

Abstract
Na+ current (INa) is determined not only by the properties of the pore-forming voltage-gated Na+ channel (VGSC) α subunit, but also by the integrated function of a molecular aggregate (the VGSC complex) that includes the VGSC β subunit family. Mutations or rare variants in Scn1b (encoding the β1 and β1B subunits) have been associated with various inherited arrhythmogenic syndromes, including cases of Brugada syndrome and sudden unexpected death in patients with epilepsy. Here, we have used Scn1b null mouse models to understand better the relation between Scn1b expression, and cardiac electrical function. Using a combination of macropatch and scanning ion conductance microscopy we show that loss of Scn1b in juvenile null animals resulted in increased tetrodotoxin-sensitive INa but only in the cell midsection, even before full T-tubule formation; the latter occurred concurrent with increased message abundance for the neuronal Scn3a mRNA, suggesting increased abundance of tetrodotoxin-sensitive NaV1.3 protein and yet its exclusion from the region of the intercalated disc. Ventricular myocytes from cardiac-specific adult Scn1b null animals showed increased Scn3a message, prolonged action potential repolarization, presence of delayed after-depolarizations and triggered beats, delayed Ca2+ transients and frequent spontaneous Ca2+ release events and at the whole heart level, increased susceptibility to polymorphic ventricular arrhythmias. Most alterations in Ca2+ homeostasis were prevented by 100 nm tetrodotoxin. Our results suggest that life-threatening arrhythmias in patients with mutations in Scn1b, a gene classically defined as ancillary to the Na+ channel α subunit, can be partly consequent to disrupted intracellular Ca2+ homeostasis in ventricular myocytes.
Key points
Na+ current (INa) results from the integrated function of a molecular aggregate (the voltage-gated Na+ channel complex) that includes the β subunit family.
Mutations or rare variants in Scn1b (encoding the β1 and β1B subunits) have been associated with various inherited arrhythmogenic syndromes, including Brugada syndrome and sudden unexpected death in patients with epilepsy.
We used Scn1b null mice to understand better the relation between Scn1b expression, and cardiac electrical function.
Loss of Scn1b caused, among other effects, increased amplitude of tetrodotoxin-sensitive INa, delayed after-depolarizations, triggered beats, delayed Ca2+ transients, frequent spontaneous calcium release events and increased susceptibility to polymorphic ventricular arrhythmias. Most alterations in Ca2+ homeostasis were prevented by 100 nm tetrodotoxin.
We propose that life-threatening arrhythmias in patients with mutations in Scn1b, a gene classically defined as ancillary to the Na+ channel α subunit, can be partly consequent to disrupted intracellular Ca2+ homeostasis.
Introduction
The rapid entry of Na+ into ventricular myocytes is a key determinant of action potential propagation. This Na+ current (INa) is determined not only by the properties of the pore-forming voltage-gated Na+ channel (VGSC) α subunit (mostly NaV1.5 in the adult ventricle), but also by the integrated function of a molecular aggregate (the VGSC complex) that includes the VGSC β subunit family. Mutations or rare variants in SCN1B (encoding the β1 and β1B subunits) have been associated with some cases of long QT syndrome and of Brugada syndrome (Hu et al. 2012; Riuro et al. 2014). Patino et al. (2009) were first to demonstrate that inheriting two loss-of-function SCN1B alleles causes Dravet syndrome (DS), a devastating paediatric epileptic encephalopathy in humans. Patients with DS exhibit developmental delay and/or regression during early childhood and frequent pharmacoresistant seizures (Guerrini & Aicardi, 2003; Dravet et al. 2005; Dravet, 2011; Genton et al. 2011; Oakley et al. 2011). A high percentage of patients with DS die during early childhood or adolescence, the majority due to SUDEP (Guerrini & Aicardi, 2003; Dravet et al. 2005; Dravet, 2011; Genton et al. 2011; Oakley et al. 2011; Sakauchi et al. 2011), defined as sudden, unexpected, witnessed or unwitnessed, non-traumatic and non-drowning death in patients with epilepsy (Hirsch et al. 2011), excluding cases of documented status epilepticus. In the most widely used definition, death may occur with or without evidence of a seizure, and post-mortem examination does not reveal a toxicological or anatomical cause of death. SUDEP accounts for 7.5–17% of all deaths in epilepsy (Schuele et al. 2007b; Shorvon & Tomson, 2011). Indirect evidence variably links SUDEP to seizure-induced apnoea, pulmonary oedema, dysregulation of cerebral circulation, autonomic dysfunction and cardiac arrhythmias (Schuele et al. 2007a,b2007b; Surges et al. 2009, 2010; Shorvon & Tomson, 2011). Arrhythmias may be primary or may occur secondary to hormonal or metabolic changes, or autonomic discharges (Schuele et al. 2007a,b2007b; Goldman et al. 2009; Surges et al. 2009, 2010).
Over the past decade, the Scn1b null mouse model has been used to understand the molecular substrate underlying DS, and its possible relation to SUDEP (Chen et al. 2004; Brackenbury et al. 2013). These mice developed severe seizures of multiple aetiologies at approximately postnatal day (P)10, and died by ∼P21. Similar to heterozygous SCN1A-R1407X knockin mice, expressing a human DS mutation (Auerbach et al. 2013), Scn1b null ventricular cardiac myocytes (CMs) had a 1.6-fold increase in transient and persistent INa density, as well as action potential prolongation. Electrocardiograms of Scn1b null mice showed increased QTc intervals (Lopez-Santiago et al. 2007). In spite of the observation that 3H-saxitoxin binding in Scn1b null cardiac membrane preparations was increased approximately two-fold, suggesting an increase in tetrodotoxin (TTX)-sensitive (TTX-S) VGSC expression (Lopez-Santiago et al. 2007), the molecular identity and subcellular location of the VGSC α subunits responsible for increased INa density, and the likelihood of arrhythmia events in these hearts were not defined. Furthermore, due to SUDEP in these mice (Chen et al. 2004), these studies were limited to juvenile animals and the phenotype was confounded by the concurrent occurrence of severe seizures consequent to Scn1b deletion in the brain.
Here, we have expanded previous studies on juvenile animals and circumvented the limitations consequent to early mortality by developing a new genetically engineered murine model with cardiac-specific Scn1b deletion. Using a combination of macropatch and scanning ion conductance microscopy (SICM) we show that loss of Scn1b in juvenile null animals results in an increase in the TTX-S INa but only in the midsection of the cell, even before full T-tubule formation; the latter occurred concurrent with an increase in message abundance for the neuronal Scn3a mRNA, suggesting an increased abundance of TTX-S NaV1.3 protein and yet its preferential exclusion from the region of the intercalated disc (ID). Ventricular myocytes from cardiac-specific adult Scn1b null animals showed a trend toward increased TTX-S INa specifically at the T-tubules. Interestingly, this was associated with increased Scn3a message, prolonged action potential repolarization, the presence of delayed after-depolarizations (DADs), disruption of intracellular Ca2+ homeostasis, and at the whole heart level, increased susceptibility to polymorphic ventricular arrhythmias. Overall, our results indicate that loss of Scn1b in the heart leads to increased expression of Scn3a and increased TTX-S INa, as well as to altered Ca2+ regulation and a proarrhythmic substrate. We postulate that life-threatening arrhythmias in patients with mutations in Scn1b, a gene classically defined as ancillary to the Na+ channel α subunit, can be partly consequent to disrupted intracellular Ca2+ homeostasis in ventricular myocytes.
Methods
All procedures were carried out in accordance with New York University and University of Michigan guidelines for animal use and care. These procedures are conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication 58–23, revised 1996).
Scn1b global null mice
Scn1b global null mice, congenic on the C57Bl/6 background, were bred from Scn1b+/− mice, as described previously (Chen et al. 2004; Brackenbury et al. 2013).
Generation and characterization of cardiac-specific Scn1b null mice
Scn1bflox/flox mice were described previously (Chen et al. 2007). Despite the earlier observation that these mice, with the neo cassette from the targeting vector intact, lived normal life spans and did not seize, we elected to cross them with FLPe transgenic mice (Jackson Laboratories, B6;SJL-Tg(ACTFLPe)9205Dym/J; Rodriguez et al. 2000) to delete the neo cassette and thus avoid any potential interference in Scn1b gene expression. Cardiac-specific Scn1b null mice were then generated by crossing the resulting Scn1bflox mice, now lacking the neo cassette, with the B6.FVB-Tg(Myh6-cre)2182Mds/J line, which expresses the α-myosin heavy chain promoter (Jackson Laboratories). This step allowed the excision of exon 2 selectively in the heart, effectively deleting Scn1b from this tissue. Mice were genotyped by polymerase chain reaction (PCR) of the genomic tail DNA using a set of primers external to the loxP sites (loxP-5′: 5′-GTT ACT CAC CAC AGT GAC ATC CTC-3′; and loxP-3′: 5′-CAT CCA GCG TAT CAC ATC CTC ATC-3′) that amplified a 720 bp band corresponding to the endogenous (‘E’) allele, a 1114 bp band corresponding to the floxed (‘Fl’) allele, or a 492 bp band corresponding to the deleted allele. In addition, Cre recombinase was detected by PCR (using the primers CreF-2: 5′-TCCAATTTACTGACCGTAC ACC-3′ and CreR: 5′-GGTATCTCTGACCAGAGTCATC-3′), which amplified an 850 bp band as described previously (Chen et al. 2007). This PCR strategy, summarized in Fig.1A, allowing for detection of the deleted allele in tail DNA, was employed as a safeguard against non-specific expression of Cre recombinase in the germline resulting in random excision of exon 2.
