Biosci. 2005;10:2257–2262. doi: 10.2741/1695. [DOI] [PubMed] [Google Scholar]
- Barrientos G, Bose DD, Feng W, Padilla I. Pessah IN. The Na+/Ca2+ exchange inhibitor 2-(2-(4-(4-nitrobenzyloxy)phenyl)ethyl)isothiourea methanesulfonate (KB-R7943) also blocks ryanodine receptors type 1 (RyR1) and type 2 (RyR2) channels. Mol Pharmacol. 2009;76:560–568. doi: 10.1124/mol.109.057265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beevers AJ. Kukol A. Secondary structure, orientation, and oligomerization of phospholemman, a cardiac transmembrane protein. Protein Sci. 2006;15:1127–1132. doi: 10.1110/ps.051899406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benziane B, Widegren U, Pirkmajer S, Henriksson J, Stepto NK. Chibalin AV. Effect of exercise and training on phospholemman phosphorylation in human skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E456–E466. doi: 10.1152/ajpendo.00533.2010. [DOI] [PubMed] [Google Scholar]
- Berry RG, Despa S, Fuller W, Bers DM. Shattock MJ. Differential distribution and regulation of mouse cardiac Na+/K+-ATPase α1 and α2 subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc Res. 2007;73:92–100. doi: 10.1016/j.cardiores.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Bers DM. Ginsburg KS. Na:Ca stoichiometry and cytosolic Ca-dependent activation of NCX in intact cardiomyocytes. Ann NY Acad Sci. 2007;1099:326–338. doi: 10.1196/annals.1387.060. [DOI] [PubMed] [Google Scholar]
- Besserer GM, Ottolia M, Nicoll DA, Chaptal V, Cascio D, Philipson KD. Abramson J. The second Ca2+-binding domain of the Na+–Ca2+ exchanger is essential for regulation: Crystal structures and mutational analysis. Proc Natl Acad Sci USA. 2007;104:18467–18472. doi: 10.1073/pnas.0707417104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best PJ, Reddan DN, Berger PB, Szczech LA, McCullough PA. Califf RM. Cardiovascular disease and chronic kidney disease: insights and an update. Am Heart J. 2004;148:230–242. doi: 10.1016/j.ahj.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Bibert S, Roy S, Schaer D, Horisberger JD. Geering K. Phosphorylation of phospholemman (FXYD1) by protein kinases A and C modulates distinct Na,K-ATPase isozymes. J Biol Chem. 2008;283:476–486. doi: 10.1074/jbc.M705830200. [DOI] [PubMed] [Google Scholar]
- Biesmans L, Macquaide N, Heinzel FR, Bito V, Smith GL. Sipido KR. Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T-tubule density. PloS One. 2011;6:e25100. doi: 10.1371/journal.pone.0025100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP. Why isn't endogenous ouabain more widely accepted? Am J Physiol Heart Circ Physiol. 2014;307:H635–H639. doi: 10.1152/ajpheart.00404.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J. Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol. 2012;302:H1031–H1049. doi: 10.1152/ajpheart.00899.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguslavskyi A, Fuller W. Shattock MJ. Phospholemman-dependent regulation of Na/K-ATPase modulates constriction and relaxation in aortic smooth muscle. Biophys J. 2014a;106:725a. [Google Scholar]
- Boguslavskyi A, Pavlovic D, Aughton K, Clark JE, Howie J, Fuller W. Shattock MJ. Cardiac hypertrophy in mice expressing unphosphorylatable phospholemman. Cardiovasc Res. 2014b;104:72–82. doi: 10.1093/cvr/cvu182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt J, Ai X, Moorman JR, Pogwizd SM. Bers DM. Expression and phosphorylation of the Na-pump regulatory subunit phospholemman in heart failure. Circ Res. 2005;97:558–565. doi: 10.1161/01.RES.0000181172.27931.c3. [DOI] [PubMed] [Google Scholar]
- Bossuyt J, Despa S, Han F, Hou Z, Robia SL, Lingrel JB. Bers DM. Isoform specificity of the Na/K-ATPase association and regulation by phospholemman. J Biol Chem. 2009;284:26749–26757. doi: 10.1074/jbc.M109.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt J, Despa S, Martin JL. Bers DM. Phospholemman phosphorylation alters its fluorescence resonance energy transfer with the Na/K-ATPase pump. J Biol Chem. 2006;281:32765–32773. doi: 10.1074/jbc.M606254200. [DOI] [PubMed] [Google Scholar]
- Bourgonje VJ, Vos MA, Ozdemir S, Doisne N, Acsai K, Varro A, Sztojkov-Ivanov A, Zupko I, Rauch E, Kattner L, Bito V, Houtman M, van der Nagel R, Beekman JD, van Veen TA, Sipido KR. Antoons G. Combined Na+/Ca2+ exchanger and L-type calcium channel block as a potential strategy to suppress arrhythmias and maintain ventricular function. Circ Arrhythm Electrophysiol. 2013;6:371–379. doi: 10.1161/CIRCEP.113.000322. [DOI] [PubMed] [Google Scholar]
- Buhagiar KA, Hansen PS, Bewick NL. Rasmussen HH. Protein kinase Cεcontributes to regulation of the sarcolemmal Na+-K+ pump. Am J Physiol Cell Physiol. 2001;281:C1059–C1063. doi: 10.1152/ajpcell.2001.281.3.C1059. [DOI] [PubMed] [Google Scholar]
- Burns WR, Cohen KD. Jackson WF. K+-induced dilation of hamster cremasteric arterioles involves both the Na+/K+-ATPase and inward-rectifier K+ channels. Microcirculation. 2004;11:279–293. doi: 10.1080/10739680490425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteels R, Kitamura K, Kuriyama H. Suzuki H. The membrane properties of the smooth muscle cells of the rabbit main pulmonary artery. J Physiol. 1977;271:41–61. doi: 10.1113/jphysiol.1977.sp011989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T. Na+-K+ pump regulation and skeletal muscle contractility. Physiol Rev. 2003;83:1269–1324. doi: 10.1152/physrev.00011.2003. [DOI] [PubMed] [Google Scholar]
- Clifford RJ. Kaplan JH. -Subunit overexpression alters the stoicheometry of assembled Na-K-ATPase subunits in MDCK cells. Am J Physiol Renal Physiol. 2008;295:F1314–F1323. doi: 10.1152/ajprenal.90406.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford RJ. Kaplan JH. Regulation of Na,K-ATPase subunit abundance by translational repression. J Biol Chem. 2009;284:22905–22915. doi: 10.1074/jbc.M109.030536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll RN, Eder P, Burr AR, Despa S, Davis J, Bers DM. Molkentin JD. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ Res. 2014;114:249–256. doi: 10.1161/CIRCRESAHA.114.302293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crambert G, Fuzesi M, Garty H, Karlish S. Geering K. Phospholemman (FXYD1) associates with Na,K-ATPase and regulates its transport properties. Proc Natl Acad Sci USA. 2002;99:11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S. Bers DM. Functional analysis of Na+/K+-ATPase isoform distribution in rat ventricular myocytes. Am J Physiol Cell Physiol. 2007;293:C321–C327. doi: 10.1152/ajpcell.00597.2006. [DOI] [PubMed] [Google Scholar]
- Despa S. Bers DM. Na+ transport in the normal and failing heart – Remember the balance. J Mol Cell Cardiol. 2013;61:2–10. doi: 10.1016/j.yjmcc.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Bossuyt J, Han F, Ginsburg KS, Jia LG, Kutchai H, Tucker AL. Bers DM. Phospholemman-phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ Res. 2005;97:252–259. doi: 10.1161/01.RES.0000176532.97731.e5. [DOI] [PubMed] [Google Scholar]
