

Abstract
Late Na+ current (INaL) contributes to action potential (AP) duration and Ca2+ handling in cardiac cells. Augmented INaL was implicated in delayed repolarization and impaired Ca2+ handling in heart failure (HF). We tested if Na+ channel (Nav) neuronal isoforms contribute to INaL and Ca2+ cycling defects in HF in 17 dogs in which HF was achieved via sequential coronary artery embolizations. Six normal dogs served as control. Transient Na+ current (INaT) and INaL in left ventricular cardiomyocytes (VCMs) were recorded by patch clamp while Ca2+ dynamics was monitored using Fluo-4. Virally delivered short interfering RNA (siRNA) ensured Nav1.1 and Nav1.5 post-transcriptional silencing. The expression of six Navs was observed in failing VCMs as follows: Nav1.5 (57.3%) > Nav1.2 (15.3%) > Nav1.1 (11.6%) > Nav2.1 (10.7%) > Nav1.3 (4.6%) > Nav1.6 (0.5%). Failing VCMs showed up-regulation of Nav1.1 expression, but reduction of Nav1.6 mRNA. A similar Nav expression pattern was found in samples from human hearts with ischaemic HF. VCMs with silenced Nav1.5 exhibited residual INaT and INaL (∼30% of control) with rightwardly shifted steady-state activation and inactivation. These currents were tetrodotoxin sensitive but resistant to MTSEA, a specific Nav1.5 blocker. The amplitude of the tetrodotoxin-sensitive INaL was 0.1709 ± 0.0299 pA pF–1 (n = 7 cells) and the decay time constant was τ = 790 ± 76 ms (n = 5). This INaL component was lacking in VCMs with a silenced Nav1.1 gene, indicating that, among neuronal isoforms, Nav1.1 provides the largest contribution to INaL. At –10 mV this contribution is ∼60% of total INaL. Our further experimental and in silico examinations showed that this new Nav1.1 INaL component contributes to Ca2+ accumulation in failing VCMs and modulates AP shape and duration. In conclusion, we have discovered an Nav1.1-originated INaL component in dog heart ventricular cells. This component is physiologically relevant to controlling AP shape and duration, as well as to cell Ca2+ dynamics.
Key points
Late Na+ current (INaL) contributes to action potential remodelling and Ca2+/Na+ changes in heart failure.
The molecular identity of INaL remains unclear.
The contributions of different Na+ channel isoforms, apart from the cardiac isoform, remain unknown.
We discovered and characterized a substantial contribution of neuronal isoform Nav1.1 to INaL.
This new component is physiologically relevant to the control of action potential shape and duration, as well as to cell Ca2+ dynamics, especially in heart failure.
Introduction
A physiological role of neuronal isoforms of voltage-sensitive Na+ channels (Navs) in heart has been suggested many years ago by Corabeouf (Coraboeuf et al. 1979) based on their result that low concentrations (33 nm) of tetrodotoxin (TTX) significantly shortened action potential (AP) duration recorded in cardiac Purkinje fibres. At this low concentration TTX blocks only so-called ‘TTX-sensitive’ Navs which are neuronal isoforms (Haufe et al. 2005a2005a). Indeed, during the past decade, besides the dominating cardiac isoform main α-subunit, Nav1.5, different transcripts of highly TTX-sensitive neuronal Nav isoforms have been found in mouse (Nav1.1, 1.3, 1.6) and dog (Nav1.1, 1.2, 1.3) heart (Maier et al. 2004; Haufe et al. 2005a2005a,b). These neuronal Navs are responsible for 10% and 20% of the peak transient Na+ current, INaT, in dog myocardial and Purkinje cells, respectively (Haufe et al. 2005b2005b). The contribution of neuronal Navs to the late Na+ current, INaL, has not been studied in cardiac cells, therefore the molecular identity of INaL needs further careful consideration. This problem is especially important in HF where the gene expression profile changes dramatically. Thus an intriguing possibility could be that the differential expression of neuronal Navs can contribute, at least in part, to INaL alterations (augmentation) (Valdivia et al. 2005; Maltsev et al. 2007) and to INaL-related electrophysiological abnormalities observed in HF (for review see Maltsev & Undrovinas, 2008).
In the present study we used a combination of molecular and pharmacological approaches, as well as numerical modelling, to test a hypothesis that neuronal Nav isoforms contribute to INaL in a physiologically significant manner in ventricular myocytes (VCMs) isolated from failing adult dog hearts. We performed post-transcriptional silencing of Nav1.1 and Nav1.5 genes by sequence-directed RNA using virally delivered short interfering RNA (siRNA). We found the expression of six Navs in both human heart and adult dog VCMs. Analysis of the expression revealed up-regulation of Nav1.1 and down-regulation of Nav1.6 in failing heart compared to normal heart. We took advantage of the method of culturing adult dog VCMs, established by our group, that preserves INaT and INaL over 5 days, i.e. sufficient time for gene silencing and membrane protein turnover (Maltsev et al. 2008a2008a; Mishra et al. 2011). In VCMs with silenced Nav1.5 we found a substantial residual INaT and INaL. Our biophysical and pharmacological examinations confirmed its neuronal isoform origin. Diastolic Ca2+ accumulation was reduced via blockade of the neuronal INaL component, indicating its physiological importance. Our numerical model simulations also demonstrated the physiological importance of the novel INaL component: the Nav1.1- associated INaL effectively modulates AP shape and duration, as well as intracellular Ca2+ homeostasis.
Methods
HF model, human heart samples
The Henry Ford Health System Institutional Review Board approved all studies using human tissue and written informed consent was obtained. Studies conducted in animals as well as the methods of killing the animals were in accordance with the Guidelines for the Care and Use of Laboratory Animals (NIH publication No. 85-23), and were approved by the Henry Ford Health System Institutional Animal Care and Use Committee.
The dog model of chronic HF is similar by a vast array of functional and pathophysiological parameters (Sabbah et al. 1992) to that in humans. HF was produced in 17 dogs by multiple sequential coronary artery microsphere embolizations as previously described (Sabbah et al. 1991). All haemodynamic data were collected during cardiac catheterization performed under general anaesthesia and sterile conditions. Anaesthesia was induced using a combination of intravenous injections of hydromorphone (0.22 mg kg−1) and diazepam (0.2–0.6 mg kg−1), and maintained throughout the procedure with 1–2% isoflurane. All dogs were intubated and ventilated with room air supplemented with oxygen. At the end of data collection, and while the animal was still under general anaesthesia, the chest was opened via a left thoracotomy and the heart rapidly removed and tissue prepared for further studies. After each coronary microembolization session, dogs were weaned off of the respirator and allowed to recover. Animals were closely monitored until the endotracheal tube was removed and the dog was in a sternal position. Animals were then placed in post-operative recovery cages with incubated cloth blankets for warmth. Animals were given analgesics for pain and/or medications for cardiovascular complications only if needed. The animals were monitored until pain medications and/or cardiovascular drugs were no longer needed. HF state was confirmed by haemodynamic and angiographic measurements. At the time of harvesting the hearts (∼3 months after last embolization), left ventricular (LV) ejection fraction was 25 ± 1.2% (n = 17).
Human heart samples were obtained from explanted hearts from patients with chronic HF.
Cell culture and transfection
VCMs were enzymatically isolated from the apical LV mid-myocardial slices as previously reported (Maltsev et al. 1998a). The yield of viable rod-shaped, Ca2+-tolerant VCMs varied from 40 to 70%. VCMs were cultured for 5 days (the time frame required for gene silencing; Maltsev et al. 2008a), as previously described (Maltsev et al. 2002). We added 10 μl of 5 × 108 virus particles ml–1 directly to the medium of the culture dish containing ∼15 × 104 cells and cultured them further during the next 4 days (5 days total). Effectiveness of transfection was visually monitored in individual cardiomyocytes (Nikon Diaphot, 200) by green fluorescent protein (GFP) fluorescence (Fig.3A inset) and by the occurrence of mRNA encoding GFP.