Figure 1.
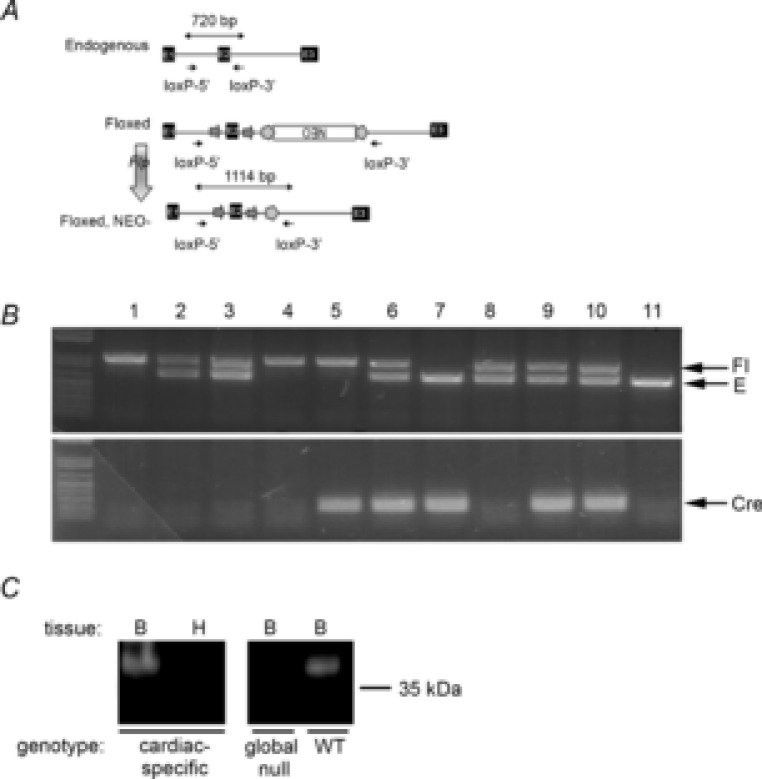
Characterization of cardiac-specific Scn1b null mice
A, schematic of breeding and genotyping strategy. The original targeting construct for generation of Scn1bflox mice contained exons 1, 2 and 3 (E1–E3) from the endogenous Scn1b allele, with E2 flanked by loxP sites (arrows) and an inverted PGK-neo-cassette flanked by FRT sites (pentagons). An inverted Neo cassette was flanked by FRT sites (pentagons). Scn1bflox mice, with the neo cassette intact, were generated previously. Crossing of Scn1bflox mice with FLPe transgenic mice (vertical arrow) resulted in removal of the Neo cassette. B, PCR of genomic DNA (obtained from mouse tails) from a typical litter was performed with two sets of primers. Top, loxP-5′ (5′-GTT ACT CAC CAC AGT GAC ATC CTC-3′) and loxP-3′ (5′-CAT CCA GCG TAT CAC ATC CTC ATC-3′) amplified a 720 bp band corresponding to the endogenousE allele, a 1114 bp band corresponding to the floxed allele, or a 492 bp band corresponding to the deleted allele. Note: none of the mice analysed in this litter contained the deleted allele in tail DNA. Bottom, PCR primers CreF-2 (5′-TCCAATTTACTGACCGTACACC-3′) and CreR-5′ (GGTATCTCTGACCAGAGTCATC-3′) amplified a 850 bp band corresponding to Cre recombinase. Lanes 1 and 4: Fl/Fl/Cre–; lanes 2, 3 and 8: Fl/E/Cre–; lane 5: Fl/Fl/Cre+; lanes 6, 9 and 10: Fl/E/Cre+; lane 7: E/E/Cre+; lane 11: E/E/Cre–. C, Western blot analysis of heart and brain membranes. Left, brain or heart membranes from cardiac-specific Scn1b null mice (65 μg protein/lane). Right, brain membranes from Scn1b global null or WT mice shown to demonstrate specificity of the anti-β1 antibody (65 μg protein/lane). Anti-β1 dilution, 1:3000. E, endogenous allele; Fl, floxed allele; WT, wild-type.
Western blot analysis of mouse brain and heart membranes
Scn1bflox/flox/Myh6-cre, Scn1bE/E/Myh6-cre or Scn1b−/− (global null) mice (Chen et al. 2004) were killed at P17–18. Brains and hearts were immediately removed and membranes were prepared from each tissue as previously described (Isom et al. 1995). Complete protease inhibitor tablets (Roche, Indianapolis, IN, USA), at twice the recommended concentration, were added to all solutions to prevent Na+ channel degradation. Western blot analysis of equal aliquots of protein was then performed as described previously (Malhotra et al. 2000). A rabbit monoclonal antibody directed to a peptide in the mouse β1 intracellular domain (TSESKENCTGVQVAE; Cell Signaling Technology, Danvers, MA, USA; 1:3000 dilution) was used to detect β1 protein.
Cardiomyocyte dissociation
Murine ventricular myocytes were obtained by enzymatic dissociation following standard procedures (Lin et al. 2011). Briefly, mice were injected with 0.1 ml heparin (500 IU ml−1 i.p.) 20 min before heart excision and anaesthetized by inhalation of isoflurane. Deep anaesthesia was confirmed by lack of response to otherwise painful stimuli. Hearts were quickly removed from the chest and placed in a Langendorff column. For cell dissociation, the isolated hearts were perfused sequentially with Ca2+-free solution containing (in mm): NaCl 113, KCl 4.7, MgSO4 1.2, Na2HPO4 0.6, KH2PO4 0.6, NaHCO3 12, KHCO3 10, Hepes 10 and taurine 30 (pH 7.45 with NaOH) and an enzyme (collagenase type II; Worthington, Lakewood, NJ, USA) solution. Ventricles were cut into small pieces, and gently minced with a Pasteur pipette. The Ca2+ concentration was increased gradually to 1.0 mm. Cardiomyocytes were kept in Tyrode's solution containing (in mm): NaCl 148, KCl 5.4, MgCl2 1.0, CaCl2 1.0, NaH2PO4 0.4, Hepes 15 and glucose 5.5; pH 7.4. Cells were used on the same day of isolation.
Electrophysiological methods
Action potential recording
Adult ventricular cardiomyocyte action potentials were obtained with whole cell current clamp mode. The recording pipette solution contained (in mm): KCl 135, MgCl2 1, EGTA 10, Hepes 10, and glucose 5, pH 7.2 with KOH. The bath solution contained (in mm): NaCl 136, KCl 4, CaCl2 1, MgCl2 2, Hepes 10 and glucose 10; pH 7.4 with NaOH. A brief current pulse (up to 3 ms duration) at approximately twice the voltage threshold for excitation was used to evoke action potentials at a cycle length of 1 s (1 Hz) (Lübkemeier et al. 2013). The action potential duration at 50% (APD50) and 70% (APD70) repolarization was measured using Clampfit version 10.0.
Whole cell patch clamp
Voltage clamp INa recordings were performed at room temperature (21–22°C) with 5 mm [Na+]o. The extracellular solution contained (mm): 5 NaCl, 1 MgCl2, 1 CaCl2, 0.1 CdCl2, 11 glucose, 132.5 CsCl and 20 Hepes. The pipette filling solution contained (mm): 5 NaCl, 135 CsF, 10 EGTA, 5 MgATP and 5 Hepes. Upon gaining access to the cell, appropriate whole cell and series resistance compensation (<70%) and leak subtraction were applied. Assessment of INa density was obtained by holding the cell at −120 mV, followed by stepping to voltages between −100 and +30 mV, in 5 mV steps, for 200 ms, with 2800 ms interpulse intervals.
Macropatch
Cell-attached (macropatch) recordings of macroscopic Na+ currents were obtained either from the end of the cell, in the area previously occupied by the ID, or from the midsection of the cell (see also Lin et al. 2011). Pipettes were filled with a solution containing (in mm): NaCl 135, KCl 5.4, CaCl2 1.0, MgCl2 1.0, CsCl 10, tetraethylammonium chloride 10, 4-aminopyridine 10, NaH2PO4 0.4, CdCl2 0.2, Hepes 5 and glucose 5.5, pH 7.4 with NaOH. Cells were maintained in a solution containing in (mm): NaH2PO4 0.33, Hepes 5, CaCl2 1.0, MgCl2 1.0, EGTA 2.0 and 140 KCl; pH 7.4. The high concentration of K+ caused depolarization of the membrane potential to a value estimated to be near zero. To determine the peak current–voltage relation, 200 ms voltage pulses were applied every 3 s from a holding potential of Vm = −120 mV to voltages ranging between −90 mV and +30 mV in 5 mV increments. The steady-state inactivation was determined by stepping Vm from −130 mV to −20 mV, followed by a 30 ms test pulse to Vm = −20 mV. The steady-state inactivation data were fitted to Boltzmann's functions. Recovery from inactivation was studied by applying paired voltage clamp steps. Two 20 ms test pulses (S1, S2) to Vm = −20 mV (holding potential = −120 mV) were separated by increments of 1 ms to a maximum S1–S2 interval of 80 ms. The S1–S1 interval was kept constant at 3 s. The recovery from inactivation data were fit with exponential functions. All recordings were obtained using an Axon multiclamp 700B Amplifier and pClamp system (versions 10.2; Axon Instruments, Foster City, CA, USA).