- Despa S, Brette F, Orchard CH. Bers DM. Na/Ca exchange and Na/K-ATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys J. 2003;85:3388–3396. doi: 10.1016/S0006-3495(03)74758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Islam MA, Pogwizd SM. Bers DM. Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J Physiol. 2002a;539:133–143. doi: 10.1113/jphysiol.2001.012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, Pogwizd SM. Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002b;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Despa S, Lingrel JB, Bers DM. Na+/K+-ATPase 2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc Res. 2012;95:480–486. doi: 10.1093/cvr/cvs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey K, Roy S, Ghosh B. Chakraborti S. Role of protein kinase C in phospholemman mediated regulation of α2β1 isozyme of Na+/K+-ATPase in caveolae of pulmonary artery smooth muscle cells. Biochimie. 2012;94:991–1000. doi: 10.1016/j.biochi.2011.12.020. [DOI] [PubMed] [Google Scholar]
- Dipla K, Mattiello JA, Margulies KB, Jeevanandam V. Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84:435–444. doi: 10.1161/01.res.84.4.435. [DOI] [PubMed] [Google Scholar]
- Dmitrieva RI, Bagrov AY, Lalli E, Sassone-Corsi P, Stocco DM. Doris PA. Mammalian bufadienolide is synthesized from cholesterol in the adrenal cortex by a pathway that is independent of cholesterol side-chain cleavage. Hypertension. 2000;36:442–448. doi: 10.1161/01.hyp.36.3.442. [DOI] [PubMed] [Google Scholar]
- Dong XH, Komiyama Y, Nishimura N, Masuda M. Takahashi H. Nanomolar level of ouabain increases intracellular calcium to produce nitric oxide in rat aortic endothelial cells. Clin Exp Pharmacol Physiol. 2004;31:276–283. doi: 10.1111/j.1440-1681.2004.03995.x. [DOI] [PubMed] [Google Scholar]
- Donohoe P, McMahon AC, Walgama OV, Bertaso F, Dockrell ME, Cramp HA, Mullen AM, Shattock MJ, Hendry BM. James AF. L-type calcium current of isolated rat cardiac myocytes in experimental uraemia. Nephrol Dial Transplant. 2000;15:791–798. doi: 10.1093/ndt/15.6.791. [DOI] [PubMed] [Google Scholar]
- Dora KA, Gallagher NT, McNeish A. Garland CJ. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res. 2008;102:1247–1255. doi: 10.1161/CIRCRESAHA.108.172379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW. Lingrel JB. The α2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol. 2005;288:H477–H485. doi: 10.1152/ajpheart.00083.2004. [DOI] [PubMed] [Google Scholar]
- Dostanic I, Schultz Jel J, Lorenz JN. Lingrel JB. The α1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J Biol Chem. 2004;279:54053–54061. doi: 10.1074/jbc.M410737200. [DOI] [PubMed] [Google Scholar]
- Dostanic-Larson I, Van Huysse JW, Lorenz JN. Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA. 2005;102:15845–15850. doi: 10.1073/pnas.0507358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Armouche A, Wittkopper K, Fuller W, Howie J, Shattock MJ. Pavlovic D. Phospholemman-dependent regulation of the cardiac Na/K-ATPase activity is modulated by inhibitor-1 sensitive type-1 phosphatase. FASEB J. 2011;25:4467–4475. doi: 10.1096/fj.11-184903. [DOI] [PubMed] [Google Scholar]
- Esler M, Lambert E. Schlaich M. Point: Chronic activation of the sympathetic nervous system is the dominant contributor to systemic hypertension. J Appl Physiol (1985) 2010;109:1996–1998. doi: 10.1152/japplphysiol.00182.2010. [DOI] [PubMed] [Google Scholar]
- Fedorova LV, Raju V, El-Okdi N, Shidyak A, Kennedy DJ, Vetteth S, Giovannucci DR, Bagrov AY, Fedorova OV, Shapiro JI. Malhotra D. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: implication of epithelial-to-mesenchymal transition. Am J Physiol Renal Physiol. 2009;296:F922–F934. doi: 10.1152/ajprenal.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova OV, Talan MI, Agalakova NI, Lakatta EG. Bagrov AY. Endogenous ligand of α1 sodium pump, marinobufagenin, is a novel mediator of sodium chloride-dependent hypertension. Circulation. 2002;105:1122–1127. doi: 10.1161/hc0902.104710. [DOI] [PubMed] [Google Scholar]
- Fedorova OV, Zhuravin IA, Agalakova NI, Yamova LA, Talan MI, Lakatta EG. Bagrov AY. Intrahippocampal microinjection of an exquisitely low dose of ouabain mimics NaCl loading and stimulates a bufadienolide Na/K-ATPase inhibitor. J Hypertens. 2007;25:1834–1844. doi: 10.1097/HJH.0b013e328200497a. [DOI] [PubMed] [Google Scholar]
- Ferrandi M, Molinari I, Barassi P, Minotti E, Bianchi G. Ferrari P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J Biol Chem. 2004;279:33306–33314. doi: 10.1074/jbc.M402187200. [DOI] [PubMed] [Google Scholar]
- Ferrari P, Ferrandi M, Valentini G. Bianchi G. Rostafuroxin: an ouabain antagonist that corrects renal and vascular Na+-K+- ATPase alterations in ouabain and adducin-dependent hypertension. AmJPhysiol Regul Integr Comp Physiol. 2006;290:R529–R535. doi: 10.1152/ajpregu.00518.2005. [DOI] [PubMed] [Google Scholar]
- Figtree GA, Liu CC, Bibert S, Hamilton EJ, Garcia A, White CN, Chia KK, Cornelius F, Geering K. Rasmussen HH. Reversible oxidative modification: a key mechanism of Na+-K+ pump regulation. Circ Res. 2009;105:185–193. doi: 10.1161/CIRCRESAHA.109.199547. [DOI] [PubMed] [Google Scholar]
- Frank JS, Mottino G, Reid D, Molday RS. Philipson KD. Distribution of the Na+-Ca2+ exchange protein in mammalian cardiac myocytes: an immunofluorescence and immunocolloidal gold-labeling study. J Cell Biol. 1992;117:337–345. doi: 10.1083/jcb.117.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzin CM, Gong XM, Teriete P. Marassi FM. Structures of the FXYD regulatory proteins in lipid micelles and membranes. J Bioenerg Biomembr. 2007a;39:379–383. doi: 10.1007/s10863-007-9105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzin CM, Gong XM, Thai K, Yu J. Marassi FM. NMR of membrane proteins in micelles and bilayers: the FXYD family proteins. Methods. 2007b;41:398–408. doi: 10.1016/j.ymeth.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller W, Eaton P, Bell JR. Shattock MJ. Ischemia-induced phosphorylation of phospholemman directly activates rat cardiac Na/K-ATPase. FASEB J. 2004;18:197–199. doi: 10.1096/fj.03-0213fje. [DOI] [PubMed] [Google Scholar]
- Fuller W, Howie J, McLatchie LM, Weber RJ, Hastie CJ, Burness K, Pavlovic D. Shattock MJ. FXYD1 phosphorylation in vitro and in adult rat cardiac myocytes: threonine 69 is a novel substrate for protein kinase C. Am J Physiol Cell Physiol. 2009;296:C1346–C1355. doi: 10.1152/ajpcell.00523.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallice PM, Kovacic HN, Brunet PJ, Berland YF. Crevat AD. A non ouabain-like inhibitor of the sodium pump in uremic plasma ultrafiltrates and urine from healthy subjects. Clin Chim Acta. 1998;273:149–160. doi: 10.1016/s0009-8981(98)00032-1. [DOI] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Wang Y, Sun X. Baldo GJ. Activation of PKC increases Na+-K+ pump current in ventricular myocytes from guinea pig heart. Pflugers Arch. 1999;437:643–651. doi: 10.1007/s004240050828. [DOI] [PubMed] [Google Scholar]