Figure 3.
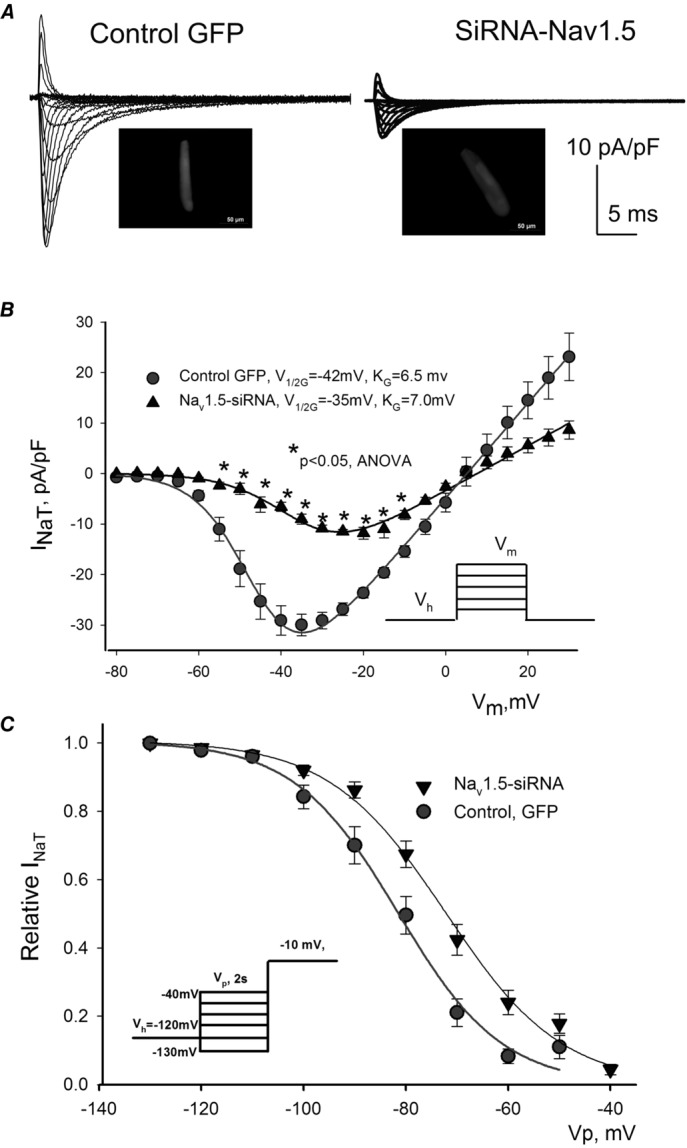
Post-transcriptional SCN5A gene silencing results in functional reduction of the transient Na+ current, INaT, and reveals non-cardiac Na+ channel component in LV cardiomyocytes from dog with chronic heart failure
A, representative raw traces of INaT were recorded at different membrane potentials (Vm) in cells 5 days after infection with virus containing control non-silencing siRNA and cDNA for GFP (control GFP, left), and siRNA-Nav1.5 (right). Cell images expressing GFP are shown in insets. B, average data from peak INaT–voltage relationship obtained in cultured cells for 5 days with virally delivered control siRNA (• control GFP, n = 6), and Nav1.5-siRNA (▴, n = 7). We found a statistically significant reduction of INaT in response to Nav1.5-siRNA in a wide range of potentials (*). Continuous lines show theoretical curves fitted to experimental current–voltage (I–V) data points to evaluate the steady-state activation (SSA) parameters which are given in the panel. •, control GFP, control siRNA (n = 18); ▴, Nav1.5-siRNA (n = 16). C, average experimental data of steady-state inactivation (SSI) data points in control (•, n = 17) and after SCN5A gene silencing by the siRNA (▴, n = 16) along with the fit to a Boltzmann function. The F test revealed a statistically significant (P < 0.05) difference between theoretical fits (B and C), and statistical significance, P < 0.05, between experimental data points presented as mean ± SEM (B) was evaluated by ANOVA followed by Bonferroni's post hoc test. Voltage clamp protocols are shown in B and C insets; Vh, holding and Vp, pre-pulse potentials, respectively.
siRNA design
Double stranded hairpin siRNAs corresponding to the previously sequenced dog Nav1.5 and Nav1.1 (GenBank DQ665939 and AY126477, respectively) and other Navs were designed as recommended (Elbashir et al. 2001) and published by our group (Mishra et al. 2011). The details and sequences of silencing and non-silencing (control) siRNAs used in this study are given in Table1. A BLAST search for these sequences against the GenBank database was performed and did not reveal matches with any other gene.
Table 1.
DNA sequences for coding siRNA designed to silence different genes that encode the Na+ channel isoform main α- and auxiliary β-subunit used in this study to make the adenovirus
| Nav1.1-A3906F | 5′-GATCATCTCTCAGGACACTAAGAGCGGAATTCGGCTCTTAGTGTCCTGAGAGATTTTTT-3′ |
| Nav1.2A-1683F | 5′-GATCCCGTAAATTGGTTCCTATCGTCTGTTGAATTCCGCAGACGATAGGAACCAATTTATTTTTTCCAAA-3′ |
| Nav1.3-B5191F | 5′-GATCCGTCAGAGCAAAGAGCAGTGTGCTTGAATTCCGGCACACTGCTCTTTGCTCTGATTTTTTCCAA-3′ |
| Nav1.5–190F | 5′-GATCAAAGCTGCCAGATCTCTATGGGATCGATGCCATAGAGATCTGGCAGCTTTTTTTT-3′ |
| Nav1.6-A1271F | 5′-GATCCGTGAGTCGGTCTAACATCTGCTTTGAATTCCGAGCAGATGTTAGACCGACTCATTTTTTCCAA-3′ |
| Nav2.1-A88F | 5′-GATCGAGTGTGGATCTTCTGGAGAAGGAATTCGTTCTCCAGAAGATCCACACTCTTTTT-3′ |
| NEG-CTRL-F | 5′-GATCCCGTCGCTTACCGATTCAGAATGGTTGATATCCGCCATTCTGAATCGGTAAGCGATTTTTTCGA-3′ |
| Navβ1–202F | 5′-GATCGAAGATCCACATTGAGGTGGTGATCGATGACCACCTCAATGTGGATCTTCTTTTT-3′ |
| Navβ2–216F | 5′-GATCCAACTGCTCTGAGGAGATGTTGATCGATGAACATCTCCTCAGAGCAGTTGTTTTT-3′ |
Real-time PCR and Western blot analysis
To quantify mRNA levels, we used both conventional gels and the fluorescence-based (SYBR green) kinetic real-time PCR performed using 7500 Fast Real-Time PCR Applied Biosystems (Life Technologies, Grand Island, NY, USA) sequence detection system. For each sodium channel isoform, several transcript-specific perfect match primer sets were designed and custom synthesized on the basis of sequences available in the NIH database. Primers were tested for production of predicted band size by RT-PCR. PCR products of correct band sizes were sequenced to check specificity. Each specific primer, which was selected after sequencing, was tested for use in real-time PCR before using in silencing experiments. Primers were also tested for amplification efficiency by standard curve analysis. Efficiencies of all the primers used in the study were between 90 and 99%. The quality of the amplified fragments was monitored using melting curve analysis and agarose gel electrophoresis. Quantitative values were obtained from the PCR quantification cycle number (Cq) at which point the increase in signal for the PCR product was exponential. The sets of primers are given in Table2.
Table 2.
