

Abstract
Aberrant diastolic calcium (Ca) release due to leaky ryanodine receptors (RyR2s) has been recently associated with atrial fibrillation (AF) and catecholaminergic polymorphic ventricular tachycardia (CPVT). However, it remains unclear how diastolic Ca release contributes to the rising of rapid repetitive focal activity, which is considered as a common AF triggering mechanism. To address this question, we conducted simultaneous voltage/Ca optical mapping in atrial tissue and one-/two-dimensional confocal imaging in atrial tissue and myocytes from wild-type (WT, n = 15) and CPVT mice lacking calsequestrin 2 (Casq2−/−, n = 45), which promotes diastolic Ca release. During β-adrenergic stimulation (100 nm isoproterenol), only Casq2−/− atrial myocytes showed pacing-induced self-sustained repetitive activity (31 ± 21 s vs. none in WT). Importantly, in atrial tissue, this repetitive activity could translate to Ca-dependent focal arrhythmia. Ectopic action potential (AP) firing during repetitive activity occurred only when diastolic Ca release achieved a sufficient level of synchronization. The AP, in turn, synchronized subsequent diastolic Ca release by temporally aligning multiple sources of Ca waves both within individual myocytes and throughout the atrial tissue. This alternating interplay between AP and diastolic Ca release perpetuates the self-sustaining repetitive activity. In fact, pharmacological disruption of synchronized diastolic Ca release (by ryanodine) prevented aberrant APs; and vice versa, the inhibition of AP (by TTX or 0 Na, 0 Ca solution) de-synchronized diastolic Ca release. Taken together, these results suggest that a cyclical interaction between synchronized diastolic Ca release and AP forms a pathological rhythm generator that is involved in Ca-dependent atrial arrhythmias in CPVT.
Key points
Atrial fibrillation is often initiated and perpetuated by abnormal electrical pulses repetitively originating from regions outside the heart's natural pacemaker.
In this study we examined the causal role of abnormal calcium releases from the sarcoplasmic reticulum in producing repetitive electrical discharges in atrial cells and tissues.
Calsequestrin2 is a protein that stabilizes the closed state of calcium release channels, i.e. the ryanodine receptors. In the atria from mice predisposed to abnormal calcium releases secondary to the absence of calsequestrin2, we observed abnormal repetitive electrical discharges that may lead to atrial fibrillation.
Here, we report a novel pathological rhythm generator. Specifically, abnormal calcium release leads to electrical activation, which in turn results in another abnormal calcium release. This process repeats itself and thus sustains the repetitive electrical discharges.
These results suggest that improving the stability of ryanodine receptors might be useful to treat atrial fibrillation.
Introduction
Atrial fibrillation (AF) is the most common form of cardiac arrhythmia, which afflicts 3 million individuals in the US alone (Go et al. 2001, 2013; Miyasaka et al. 2006). However, despite extensive research, the underlying mechanisms responsible for the induction and perpetuation of AF remain incompletely understood. AF is often initiated and perpetuated by repetitive focal discharges (Wit & Boyden, 2007; Dobrev & Nattel, 2010). Ablation of such sources of focal activity has often proven to be an effective intervention venue for the treatment of AF (Jais et al. 1997; Haissaguerre et al. 1998). Repetitive focal discharges could either directly drive high frequency electrical oscillations that mark AF or result in AF through initiation of re-entry circuits (Wakili et al. 2011). However, the precise mechanisms underlying how regions outside the specialized normal pacemaker sites (e.g. sinoatrial node and latent atrial pacemaker cells) can generate repetitive firing remains elusive.