Scanning ion conductance microscopy
SICM is a non-contact scanning probe microscopy technique based on the principle that the flow of ions through the tip of a nanopipette filled with electrolytes decreases when the pipette approaches the surface of the sample. The result is a three-dimensional topography image of live cells with resolution of up to ≤50 nm. All topographical images in this study were recorded using a variant of SICM called hopping probe ion conductance microscopy, implemented on a software platform that controls the ICnano sample scan system (Ionscope Ltd, Melbourn, UK). A detailed description of this technique can be found in our previous publication (Bhargava et al. 2013).
Super-resolution scanning patch clamp
This method combines SICM with cell-attached patch clamp technology for recording of ion channels at a particular subcellular location. A detailed description of this technique can be found in Bhargava et al. (2013). Briefly, after generating the topographical image of the cardiomyocyte surface with SICM, the pipette was moved to an area clear of cells or debris. At that coordinate, a custom-built program was used to clip the tip of the pipette against the bottom of the dish. The pipette resistance was continuously monitored and the clipping manoeuvre stopped once the current through the pipette reached the desired level. At that point, the pipette was repositioned to spatial coordinates that were selected based on the topography image recorded with the sharp pipette. The repositioned, clipped pipette was lowered at the chosen subcellular location to record Na+ channels in the cell-attached configuration.
Ca2+ imaging
Isolated ventricular myocytes were loaded for 12 min with x-Rhod-1/AM (Invitrogen Inc., Eugene, OR, USA) in Tyrode's solution containing (in mm): 140 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 Hepes and 5.6 glucose followed by a 30 min wash in Tyrode's solution. Fluorescent signals were acquired using a 40× UVF objective (numerical aperture 1.0; Nikon, Tokyo, Japan), and single excitation wavelength microfluorimetry was performed using a PMT system (IonOptix Corp., Milton, MA, USA). Cells were paced at 1 Hz to achieve steady state, then paced either for 10 beats at 0.5 Hz and 1 Hz, or for 3 min at 1 Hz. Following background subtractions, data were calculated as the ratio of fluorescence intensity of x-Rhod-1 over baseline (F/F0).
Real-time polymerase chain reaction
Total RNA was extracted using RNeasy Mini Kit (QIAGEN, Hilden, Germany). The cDNA was generated by reverse transcription (RT)-PCR with SuperScript VILO cDNA Synthesis Kit. Real-time PCR analysis was performed with TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) applied on a StepOnePlus Real Time PCR System. Cycle threshold (CT) was calculated during the exponential phase at identical threshold values for all runs. Each gene expression was normalized to CT of GAPDH. Normalized expression relative quantification (RQ) was calculated as RQ = 2−ΔΔCT.
Optical mapping
Optical recordings, with high spatial and temporal resolution, were acquired from heparinized (5 U g−1 i.p.) Langendorff perfused hearts excised from 3–6 month old mice. Continuous volume conducted ECG recordings were acquired throughout the experiments. Hearts were allowed to stabilize for 15 min while being perfused with 37°C oxygenated Tyrode's solution and 10 μm blebbistatin (Enzo Life Sciences, Farmingdale, NY, USA) (Mironov et al. 2006; Noujaim et al. 2007b; Glukhov et al. 2010). The hearts were then stained with the voltage sensitive dye, Di-4-ANNEPS (Sigma-Aldrich, St Louis, MO, USA). The dye was excited using a laser (1 W; Shanghai Dream Laser Technology, Shanghai, China) connected to three liquid-light guides to create uniform excitation of the epicardial wall of the heart. Images were acquired from the anterior epicardial ventricular surface of the heart using a CCD camera (80 × 80 pixel, ∼80 μm per pixel, 1 kHz; Sci Measure, Decatur, GA, USA). Optical signals and volume conducted ECGs were acquired during sinus rhythm. Next, 3 ms pulses of 2× diastolic threshold intensity were applied from a thin bipolar electrode positioned on the epicardial surface of the right ventricular apex. Optical movies were analysed for action potential morphology and conduction velocity differences between genotypes at decreasing pacing cycle lengths (175–70 ms). Arrhythmia induction was performed using burst pacing (30–60 ms cycle length). Optical signals were digitized, amplified, and spatial and temporal filtration was applied (Mironov et al. 2006; Zarzoso et al. 2013). All in-house developed analytical tools were previously described (Gray et al. 1995).
Statistics
The Na+ current density/amplitude and Ca2+ transients kinetics were compared using Student's unpaired t test. The proportion of cells displaying DADs or Ca2+ oscillation was compared using Fisher's exact test. Probability values of P < 0.05 were considered significant.
Results
Generation of cardiac-specific Scn1b null mice
We generated cardiac-specific Scn1b null mice by first crossing Scn1bflox mice (Chen et al. 2007) with FLPe transgenic mice to delete the Neo cassette and then crossed these progeny with the B6.FVB-Tg(Myh6-cre)2182Mds/J line, which expresses Cre recombinase under the transcriptional control of the α-myosin heavy chain promoter, (Jackson Laboratories). Following intercrossing of B6.FVB-Tg(Myh6-cre)2182Mds/J with Scn1bflox/flox mice, excision of exon 2 occurred selectively in the heart, effectively deleting Scn1b (Fig.1). Scn1bflox/flox/Myh6-cre mice had normal lifespans with no evidence of seizures. Throughout the paper, Scn1bflox/flox/Myh6-cre mice are referred to as cardiac-specific null mice and Scn1bE/E/Myh6-cre littermates are referred to as control mice.
Location-specific changes in INa in juvenile Scn1b global null hearts
Previous studies have shown that VGSCs in adult myocytes segregate into distinct location-specific pools (Lin et al. 2011; Petitprez et al. 2011; Bhargava et al. 2013). As a first step, we explored whether the reported increase in INa in juvenile global Scn1b null ventricular myocytes was location-specific. Data are shown in Fig.2 and Table1. Macropatch recordings obtained from the cell midsection of P15 myocytes revealed that the INa amplitude was larger in cells obtained from Scn1b null hearts (red symbols) than from control littermates (black symbols). In contrast, average peak INa recorded from the region previously occupied by the ID was unaffected by Scn1b deletion. Recordings at either location demonstrated no differences in the voltage dependence of steady-state inactivation (Fig.2B and E) or recovery from inactivation (Fig.2C and F) associated with a given genotype. Addition of 100 nm TTX to Scn1b null myocytes caused a decrease in INa recorded in the midsection region, but no change was observed at the cell end (ID; Fig.3 and Table2). The reduced amplitude coincided with a negative shift in the steady-state inactivation (Fig.3C). The amplitude of the remaining (TTX-resistant) current in Scn1b null cells was not statistically different from that observed in cells from wild-type animals (Fig.4 and Table3). qtRT-PCR analysis revealed an increase in the transcript abundance of Scn3a in mRNA prepared from Scn1b null hearts, in addition to the minor increase in Scn5a that has been previously reported (Lopez-Santiago et al. 2007) (Fig.5). Overall, these results strongly suggest that global loss of Scn1b causes an increase in the transcription of Scn3a in juvenile hearts, as well as an increase in TTX-S INa in the cell midsection. Interestingly, SICM recordings showed minor grooves indicative of only a nascent invagination preceding T-tubule formation in juvenile Scn1b null myocytes (Fig.6A and C), consistent with the finding of underdeveloped T-tubules in early development (Haddock et al. 1999). As shown by the height profile in Fig.6C, contours could already be detected, repeating with a periodicity of approximately 2 μm. Quantitative analysis showed that the topology of Scn1b null myocytes was not different from the control (Fig.6E and G). These results indicate that segregation of TTX-S channels away from the ID precedes the development of a full T-tubular structure.
Figure 2.
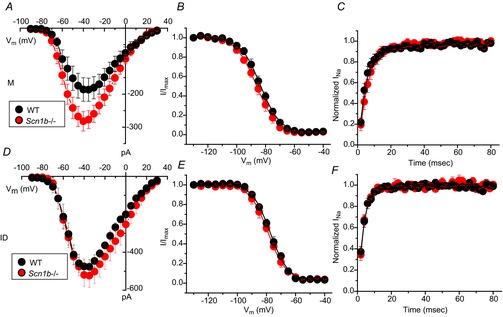
Region-specific recordings of Na+ current in juvenile cardiac ventricular myocytes
Cells were obtained from mice with global knockout of the Scn1b gene (Scn1b–/–; red circles) or littermate controls (WT; black circles), Data collected from macropatch recordings of the cell midsection (A–C) or from the cell end, in the region previously occupied by the ID (D–F). Peak current–voltage relations (A and D), voltage dependence of steady-state inactivation (B and E) and time course of recovery from inactivation (C and F). Quantitative analysis reported in Table1. ID, intercalated disc; M, midsection; WT, wild-type.
Table 1.
INa recorded from M or ID region of juvenile ventricular myocytes
| INa, peak (pA) | V1/2, inactivation (mV) | Recovery time constant (ms) | ||||
|---|---|---|---|---|---|---|
| WT | Scn1b–/– | WT | Scn1b–/– | WT | Scn1b–/– | |
| M | −185.8 ± 35.7 (9) | −282.2 ± 29.3 (12)* | −83.1 ± 1.3 (9) | −86.1 ± 1.7 (12) | 5.1 ± 1.0 (8) | 6.2 ± 1.1 (9) |
| ID | −475.0 ± 32.9 (6) | −519.2 ± 59.6 (7) | −77.4 ± 0.7 (6) | −78.2 ± 1.4 (7) | 3.3 ± 0.4 (6) | 3.6 ± 0.3 (7) |
P < 0.05, Scn1b–/– Vs WT. Abbreviations: ID, intercalated disc; M, midsection; WT, wild-type.