- Gao J, Sun X, Potapova IA, Cohen IS, Mathias RT. Kim JH. Autocrine A2 in the T-system of ventricular myocytes creates transmural gradients in ion transport: a mechanism to match contraction with load. Biophys J. 2014;106:2364–2374. doi: 10.1016/j.bpj.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Rasmussen TP, Li Y, Kutschke W, Koval OM, Wu Y, Wu Y, Hall DD, Joiner ML, Wu XQ, Swaminathan PD, Purohit A, Zimmerman K, Weiss RM, Philipson KD, Song LS, Hund TJ. Anderson ME. Genetic inhibition of Na+-Ca2+ exchanger current disables fight or flight sinoatrial node activity without affecting resting heart rate. Circ Res. 2013;112:309–317. doi: 10.1161/CIRCRESAHA.111.300193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi M, Boyman L, Mikhasenko H, Hiller R. Khananshvili D. Essential role of the CBD1-CBD2 linker in slow dissociation of Ca2+ from the regulatory two-domain tandem of NCX1. J Biol Chem. 2010;285:28117–28125. doi: 10.1074/jbc.M110.127001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg KS, Weber CR. Bers DM. Cardiac Na+–Ca2+ exchanger: dynamics of Ca2+-dependent activation and deactivation in intact myocytes. J Physiol. 2013;591:2067–2086. doi: 10.1113/jphysiol.2013.252080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldhaber JI, Lamp ST, Walter DO, Garfinkel A, Fukumoto GH. Weiss JN. Local regulation of the threshold for calcium sparks in rat ventricular myocytes: role of sodium-calcium exchange. J Physiol. 1999;520:431–438. doi: 10.1111/j.1469-7793.1999.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonick HC, Ding Y, Vaziri ND, Bagrov AY. Fedorova OV. Simultaneous measurement of marinobufagenin, ouabain, and hypertension-associated protein in various disease states. Clin Exp Hypertens. 1998;20:617–627. doi: 10.3109/10641969809053240. [DOI] [PubMed] [Google Scholar]
- Grandi E, Pasqualini FS. Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48:112–121. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenke S, Larson ED, Alber S, Zhang R, Lamp ST, Ren X, Nakano H, Jordan MC, Karagueuzian HS, Roos KP, Nakano A, Proenza C, Philipson KD. Goldhaber JI. Complete atrial-specific knockout of sodium-calcium exchange eliminates sinoatrial node pacemaker activity. PloS One. 2013;8:e81633. doi: 10.1371/journal.pone.0081633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, McArthur C, Grady C. Ruderman NB. Stimulation of vascular Na+-K+-ATPase activity by nitric oxide: a cGMP-independent effect. Am J Physiol Heart Circ Physiol. 1994;266:H2146–H2151. doi: 10.1152/ajpheart.1994.266.5.H2146. [DOI] [PubMed] [Google Scholar]
- Haddock PS, Shattock MJ. Hearse DJ. Modulation of cardiac Na+-K+ pump current: role of protein and nonprotein sulfhydryl redox status. Am J Physiol Heart Circ Physiol. 1995;38:H297–H319. doi: 10.1152/ajpheart.1995.269.1.H297. [DOI] [PubMed] [Google Scholar]
- Hamlyn JM, Ringel R, Schaeffer J, Levinson PD, Hamilton BP, Kowarski AA. Blaustein MP. A circulating inhibitor of (Na+ + K+)ATPase associated with essential hypertension. Nature. 1982;300:650–652. doi: 10.1038/300650a0. [DOI] [PubMed] [Google Scholar]
- Han F, Bossuyt J, Despa S, Tucker AL. Bers DM. Phospholemman phosphorylation mediates the protein kinase C-dependent effects on Na+/K+ pump function in cardiac myocytes. Circ Res. 2006;99:1376–1383. doi: 10.1161/01.RES.0000251667.73461.fb. [DOI] [PubMed] [Google Scholar]
- Harrison SM, McCall E. Boyett MR. The relationship between contraction and intracellular sodium in rat and guinea pig ventricular myocytes. J Physiol. 1992;449:517–550. doi: 10.1113/jphysiol.1992.sp019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood S, Mullen AM, McMahon AC. Dawnay A. Plasma OLC is elevated in mild experimental uremia but is not associated with hypertension. Am J Hypertens. 2001;14:1112–1115. doi: 10.1016/s0895-7061(01)02219-1. [DOI] [PubMed] [Google Scholar]
- He S, Shelly DA, Moseley AE, James PF, James JH, Paul RJ. Lingrel JB. The α1- and α2-isoforms of Na-K-ATPase play different roles in skeletal muscle contractility. AmJPhysiol Regul Integr Comp Physiol. 2001;281:R917–R925. doi: 10.1152/ajpregu.2001.281.3.R917. [DOI] [PubMed] [Google Scholar]
- Heiny JA, Kravtsova VV, Mandel F, Radzyukevich TL, Benziane B, Prokofiev AV, Pedersen SE, Chibalin AV. Krivoi II. The nicotinic acetylcholine receptor and the Na,K-ATPase α2 isoform interact to regulate membrane electrogenesis in skeletal muscle. J Biol Chem. 2010;285:28614–28626. doi: 10.1074/jbc.M110.150961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CB, Azuma KK, Tang MJ. McDonough AA. Thyroid hormone induction of rat myocardial Na+-K+-ATPase: alpha 1-, alpha 2-, and beta 1-mRNA and -protein levels at steady state. Am J Physiol Cell Physiol. 1992;262:C484–C492. doi: 10.1152/ajpcell.1992.262.2.C484. [DOI] [PubMed] [Google Scholar]
- Herrmann S, Lipp P, Wiesen K, Stieber J, Nguyen H, Kaiser E. Ludwig A. The cardiac sodium-calcium exchanger NCX1 is a key player in the initiation and maintenance of a stable heart rhythm. Cardiovasc Res. 2013;99:780–788. doi: 10.1093/cvr/cvt154. [DOI] [PubMed] [Google Scholar]
- Hilge M, Aelen J. Vuister GW. Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol Cell. 2006;22:15–25. doi: 10.1016/j.molcel.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW, Collins A. Matsuoka S. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Secondary modulation by cytoplasmic calcium and ATP. J Gen Physiol. 1992;100:933–961. doi: 10.1085/jgp.100.6.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobai IA. O'Rourke B. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- Hobbs RE. Digoxin's effect on mortality and hospitalization in heart failure: implications of the DIG study. Digitalis Investigation Group. Cleve Clin J Med. 1997;64:234–237. doi: 10.3949/ccjm.64.5.234. [DOI] [PubMed] [Google Scholar]
- Horvat D, Severson J, Uddin MN, Mitchell B. Puschett JB. Resibufogenin prevents the manifestations of preeclampsia in an animal model of the syndrome. Hypertens Pregnancy. 2010;29:1–9. doi: 10.3109/10641950802629709. [DOI] [PubMed] [Google Scholar]
- Howie J, Tulloch LB, Shattock MJ. Fuller W. Regulation of the cardiac Na+ pump by palmitoylation of its catalytic and regulatory subunits. Biochem Soc Trans. 2013;41:95–100. doi: 10.1042/BST20120269. [DOI] [PubMed] [Google Scholar]
- Huang BS, Amin MS. Leenen FH. The central role of the brain in salt-sensitive hypertension. Curr Opin Cardiol. 2006;21:295–304. doi: 10.1097/01.hco.0000231398.64362.94. [DOI] [PubMed] [Google Scholar]
- Huang BS, Van Vliet BN. Leenen FH. Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am J Physiol Heart Circ Physiol. 2004;287:H1160–H1166. doi: 10.1152/ajpheart.00126.2004. [DOI] [PubMed] [Google Scholar]
- Huang BS, Veerasingham SJ. Leenen FH. Brain ‘ouabain,’ ANG II, and sympathoexcitation by chronic central sodium loading in rats. Am J Physiol Heart Circ Physiol. 1998;274:H1269–H1276. doi: 10.1152/ajpheart.1998.274.4.H1269. [DOI] [PubMed] [Google Scholar]
- Huang BS, Wang H. Leenen FH. Enhanced sympathoexcitatory and pressor responses to central Na+ in Dahl salt-sensitive vs. -resistant rats. Am J Physiol Heart Circ Physiol. 2001;281:H1881–H1889. doi: 10.1152/ajpheart.2001.281.5.H1881. [DOI] [PubMed] [Google Scholar]
- Hundal HS, Marette A, Mitsumoto Y, Ramlal T, Blostein R. Klip A. Insulin induces translocation of the α2 and β1 subunits of the Na+/K+-ATPase from intracellular compartments to the plasma membrane in mammalian skeletal muscle. J Biol Chem. 1992;267:5040–5043. [PubMed] [Google Scholar]