Sequences of primers used in this study for the real-time PCR
| Isoform | Sequences | Product size (bp) | |
|---|---|---|---|
| Nav1.1A | F | 5′-CCTGGTGTTCATTGTGTTGTTC | 491 |
| R | 5′-AATCGGGTGGTTTACTGTTGAG | ||
| Nav1.2A | F | 5′-TCAAGAGAGTTCAGTGGTGCTG | 418 |
| R | 5′-AGTCTCTCCGGCTGTCATTGT | ||
| Nav1.3A | F | 5′-CTACCACATCTCCTCCTTCCT | 189 |
| R | 5′-TCCAGTTGACACATAGACTTTACCA | ||
| Nav1.5A | F | 5′-CCCAGAAGCAGGATGAGAAG | 516 |
| R | 5′-CGGTGAAGGTGTACTCGACA | ||
| Nav1.6A | F | 5′-CAGCAGATGTTAGACCGACTCA | 155 |
| R | 5′-CTCCTTGGCACTCTTTGAGC | ||
| Nav2.1A | F | 5′-TATTTGCTGGTTGGGATGG | 586 |
| R | 5′-ACTGCCTCTTGTTTTCGTTTCA | ||
| Navβ1 | F | 5′-TATGAGAATGAGGTGTTGCAGCTGG-3′ | 482 |
| R | 5′-ATACGGCTGGCTCTTCCTTGAGG-3′ | ||
| Navβ2 | F | 5′-CCGACTAACATCTCAGTCTC-3′ | 211 |
| R | 5′-CTGTTTGTGGTTCACTGTG-3′ |
The relative mRNA abundance expressed as arbitrary units was calculated using expression levels of all transcripts normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. This value was then normalized to mRNA levels measured from VCMs transfected with the non-silencing siRNA for the appropriate transcript (2−ΔΔCt method). Membrane protein preparations were obtained from approximately 3 × 106 myocytes as previously described (Zicha et al. 2004; Mishra et al. 2011). Antibody was detected with Western blot chemo-luminescence reagent (NEN Life Science Products, Boston, MA, USA). Western blots were considered specific if the peptide epitope reduced the band intensity. The resulting images of Western blots and PCR gels (See Fig.2A and B) were scanned and the relative densities of bands were quantified using SigmaGel (Sigma-Aldrich, USA) or ImageJ (National Institute of Health free software) software. The primary polyclonal anti-Nav1.5 and anti-Nav1.1 antibodies were obtained from Alomone Labs (Jerusalem, Israel). Polyclonal calsequestrin antibody (Abcam Inc., Cambridge, MA, USA) was used for the protein loading control.
Figure 2.

Silencing of SCN5A gene that encodes Nav1.5 in LV cardiomyocytes of dog with chronic HF by virally delivered siRNA
A, RT-PCR analysis of mRNA abundance in control (fresh isolated cells), in cells infected with virus that contains cDNA for Nav1.5 siRNA + GFP (Ad-GFP-Nav1.5) and GFP alone (Ad-GFP). Upper panel: representative gel of such analysis performed at different times after the virus infection. Lower panel: summary data with statistical analysis of experiments similar to that shown in the upper panel obtained in 5–7 cells. Relative Nav1.5 mRNA is given as relative to control density of bands. *P < 0.05, vs. control and GFP. B, Western blot analysis of Nav1.5 protein abundance in control, and in Ad-GFP-Nav1.5- and Ad-GFP-treated cells. *P < 0.001, vs. control and Ad-GFP. C, RT-PCR analysis of different sodium channel isoforms after transfection with Nav1.5-siRNA (from 4 to 8 samples). *P < 0.05. Statistically significant changes in A, B and C, were evaluated by the ANOVA test followed by Bonferroni's post hoc test. Data are mean ± SEM.
Electrophysiology
INa was recorded using conventional whole-cell patch-clamp techniques. Experimental and theoretical evaluation of the decay kinetics of INa and the steady-state activation (SSA) and inactivation (SSI) voltage dependency of INa were the same as we reported previously (Mishra et al. 2011). The decays of both INaT and INaL were approximated by double exponential fits, but the SSA and SSI were approximated by respective Boltzmann functions. Ca2+ signals were measured at 35 °C and 1.8 mm [Ca2+]o using a photo-multiplier in field-stimulated single VCMs loaded with the acetoxymethyl ester form of Fluo-4, Fluo-4 AM (Undrovinas et al. 2010). INaT and INaL were measured at room temperature (21–23 °C).
Numerical excitation–contraction coupling model
To address the physiological significance of the neuronal INaL component, we used a modified excitation–contraction (E–C) coupling model of a failing canine ventricular myocyte developed previously by Winslow et al. (1999). The original model was modified to include INaL equations as we previously reported (Undrovinas et al. 2010; Mishra et al. 2011). In this study we further modified INaL formulations to include two separate INaL components for cardiac and neuronal isoforms, respectively. Each component has its own activation and inactivation gating variables.
Statistical analysis
Statistics are reported as mean ± SEM with ‘n’ representing the number of cells. Multiple comparisons between treatment groups were made using one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test. The significance of changes of theoretical SSA and SSI fits was evaluated with an F test (StatMost, Data-Most, Salt Lake City, UT, USA) for tabulated values predicted by the Boltzmann model at a confidence level of 0.95. Differences for both experimental data and model predictions were considered statistically significant for P < 0.05.
Results
Molecular biology data
We observed HF-related changes in mRNA levels of Nav1.1 and Nav1.6 in human and dog VCMs, whereas bands corresponding to Nav1.2 were strong in both samples shown in Fig.1A and B. The relative level of main α-subunit Nav expression was compared by real-time PCR. We found transcripts of six genes encoding the Na+ channel main α-subunit expressed in failing VCMs in the following order: Nav1.5 (57.3%) > Nav1.2 (15.3%) > Nav1.1 (11.6%) > Nav2.1 (10.7%) > Nav1.3 (4.6%) > Nav1.6 (0.5%). Statistical analysis in normal and failing hearts of both human and dog revealed up-regulation of Nav1.1 expression, but reduction of Nav1.6 mRNA levels (Fig.1C and D).
Figure 1.

Expression of genes encoding different Na+ channel isoforms Navx
RT-PCR analysis of Nav transcripts in normal and failing human (A) and dog (B) left ventricular tissue samples. A, from left to right: Nav1.1 isoform obtained in normal heart (NH), in explanted heart from patients with ischaemic (IHF), and non-ischaemic (NIHF) heart failure, respectively. The expected 491 bp band was not detected in NH and NIHF, Nav1.2 isoform (417 bp), Nav1.3 isoform (189 bp), Nav1.6 isoform. A specific band of 155 bp is missing in IHF; a non-specific smaller band is present instead. Nav2.1 isoform (586 bp). B, the same Na+ channel isoforms as shown in A were obtained in dog normal (NH) and failing (HF) hearts. Last gel lane on right represents standard bp marker. C, summary data with statistics for Nav isoforms expressed in human heart. RT-PCR gels were analysed in 5–7 tissue samples from left ventricles, D, summary data with statistics for Nav isoforms expressed in dog heart. RT-PCR gels were analysed in 5 samples of ventricular cardiac myocytes from left ventricles of normal dog hearts and failing dog hearts, respectively. *P < 0.05, ANOVA, data are mean ± SEM for both C and D.
The adenovirus-delivered Nav1.5-siRNA reduced (in time-dependent fashion) the numbers of transcripts of the main α-subunit of the cardiac Na+ channel isoform (Fig.2A), as well as its protein level (Fig.2B). This inhibition of gene expression is specific to Nav1.5, without a cross-reaction with the genes expressing auxiliary β-subunits (not shown) or other Navs (Fig.2C).