Figure 3.
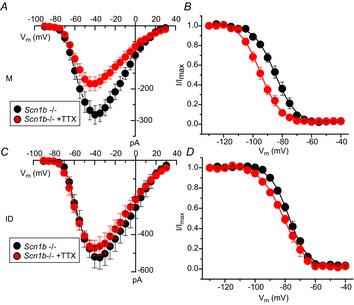
Effect of 100 nm TTX on region-specific INa recorded from cardiac ventricular myocytes dissociated from juvenile global Scn1b null mice (Scn1b–/–)
Average peak current–voltage relations (A and C) and voltage dependence of steady-state inactivation curves (B and D). Recordings were obtained from the cell midsection (A and B) or from the region previously occupied by the ID. Quantitative analysis reported in Table2. ID, intercalated disc; M, midsection; TTX, tetrodotoxin.
Table 2.
INa recorded from M or ID regions of juvenile Scn1b null ventricular myocytes, in the presence or absence of 100 nm TTX
| INa, peak (pA) | V1/2, inactivation (mV) | |||
|---|---|---|---|---|
| Scn1b–/– | Scn1b–/– +TTX | Scn1b–/– | Scn1b–/– +TTX | |
| M | −282.2 ± 29.3 (12) | −186.3 ± 18.5 (8)* | −86.1 ± 1.7 (12) | −92.1 ± 1.3 (8) |
| ID | −519.2 ± 59.6 (7) | −472.2 ± 42.3 (6) | −78.2 ± 1.4 (7) | −81.3 ± 1.2 (8) |
P < 0.05, Scn1b–/–+TTX vs Scn1b–/–. Abbreviations: ID, intercalated disc; M, midsection; TTX, tetrodotoxin.
Figure 4.
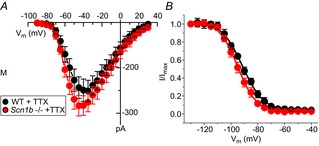
TTX-resistant INa recorded from the midsection of juvenile cardiac ventricular myocytes
Cells were dissociated from global Scn1b null mice (Scn1b–/–; data in red symbols) or littermate controls (WT; black symbols). Peak current–voltage relations (A) and voltage dependence of steady-state inactivation (B) recorded in the presence of 100 nm TTX. Quantitative analysis reported in Table3. M, midsection; TTX, tetrodotoxin; WT, wild-type.
Table 3.
TTX-resistant INa recorded from the M region of WT or Scn1b null juvenile ventricular myocytes
| INa, peak (pA) | V1/2, inactivation (mV) | |||
|---|---|---|---|---|
| WT+TTX | Scn1b–/– +TTX | WT+TTX | Scn1b–/– +TTX | |
| M | −149.2 ± 24.6 (8) | −186.3 ± 18.5 (8) | −94.8 ± 1.4 (8) | −92.1 ± 1.3 (8) |
Abbreviations: M, midsection; TTX, tetrodotoxin; WT, wild-type.
Figure 5.
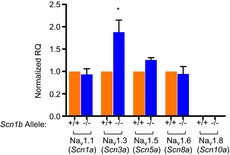
Gene expression levels in juvenile hearts
Transcript levels for Scn1a, Scn3a, Scn5a, Scn8a and Scn10a were examined in hearts of control animals (Scn1b+/+) or Scn1b nulls (Scn1b–/–). Each gene expression was normalized to the CT of GAPDH. ‘Normalized RQ’ refers to the normalized value of expression of a given gene, estimated from 2−ΔΔCT. Normalized RQ values were 1.88 ± 0.27 for Scn3a (P = 0.03 vs. wild-type) and 1.26 ± 0.05 for Scn5a (P = 0.008 vs. wild-type). CT, threshold cycle.
Figure 6.
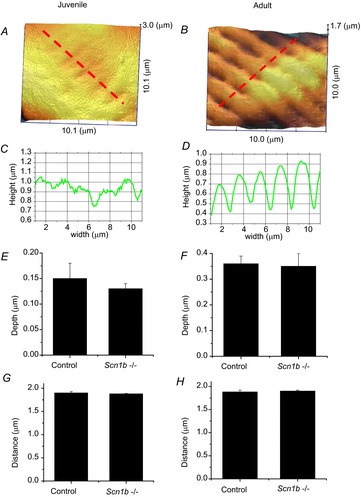
Surface topology characteristics of cardiac myocytes resolved by scanning ion conductance microscopy
Surface of a juvenile (A) and of an adult (B) Scn1b null myocyte. C and D, topology profiles along the red dotted lines in (A) and (B), respectively. Average depths (measured from top of crest to bottom of valley) in juvenile cells (E) or in adult cells (F) either control or null for Scn1b (Scn1b–/–). G and F, distance between valleys. Notice that juvenile cells have a rather smooth topology, with minimum variations in depth, but the periodicity of the ridges is the same as that observed in adult cells.
Regional distribution of INa in adult cardiac-specific null ventricular myocytes
We generated cardiac-specific Scn1b null animals to circumvent the issue of the SUDEP phenotype at ∼P21 observed in the global null animals (Chen et al. 2004). These animals lived normal life spans and did not present with seizures or any other apparent neurological disorder. Surface topology of adult ventricular myocytes was similar between control and cardiac-specific null cells (Fig.6B,D,F,H). In contrast to juvenile cells, adult ventricular myocytes showed a highly organized topology, with deep, large invaginations (T-tubules) spaced at ∼2 μm intervals, separated by crests and aligned with z-grooves. An example of an SICM image of a cardiac-specific Scn1b null myocyte is shown in Fig.6B; the corresponding topology profile is shown in Fig.6D; quantitative comparisons to data obtained from control cells are shown in Fig.6F and H. The results indicate that loss of Scn1b expression did not modify the pattern of T-tubule formation (see also Bhargava et al. 2013). Yet, functional differences were observed. Using super-resolution scanning patch clamp, which allows us to position the recording pipette with nanometric precision (Bhargava et al. 2013), we determined that the organization of VGSCs varied with the specific location. Figure7 shows histograms depicting the percentage of SICM-guided recordings in which we detected the range of channel numbers expressed in the abscissae. Figure7A corresponds to recordings taken from the crest and Fig.7B, from the T-tubules. As in control cells (Fig.7Aa) (Bhargava et al. 2013), recordings made in the crest region of cardiac-specific null cells (Fig.7Ab) yielded an almost bimodal distribution: either no channels were detected at the site of recording, or we found large clusters with more than 20 channels opened simultaneously within the area of the patch. The average number of opened channels was 38.9 ± 8.3 (n = 27) in cardiac-specific null cells; this number represents a slight, though not statistically significant increase over the results obtained in control cells (30.0 ± 8.3; n = 23; see also Bhargava et al. 2013). In contrast, channels recorded at the T-tubules had a Gaussian frequency distribution (Fig.7B). The average number of channels recorded from the T-tubule in cardiac-specific null cells was 7.3 ± 1.4 (n = 17). This number was smaller than the one recorded in the crest, and larger (though not reaching statistical significance) when compared to the average number of channels recorded from T-tubules in control myocytes (5.4 ± 1.2; n = 17; see also Bhargava et al. 2013). Interestingly, addition of 100 nm TTX to the extracellular pipette solution in cardiac-specific null cells drastically changed the frequency histogram for T-tubule recordings (Fig.7Bc), but only a minor change was seen in crest recordings (Fig.7Ac). Indeed, in the presence of TTX, almost 50% of T-tubule recordings detected no channels, and the average number of open channels dropped to 2.4 ± 1.0 (n = 12; Fig.7Bc). These results strongly suggest an abundance of TTX-S VGSCs in the T-tubule region of cardiac-specific null cells. Consistent with the difference in the number of VGSCs detected by super-resolution scanning patch clamp, we observed a small increase in whole cell peak average INa density when comparing cardiac-specific null cells with their controls (control INa density: −50.7 ± 4.6 pA/pF, n = 22 cells from four mice; cardiac-specific null: –64.4 ± 6.3 pA/pF, n = 19 cells from four mice; P = 0.089), with no change in the voltage dependence of activation or inactivation of VGSC. Furthermore, qtRT-PCR analysis revealed a 1.44-fold increase in Scn3a message in cardiac-specific Scn1b null hearts compared to their control.
Figure 7.

Fraction of super-resolution scanning patch clamp seals in which we detected the number of active channels expressed in the abscissae
The number of separate recordings reported in each histogram is indicated in the top left of each panel. The pipette was placed on the crest (A) or on the opening of the T-tubules (B). Adult ventricular myocytes were obtained from cardiac-specific Scn1b null animals (Scn1b–/–) or from its control. In a separate group, Na+ channels in cardiac-specific Scn1b null cells were recorded in the presence of 100 nm TTX in the internal pipette solution. TTX, tetrodotoxin.