- Iwamoto T, Nakamura TY, Pan Y, Uehara A, Imanaga I. Shigekawa M. Unique topology of the internal repeats in the cardiac Na+/Ca2+ exchanger. FEBS Lett. 1999;446:264–268. doi: 10.1016/s0014-5793(99)00218-5. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Uehara A, Imanaga I. Shigekawa M. The Na+/Ca2+ exchanger NCX1 has oppositely oriented reentrant loop domains that contain conserved aspartic acids whose mutation alters its apparent Ca2+ affinity. J Biol Chem. 2000;275:38571–38580. doi: 10.1074/jbc.M003788200. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Watanabe Y, Kita S. Blaustein MP. Na+/Ca2+ exchange inhibitors: a new class of calcium regulators. Cardiovasc Hematol Disord Drug Targets. 2007;7:188–198. doi: 10.2174/187152907781745288. [DOI] [PubMed] [Google Scholar]
- James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA. Lingrel JB. Identification of a specific role for the Na,K-ATPase α2 isoform as a regulator of calcium in the heart. Mol Cell. 1999;3:555–563. doi: 10.1016/s1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- Jayasinghe ID, Cannell MB. Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys J. 2009;97:2664–2673. doi: 10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John SA, Liao J, Jiang Y. Ottolia M. The cardiac Na+-Ca2+ exchanger has two cytoplasmic ion permeation pathways. Proc Natl Acad Sci USA. 2013;110:7500–7505. doi: 10.1073/pnas.1218751110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John SA, Ribalet B, Weiss JN, Philipson KD. Ottolia M. Ca2+-dependent structural rearrangements within Na+-Ca2+ exchanger dimers. Proc Natl Acad Sci USA. 2011;108:1699–1704. doi: 10.1073/pnas.1016114108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen PL, Hakansson KO. Karlish SJ. Structure and mechanism of Na,K-ATPase: functional sites and their interactions. Annu Rev Physiol. 2003;65:817–849. doi: 10.1146/annurev.physiol.65.092101.142558. [DOI] [PubMed] [Google Scholar]
- Juel C, Pilegaard H, Nielsen JJ. Bangsbo J. Interstitial K+ in human skeletal muscle during and after dynamic graded exercise determined by microdialysis. Am J Physiol Regul Integr Comp Physiol. 2000;278:R400–R406. doi: 10.1152/ajpregu.2000.278.2.R400. [DOI] [PubMed] [Google Scholar]
- Juhaszova M. Blaustein MP. Na+ pump low and high ouabain affinity α subunit isoforms are differently distributed in cells. Proc Natl Acad Sci USA. 1997;94:1800–1805. doi: 10.1073/pnas.94.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai R, Ogawa H, Vilsen B, Cornelius F. Toyoshima C. Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature. 2013;502:201–206. doi: 10.1038/nature12578. [DOI] [PubMed] [Google Scholar]
- Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem. 2002;71:511–535. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- Karim CB, Zhang Z, Howard EC, Torgersen KD. Thomas DD. Phosphorylation-dependent conformational switch in spin-labeled phospholamban bound to SERCA. J Mol Biol. 2006;358:1032–1040. doi: 10.1016/j.jmb.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Kennedy DJ, Elkareh J, Shidyak A, Shapiro AP, Smaili S, Mutgi K, Gupta S, Tian J, Morgan E, Khouri S, Cooper CJ, Periyasamy SM, Xie Z, Malhotra D, Fedorova OV, Bagrov AY. Shapiro JI. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol. 2008;294:F450–F454. doi: 10.1152/ajprenal.00472.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY. Shapiro JI. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47:488–495. doi: 10.1161/01.HYP.0000202594.82271.92. [DOI] [PubMed] [Google Scholar]
- Khafaga M, Bossuyt J, Mamikonian L, Li JC, Lee LL, Yarov-Yarovoy V, Despa S. Bers DM. Na+/K+-ATPase E960 and phospholemman F28 are critical for their functional interaction. Proc Natl Acad Sci USA. 2012;109:20756–20761. doi: 10.1073/pnas.1207866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Manunta P, Hamilton BP. Hamlyn JM. Different effects of in vivo ouabain and digoxin on renal artery function and blood pressure in the rat. Hypertens Res. 2000;23(Suppl):S67–76. doi: 10.1291/hypres.23.supplement_s67. [DOI] [PubMed] [Google Scholar]
- Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O'Rourke B. Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M. Takahashi H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem. 2005;38:36–45. doi: 10.1016/j.clinbiochem.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Larbig R, Torres N, Bridge JH, Goldhaber JI. Philipson KD. Activation of reverse Na+–Ca2+ exchange by the Na+ current augments the cardiac Ca2+ transient: evidence from NCX knockout mice. J Physiol. 2010;588:3267–3276. doi: 10.1113/jphysiol.2010.187708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laredo J, Hamilton BP. Hamlyn JM. Secretion of endogenous ouabain from bovine adrenocortical cells: role of the zona glomerulosa and zona fasciculata. Biochem Biophys Res Commun. 1995;212:487–493. doi: 10.1006/bbrc.1995.1996. [DOI] [PubMed] [Google Scholar]
- Lavoie L, Levenson R, Martin-Vasallo P. Klip A. The molar ratios of αand βsubunits of the Na+–K+-ATPase differ in distinct subcellular membranes from rat skeletal muscle. Biochemistry. 1997;36:7726–7732. doi: 10.1021/bi970109s. [DOI] [PubMed] [Google Scholar]
- Lee C, Dhalla NS. Hryshko LV. Therapeutic potential of novel Na+-Ca2+ exchange inhibitors in attenuating ischemia-reperfusion injury. Can J Cardiol. 2005;21:509–516. [PubMed] [Google Scholar]
- Lee MY, Song H, Nakai J, Ohkura M, Kotlikoff MI, Kinsey SP, Golovina VA. Blaustein MP. Local subplasma membrane Ca2+ signals detected by a tethered Ca2+ sensor. Proc Natl Acad Sci USA. 2006;103:13232–13237. doi: 10.1073/pnas.0605757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung HS, Yung LM, Leung FP, Yao X, Chen ZY, Ko WH, Laher I. Huang Y. Tamoxifen dilates porcine coronary arteries: roles for nitric oxide and ouabain-sensitive mechanisms. Br J Pharmacol. 2006;149:703–711. doi: 10.1038/sj.bjp.0706921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Grosdidier A, Crambert G, Horisberger JD, Michielin O. Geering K. Structural and functional interaction sites between Na,K-ATPase and FXYD proteins. J Biol Chem. 2004;279:38895–38902. doi: 10.1074/jbc.M406697200. [DOI] [PubMed] [Google Scholar]
- Li S, Liu G, Jia J, Miao Y, Gu S, Miao P, Shi X, Wang Y. Yu C. Therapeutic monitoring of serum digoxin for patients with heart failure using a rapid LC-MS/MS method. Clin Biochem. 2010;43:307–313. doi: 10.1016/j.clinbiochem.2009.09.025. [DOI] [PubMed] [Google Scholar]
- Liang M, Tian J, Liu L, Pierre S, Liu J, Shapiro J. Xie ZJ. Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem. 2007;282:10585–10593. doi: 10.1074/jbc.M609181200. [DOI] [PubMed] [Google Scholar]
- Liao J, Li H, Zeng W, Sauer DB, Belmares R. Jiang Y. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science. 2012;335:686–690. doi: 10.1126/science.1215759. [DOI] [PubMed] [Google Scholar]
- Linde CI, Antos LK, Golovina VA. Blaustein MP. Nanomolar ouabain increases NCX1 expression and enhances Ca2+ signaling in human arterial myocytes: a mechanism that links salt to increased vascular resistance. Am J Physiol Heart Circ Physiol. 2012;303:H784–H794. doi: 10.1152/ajpheart.00399.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindzen M, Gottschalk KE, Fuzesi M, Garty H. Karlish SJ. Structural interactions between FXYD proteins and Na+,K+-ATPase: α/β/FXYD subunit stoichiometry and cross-linking. J Biol Chem. 2006;281:5947–5955. doi: 10.1074/jbc.M512063200. [DOI] [PubMed] [Google Scholar]