Patch-clamp data
Patch-clamp data experiments were performed in HF dog VCMs infected with adenovirus-Nav1.5-siRNA. Our previous studies with specific oligonucleotides (Maltsev et al. 2008a) have shown that Nav1.5 provides a dominant contribution to INaL. Indeed, silencing of the Nav1.5 gene in the present study also resulted in a significant and substantial reduction (by about 70%) of the peak transient Na+ current, INaT, shown in Fig.3. Furthermore, the results of our patch clamp experiments in Fig.3B and C show that there was a prominent and statistically significant rightward shift of the SSA (V1/2G = −40.9 ± 1.7 mV, KG = 6.5 mV, n = 8 in control vs. 36.8 ± 1.6 mV, KG = 6.9 mV, n = 5 in Nav1.5-siRNA, P < 0.05 when V1/2 G was compared) and SSI (V1/2A = 81.3 ± 1.8 mV, KA = −6.6 mV, n = 8 in control vs. V1/2A = 70.3 ± 2.3 mV, KA = −6.4 mV, n = 5, P < 0.05 when V1/2A was compared). G and A stands for activation and inactivation, respectively. These results illustrate that the remaining pool (whole or part) of Na+ channels demonstrates activation and inactivation characteristics different from that for Nav1.5. It is known that the neuronal Na+ channels activate at more depolarized potentials (Haufe et al. 2005b). Therefore these results support the idea that neuronal channels may contribute to the remaining INaT in these failing VCMs.
To further explore the idea of the neuronal isoforms’ contribution to INaT, we used 2-aminoethyl methanethiosulfonate hydrobromide (MTSEA), which is known to block only cardiac and not neuronal isoforms of the Na+ channel (Haufe et al. 2005b). In the freshly isolated failing VCMs we found a substantial portion of INaT (∼20%) that was MTSEA resistant (Fig.4). While this component was smaller than the remaining INaT after Nav1.5 post-transcriptional silencing (∼30%) (Fig.4C), it can still be physiologically significant (as we show below).
Figure 4.

Contribution of non-cardiac Na+ channel isoforms to INaT in LV cardiomyocytes of dogs with HF
A, consecutive raw INaT traces recorded before (control) and during MTSEA application in freshly isolated cells. B, time course of MTSEA effect on the peak INaT recorded in the same cell as shown in A. C, summary data and statistical analysis of INaT density in freshly isolated control and MTSEA-treated cells and infected cells with virus containing non-silencing siRNA or Nav1.5-siRNA. Statistically significant changes, P < 0.05, were found in both groups evaluated by the ANOVA test followed by Bonferroni's post hoc test (*,** Nav1.5-siRNA vs. freshly isolated control and GFP control, respectively, § MTSEA vs. freshly isolated control) INaT was recorded at –10 mV. Data are mean ± SEM.
In the next series of experiments we tested if these channels with different SSA and SSI characteristics contribute to INaL. To unmask the contribution of neuronal Navs to INaL we deliberately silenced Nav1.5 gene expression by the virally delivered Nav1.5-siRNA in these cells. Nav1.5-siRNA caused a substantial INaL reduction with an apparent slower decay of the remaining INaL (Fig.5A). The density of INaL evaluated at –30 mV significantly decreased in cells infected with the virus containing Nav1.5-siRNA compared to freshly isolated cells and to cells infected with the non-silencing siRNA expressing GFP (Fig.5B). Analysis of SSA and SSI in these cells clearly shows a rightward shift of the V1/2 (Fig.5C and D) as we found for INaT. We then fitted this INaL component to a two exponential decay function (yielding ‘slow’ and ‘fast’ sub-components, respectively). The slow sub-component of INaL decay (probably corresponding to the late scattered mode; Maltsev & Undrovinas, 2006) of the remaining INaL was significantly slower (Fig.5F). The first (i.e. fast) exponent that corresponds to the burst mode (Maltsev & Undrovinas, 2006) remained unchanged (not shown). Therefore these data reveal a different Nav contribution to INaL in failing VCMs.
Figure 5.

Post-transcriptional SCN5A gene silencing results in functional reduction of the late Na+ current, INaL, and unmasks non-cardiac Na+ channel component in LV cardiomyocytes from dog with chronic heart failure
A, representative raw traces of INaL were recorded at membrane potentials Vm = −10 mV in freshly isolated cells, and in cells 5 days after infection with virus containing control non-silencing siRNA and cDNA for GFP and siRNA-Nav1.5 (indicated by the arrows). Exponential fits to INaL decay are shown by continuous lines and are superimposed with traces. Values of slow exponent time constant, τ, are given in the panel. B, summary data with statistics for INaL density. C, representative INaL–voltage relationship obtained in freshly isolated (•) and cultured cells for 5 days with virally delivered control siRNA (▴ control GFP), and Nav1.5-siRNA (▪). Continuous lines show theoretical curves fitted to experimental current–voltage (I–V) data points to evaluate steady-state activation (SSA) parameters which are given in the panel. D, average experimental data of steady-state inactivation (SSI) data points in control (•, n = 17) and after SCN5A gene silencing by the siRNA (▪, n = 16) along with the fit to a Boltzmann function. E and F, statistical data for INaL decay time course evaluated by the double exponent fit. Difference between theoretical fits (C and D) was statistically significant (P < 0.05) evaluated by F test. Statistical significance, P < 0.05, between experimental data points presented as mean ± SEM (B, E and F) were evaluated by ANOVA followed by Bonferroni's post hoc test.
To further study the neuronal isoform-generated INaL component, we used low concentrations (100 nm) of TTX in VCMs with silenced Nav1.5 expression (results of these experiments are shown in Fig.6A and B). Under these conditions the contribution of the cardiac (TTX-resistant) Na+ channel must remain almost unchanged. However, application of TTX to these myocytes caused a marked reduction of INaL (Fig.6A). We then evaluated the revealed TTX-sensitive current as the difference between the recordings before and after TTX application (Fig.6B). This TTX-sensitive current, presumably carried by the neuronal Navs, was slowly inactivating in a single-exponential manner. The amplitude of the TTX-sensitive INaL was 0.1709 ± 0.0299 pA pF−1 (n = 7 cells) and the decay time constant was τ = 790 ± 76 ms (n = 5 cells) recorded at Vm = −10 mV. These values we used in our numerical model simulations to evaluate the physiological significance of the neuronal INaL component (see below). The experimental current-to-voltage relation (I–V curve) for such experiments and theoretical (Boltzmann function) fits are shown in Fig.6C. The I–V for the TTX-sensitive current was calculated (squares) as a subtraction of two I–V curves (circles and triangles). It is clearly seen that the difference I–V is shifted rightwards and we found the average SSA parameters for this component to be as follows: V1/2G = −24.1 ± 1.0 mV, and slope KG = 5.7 ± 0.3 mV vs. control V1/2G = –38.5 ± 1.2 mV, KG = 5.6 ± 0.9 mV. The I–V relationship analysis revealed a significant neuronal INaL component within the range of the AP plateau (Fig.6C). These parameters of TTX-sensitive INaL were also used in our numerical modelling.
Figure 6.

Contribution of tetrodotoxin (TTX)-sensitive neuronal Navs to INaL in failing dog ventricular cardiomyocytes
A, raw current traces recorded in cultured cells in which SCN5A was silenced by the virally delivered Nav1.5-siRNA before and after TTX (100 nm). B, difference current (Control-TTX), or neuronal INaL component along with the best single-exponent fit. Zero current represents difference between control recording and after TTX washout. Fit parameters and voltage-clamp protocol (same for A) is given in the panel. C, normalized I–V relationship of INaL before and after TTX recorded in freshly isolated cells. Current density was evaluated as an average within 200–220 ms after depolarization pulse for 2 s, Vh = −120 mV. Continuous lines are theoretical curves of steady-state activation. Note that TTX produced SSA shift to left (V1/2 = −43 mV in control vs. –45 mV with TTX). Data are mean ± SEM.