Action potential duration in adult cardiac-specific Scn1b null ventricular myocytes
Studies in hearts from juvenile animals revealed that global loss of Scn1b led to a prolongation in action potential duration in ventricular myocytes (Lopez-Santiago et al. 2007). We examined whether a similar effect was observed in adult cells from cardiac-specific null mice. As shown in Fig.8C and D, APD50 was increased in cardiac-specific null cells, though APD70 was not different between the groups. Interestingly, we observed a high incidence (13 of 18 cells) of DADs in the cardiac-specific null group (see Fig.8A), whereas no DADs were observed in control cells (n = 12; Fisher's exact test, P = 0.000072). In eight Scn1b null cells, a DAD eventually gave rise to a triggered action potential, followed by cell hypercontracture. None of the control cells recorded displayed a similar phenomenon. These results led us to investigate whether loss of Scn1b in the heart can alter intracellular Ca2+ homeostasis in adult ventricular myocytes.
Figure 8.
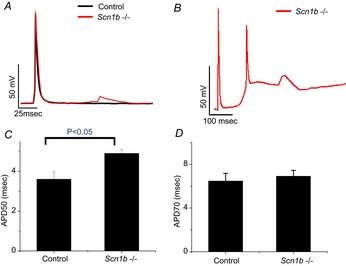
Action potential duration and delayed after-depolarizations in cardiac-specific Scn1b null adult ventricular myocytes
Action potentials recorded from ventricular myocytes isolated from control (black trace in A), and from cardiac-specific Scn1b null (Scn1b–/–) mice (red traces in A and in B). Notice the presence of a delayed after-depolarization recorded from the Scn1b null cell (A) and a triggered action potential (B). Average measurements of APD50 (C) and APD70 (D). Number of cells studied were 18 for Scn1b null and 12 for wild-type.
Intracellular Ca2+ measurements in adult cardiac-specific null ventricular myocytes
Figure9A shows intracellular Ca2+ transients measured during repetitive activity in single adult ventricular myocytes obtained from control (top trace), cardiac-specific null (middle trace) and cardiac-specific null exposed to 100 nm of TTX (bottom). The region demarcated by a green box (labelled ‘1’ in Fig.9A) is shown at an expanded time scale in Fig.9B. We observed a cardiac-specific loss of Scn1b expression associated with an activity-dependent accumulation of cytosolic Ca2+ at stimulation frequencies of 1 Hz, which was blunted by the block of TTX-S VGSCs (see also bar graph in Fig.9C). Transient oscillations following the relaxation of the Ca2+ transient (red arrows in Fig.9B) were detected in 13 of 14 cardiac-specific null cells during 180 s of recording and, on average, the first oscillation was detected after ∼70 s. The amplitude of these Ca2+ transients increased in the course of the stimulation period (box labelled ‘2’ in Fig.9A; expanded in time as trace ‘2’ in Fig.9B). In contrast, in control cells these oscillations were not present after 180 s of recording in 11 of 14 cells (P = 0.0003 by Fisher's exact test; see also bar graph in Fig.9D). Frequent spontaneous Ca2+ release events were observed following the end of the stimulation period in 13 of 14 Scn1b null cardiomyocytes, and only in one of 14 control cells (P = 4.9 × 10−6 by Fisher's exact test; see example in box ‘3’ in Fig.9A, and trace ‘3’ in Fig.9B). As in the case of the increase in diastolic Ca2+, 100 nm TTX drastically delayed or prevented the occurrence of Ca2+ oscillations in cardiac-specific null cells (bottom trace in Fig.9A). Finally, as shown in Fig.10, the time course of Ca2+ decay and the relative amplitude of the Ca2+ transient were increased in cardiac-specific null cells when compared to control (Fig.10A–C; each data point obtained from the average of events 6–10 in a train of 10 stimuli, delivered with a frequency of either 0.5 or 1 Hz); these effects were significantly blunted by addition of 100 nm TTX. Time to peak was reduced (Fig.10D) in cardiac-specific null cells compared to control but in this case, the effect was not sensitive to TTX. Overall, the data show that cardiac-specific loss of Scn1b expression leads to disruption of intracellular Ca2+ homeostasis that is mostly reversed by addition of 100 nm TTX.
Figure 9.
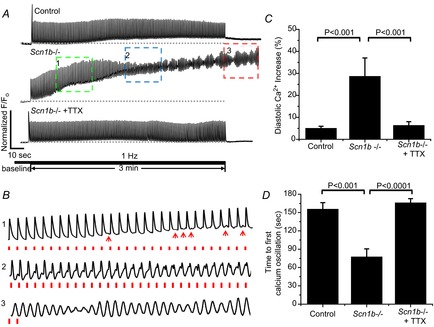
Ca2+ transients in adult ventricular myocytes dissociated from cardiac-specific Scn1b null mice, or control
A, Ca2+ transients recorded from a control adult myocyte (top), from a cardiac-specific Scn1b null (Scn1b–/–) myocyte (middle) and from an Scn1b–/– myocyte in the presence of 100 nm TTX (bottom). The cells were paced for 3 min at a rate of 1 Hz. The traces inside the boxed insets labelled ‘1’, ‘2’ and ‘3’ are displayed at an expanded time scale in (B). Red dots in the bottom traces in (B) mark the pacing event. Red arrowheads in trace ‘1’ point to the presence of Ca2+ oscillations occurring after the relaxation of the Ca2+ spike. C, average fractional increase in diastolic Ca2+ (percentage from baseline) after 3 min of regular pacing at a frequency of 1 Hz. D, time to first Ca2+ oscillation recorded from control cells, those from cardiac-specific Scn1b null mice (Scn1b–/–) and Scn1b–/– cells exposed to 100 nm TTX. Number of cells studied was 14, 14 and 11 for wild-type, Scn1b null and Scn1b null with TTX, respectively. TTX, tetrodotoxin.
Figure 10.
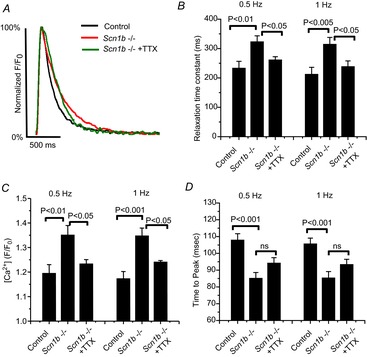
Characteristics of Ca2+ transients recorded from cells dissociated from cardiac-specific Scn1b null (Scn1b–/–) hearts, or their control
‘Scn1b–/– + TTX’ refers to data obtained from Scn1b–/– cells in the presence of 100 nm TTX. A, example of Ca2+ transients recorded under the three different conditions. The fluorescent transient was normalized to its maximum amplitude for comparison. B–D, each presents average data obtained at pacing frequencies of 0.5 and 1 Hz. Significance (P) values indicated for the respective brackets. Variables measured were relaxation time constant (B), total amplitude of the Ca2+ transient (C) and time to peak (D). Number of cells studied was 15, 19 and 11 for WT, Scn1b null and Scn1b null with TTX, respectively. TTX, tetrodotoxin.
Optical mapping of action potential propagation in cardiac-specific null hearts
A number of studies have shown an association between Ca2+ dysregulation and arrhythmogenicity. We utilized optical mapping methods to study action potential propagation in isolated Langendorff-perfused hearts from control and cardiac-specific null hearts (Vaidya et al. 1999; Morley & Jalife, 2000; Noujaim et al. 2012). We assessed the dynamics of electrical wave propagation on the anterior ventricular epicardial surface and observed that the null hearts were prone to arrhythmogenesis. On ECG, two of three null hearts developed premature ventricular complexes (PVCs) followed by the spontaneous initiation of non-sustained ventricular tachycardia (NSVT). The third null heart developed PVCs and ultimately VT upon the creation of severe bradycardia (by crushing the SA node). Spontaneous ventricular arrhythmias were not observed in two control hearts, in agreement with previous studies (Mironov et al. 2006; Noujaim et al. 2007b, 2012; Glukhov et al. 2010). In all hearts we observed normal baseline sinus ECG patterns and regular sinus activation with dual breakthrough in the right and left ventricles followed by fast uniform spread of the impulse throughout the ventricles. FigureA and B show a clear example of a single spontaneous PVC leading to the initiation of NSVT (R1…Rn). Sinus activation of the entire ventricles (Figure1C, CV 1.26 m s−1, with dual breakthroughs) was followed by re-excitation, conduction block and initiation of re-entry. In Fig.1D, during the action potential plateau a PVC emerged at the base of the left ventricle (asterisk), which resulted in unidirectional block (wavebreak) and formation of a rotor with counter-clockwise rotational spread (R1) around the line of block, setting up conditions for the initiation of VT. The rotor then meandered toward the left ventricular apex (Fig.1E, R2), resulting in an ECG pattern of non-sustained polymorphic VT (Fig.1F) with progressive changes in the QRS amplitude and morphology and a frequency of 20.8 Hz. This pattern is consistent with previous publications (Noujaim et al. 2007a,b), indicative of the viability and health of the preparation. A second cardiac-specific null heart spontaneously developed NSVT with an ECG pattern of Torsades de Pointes at 21.9 Hz (Fig.1G). This heart later developed sustained ventricular tachycardia/fibrillation, which lasted 24.12 s at 27.5 Hz. To assess whether the frequency of the ventricular arrhythmias differed between the mice, ventricular tachycardia/fibrillation was initiated by burst pacing in wild-type mice. The dominant frequencies of the arrhythmias were similar in wild-type and cardiac-specific null mouse hearts (21.1–27.6 Hz). These results demonstrate increased arrhythmia susceptibility in mice deficient in Scn1b.
Figure 11.