- Lingrel JB. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Ann Rev Physiol. 2010;72:395–412. doi: 10.1146/annurev-physiol-021909-135725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingrel JB. Kuntzweiler T. Na+,K+-ATPase. J Biol Chem. 1994;269:19659–19662. [PubMed] [Google Scholar]
- Liu J, Kesiry R, Periyasamy SM, Malhotra D, Xie Z. Shapiro JI. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 2004;66:227–241. doi: 10.1111/j.1523-1755.2004.00723.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Liang M, Liu L, Malhotra D, Xie Z. Shapiro JI. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 2005;67:1844–1854. doi: 10.1111/j.1523-1755.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Periyasamy SM, Gunning W, Fedorova OV, Bagrov AY, Malhotra D, Xie Z. Shapiro JI. Effects of cardiac glycosides on sodium pump expression and function in LLC-PK1 and MDCK cells. Kidney Int. 2002;62:2118–2125. doi: 10.1046/j.1523-1755.2002.00672.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Tian J, Haas M, Shapiro JI, Askari A. Xie Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem. 2000;275:27838–27844. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- Liu T, Brown DA. O'Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol. 2010;49:728–736. doi: 10.1016/j.yjmcc.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. O'Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz JN, Loreaux EL, Dostanic-Larson I, Lasko V, Schnetzer JR, Paul RJ. Lingrel JB. ACTH-induced hypertension is dependent on the ouabain-binding site of the α2-Na+-K+-ATPase subunit. Am J Physiol Heart Circ Physiol. 2008;295:H273–H280. doi: 10.1152/ajpheart.00183.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K. Sipido KR. Reduced synchrony of Ca2+ release with loss of T-tubules–a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- Lubarski I, Pihakaski-Maunsbach K, Karlish SJ, Maunsbach AB. Garty H. Interaction with the Na,K-ATPase and tissue distribution of FXYD5 (related to ion channel) J Biol Chem. 2005;280:37717–37724. doi: 10.1074/jbc.M506397200. [DOI] [PubMed] [Google Scholar]
- Lundmark JL, Ramasamy R, Vulliet PR. Schaefer S. Chelerythrine increases Na-K-ATPase activity and limits ischemic injury in isolated rat hearts. Am J Physiol Heart Circ Physiol. 1999;277:H999–H1006. doi: 10.1152/ajpheart.1999.277.3.H999. [DOI] [PubMed] [Google Scholar]
- Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406:365–382. doi: 10.1042/BJ20070619. [DOI] [PubMed] [Google Scholar]
- McDonough AA, Zhang Y, Shin V. Frank JS. Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am J Physiol Cell Physiol. 1996;270:C1221–C1227. doi: 10.1152/ajpcell.1996.270.4.C1221. [DOI] [PubMed] [Google Scholar]
- McMahon AC, Greenwald SE, Dodd SM, Hurst MJ. Raine AE. Prolonged calcium transients and myocardial remodelling in early experimental uraemia. Nephrol Dial Transplant. 2002;17:759–764. doi: 10.1093/ndt/17.5.759. [DOI] [PubMed] [Google Scholar]
- McMahon AC, Naqvi RU, Hurst MJ, Raine AE. MacLeod KT. Diastolic dysfunction and abnormality of the Na+/Ca2+ exchanger in single uremic cardiac myocytes. Kidney Int. 2006;69:846–851. doi: 10.1038/sj.ki.5000193. [DOI] [PubMed] [Google Scholar]
- McMahon AC, Vescovo G, Dalla Libera L, Wynne DG, Fluck RJ, Harding SE. Raine AE. Contractile dysfunction of isolated ventricular myocytes in experimental uraemia. Exp Nephrol. 1996;4:144–150. [PubMed] [Google Scholar]
- Manunta P, Hamilton BP. Hamlyn JM. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am J Physiol Regul Integr Comp Physiol. 2006;290:R553–R559. doi: 10.1152/ajpregu.00648.2005. [DOI] [PubMed] [Google Scholar]
- Manunta P, Hamilton J, Rogowski AC, Hamilton BP. Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res. 2000;23(Suppl):S77–85. doi: 10.1291/hypres.23.supplement_s77. [DOI] [PubMed] [Google Scholar]
- Manunta P, Stella P, Rivera R, Ciurlino D, Cusi D, Ferrandi M, Hamlyn JM. Bianchi G. Left ventricular mass, stroke volume, and ouabain-like factor in essential hypertension. Hypertension. 1999;34:450–456. doi: 10.1161/01.hyp.34.3.450. [DOI] [PubMed] [Google Scholar]
- Matchkov VV, Gustafsson H, Rahman A, Briggs Boedtkjer DM, Gorintin S, Hansen AK, Bouzinova EV, Praetorius HA, Aalkjaer C. Nilsson H. Interaction between Na+/K+-pump and Na+/Ca2+-exchanger modulates intercellular communication. Circ Res. 2007;100:1026–1035. doi: 10.1161/01.RES.0000262659.09293.56. [DOI] [PubMed] [Google Scholar]
- Matchkov VV, Moeller-Nielsen N, Dam VS, Nourian Z, Briggs Boedtkjer DM. Aalkjaer C. The α2 isoform of the Na,K-pump is important for intercellular communication, agonist-induced contraction, and EDHF-like response in rat mesenteric arteries. Am J Physiol Heart Circ Physiol. 2012;303:H36–H46. doi: 10.1152/ajpheart.00673.2011. [DOI] [PubMed] [Google Scholar]
- Matsuoka S. Hilgemann DW. Inactivation of outward Na+-Ca2+ exchange current in guinea-pig ventricular myocytes. J Physiol. 1994;476:443–458. doi: 10.1113/jphysiol.1994.sp020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Nicoll DA, Hryshko LV, Levitsky DO, Weiss JN. Philipson KD. Regulation of the cardiac Na+-Ca2+ exchanger by Ca2+. Mutational analysis of the Ca2+-binding domain. J Gen Physiol. 1995;105:403–420. doi: 10.1085/jgp.105.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medbo JI. Sejersted OM. Plasma potassium changes with high intensity exercise. J Physiol. 1990;421:105–122. doi: 10.1113/jphysiol.1990.sp017935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler PJ, Davis JQ. Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin JC, Sguilla FS. Bendhack LM. Decreased contraction to phenylephrine by ouabain in 2K-1C rat aorta is modulated by the endothelium. Eur J Pharmacol. 2005;522:94–99. doi: 10.1016/j.ejphar.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Cassell MD, Beltz TG, Johnson AK, Davisson RL. Sigmund CD. Elevated blood pressure in transgenic mice with brain-specific expression of human angiotensinogen driven by the glial fibrillary acidic protein promoter. Circ Res. 2001;89:365–372. doi: 10.1161/hh1601.094988. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Cassell MD. Sigmund CD. Glia- and neuron-specific expression of the renin-angiotensin system in brain alters blood pressure, water intake, and salt preference. J Biol Chem. 2002;277:33235–33241. doi: 10.1074/jbc.M204309200. [DOI] [PubMed] [Google Scholar]
- Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TL, Petersen J, Andersen JP, Vilsen B. Nissen P. Crystal structure of the sodium-potassium pump. Nature. 2007;450:1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- Murrell JR, Randall JD, Rosoff J, Zhao JL, Jensen RV, Gullans SR. Haupert GT., Jr Endogenous ouabain: upregulation of steroidogenic genes in hypertensive hypothalamus but not adrenal. Circulation. 2005;112:1301–1308. doi: 10.1161/CIRCULATIONAHA.105.554071. [DOI] [PubMed] [Google Scholar]
- Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD. Goldhaber JI. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J. 2010;99:755–764. doi: 10.1016/j.bpj.2010.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer S, Newell JB. Ingwall JS. Metabolic consequences and predictability of ventricular fibrillation in hypoxia. A 31P- and 23Na-nuclear magnetic resonance study of the isolated rat heart. Circulation. 1992;86:302–310. doi: 10.1161/01.cir.86.1.302. [DOI] [PubMed] [Google Scholar]