To further study the molecular identity of the TTX-sensitive INaL, we made adenovirus-based constructs containing cDNA templates for endogenous transcription of the GFP reporter gene, and cognitive siRNA, targeting different Nav isoforms that are expressed in human and dog LV cardiomyocytes according to our data (Fig.1). As shown in Fig.7, transfection of failing dog VCMs with adenovirus containing Nav1.1-siRNA significantly reduced both mRNA abundance (Fig.7A) and Nav1.1 protein level (Fig.7B) without cross-reacting with other Na+ channel isoforms (Fig.7C). Furthermore, we found that application of MTSEA completely blocked INaL in failing VCMs (shown in Fig.8A and B) with silenced Nav1.1. At the same time, application of 100 nm TTX was not effective in VCMs with the silenced expression of Nav1.1 (Fig.8C). Figure8D shows evaluated INaL densities measured at –10 mV in GFP control (0.3387 ± 0.06 pA pF−1), Nav1.5-siRNA (0.2123 ± 0.023 pA pF−1) and Nav1.1-siRNA (0.1737 ± 0.018 pA pF−1). Therefore at this membrane potential the Nav1.1-originated INaL component (∼60% of total INaL) is comparable with Nav1.5-originated INaL (i.e. remaining ∼40% of total INaL). Furthermore, in our in silico simulations of APs we compared the dynamic contributions of these components and confirmed that Nav1.1 provides a major contribution to total INaL during the AP plateau (see below). Based on these data, further studies with other Navs contributions to INaL seemed to be irrelevant and results are not shown. These data support the idea that Nav1.1 is the major Na+ channel isoform underlying the TTX-sensitive component of INaL in failing VCMs.
Figure 7.
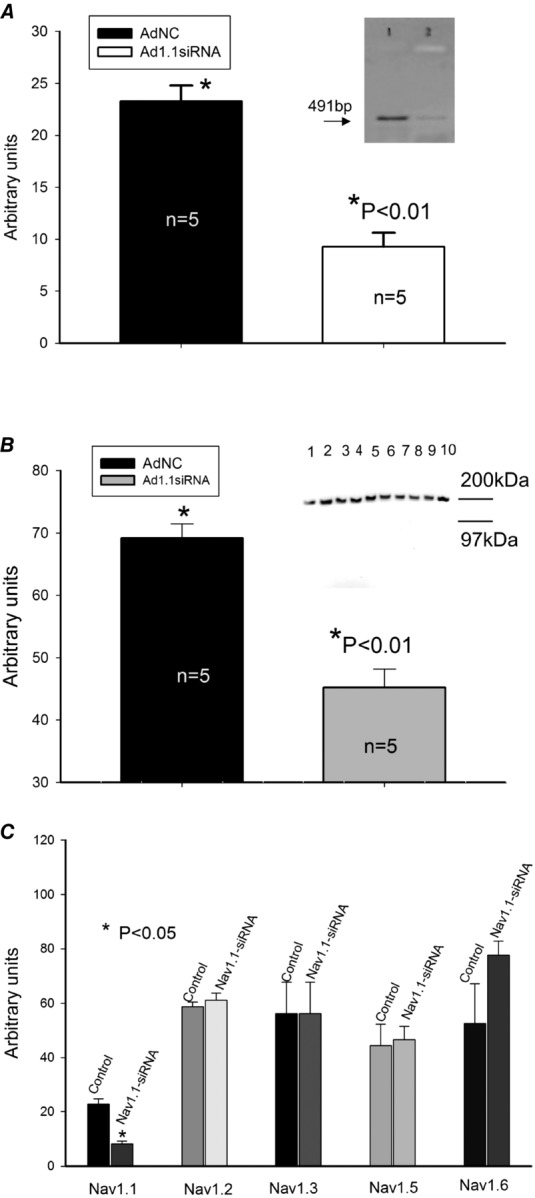
Silencing of SCN1A gene that encodes Nav1.1 with adenovirus containing coding sequences for siRNA in cardiomyocytes from dogs with chronic HF
A, summary data of RT-PCR analysis of mRNA abundance (representative gels are shown in the inset at the upper right corner of the panel) in cultured dog cardiomyocytes. AdNC, adenovirus expressing non-specific siRNA as a control, Ad1.1siRNA, adenoviral construct expressing Nav1.1 silencing sequences. B, summary data of Western blot analysis of Nav1.1 protein abundance in cells treated with control virus (lanes 2–5 and 10 in the representative gels shown in the inset at the right upper corner of the panel) or Nav1.1-siRNA (lanes 1 and 6–9, see inset). C, summary data of RT-PCR analysis of different Na+ channel isoforms after transfection with Nav1.1-siRNA (from 4 to 8 samples). *Statistically significant changes in A, B and C were evaluated by the ANOVA test followed by Bonferroni's post hoc test. Data are mean ± SEM.
Figure 8.

Contribution of neuronal Nav1.1 to INaL in failing dog ventricular cardiomyocytes
In these cells expression of Nav1.1 was silenced by virally delivered Nav1.1-siRNA. A, raw current traces recorded before (Nav1.1-siRNA, control) and after MTSEA application. Continuous line represents single exponent fit to INaL decay, and value of time constant, τ, is given at the trace. B, time course of INaL amplitude, evaluated between 200 and 220 ms after depolarization onset, during MTSEA application. C, raw current traces recorded before (Nav1.1-siRNA, control) and after 100 nm TTX application (Nav1.1-siRNA + TTX). The difference current was obtained by subtracting currents before and after TTX. D, summary data and statistical analysis of INaL current densities obtained in cells infected with virus that contains cDNA for GFP (GFP Control) for Nav1.5-siRNA + GFP (Nav1.5-siRNA) and for Nav1.1-siRNA + GFP (Nav1.1-siRNA). Current densities were measured 200 ms after depolarization to –10 mV onset. Statistical significance, P < 0.01, between experimental data points presented as mean ± SEM were evaluated by ANOVA followed by Bonferroni's post hoc test.
Intracellular Ca2+ dynamics data
In the next series of experiments we addressed the question of the physiological importance of the neuronal INaL component in failing heart. Indeed, we found that blockade of neuronal INaL by low concentrations of TTX reduced diastolic Ca2+ accumulation in response to a 1.5 Hz pulse train in freshly isolated failing VCMs (Fig.9).
Figure 9.

Blockade of neuronal Na+ channels by 100 nm TTX reversibly attenuates diastolic intracellular Ca2+ elevation in ventricular cardiomyocytes of dogs with chronic heart failure
Intracellular Ca2+ dynamics recorded during 1.5 Hz pulse train in control (A), in the presence of 100 nm TTX (B) and after the toxin washout (C). Dotted lines in A, B and C refer to average diastolic Ca2+ levels before and during pulse train. D, data summary for diastolic Ca2+ (CaD) changes (ΔCaD) in response to the pulse train. Data are mean ± SEM, cell numbers are given above the bars. *P < 0.05, ANOVA followed by Bonferroni's post hoc test.
Numerical model simulations of the neuronal component of INaL
To further address the physiological significance of the neuronal INaL component, we used a modified E–C coupling model in which we carefully introduced experimentally obtained INaL density, decay kinetics, as well as SSA and SSI parameters. The model predicted INaL traces and I–V relationship in good agreement with those obtained in our voltage-clamp experiments (compare Figs5,6 and8 with Fig.10A). We tested the physiological importance of the novel neuronal type INaL by numerical simulations of INaL dynamics during APs (Fig.10B). Our simulations showed that the neuronal INaL lasts during the whole AP plateau and its amplitude (especially at the late phase of the plateau where it amounted to 60% of cardiac INaL), indicating that the neuronal component of INaL is a major player in the ion current balance during the AP plateau. Indeed, this new INaL neuronal component can significantly modulate (prolong) AP. In concert with the cardiac INaL, neuronal INaL leads to AP prolongation and to a higher and longer plateau of the AP (Fig.10C), i.e. reproducing well-known features of failing myocardium. Next we numerically evaluated cytosolic Ca2+ dynamics during a 1.5 Hz pulse train. Introduction of the neuronal INaL component significantly increased diastolic Ca2+ accumulation at the end of the pulse train (Fig.10D), which is in a good agreement with the experimental data (Fig.9).