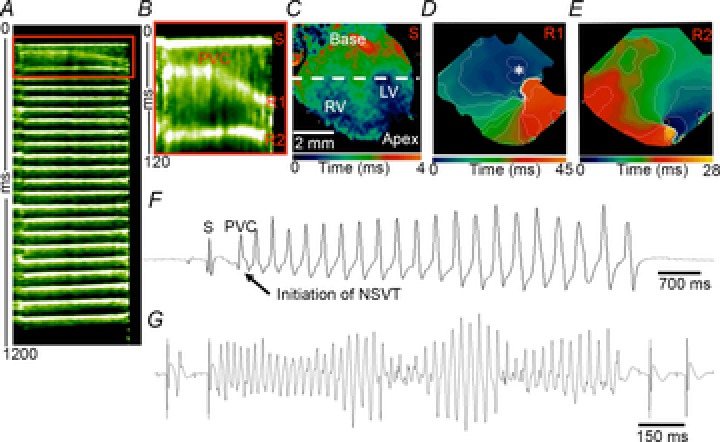
Arrhythmia events in cardiac-specific Scn1b null hearts
Spontaneous initiation of NSVT in an isolated cardiac-specific Scn1b null heart. A and B, time–space plot showing the sinus (S) activation followed by a PVC and the initiation of NSVT (R, re-entry 21.1 Hz). B, time–space plot zoom of the red box in (A) shows in detail the rapid propagation of the sinus impulse followed by a PVC leading to the first (R1) and second (R2) re-entrant impulses. C, activation map of sinus activation of the ventricles (dual ventricular breakthrough in the RV and LV). D, Early afterdepolarization formation in the LV (*), leading to a propagated response. The impulse propagated fast through the RV, while it propagated slowly and eventually resulted in a wavebreak at the LV, leading to the formation of a rotor (R1). E, second rotation indicates that the rotor meandered, with subsequent changes in the direction of propagation. F, volume-conducted ECG recording showing the sinus activation followed by a PVC and the initiation of NSVT. G, Torsade de pointes ECG morphology (21.9 Hz) during NSVT in a volume-conducted ECG from a second Langendorff-perfused Scn1b-MHC heart. LV, left ventricle; NSVT, non-sustained ventricular tachycardia; PVC, premature ventricular complex; RV, right ventricle; S, sinus.
Discussion
We showed previously that ventricular myocytes from juvenile Scn1b null hearts have increased transient and persistent INa and 3H-saxitoxin binding (Lopez-Santiago et al. 2007). Here, using methods that are more sensitive, we show increased TTX-S INa in the midsection of ventricular myocytes isolated from juvenile mice. Consistent with this, we also observed increased levels of Scn3a mRNA in heart, leading to the hypothesis that, similar to the previously observed increase in NaV1.3 in Scn1b null hippocampus (Chen et al. 2004), these animals also have increased expression of NaV1.3, a TTX-S VGSC, in CMs. Furthermore, we observed that TTX-S VGSCs in Scn1b null myocytes were excluded from the ID region even before the formation of the T-tubular system. In adult ventricular myocytes from cardiac-specific Scn1b null mice, we found that TTX-S VGSCs were detected primarily at the T-tubules. Interestingly, cardiac-specific Scn1b null adult ventricular myocytes also showed dysfunctional Ca2+ homeostasis that was corrected by low concentrations of TTX, suggesting a direct link between the increased expression of TTX-S VGSCs and the control of intracellular Ca2+. Finally, adult cardiac-specific Scn1b null hearts showed an arrhythmogenic phenotype. Taken together, our results lead us to speculate that loss of expression of β1/β1B subunits, critical members of cardiac VGSC signalling complexes, can result in life-threatening arrhythmias via a mechanism commonly associated with molecules that regulate the intracellular Ca2+ concentration.
It is possible for Na+ entry through VGSCs to cause Ca2+ release from internal stores at the sarcoplasmic reticulum (SR), even in the absence of Ca2+ entry through voltage-dependent Ca2+ channels (Leblanc & Hume, 1990). The underlying mechanism for this is thought to be Ca2+ influx through the Na+/Ca2+ exchanger (NCX1), triggered by a transient elevation of intracellular Na+ in a submembrane ‘fuzzy space’ (Lederer et al. 1990; Verdonck et al. 2004). Functional coupling of Na+ and Ca2+ currents within the confines of the T-tubule subdomain has also been proposed by others (see, e.g. Scriven et al. 2000; Gershome et al. 2011; Noujaim et al. 2012). This coupling may also involve NCX1, which is present in T-tubules and participates in Ca2+ dynamics (Thomas et al. 2003). Furthermore, Ramirez et al. (2011) have shown that Ca2+ entry via reverse mode NCX activity can facilitate SR-Ca2+ release triggered by ICaL during a typical cardiac action potential, particularly at elevated levels of intracellular Na+. The presence of TTX-S VGSCs (including NaV1.3) near T-tubules has been previously documented (Maier et al. 2004). We speculate that the enhanced Na+ influx via TTX-S VGSCs at the T-tubules might increase the local Na+ ion activity in the dyadic cleft, which then stimulates reverse Ca2+ influx via NCX1, and in turn sensitizes the coupling between Ca2+ channels and SR-Ca2+ release. A similar model has been proposed for demyelinated brain axons in multiple sclerosis, in which upregulated TTX-S NaV1.6 VGSCs leads to increased local Na+ ion concentrations, reverse Ca2+ influx and subsequent activation of neurodegenerative pathways (Waxman, 2006).
Experimental evaluation of each step in the hypothetical sequence of events postulated above is limited by a shortage of methods to test, with nanometer accuracy and with sufficient time resolution, the location of NaV1.3 in relation to NCX or the concentration of intracellular Na+ at the dyadic cleft. [Probes have recently been generated to measure Ca2+ in this location (Despa et al. 2014), but Na+-sensitive, cleft-confined probes are not available.] Of note, Lopez-Santiago et al. (2007) previously found increased persistent Na+ current in ventricular myocytes of Scn1b null mice. This observation fits within a scheme where increased intracellular Na+ concentration can shift the equilibrium potential of NCX1, thus reducing Ca2+ efflux and promoting Ca2+ overload (see, e.g. Fig.4 of Zaza et al. 2008). Furthermore, diastolic Na+ loading is an additional possibility. Yet, it should be noted that Despa et al. (2002) showed that diastolic Na+ loading was not TTX-dependent under control conditions. The latter departs from our observation of a phenomenon that can be partly reversed by small concentrations of TTX.
The hypothesis proposed above is supported by our scanning patch clamp data, which place TTX-S VGSCs primarily at the T-tubules. Our results extend the early observations of Duclohier (2005) and others (Maier et al. 2004; Brette & Orchard, 2006; Westenbroek et al. 2013) and predict that SCN1B null or loss-of-function mutations in human patients can lead to significant, TTX-dependent changes in intracellular Ca2+ homeostasis in the heart. Our observation of increased Scn3a mRNA in Scn1b null hearts further supports the notion that NaV1.3 channels at the T-tubules serve as an important link in excitation–contraction coupling. Yet, it is also possible that the effects mediated by TTX-S VGSCs are compounded by a certain degree of remodelling of Ca2+ handling proteins in the Scn1b null mice. For example, the time-to-peak of the Ca2+ transient is smaller in Scn1b null myocytes compared to controls, but this is only partially restored by TTX. This result may be consequent to changes in Ca2+ channel function, RyR2, the coupling between the latter, or an increase in the SR-Ca2+ content. In addition, the relaxation time constant in Scn1b nulls does not return completely to control levels in response to TTX. The slowing of the Ca2+ decline is probably due to either slowed SR-Ca2+ uptake (by SERCA2) or prolonged SR-Ca2+ release. We cannot rule out, however, the possibility that Ca2+ efflux via NCX1 is reduced, which might also contribute to the slower decline. Whichever the mechanism, the impaired removal of cytosolic Ca2+ following a twitch is probably responsible for the Ca2+ overload that occurs with rapid stimulation we observed in cardiac-specific Scn1b null ventricular myocytes. The extent to which deletion of Scn1b causes the observed functional phenotype by changes in transcription of relevant molecular players, in addition to the observed Scn3a effect, remains to be determined. The detailed mechanism of this regulatory function of Scn1b (or of the encoded proteins, VGSC β1/β1B subunits) deserves future investigation. VGSC β subunits have been shown previously to be substrates for successive cleavage by β- and γ-secretases (Wong et al. 2005). In the case of β2, this cleavage results in translocation of a C-terminal fragment to the nucleus and subsequent regulation of VGSC α subunit transcription (Kim et al. 2007). A similar mechanism for β1 may normally occur in CMs, with the absence of these important regulatory events in Scn1b null animals resulting in misregulated VGSC α subunit gene transcription.