- Nicoll DA, Hryshko LV, Matsuoka S, Frank JS. Philipson KD. Mutation of amino acid residues in the putative transmembrane segments of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem. 1996;271:13385–13391. doi: 10.1074/jbc.271.23.13385. [DOI] [PubMed] [Google Scholar]
- Nicoll DA, Sawaya MR, Kwon S, Cascio D, Philipson KD. Abramson J. The crystal structure of the primary Ca2+ sensor of the Na+/Ca2+ exchanger reveals a novel Ca2+ binding motif. J Biol Chem. 2006;281:21577–21581. doi: 10.1074/jbc.C600117200. [DOI] [PubMed] [Google Scholar]
- Nielsen OB. Clausen T. The significance of active Na+,K+ transport in the maintenance of contractility in rat skeletal muscle. Acta Physiol Scand. 1996;157:199–209. doi: 10.1046/j.1365-201X.1996.d01-748.x. [DOI] [PubMed] [Google Scholar]
- Nielsen OB. de Paoli FV. Regulation of Na+-K+ homeostasis and excitability in contracting muscles: implications for fatigue. Appl Physiol Nutr Metab. 2007;32:974–984. doi: 10.1139/H07-099. [DOI] [PubMed] [Google Scholar]
- Niu CF, Watanabe Y, Ono K, Iwamoto T, Yamashita K, Satoh H, Urushida T, Hayashi H. Kimura J. Characterization of SN-6, a novel Na+/Ca2+ exchange inhibitor in guinea pig cardiac ventricular myocytes. Eur J Pharmacol. 2007;573:161–169. doi: 10.1016/j.ejphar.2007.06.033. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Shinoda T, Cornelius F. Toyoshima C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc Natl Acad Sci USA. 2009;106:13742–13747. doi: 10.1073/pnas.0907054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolia M, Nicoll DA. Philipson KD. Mutational analysis of the α-1 repeat of the cardiac Na+-Ca2+ exchanger. J Biol Chem. 2005;280:1061–1069. doi: 10.1074/jbc.M411899200. [DOI] [PubMed] [Google Scholar]
- Ottolia M, Nicoll DA. Philipson KD. Roles of two Ca2+-binding domains in regulation of the cardiac Na+-Ca2+ exchanger. J Biol Chem. 2009;284:32735–32741. doi: 10.1074/jbc.M109.055434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir S, Bito V, Holemans P, Vinet L, Mercadier JJ, Varro A. Sipido KR. Pharmacological inhibition of Na/Ca exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ Res. 2008;102:1398–1405. doi: 10.1161/CIRCRESAHA.108.173922. [DOI] [PubMed] [Google Scholar]
- Palmer CJ, Scott BT. Jones LR. Purification and complete sequence determination of the major plasma membrane substrate for cAMP-dependent protein kinase and protein kinase C in myocardium. J Biol Chem. 1991;266:11126–11130. [PubMed] [Google Scholar]
- Pamnani MB, Chen S, Yuan CM. Haddy FJ. Chronic blood pressure effects of bufalin, a sodium-potassium ATPase inhibitor, in rats. Hypertension. 1994;23:I106–109. doi: 10.1161/01.hyp.23.1_suppl.i106. [DOI] [PubMed] [Google Scholar]
- Pavlovic D. The role of cardiotonic steroids in the pathogenesis of cardiomyopathy in chronic kidney disease. Nephron Clin Pract. 2014 doi: 10.1159/000363301. (in press); DOI: 10.1159/000363301. [DOI] [PubMed] [Google Scholar]
- Pavlovic D, Aksentijevic D, Seymour AL, Fuller W. Shattock MJ. A soluble inhibitor depresses Na/K ATPase activity in cardiomyopathic hearts from uremic rats. Circulation. 2010;122:A16016. [Google Scholar]
- Pavlovic D, Fuller W. Shattock MJ. The intracellular region of FXYD1 is sufficient to regulate cardiac Na/K ATPase. FASEB J. 2007;21:1539–1546. doi: 10.1096/fj.06-7269com. [DOI] [PubMed] [Google Scholar]
- Pavlovic D, Hall AR, Kennington EJ, Aughton K, Boguslavskyi A, Fuller W, Despa S, Bers DM. Shattock MJ. Nitric oxide regulates cardiac intracellular Na+ and Ca2+ by modulating Na/K ATPase via PKCεand phospholemman-dependent mechanism. J Mol Cell Cardiol. 2013;61:164–171. doi: 10.1016/j.yjmcc.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Huang L, Xie Z, Huang WH. Askari A. Partial inhibition of Na+/K+-ATPase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J Biol Chem. 1996;271:10372–10378. doi: 10.1074/jbc.271.17.10372. [DOI] [PubMed] [Google Scholar]
- Periyasamy SM, Chen J, Cooney D, Carter P, Omran E, Tian J, Priyadarshi S, Bagrov A, Fedorova O, Malhotra D, Xie Z. Shapiro JI. Effects of uremic serum on isolated cardiac myocyte calcium cycling and contractile function. Kidney Int. 2001;60:2367–2376. doi: 10.1046/j.1523-1755.2001.00053.x. [DOI] [PubMed] [Google Scholar]
- Philipson KD, Nicoll DA, Ottolia M, Quednau BD, Reuter H, John S. Qiu Z. The Na+/Ca2+ exchange molecule: an overview. Ann NY Acad Sci. 2002;976:1–10. doi: 10.1111/j.1749-6632.2002.tb04708.x. [DOI] [PubMed] [Google Scholar]
- Pierre SV. Xie Z. The Na,K-ATPase receptor complex: its organization and membership. Cell Biochem Biophys. 2006;46:303–316. doi: 10.1385/cbb:46:3:303. [DOI] [PubMed] [Google Scholar]
- Pieske B. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM. Clinical potential of sodium-calcium exchanger inhibitors as antiarrhythmic agents. Drugs. 2003;63:439–452. doi: 10.2165/00003495-200363050-00001. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM. Bers DM. Na/Ca exchange in heart failure: contractile dysfunction and arrhythmogenesis. Ann NY Acad Sci. 2002;976:454–465. doi: 10.1111/j.1749-6632.2002.tb04775.x. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Qi M, Yuan W, Samarel AM. Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Sipido KR, Verdonck F. Bers DM. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res. 2003;57:887–896. doi: 10.1016/s0008-6363(02)00735-6. [DOI] [PubMed] [Google Scholar]
- Pott C, Philipson KD. Goldhaber JI. Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: reduced transsarcolemmal Ca2+ flux. Circ Res. 2005;97:1288–1295. doi: 10.1161/01.RES.0000196563.84231.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price EM. Lingrel JB. Structure-function relationships in the Na,K-ATPase αsubunit: site-directed mutagenesis of glutamine-111 to arginine and asparagine-122 to aspartic acid generates a ouabain-resistant enzyme. Biochemistry. 1988;27:8400–8408. doi: 10.1021/bi00422a016. [DOI] [PubMed] [Google Scholar]
- Pritchard TJ, Bowman PS, Jefferson A, Tosun M, Lynch RM. Paul RJ. Na+-K+-ATPase and Ca2+ clearance proteins in smooth muscle: a functional unit. Am J Physiol Heart Circ Physiol. 2010;299:H548–H556. doi: 10.1152/ajpheart.00527.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard TJ, Parvatiyar M, Bullard DP, Lynch RM, Lorenz JN. Paul RJ. Transgenic mice expressing Na+-K+-ATPase in smooth muscle decreases blood pressure. Am J Physiol Heart Circ Physiol. 2007;293:H1172–H1182. doi: 10.1152/ajpheart.00279.2007. [DOI] [PubMed] [Google Scholar]
- Pulina MV, Zulian A, Baryshnikov SG, Linde CI, Karashima E, Hamlyn JM, Ferrari P, Blaustein MP. Golovina VA. Cross talk between plasma membrane Na+/Ca2+ exchanger-1 and TRPC/Orai-containing channels: key players in arterial hypertension. Adv Exp Med Biol. 2013;961:365–374. doi: 10.1007/978-1-4614-4756-6_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen MA, Brooks DP. Edwards RM. Characterization of the neutralizing activity of digoxin-specific Fab toward ouabain-like steroids. J Pharmacol Exp Ther. 2004;310:319–325. doi: 10.1124/jpet.104.065250. [DOI] [PubMed] [Google Scholar]
- Quinn K, Guibert C. Beech DJ. Sodium-potassium-ATPase electrogenicity in cerebral precapillary arterioles. Am J Physiol Heart Circ Physiol. 2000;279:H351–H360. doi: 10.1152/ajpheart.2000.279.1.H351. [DOI] [PubMed] [Google Scholar]