Figure 10.

In silico investigation on neuronal and cardiac INaL components in HF dog ventricular cell
A, predicted current–voltage relationships. The upper panels show families of predicted current traces at different membrane voltages for the cardiac and neuronal Nav components. The lower panels show plotted amplitude values of these predictions. Left lower sub-panel shows the peak INaL (at zero time after the depolarization onset) and right lower sub-panel shows the current amplitudes 200 ms after depolarization onset. Holding potential was Vh = −120 mV, test pulses were applied from –70 mV to +70 mV. B, predicted profile of INaL generated by cardiac and neuronal Navs in numerical model simulations of AP. C and D, predicted differential contribution of INaL by the neuronal and cardiac Navs to shape and duration of AP and intracellular Ca2+ dynamics, respectively, at the pacing rate of 1 Hz. Complete INaL elimination from simulations (INaL = 0) reduces AP duration and eliminates diastolic Ca2+ accumulation. Introduction of only the cardiac or neuronal component of INaL increases AP duration and leads to diastolic Ca2+ accumulation, whereas incorporation of both INaL components, which reflects the situation in situ, affects these physiological responses dramatically. All numerical simulations were performed at a physiological temperature of 37 °C using our modification of the Winslow E–C coupling model of canine failing VCMs (Winslow et al. 1999) with formulations for INaL which were previously developed based on our experimental voltage-clamp data (Undrovinas et al. 2010; Mishra et al. 2011). In the present study INaL formulations included two separate INaL components for cardiac and neuronal isoforms, respectively. Each component has its own activation and inactivation gating variables evaluated from patch-clamp data obtained in the present study (see section ‘Patch-clamp data’).
Discussion
Using a combination of post-transcriptional gene silencing by siRNA interference with the pharmacological approaches, cell electrophysiology (patch-clamping), intracellular Ca2+ monitoring, and numerical modelling, we discovered and characterized a novel neuronal component of INaL in dog ventricular cardiomyocytes. We also found that expression of the Nav1.1 gene is responsible for the molecular identity of the neuronal INaL component. This component seems to be involved in E–C coupling by affecting AP shape and duration as well as intracellular Ca2+ dynamics. Furthermore, as discussed below in detail, the neuronal INaL component may provide a novel cellular and molecular mechanism contributing to major abnormalities associated with chronic HF, such as the impaired, rate-dependent (Rao et al. 2007; Logeart et al. 2009; Undrovinas et al. 2010) diastolic function and repolarization (for review see Maltsev & Undrovinas, 2008).
After INaL had been discovered in human hearts (Maltsev et al. 1998a) it was implicated in HF-related rhythm and contractility deficiencies (for review see Maltsev & Undrovinas, 2008). The importance of the augmented INaL contribution to HF mechanisms has been demonstrated in a series of experiments in which ‘correction’ of INaL in failing cardiomyocytes resulted in: (1) rescue of normal repolarization, (2) improvement (decreasing) of repolarization beat-to-beat APD variability, and (3) improvement of Ca2+ handling and contractility (Maltsev et al. 1998b1998b; Undrovinas et al. 1999, 2006; Maltsev et al. 2007). Accordingly INaL has emerged as a novel possible target for cardioprotection to treat the failing heart (Maltsev et al. 2001, 2007; Noble & Noble, 2006; Maltsev & Undrovinas, 2008; Undrovinas & Undrovinas & Maltsev, 2008).
Therefore, the study of modulatory factors and the molecular identity of INaL is of obvious importance from both therapeutic and scientific standpoints. Modulatory mechanisms leading to INaL augmentation and decay slowing include: (1) the effects of the Na+ channel protein microenvironment, such as sub-sarcolemmal cytoskeleton (Undrovinas et al. 1995; Maltsev & Undrovinas, 1997), lipid bilayer composition (Undrovinas et al. 1992) and auxiliary β-subunits (Mishra et al. 2011); and (2) the Ca2+/calmodulin/calmodulin kinase signalling pathway (Maltsev et al. 2008a). Recent discoveries of the expression of array of different Na+ channel isoforms in hearts indicate a possibility for new molecular mechanisms for INaL modulation, i.e. the different Na+ channel isoform contributions to INaL that were the main objective of the present study.
Molecular identity
We found transcripts of six genes encoding the Na+ channel main α-subunit expressed in both human and dog failing hearts and the abundance of mRNA was in the following order: Nav1.5 (57.3%) > Nav1.2 (15.3%) > Nav1.1 (11.6%) > Nav2.1 (10.7%) > Nav1.3 (4.6%) > Nav1.6 (0.5%). It is, in part, in line with the results of previous studies. In rodents, Nav1.1, Nav1.3 and Nav1.6 were initially found in the T tubules (Maier et al. 2002). In dog VCMs and Purkinje fibres, Nav1.1 and both Nav1.2 and Nav1.6 were localized in the intercalated discs and at Z-lines, respectively (Haufe et al. 2005a2005a,b). Therefore these studies suggest species-specific expression and protein localization within compartments of the sarcolemma. Immunoblot data exclude a membrane location for Nav1.3, despite an abundant amount of mRNA in dog heart (Haufe et al. 2005b). We found expression of Nav2.1 that has been previously discovered in uterus and human hearts (hNav2.1) and may represent a secondary Nav subfamily (George et al. 1992; Felipe et al. 1994). Its function as a voltage-gated Na+ channel has not been confirmed; therefore it is not likely to be a candidate for the INaL (Trimmer & Agnew, 1989; Watanabe et al. 2000). Further analysis of expression of other genes as possible candidates for INaL revealed HF-related changes in the expression of Nav1.1 (up-regulation) and Nav1.6 (down-regulation): at the same time no significant changes in transcripts of other Na+ channel isoforms were detected. Therefore our focus was on Nav1.1 as a plausible contributor to INaL. Our dog model is an ischaemic model of heart failure. We show that increased expression of Nav1.1 in failing VCMs from dogs correlates with human ischaemic HF (IHF) (Fig.1A). At the same time we did not find increased Nav1.1 expression in ventricle tissue samples from human hearts with non-ischaemic HF.
The contributions of these isoforms to INaL under normal conditions and the changes of these contributions in different pathological conditions remain unknown. The molecular identity of INaL needs careful consideration especially for failing hearts where the gene expression profile changes dramatically. Using antisense inhibition by oligonucleotides, we have previously demonstrated that Nav1.5 is a major contributor to INaL in normal dog heart (Maltsev et al. 2008a) and this finding was confirmed in the present study. At least 70% of INaT and INaL are related to this cardiac Na+ channel isoform (see Figs6). The late Na+ channel openings are expected for at least Nav1.6 (Welch et al. 2008) and have been reported also for Nav1.1 (Aman et al. 2009). Therefore, the hypothesis of possible roles of the neuronal isoforms of Na+ channel in Na+-related Ca2+ signals in HF makes sense based on these previous data and is supported, in part, by our findings reported here. A contribution of Nav1.6 to the mechanism of augmented INaL in HF is unlikely because it is down-regulated as we have discussed above. A separate study focusing on Nav1.6 is required to understand its changes in failing hearts. The Nav1.1 has been widely studied because of its involvement in the genetic epilepsy syndrome (Catterall et al. 2010). Not only mutant but also wild-type (to a lesser extent though) forms of these channels exhibit slow inactivation and persistent INa, which can be modulated by auxiliary β-subunits (Spampanato et al. 2004) similar to what we reported for Nav1.5 (Maltsev et al. 2009; Mishra et al. 2011).