To our knowledge, this is the first study to provide experimental data in favour of the hypothesis that loss of Scn1b can be arrhythmogenic due to alterations not only of INa, but of intracellular Ca2+ homeostasis. The notion that proteins classically defined as belonging to one particular molecular complex can affect the function of others has been proposed and documented before. In brain, for example, VGSC β1 associates with and modulates Kv4.2 in addition to VGSCs (Marionneau et al. 2012). At the cardiac ID, loss or mutations in connexin43 can change INa density (Desplantez et al. 2012; Jansen et al. 2012; Lübkemeier et al. 2013). Conversely, loss of ankyrin-G, a scaffolding protein commonly associated with the VGSC complex, decreases gap junctional conductance in the heart (Sato et al. 2011). The regulation of INa by molecules ‘of the desmosome’ is well documented. Separate studies have shown a reciprocal modulation of INa and the inward rectifier current IK1 (Milstein et al. 2012). At the T-tubule, TTX-S VGSCs probably coexist with NCX and with ankyrin-B, complexed with VGSC β1 in a manner that is regulated by intracellular tyrosine phosphorylation of β1 (Malhotra et al. 2002). Ankyrin-B has been shown to coordinate NCX at the cardiac T-tubules (Mohler et al. 2005) and loss of ankyrin-B function is linked to cardiac arrhythmia (Mohler et al. 2004). In the case presented in this paper, the relation between the VGSC complex and the regulation of intracellular Ca2+ does not seem to be consequent to protein–protein interactions within a functional domain, but to (a) changes in the transcription of a TTX-S VGSC, and (b) functional coupling, perhaps resulting from the changes in ionic concentrations in the cleft, among other possibilities. Overall, the present study further emphasizes that ion channel complexes are not molecular silos, but aggregates of multifunctional molecules capable of interacting within confined locations to achieve a common function (Sato et al. 2011; Milstein et al. 2012; Agullo-Pascual et al. 2014).
As molecules play a multiplicity of roles, their dysfunction can cause diseases by more than one mechanism (or be causative of more than one disease; see, e.g. Cerrone & Delmar, 2014; Cerrone et al. 2014). Mutations or rare variants in SCN1B have been linked to cases of Brugada and of long QT syndrome. Furthermore, functional loss of SCN1B leads to DS (Patino et al. 2009), a paediatric epileptic encephalopathy often associated with SUDEP. Because of its well-known association with Nav1.5, Scn1b-related arrhythmias are often interpreted as consequent to a reduction in INa. However, here and previously (Lopez-Santiago et al. 2007), we show that Scn1b null ventricular myocytes have increased INa. These results are similar to another mouse model of DS, SCN1AR1407X/+ knockin mice, which have increased cardiac INa (Auerbach et al. 2013). Here, we propose that life-threatening arrhythmias in patients with DS might be partly consequent to triggered activity, resulting from an overload of intracellular Ca2+. These observations may help guide a therapeutic approach to patients with inherited arrhythmia diseases associated with loss of expression of the Scn1b gene.
Glossary
- APD50
action potential duration at 50% repolarization
- APD70
action potential duration at 70% repolarization
- CMs
cardiac myocytes
- CT
cycle threshold
- DAD
delayed after-depolarizations
- DS
Dravet syndrome
- ID
intercalated disc
- INa
Na+ current
- NCX1
Na+/Ca2+ exchanger
- NSVT
non-sustained ventricular tachycardia
- PVCs
premature ventricular complexes
- RQ
relative quantification
- SICM
scanning ion conductance microscopy
- SR
sarcoplasmic reticulum
- SUDEP
sudden unexpected death in patients with epilepsy
- TTX-S
tetrodotoxin-sensitive
- VGSC
voltage-gated Na+ channel
Additional information
Competing interests
None
Author contributions
X.L. carried out all cell electrophysiological experiments, including SICM studies. He contributed to overall experimental design, manuscript preparation and submission. The murine models were generated by H.O'M. and C.C., under the advice of L.L.I. A.S. carried out the intracellular calcium measurements, under the advice and supervision of G.I.F. M.Z. conducted the qtRT-PCR experiments, with advice from W.C. and with assistanceof M.F. Optical mapping experiments were carried out by D.A. under the advice of J.J. All authors contributed to data analysis. J.J, W.C. and G.I.F. contributed directly to data interpretation and manuscript preparation. M.D. and L.L.I. contributed to the overall experimental design, study co-ordination and data interpretation. They drafted the manuscript and organized its final preparation and submission.
Funding
This work was supported by grants from the US National Institutes of Health (R01-HL106632 and R01-GM57691 to M.D.; NIH-R01 NS076752 to L.L.I.; T32-GM066704 (Bach) to A.S.) and by a Leducq Foundation Transatlantic Network (J.J. and M.D.).
References
- Agullo-Pascual E, Cerrone M. Delmar M. Arrhythmogenic cardiomyopathy and Brugada syndrome: Diseases of the connexome. FEBS Lett. 2014;588:1322–1330. doi: 10.1016/j.febslet.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach DS, Jones J, Clawson BC, Offord J, Lenk GM, Ogiwara I, Yamakawa K, Meisler MH, Parent JM. Isom LL. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One. 2013;8:e77843. doi: 10.1371/journal.pone.0077843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava A, Lin X, Novak P, Mehta K, Korchev Y, Delmar M. Gorelik J. Super-resolution scanning patch clamp reveals clustering of functional ion channels in adult ventricular myocyte. Circ Res. 2013;112:1112–1120. doi: 10.1161/CIRCRESAHA.111.300445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Yuan Y, O'Malley HA, Parent JM. Isom LL. Abnormal neuronal patterning occurs during early postnatal brain development of Scn1b-null mice and precedes hyperexcitability. Proc Natl Acad Sci U S A. 2013;110:1089–1094. doi: 10.1073/pnas.1208767110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brette F. Orchard CH. Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem Biophys Res Commun. 2006;348:1163–1166. doi: 10.1016/j.bbrc.2006.07.189. [DOI] [PubMed] [Google Scholar]
- Cerrone M. Delmar M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc Med. 2014;24:184–190. doi: 10.1016/j.tcm.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori SG. Delmar M. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 2014;129:1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O'Malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilaythong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA. Isom LL. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci. 2004;24:4030–4042. doi: 10.1523/JNEUROSCI.4139-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Dickendesher TL, Oyama F, Miyazaki H, Nukina N. Isom LL. Floxed allele for conditional inactivation of the voltage-gated sodium channel beta1 subunit Scn1b. Genesis. 2007;45:547–553. doi: 10.1002/dvg.20324. [DOI] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, Pogwizd SM. Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Despa S, Shui B, Bossuyt J, Lang D, Kotlikoff MI. Bers DM. Junctional cleft [Ca2+]i measurements using novel cleft-targeted Ca2+ sensors. Circ Res. 2014;115:339–347. doi: 10.1161/CIRCRESAHA.115.303582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplantez T, McCain ML, Beauchamp P, Rigoli G, Rothen-Rutishauser B, Parker KK. Kleber AG. Connexin43 ablation in foetal atrial myocytes decreases electrical coupling, partner connexins, and sodium current. Cardiovasc Res. 2012;94:58–65. doi: 10.1093/cvr/cvs025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52(Suppl 2):3–9. doi: 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- Dravet C, Bureau M, Oguni H, Fukuyama Y. Cokar O. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv Neurol. 2005;95:71–102. [PubMed] [Google Scholar]
- Duclohier H. Neuronal sodium channels in ventricular heart cells are localized near T-tubules openings. Biochem Biophys Res Commun. 2005;334:1135–1140. doi: 10.1016/j.bbrc.2005.06.203. [DOI] [PubMed] [Google Scholar]
- Genton P, Velizarova R. Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. 2011;52(Suppl 2):44–49. doi: 10.1111/j.1528-1167.2011.03001.x. [DOI] [PubMed] [Google Scholar]
- Gershome C, Lin E, Kashihara H, Hove-Madsen L. Tibbits GF. Colocalization of voltage-gated Na+ channels with the Na+/Ca2+ exchanger in rabbit cardiomyocytes during development. Am J Physiol Heart Circ Physiol. 2011;300:H300–H311. doi: 10.1152/ajpheart.00798.2010. [DOI] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Anderson ME, Mohler PJ. Efimov IR. Functional anatomy of the murine sinus node: high-resolution optical mapping of ankyrin-B heterozygous mice. Am J Physiol Heart Circ Physiol. 2010;299:H482–491. doi: 10.1152/ajpheart.00756.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL. Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray RA, Jalife J, Panfilov A, Baxter WT, Cabo C, Davidenko JM. Pertsov AM. Nonstationary vortexlike reentrant activity as a mechanism of polymorphic ventricular tachycardia in the isolated rabbit heart. Circulation. 1995;91:2454–2469. doi: 10.1161/01.cir.91.9.2454. [DOI] [PubMed] [Google Scholar]
- Guerrini R. Aicardi J. Epileptic encephalopathies with myoclonic seizures in infants and children (severe myoclonic epilepsy and myoclonic-astatic epilepsy) J Clin Neurophysiol. 2003;20:449–461. doi: 10.1097/00004691-200311000-00007. [DOI] [PubMed] [Google Scholar]
- Haddock PS, Coetzee WA, Cho E, Porter L, Katoh H, Bers DM, Jafri MS. Artman M. Subcellular [Ca2+]i gradients during excitation-contraction coupling in newborn rabbit ventricular myocytes. Circ Res. 1999;85:415–427. doi: 10.1161/01.res.85.5.415. [DOI] [PubMed] [Google Scholar]