- Radzyukevich TL, Lingrel JB. Heiny JA. The cardiac glycoside binding site on the Na,K-ATPase α2 isoform plays a role in the dynamic regulation of active transport in skeletal muscle. Proc Natl Acad Sci USA. 2009;106:2565–2570. doi: 10.1073/pnas.0804150106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzyukevich TL, Neumann JC, Rindler TN, Oshiro N, Goldhamer DJ, Lingrel JB. Heiny JA. Tissue-specific role of the Na,K-ATPase α2 isozyme in skeletal muscle. J Biol Chem. 2013;288:1226–1237. doi: 10.1074/jbc.M112.424663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HH. Figtree G. ‘Don't flog the heart!’ – development of specific drug therapies for heart failure. Crit Care Resusc. 2007;9:364–369. [PubMed] [Google Scholar]
- Reeves JP. Condrescu M. Allosteric activation of sodium-calcium exchange activity by calcium: persistence at low calcium concentrations. J Gen Physiol. 2003;122:621–639. doi: 10.1085/jgp.200308915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM, Ripley ML, Meeks MK, Geddis LM, Kutchai HC, Marassi FM, Cheung JY. Moorman JR. Serine 68 phospholemman phosphorylation during forskolin-induced swine carotid artery relaxation. J Vasc Res. 2005;42:483–491. doi: 10.1159/000088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Nicoll DA, Galang G. Philipson KD. Intermolecular cross-linking of Na+-Ca2+ exchanger proteins: evidence for dimer formation. Biochemistry. 2008;47:6081–6087. doi: 10.1021/bi800177t. [DOI] [PubMed] [Google Scholar]
- Ren X. Philipson KD. The topology of the cardiac Na+/Ca2+ exchanger, NCX1. J Mol Cell Cardiol. 2013;57:68–71. doi: 10.1016/j.yjmcc.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter H, Henderson SA, Han T, Matsuda T, Baba A, Ross RS, Goldhaber JI. Philipson KD. Knockout mice for pharmacological screening: testing the specificity of Na+-Ca2+ exchange inhibitors. Circ Res. 2002;91:90–92. doi: 10.1161/01.res.0000027529.37429.38. [DOI] [PubMed] [Google Scholar]
- Rindler TN, Dostanic I, Lasko VM, Nieman ML, Neumann JC, Lorenz JN. Lingrel JB. Knockout of the Na,K-ATPase α2-isoform in the cardiovascular system does not alter basal blood pressure but prevents ACTH-induced hypertension. Am J Physiol Heart Circ Physiol. 2011;301:H1396–H1404. doi: 10.1152/ajpheart.00121.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathishkumar K, Ross RG, Bawankule DU, Sardar KK, Prakash VR. Mishra SK. Segmental heterogeneity in the mechanism of sodium nitroprusside-induced relaxation in ovine pulmonary artery. J Cardiovasc Pharmacol. 2005;45:491–498. doi: 10.1097/01.fjc.0000159043.50488.ac. [DOI] [PubMed] [Google Scholar]
- Schinke M, Baltatu O, Bohm M, Peters J, Rascher W, Bricca G, Lippoldt A, Ganten D. Bader M. Blood pressure reduction and diabetes insipidus in transgenic rats deficient in brain angiotensinogen. Proc Natl Acad Sci USA. 1999;96:3975–3980. doi: 10.1073/pnas.96.7.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoner W. Endogenous cardiac glycosides, a new class of steroid hormones. Eur J Biochem. 2002;269:2440–2448. doi: 10.1046/j.1432-1033.2002.02911.x. [DOI] [PubMed] [Google Scholar]
- Schwarz EM. Benzer S. Calx, a Na-Ca exchanger gene of Drosophila melanogaster. Proc Natl Acad Sci USA. 1997;94:10249–10254. doi: 10.1073/pnas.94.19.10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JR, Laredo J, Hamilton BP. Hamlyn JM. Effects of angiotensin II on sodium potassium pumps, endogenous ouabain, and aldosterone in bovine zona glomerulosa cells. Hypertension. 1999;33:373–377. doi: 10.1161/01.hyp.33.1.373. [DOI] [PubMed] [Google Scholar]
- Shattock MJ. Phospholemman: its role in normal cardiac physiology and potential as a druggable target in disease. Curr Opin Pharmacol. 2009;9:160–166. doi: 10.1016/j.coph.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Shattock MJ. Bers DM. Rat vs. rabbit ventricle: Ca flux and intracellular Na assessed by ion-selective microelectrodes. Am J Physiol Cell Physiol. 1989;256:C813–C822. doi: 10.1152/ajpcell.1989.256.4.C813. [DOI] [PubMed] [Google Scholar]
- Shattock MJ. Matsuura H. Measurement of Na+-K+ pump current in isolated rabbit ventricular myocytes using the whole-cell voltage-clamp technique. Inhibition of the pump by oxidant stress. Circ Res. 1993;72:91–101. doi: 10.1161/01.res.72.1.91. [DOI] [PubMed] [Google Scholar]
- Shelly DA, He S, Moseley A, Weber C, Stegemeyer M, Lynch RM, Lingrel J. Paul RJ. Na+ pump α2-isoform specifically couples to contractility in vascular smooth muscle: evidence from gene-targeted neonatal mice. Am J Physiol Cell Physiol. 2004;286:C813–C820. doi: 10.1152/ajpcell.00389.2003. [DOI] [PubMed] [Google Scholar]
- Shigekawa M, Iwamoto T, Uehara A. Kita S. Probing ion binding sites in the Na+/Ca2+ exchanger. Ann NY Acad Sci. 2002;976:19–30. doi: 10.1111/j.1749-6632.2002.tb04710.x. [DOI] [PubMed] [Google Scholar]
- Shinoda T, Ogawa H, Cornelius F. Toyoshima C. Crystal structure of the sodium-potassium pump at 2.4 Å resolution. Nature. 2009;459:446–450. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- Silverman B, Fuller W, Eaton P, Deng J, Moorman JR, Cheung JY, James AF. Shattock MJ. Serine 68 phosphorylation of phospholemman: acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc Res. 2005;65:93–103. doi: 10.1016/j.cardiores.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Volders PG, Vos MA. Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy. Cardiovasc Res. 2002;53:782–805. doi: 10.1016/s0008-6363(01)00470-9. [DOI] [PubMed] [Google Scholar]
- Sobie EA, Cannell MB. Bridge JH. Allosteric activation of Na+-Ca2+ exchange by L-type Ca2+ current augments the trigger flux for SR Ca2+ release in ventricular myocytes. Biophys J. 2008;94:L54–56. doi: 10.1529/biophysj.107.127878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma S, Kuwashima H, Matsumura C. Kimura T. Inhibition by SEA0400, a selective inhibitor of Na+/Ca2+ exchanger, of Na+-dependent Ca2+ uptake and catecholamine release in bovine adrenal chromaffin cells. J Pharmacol Sci. 2006;102:88–95. doi: 10.1254/jphs.fpj06006x. [DOI] [PubMed] [Google Scholar]
- Song H, Karashima E, Hamlyn JM. Blaustein MP. Ouabain-digoxin antagonism in rat arteries and neurones. J Physiol. 2014;592:941–969. doi: 10.1113/jphysiol.2013.266866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q, Pallikkuth S, Bossuyt J, Bers DM. Robia SL. Phosphomimetic mutations enhance oligomerization of phospholemman and modulate its interaction with the Na/K-ATPase. J Biol Chem. 2011;286:9120–9126. doi: 10.1074/jbc.M110.198036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella P, Manunta P, Mallamaci F, Melandri M, Spotti D, Tripepi G, Hamlyn JM, Malatino LS, Bianchi G. Zoccali C. Endogenous ouabain and cardiomyopathy in dialysis patients. J Intern Med. 2008;263:274–280. doi: 10.1111/j.1365-2796.2007.01883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J. Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- Sweadner KJ. Donnet C. Structural similarities of Na,K-ATPase and SERCA, the Ca2+-ATPase of the sarcoplasmic reticulum. Biochem J. 2001;356:685–704. doi: 10.1042/0264-6021:3560685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweadner KJ, Herrera VLM, Amato S, Moellmann A, Gibbons DK. Repke KRH. Immunologic identification of Na+,K+-ATPase isoforms in myocardium. Isoforms change in deoxycorticosterone acetate-salt hypertension. Circ Res. 1994;74:669–678. doi: 10.1161/01.res.74.4.669. [DOI] [PubMed] [Google Scholar]