We demonstrated that β1 and β2 subunits act in opposite directions by enhancing or reducing INaL in normal and failing dog hearts. It is interesting to note that, similar to Nav1.5 (Undrovinas et al. 2002; Maltsev & Undrovinas, 2006), the Nav1.1 also demonstrates modal gating behaviour and can enter a slow mode that generates late openings (Vanoye et al. 2006). This modal shift can be enhanced or attenuated by β-auxiliary subunits (Spampanato et al. 2004).
Physiological importance of neuronal Navs in heart
The neuronal component of INaL is a significant contributor to AP duration and shape because of its biophysical properties such as a relatively high level of midpoint for steady-state activation and inactivation and a relatively slow decay time course (vs. cardiac type INaL). Our numerical model simulations demonstrated that, despite a smaller initial density of the neuronal component (after membrane depolarization) vs. cardiac INaL, its ratio to the cardiac INaL increases during AP plateau and becomes comparable to the cardiac component at the end of the AP plateau (see Fig.10B). Therefore targeting the neuronal component of INaL can be an effective strategy to reduce AP duration and its variability in HF (Fig.10C). We demonstrate that blockade of the neuronal INaL component by a low TTX concentration reduces diastolic Ca2+ accumulation in both experiments (Fig.9) and in silico simulations (Fig.10D). Therefore this neuronal INaL component is probably involved in abnormalities of E–C coupling in failing heart, such as diastolic dysfunction, i.e. the reduced ability of the myocardium to relax during diastole. In contrast, TTX-sensitive INa was not implicated in triggered Ca2+ release from sarcoplasmic reticulum (SR) in rodent VCMs and the authors claimed no apparent commitment for neuronal Navs in E–C coupling (Brette & Orchard, 2006). This discrepancy can be explained in two ways: (1) E–C coupling in rodents versus large animals, such as dog, as well as in humans, is different in the way that the source of Ca2+ involved in contraction is mainly stored in SR and is less (or almost not) dependent on transmembrane ion fluxes during AP development, and (2) in rodents, heart rate is very high and APs are very short, therefore INaL does not play the same role as in humans or dogs. Since late openings of Na+ channels generate both electric current and Na+ influx during the AP plateau, INaL is expected to contribute to at least two known HF cellular mechanisms: (1) electrophysiological remodelling, and (2) altered cell Na+ cycling. The latter mechanism is tightly integrated with Ca2+ cycling, as Na+ modulates the Na+–Ca2+ exchanger operation (Bers et al. 2006). Therefore the neuronal component of INaL can also play a role in the impaired E–C coupling in HF via Na+ balance and its interplay with Ca2+ handling and signalling. Haufe et al. (2005b) suggested that neuronal Navs might act as a safety mechanism. Because of the biophysical properties of neuronal Navs (SSI and SSA are shifted to more depolarized potentials compared to that for Nav1.5) this safety mechanism can be recruited under pathological conditions, when the resting membrane potential is depolarized.
Study limitations
The relative mRNA abundance expressed as arbitrary units was calculated using the expression levels of all transcripts normalized to GAPDH mRNA. There are some indications that GAPDH expression can change in atria vs. ventricles in patients with HF. Since in this study we did not compare atria vs. ventricles in humans we assume that GAPDH can serve as a valid marker. According to previous studies by our group and others, levels of mRNA for this marker gene do not change in the left ventricle dog or mini pig HF model (Mishra et al. 2005; Imai et al. 2007; Martino et al. 2011). Therefore we are confident that our data are not affected because of this marker choice.
The human data are based on ventricular tissue mRNA analysis, rather than isolated myocytes. It could be, in part, contaminated with mRNA from neurons that can be found in ventricles. We found a very similar pattern of Nav expression in these samples when compared with samples from isolated myocytes of dog. Therefore we believe that our conclusions are valid.
New strategy to treat HF-related deficiencies
The paradigm in HF for the Na+ current is that INaT, responsible for the AP upstroke and propagation, is down-regulated, whereas INaL is up-regulated and involved in repolarization and Ca2+-handling abnormalities. A direct blockade of Na+ channels by Class I drugs is not acceptable as a therapeutic strategy in HF, because it leads to further reduction in propagation velocity and increases the probability of excitation propagation blockade or re-entrant arrhythmias. The discovery of a new neuronal component of INaL allows a new differential treatment of rhythm abnormalities and contractility deficiency as discussed above. The treatment can include a specific neuronal channel blocker or genetic approaches to specifically reduce the neuronal INaL component in heart that is especially important for the late AP plateau phase (Fig.10C) (due to its relatively slow decay, see Fig.6A) but is probably less important for cell excitability (due to its relatively higher activation voltage).
In conclusion we have discovered an SCN1A-related Nav1.1 component of INaL in ventricular myocytes of dog with heart failure. This component is physiologically relevant to controlling AP shape and duration as well as Ca2+ dynamics.
Glossary
- AP
action potential
- E–C
excitation–contraction
- GFP
green fluorescent protein
- HF
heart failure
- INaL
late Na+ current
- INaT
transient Na+ current
- LV
left ventricular
- MTSEA
2-aminoethyl methanethiosulfonate hydrobromide
- Nav
sodium channel
- siRNA
short interfering RNA
- SSA
steady-state activation
- SSI
steady-state inactivation
- VCM
ventricular cardiomyocyte
Additional information
Competing interests
The authors have no competing interests to declare.
Author contributions
Conception, analysis and design of the experiments: A.U. Collection, analysis and interpretation of data: S.M., V.R., V.A.M., N.A.U. and A.U. Dog chronic heart failure model and mathematical modelling: H.N.S. and V.A.M. Drafting the article or reviewing it critically for important intellectual content: A.U., V.A.M., H.N.S. and S.M. All authors approved the final version of the manuscript.
Funding
This study was supported by National Heart, Lung, and Blood Institute Grants HL-53819 and HL-074238, by a grant-in-aid from the American Heart Association (0350472Z; to A. Undrovinas), and, in part, by the Intramural Research Program of the National Institute on Aging (to V. A. Maltsev; the numerical modelling part).
Author's present address
S. Mishra: Department of Translational Science and Molecular Medicine, MSU College of Human Medicine, Grand Rapids, MI, USA.