- Hirsch LJ, Donner EJ, So EL, Jacobs M, Nashef L, Noebels JL. Buchhalter JR. Abbreviated report of the NIH/NINDS workshop on sudden unexpected death in epilepsy. Neurology. 2011;76:1932–1938. doi: 10.1212/WNL.0b013e31821de7de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Barajas-Martinez H, Medeiros-Domingo A, Crotti L, Veltmann C, Schimpf R, Urrutia J, Alday A, Casis O, Pfeiffer R, Burashnikov E, Caceres G, Tester DJ, Wolpert C, Borggrefe M, Schwartz P, Ackerman MJ. Antzelevitch C. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Nav1.5 and Kv4.3 channel currents. Heart Rhythm. 2012;9:760–769. doi: 10.1016/j.hrthm.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ. Catterall WA. Functional co-expression of the β1 and type IIA α subunits of sodium channels in a mammalian cell line. J Biol Chem. 1995;270:3306–3312. doi: 10.1074/jbc.270.7.3306. [DOI] [PubMed] [Google Scholar]
- Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M. van Rijen HV. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2012;9:600–607. doi: 10.1016/j.hrthm.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ. Kovacs DM. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol. 2007;9:755–764. doi: 10.1038/ncb1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc N. Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science. 1990;248:372–376. doi: 10.1126/science.2158146. [DOI] [PubMed] [Google Scholar]
- Lederer WJ, Niggli E. Hadley RW. Sodium-calcium exchange in excitable cells: fuzzy space. Science. 1990;248:283. doi: 10.1126/science.2326638. [DOI] [PubMed] [Google Scholar]
- Lin X, Liu N, Lu J, Zhang J, Anumonwo JM, Isom LL, Fishman GI. Delmar M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011;8:1923–1930. doi: 10.1016/j.hrthm.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Meadows LS, Ernst SJ, Chen C, Malhotra JD, McEwen DP, Speelman A, Noebels JL, Maier SK, Lopatin AN. Isom LL. Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J Mol Cell Cardiol. 2007;43:636–647. doi: 10.1016/j.yjmcc.2007.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübkemeier I, Requardt RP, Lin X, Sasse P, Andrie R, Schrickel JW, Chkourko H, Bukauskas FF, Kim JS, Frank M, Malan D, Zhang J, Wirth A, Dobrowolski R, Mohler PJ, Offermanns S, Fleischmann BK, Delmar M. Willecke K. Deletion of the last five C-terminal amino acid residues of connexin43 leads to lethal ventricular arrhythmias in mice without affecting coupling via gap junction channels. Basic Res Cardiol. 2013;108:348–364. doi: 10.1007/s00395-013-0348-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T. Catterall WA. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation. 2004;109:1421–1427. doi: 10.1161/01.CIR.0000121421.61896.24. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kazen-Gillespie K, Hortsch M. Isom LL. Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem. 2000;275:11383–11388. doi: 10.1074/jbc.275.15.11383. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Koopmann MC, Kazen-Gillespie KA, Fettman N, Hortsch M. Isom LL. Structural requirements for interaction of sodium channel beta 1 subunits with ankyrin. J Biol Chem. 2002;277:26681–26688. doi: 10.1074/jbc.M202354200. [DOI] [PubMed] [Google Scholar]
- Marionneau C, Carrasquillo Y, Norris AJ, Townsend RR, Isom LL, Link AJ. Nerbonne JM. The sodium channel accessory subunit Navbeta1 regulates neuronal excitability through modulation of repolarizing voltage-gated K+ channels. J Neurosci. 2012;32:5716–5727. doi: 10.1523/JNEUROSCI.6450-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR. Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 2012;109:E2134–2143. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SF, Vetter FJ. Pertsov AM. Fluorescence imaging of cardiac propagation: spectral properties and filtering of optical action potentials. Am J Physiol Heart Circ Physiol. 2006;291:H327–335. doi: 10.1152/ajpheart.01003.2005. [DOI] [PubMed] [Google Scholar]
- Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT. Bennett V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101:9137–9142. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler PJ, Davis JQ. Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley GE. Jalife J. Cardiac gap junction remodeling by stretch: is it a good thing. Circ Res. 2000;87:272–274. doi: 10.1161/01.res.87.4.272. [DOI] [PubMed] [Google Scholar]
- Noujaim SF, Auerbach DS. Jalife J. Ventricular fibrillation: dynamics and ion channel determinants. Circ J. 2007a;71(Suppl A):A1–11. doi: 10.1253/circj.71.a1. [DOI] [PubMed] [Google Scholar]
- Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN. Jalife J. Up-regulation of the inward rectifier K+ current (IK1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007b;578:315–326. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noujaim SF, Kaur K, Milstein M, Jones JM, Furspan P, Jiang D, Auerbach DS, Herron T, Meisler MH. Jalife J. A null mutation of the neuronal sodium channel NaV1.6 disrupts action potential propagation and excitation-contraction coupling in the mouse heart. FASEB J. 2012;26:63–72. doi: 10.1096/fj.10-179770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley JC, Kalume F. Catterall WA. Insights into pathophysiology and therapy from a mouse model of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):59–61. doi: 10.1111/j.1528-1167.2011.03004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino GA, Claes LR, Lopez-Santiago LF, Slat EA, Dondeti RS, Chen C, O'Malley HA, Gray CB, Miyazaki H, Nukina N, Oyama F, De Jonghe P. Isom LL. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci. 2009;29:10764–10778. doi: 10.1523/JNEUROSCI.2475-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A. Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304. doi: 10.1161/CIRCRESAHA.110.228312. [DOI] [PubMed] [Google Scholar]
- Ramirez RJ, Sah R, Liu J, Rose RA. Backx PH. Intracellular [Na+] modulates synergy between Na+/Ca2+ exchanger and L-type Ca2+ current in cardiac excitation–contraction coupling during action potentials. Basic Res Cardiol. 2011;106:967–977. doi: 10.1007/s00395-011-0202-z. [DOI] [PubMed] [Google Scholar]
- Riuro H, Campuzano O, Arbelo E, Iglesias A, Batlle M, Perez-Villa F, Brugada J, Perez GJ, Scornik FS. Brugada R. A missense mutation in the sodium channel beta1b subunit reveals SCN1B as a susceptibility gene underlying LQT Syndrome. Heart Rhythm. 2014 doi: 10.1016/j.hrthm.2014.03.044. [DOI] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF. Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Sakauchi M, Oguni H, Kato I, Osawa M, Hirose S, Kaneko S, Takahashi Y, Takayama R. Fujiwara T. Retrospective multiinstitutional study of the prevalence of early death in Dravet syndrome. Epilepsia. 2011;52:1144–1149. doi: 10.1111/j.1528-1167.2011.03053.x. [DOI] [PubMed] [Google Scholar]
- Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM. Delmar M. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201. doi: 10.1161/CIRCRESAHA.111.247023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuele SU, Bermeo AC, Alexopoulos AV, Locatelli ER, Burgess RC, Dinner DS. Foldvary-Schaefer N. Video-electrographic and clinical features in patients with ictal asystole. Neurology. 2007a;69:434–441. doi: 10.1212/01.wnl.0000266595.77885.7f. [DOI] [PubMed] [Google Scholar]
- Schuele SU, Widdess-Walsh P, Bermeo A. Luders HO. Sudden unexplained death in epilepsy: the role of the heart. Cleve Clin J Med. 2007b;74(Suppl 1):S121–127. doi: 10.3949/ccjm.74.suppl_1.s121. [DOI] [PubMed] [Google Scholar]
- Scriven DR, Dan P. Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorvon S. Tomson T. Sudden unexpected death in epilepsy. Lancet. 2011;378:2028–2038. doi: 10.1016/S0140-6736(11)60176-1. [DOI] [PubMed] [Google Scholar]
- Surges R, Thijs RD, Tan HL. Sander JW. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol. 2009;5:492–504. doi: 10.1038/nrneurol.2009.118. [DOI] [PubMed] [Google Scholar]
- Surges R, Adjei P, Kallis C, Erhuero J, Scott CA, Bell GS, Sander JW. Walker MC. Pathologic cardiac repolarization in pharmacoresistant epilepsy and its potential role in sudden unexpected death in epilepsy: a case-control study. Epilepsia. 2010;51:233–242. doi: 10.1111/j.1528-1167.2009.02330.x. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Sjaastad I, Andersen K, Helm PJ, Wasserstrom JA, Sejersted OM. Ottersen OP. Localization and function of the Na+/Ca2+-exchanger in normal and detubulated rat cardiomyocytes. J Mol Cell Cardiol. 2003;35:1325–1337. doi: 10.1016/j.yjmcc.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Vaidya D, Morley GE, Samie FH. Jalife J. Reentry and fibrillation in the mouse heart. A challenge to the critical mass hypothesis. Circ Res. 1999;85:174–181. doi: 10.1161/01.res.85.2.174. [DOI] [PubMed] [Google Scholar]
- Verdonck F, Mubagwa K. Sipido KR. [Na+] in the subsarcolemmal ‘fuzzy’ space and modulation of [Ca2+]i and contraction in cardiac myocytes. Cell Calcium. 2004;35:603–612. doi: 10.1016/j.ceca.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci. 2006;7:932–941. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Bischoff S, Fu Y, Maier SK, Catterall WA. Scheuer T. Localization of sodium channel subtypes in mouse ventricular myocytes using quantitative immunocytochemistry. J Mol Cell Cardiol. 2013;64:69–78. doi: 10.1016/j.yjmcc.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, De Strooper B, Saftig P. Nukina N. beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem. 2005;280:23009–23017. doi: 10.1074/jbc.M414648200. [DOI] [PubMed] [Google Scholar]
- Zarzoso M, Rysevaite K, Milstein ML, Calvo CJ, Kean AC, Atienza F, Pauza DH, Jalife J. Noujaim SF. Nerves projecting from the intrinsic cardiac ganglia of the pulmonary veins modulate sinoatrial node pacemaker function. Cardiovasc Res. 2013;99:566–575. doi: 10.1093/cvr/cvt081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaza A, Belardinelli L. Shryock JC. Pathophysiology and pharmacology of the cardiac ‘late sodium current. Pharmacol Ther. 2008;119:326–339. doi: 10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]