- Swift F, Tovsrud N, Enger UH, Sjaastad I. Sejersted OM. The Na+/K+-ATPase α2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc Res. 2007;75:109–117. doi: 10.1016/j.cardiores.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Tani M. Neely JR. Role of intracellular Na in Ca overload and depressed recovery of ventricular function of reperfused ischemic rat hearts. Circ Res. 1989;65:1045–1056. doi: 10.1161/01.res.65.4.1045. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Kaya S, Abe K. Mardh S. The oligomeric nature of Na/K-transport ATPase. J Biochem. 2001;129:335–342. doi: 10.1093/oxfordjournals.jbchem.a002862. [DOI] [PubMed] [Google Scholar]
- Teriete P, Franzin CM, Choi J. Marassi FM. Structure of the Na,K-ATPase regulatory protein FXYD1 in micelles. Biochemistry. 2007;46:6774–6783. doi: 10.1021/bi700391b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomassen M, Christensen PM, Gunnarsson TP, Nybo L. Bangsbo J. Effect of 2-wk intensified training and inactivity on muscle Na+-K+ pump expression, phospholemman (FXYD1) phosphorylation, and performance in soccer players. J Appl Physiol (1985) 2010;108:898–905. doi: 10.1152/japplphysiol.01015.2009. [DOI] [PubMed] [Google Scholar]
- Thomassen M, Murphy RM. Bangsbo J. Fibre type-specific change in FXYD1 phosphorylation during acute intense exercise in humans. J Physiol. 2013;591:1523–1533. doi: 10.1113/jphysiol.2012.247312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomassen M, Rose AJ, Jensen TE, Maarbjerg SJ, Bune L, Leitges M, Richter EA, Bangsbo J. Nordsborg NB. Protein kinase Cαactivity is important for contraction-induced FXYD1 phosphorylation in skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1808–R1814. doi: 10.1152/ajpregu.00066.2011. [DOI] [PubMed] [Google Scholar]
- Tian J, Haller S, Periyasamy S, Brewster P, Zhang H, Adlakha S, Fedorova OV, Xie ZJ, Bagrov AY, Shapiro JI. Cooper CJ. Renal ischemia regulates marinobufagenin release in humans. Hypertension. 2010;56:914–919. doi: 10.1161/HYPERTENSIONAHA.110.155564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres NS, Larbig R, Rock A, Goldhaber JI. Bridge JH. Na+ currents are required for efficient excitation-contraction coupling in rabbit ventricular myocytes: a possible contribution of neuronal Na+ channels. J Physiol. 2010;588:4249–4260. doi: 10.1113/jphysiol.2010.194688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima C, Kanai R. Cornelius F. First crystal structures of Na+,K+-ATPase: new light on the oldest ion pump. Structure. 2011;19:1732–1738. doi: 10.1016/j.str.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Diaz ME, O'Neill SC. Eisner DA. Comparison of subsarcolemmal and bulk calcium concentration during spontaneous calcium release in rat ventricular myocytes. J Physiol. 1995;488:577–586. doi: 10.1113/jphysiol.1995.sp020991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyralla K. Amann K. Cardiovascular changes in renal failure. Blood Purif. 2002;20:462–465. doi: 10.1159/000063551. [DOI] [PubMed] [Google Scholar]
- Van Huysse JW, Dostanic I, Lingrel JB, Hou X. Wu H. Hypertension from chronic central sodium chloride in mice is mediated by the ouabain-binding site on the Na,K-ATPase α2-isoform. Am J Physiol Heart Circ Physiol. 2011;301:H2147–H2153. doi: 10.1152/ajpheart.01216.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonck F. Intracellular Na+ and altered Na+ transport mechanisms in cardiac hypertrophy and failure. J Mol Cell Cardiol. 2003;35:5–25. doi: 10.1016/s0022-2828(02)00280-8. [DOI] [PubMed] [Google Scholar]
- Verdonck F, Volders PG, Vos MA. Sipido KR. Increased Na+ concentration and altered Na/K pump activity in hypertrophied canine ventricular cells. Cardiovasc Res. 2003a;57:1035–1043. doi: 10.1016/s0008-6363(02)00734-4. [DOI] [PubMed] [Google Scholar]
- Verdonck F, Volders PG, Vos MA. Sipido KR. Intracellular Na+ and altered Na+ transport mechanisms in cardiac hypertrophy and failure. J Mol Cell Cardiol. 2003b;35:5–25. doi: 10.1016/s0022-2828(02)00280-8. [DOI] [PubMed] [Google Scholar]
- Wang H, Haas M, Liang M, Cai T, Tian J, Li S. Xie Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem. 2004;279:17250–17259. doi: 10.1074/jbc.M313239200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gao J, Mathias RT, Cohen IS, Sun X. Baldo GJ. -Adrenergic effects on Na+-K+ pump current in guinea-pig ventricular myocytes. J Physiol. 1998;509:117–128. doi: 10.1111/j.1469-7793.1998.117bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CR, Piacentino V, 3rd, Ginsburg KS, Houser SR. Bers DM. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- Weston AH, Richards GR, Burnham MP, Félétou M, Vanhoutte PM. Edwards G. K+-induced hyperpolarization in rat mesenteric artery: identification, localization and role of Na+/K+-ATPases. Br J Pharmacol. 2002;136:918–926. doi: 10.1038/sj.bjp.0704787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CN, Figtree GA, Liu CC, Garcia A, Hamilton EJ, Chia KK. Rasmussen HH. Angiotensin II inhibits the Na+-K+ pump via PKC-dependent activation of NADPH oxidase. Am J Physiol Cell Physiol. 2009;296:C693–C700. doi: 10.1152/ajpcell.00648.2008. [DOI] [PubMed] [Google Scholar]
- Williams MW, Resneck WG, Kaysser T, Ursitti JA, Birkenmeier CS, Barker JE. Bloch RJ. Na,K-ATPase in skeletal muscle: two populations of β-spectrin control localization in the sarcolemma but not partitioning between the sarcolemma and the transverse tubules. J Cell Sci. 2001;114:751–762. doi: 10.1242/jcs.114.4.751. [DOI] [PubMed] [Google Scholar]
- Wu M, Le HD, Wang M, Yurkov V, Omelchenko A, Hnatowich M, Nix J, Hryshko LV. Zheng L. Crystal structures of progressive Ca2+ binding states of the Ca2+ sensor Ca2+ binding domain 1 (CBD1) from the CALX Na+/Ca2+ exchanger reveal incremental conformational transitions. J Biol Chem. 2009a;285:2554–2561. doi: 10.1074/jbc.M109.059162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Tong S, Waltersperger S, Diederichs K, Wang M. Zheng L. Crystal structure of Ca2+/H+ antiporter protein YfkE reveals the mechanisms of Ca2+ efflux and its pH regulation. Proc Natl Acad Sci USA. 2013;110:11367–11372. doi: 10.1073/pnas.1302515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Wang M, Nix J, Hryshko LV. Zheng L. Crystal structure of CBD2 from the Drosophila Na+/Ca2+ exchanger: diversity of Ca2+ regulation and its alternative splicing modification. J Mol Biol. 2009b;387:104–112. doi: 10.1016/j.jmb.2009.01.045. [DOI] [PubMed] [Google Scholar]
- Wypijewski KJ, Howie J, Reilly L, Tulloch LB, Aughton KL, McLatchie LM, Shattock MJ, Calaghan SC. Fuller W. A separate pool of cardiac phospholemman that does not regulate or associate with the sodium pump: multimers of phospholemman in ventricular muscle. J Biol Chem. 2013;288:13808–13820. doi: 10.1074/jbc.M113.460956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG. Blaustein MP. Sodium pump α2 subunits control myogenic tone and blood pressure in mice. J Physiol. 2005;569:243–256. doi: 10.1113/jphysiol.2005.091801. [DOI] [PMC free article] [PubMed] [Google Scholar]