References
- Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL. Raman IM. Regulation of persistent Na current by interactions between β subunits of voltage-gated Na channels. J Neurosci. 2009;29:2027–2042. doi: 10.1523/JNEUROSCI.4531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Despa S. Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- Brette F. Orchard CH. No apparent requirement for neuronal sodium channels in excitation-contraction coupling in rat ventricular myocytes. Circ Res. 2006;98:667–674. doi: 10.1161/01.RES.0000209963.02720.70. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Kalume F. Oakley JC. NaV1.1 channels and epilepsy. J Physiol. 2010;588:1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coraboeuf E, Deroubaix E. Coulombe A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am J Physiol Heart Circ Physiol. 1979;236:H561–H567. doi: 10.1152/ajpheart.1979.236.4.H561. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W. Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felipe A, Knittle TJ, Doyle KL. Tamkun MM. Primary structure and differential expression during development and pregnancy of a novel voltage-gated sodium channel in the mouse. J Biol Chem. 1994;269:30125–30131. [PubMed] [Google Scholar]
- George AL, Jr, Knittle TJ. Tamkun MM. Molecular cloning of an atypical voltage-gated sodium channel expressed in human heart and uterus: evidence for a distinct gene family. Proc Natl Acad Sci U S A. 1992;89:4893–4897. doi: 10.1073/pnas.89.11.4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haufe V, Camacho JA, Dumaine R, Gunther B, Bollensdorff C, von Banchet GS, Benndorf K. Zimmer T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol. 2005a;564:683–696. doi: 10.1113/jphysiol.2004.079681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K. Dumaine R. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. 2005b;65:117–127. doi: 10.1016/j.cardiores.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Imai M, Rastogi S, Gupta RC, Mishra S, Sharov VG, Stanley WC, Mika Y, Rousso B, Burkhoff D, Ben-Haim S. Sabbah HN. Therapy with cardiac contractility modulation electrical signals improves left ventricular function and remodeling in dogs with chronic heart failure. J Am Coll Cardiol. 2007;49:2120–2128. doi: 10.1016/j.jacc.2006.10.082. [DOI] [PubMed] [Google Scholar]
- Logeart D, Gueffet JP, Rouzet F, Pousset F, Chavelas C, Solal AC. Jondeau G. Heart rate per se impacts cardiac function in patients with systolic heart failure and pacing: a pilot study. Eur J Heart Fail. 2009;11:53–57. doi: 10.1093/eurjhf/hfn016. [DOI] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T. Catterall WA. Distinct subcellular localization of different sodium channel α and β subunits in single ventricular myocytes from mouse heart. Circulation. 2004;109:1421–1427. doi: 10.1161/01.CIR.0000121421.61896.24. [DOI] [PubMed] [Google Scholar]
- Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T. Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–4078. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Kyle JW, Mishra S. Undrovinas A. Molecular identity of the late sodium current in adult dog cardiomyocytes identified by Nav1.5-antisense inhibition. Am J Physiol Heart Circ Physiol. 2008a;295:H667–H676. doi: 10.1152/ajpheart.00111.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Kyle JW. Undrovinas A. Late Na+ current produced by human cardiac Na+ channel isoform Nav1.5 is modulated by its β 1 subunit. J Physiol Sci. 2009;59:217–225. doi: 10.1007/s12576-009-0029-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN. Undrovinas A. Modulation of the late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008b;294:H1597–H1608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Higgins RSD, Silverman N, Lesch M. Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998a;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Tanimura M, Lesch M, Goldstein S. Undrovinas AI. Relationship between action potential, contraction-relaxation pattern, and intracellular Ca2+ transient in cardiomyocytes of dogs with chronic heart failure. Cell Mol Life Sci. 1998b;54:597–605. doi: 10.1007/s000180050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN. Undrovinas AI. Late sodium current is a novel target for amiodarone: Studies in failing human myocardium. J Mol Cell Cardiol. 2001;33:923–932. doi: 10.1006/jmcc.2001.1355. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN. Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effects of long-term therapy with carvedilol. Cell Mol Life Sci. 2002;59:1561–1568. doi: 10.1007/s00018-002-8529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Silverman N, Sabbah HN. Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA. Undrovinas A. Late sodium current in failing heart: Friend or foe. Prog Biophys Mol Biol. 2008;96:421–451. doi: 10.1016/j.pbiomolbio.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA. Undrovinas AI. Cytoskeleton modulates coupling between availability and activation of cardiac sodium channel. Am J Physiol Heart Circ Physiol. 1997;273:H1832–H1840. doi: 10.1152/ajpheart.1997.273.4.H1832. [DOI] [PubMed] [Google Scholar]
- Maltsev VA. Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116–127. doi: 10.1016/j.cardiores.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino A, Cabiati M, Campan M, Prescimone T, Minocci D, Caselli C, Rossi AM, Giannessi D. Del Ry S. Selection of reference genes for normalization of real-time PCR data in minipig heart failure model and evaluation of TNF- α mRNA expression. J Biotechnol. 2011;153:92–99. doi: 10.1016/j.jbiotec.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Mishra S, Sabbah HN, Rastogi S, Imai M. Gupta RC. Reduced sarcoplasmic reticulum Ca2+ uptake and increased Na+-Ca2+ exchanger expression in left ventricle myocardium of dogs with progression of heart failure. Heart Vessels. 2005;20:23–32. doi: 10.1007/s00380-004-0792-6. [DOI] [PubMed] [Google Scholar]
- Mishra S, Undrovinas NA, Maltsev VA, Reznikov V, Sabbah HN. Undrovinas A. Post-transcriptional silencing of SCN1B and SCN2B genes modulates late sodium current in cardiac myocytes from normal dogs and dogs with chronic heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H1596–H1605. doi: 10.1152/ajpheart.00948.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D. Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92:iv1–iv5. doi: 10.1136/hrt.2005.078782. Suppl. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao K, Fisher ML, Robinson S, Shorofsky S. Gottlieb SS. Effect of chronic changes in heart rate on congestive heart failure. J Card Fail. 2007;13:269–274. doi: 10.1016/j.cardfail.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Goldberg AD, Schoels W, Kono T, Webb C, Brachmann J. Goldstein S. Spontaneous and inducible ventricular arrhythmias in a canine model of chronic heart failure: relation to haemodynamics and sympathoadrenergic activation. Eur Heart J. 1992;13:1562–1572. doi: 10.1093/oxfordjournals.eurheartj.a060102. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Stein PD, Kono T, Gheorghiade M, Levine TB, Jafri S, Hawkins ET. Goldstein S. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am J Physiol Heart Circ Physiol. 1991;260:H1379–H1384. doi: 10.1152/ajpheart.1991.260.4.H1379. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Kearney JA, de Haan G, McEwen DP, Escayg A, Aradi I, MacDonald BT, Levin SI, Soltesz I, Benna P, Montalenti E, Isom LL, Goldin AL. Meisler MH. A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for β subunit interaction. J Neurosci. 2004;24:10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS. Agnew WS. Molecular diversity of voltage-sensitive Na channels. Annu Rev Physiol. 1989;51:401–418. doi: 10.1146/annurev.ph.51.030189.002153. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Belardinelli L, Undrovinas NA. Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas AI, Fleidervish IA. Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231–1241. doi: 10.1161/01.res.71.5.1231. [DOI] [PubMed] [Google Scholar]
- Undrovinas A. Maltsev VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem. 2008;6:348–359. doi: 10.2174/187152508785909447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas NA, Maltsev VA, Belardinelli L, Sabbah HN. Undrovinas A. Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci. 2010;60:245–257. doi: 10.1007/s12576-010-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas AI, Maltsev VA, Kyle JW, Silverman NA. Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Maltsev VA. Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: Role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas AI, Shander GS. Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. Am J Physiol Heart Circ Physiol. 1995;269:H203–H214. doi: 10.1152/ajpheart.1995.269.1.H203. [DOI] [PubMed] [Google Scholar]
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ. Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Vanoye CG, Lossin C, Rhodes TH. George AL., Jr Single-channel properties of human NaV1.1 and mechanism of channel dysfunction in SCN1A-associated epilepsy. J Gen Physiol. 2006;127:1–14. doi: 10.1085/jgp.200509373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe E, Fujikawa A, Matsunaga H, Yasoshima Y, Sako N, Yamamoto T, Saegusa C. Noda M. Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J Neurosci. 2000;20:7743–7751. doi: 10.1523/JNEUROSCI.20-20-07743.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch NC, Lin W, Juranka PF, Morris CE. Stys PK. Traditional AMPA receptor antagonists partially block Nav1.6-mediated persistent current. Neuropharmacology. 2008;55:1165–1171. doi: 10.1016/j.neuropharm.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Winslow RL, Rice J, Jafri S, Marbán E. O'Rourke B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ Res. 1999;84:571–586. doi: 10.1161/01.res.84.5.571. [DOI] [PubMed] [Google Scholar]
- Zicha S, Maltsev VA, Nattel S, Sabbah HN. Undrovinas AI. Post-transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol. 2004;37:91–100. doi: 10.1016/j.yjmcc.2004.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]