In recent years AF has been linked to abnormal Ca release from the sarcoplasmic reticulum (SR) via cardiac ryanodine receptors (RyR2s). Specifically, genetic mutations that result in ‘leaky’ RyR2s (Capes et al. 2011) and ventricular tachyarrhythmias (i.e. catecholaminergic polymorphic ventricular tachycardia (CPVT)) have also been associated with AF (Bhuiyan et al. 2007; Sumitomo et al. 2007; Pizzale et al. 2008; Kazemian et al. 2011). Moreover, increased susceptibility to AF was found in genetically modified mice harbouring mutations in the RyR2 complex (Chelu et al. 2009; Shan et al. 2012; Glukhov et al. 2013; Faggioni et al. 2014). It has been suggested (Chelu et al. 2009; Shan et al. 2012; Faggioni et al. 2014) that aberrant diastolic SR Ca release (DCR) due to abnormal gating of RyR2 can activate transient inward current and thereby result in an ectopic depolarization (Kass et al. 1978; Berlin et al. 1989; Venetucci et al. 2008), also known as the delayed afterdepolarization (DAD) (January & Fozzard, 1988; Ter Keurs & Boyden, 2007). However, it is not clear whether and how DCR can repetitively generate discharges that result in repetitive DAD-mediated firing (triggered activity) in atrial cells. Moreover, it is uncertain whether such DCR and DADs potentially arising in individual atrial cells could synchronize across atrial myocardium leading to atrial arrhythmia. Indeed, such synchronization of aberrant Ca signalling and electrical activity is thought to be required to overcome the source–sink mismatch, thus resulting in ectopic drives (Houser, 2000; Xie et al. 2010; Myles et al. 2012, 2014).
In this study, we used calsequestrin 2 null (Casq2−/−) mice, which are susceptible to aberrant DCR not only in the ventricular (Knollmann et al. 2006) but also in atrial myocytes (Faggioni et al. 2014), potentially resulting in AF in vivo (Faggioni et al. 2014). Here, we demonstrate that aberrant DCR in isolated atrial myocytes can result in self-sustained repetitive activity, which leads to focal arrhythmia in atrial tissue. We applied an integrative imaging approach to reveal specifically how DCR leads to repetitive activity at both cellular and tissue level in Casq2−/− mice. Our results demonstrated a new rhythm generating system that relies on an alternating interplay between membrane potential (Vm) and Ca for generation of tissue-wide focal arrhythmia.
Methods
Mice
We conducted experiments in 3- to 5-month old wild-type (WT) and Casq2−/− mice (Knollmann et al. 2006; Faggioni et al. 2014). All animal procedures were in compliance with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1996). Animal protocols were approved by The Ohio State University Institutional Animal Care and Use Committee.
Atrial myocytes
Atrial myocytes were isolated from both WT (n = 10) and Casq2−/− mice (n = 32) using a protocol adapted from ventricular myocyte isolation (Ho et al. 2014; Liu et al. 2014). Briefly, mice were anaesthetized with 3% isoflurane. Response to toe-pinch reflex was examined to ensure the proper level of anaesthesia. The mouse heart was quickly excised, cannulated, Langendorff-perfused using Ca-free Tyrode's solution (containing, in mm: 140 NaCl, 5.4 KCl, 0.5 MgCl2, 10 Hepes and 5.6 glucose; pH 7.3) at 37 °C for 3 min. The perfusate was then switched to Ca-free Tyrode's solution containing Liberase TH Research Grade enzyme (2.22 mg/50 ml; Roche, Indianapolis, IN, USA) for digestion. After 10–15 min digestion, atrial tissue was placed in BSA containing Tyrode's solution, cut into pieces and then gently triturated until single atrial myocytes were released into the solution. The Ca concentration of the Tyrode's solution containing the myocytes was slowly brought up to 1.3 mm. Experiments with single atrial myocytes were conducted in solution with 1.3 mm Ca unless stated otherwise. To fully characterize DCR synchronicity in isolated atrial myocytes, cells were paced (10 stimuli) at 1 Hz. Arrhythmic potential was assessed by applying 10 stimuli at 2 Hz. SR Ca content was assessed by rapid caffeine (20 mm) application 5 s after a train of five stimuli at 1 Hz.
Two-dimensional (2D) atrial tissue preparation
Isolated mouse atrial preparations were obtained from both WT (n = 10) and Casq2−/− mice (n = 13) as described previously (Glukhov et al. 2013). Specifically, the heart was quickly cannulated and Langendorff-perfused with oxygenated Ca-containing Tyrode's solution (containing, in mm: 128.2 NaCl, 4.7 KCl, 1.19 NaH2PO4, 1.05 MgCl2, 1.3 CaCl2, 20.0 NaHCO3 and 11.1 glucose; pH 7.3–7.35) at room temperature. The fat and lung connected to the atria were carefully trimmed away, and the ventricle was removed. Both atrial chambers were then surgically opened, and pinned to be a 2D preparation, which was superfused by Tyrode's solution. Right and left atrial appendages were separated from the SAN pacemaker complex (intercaval region, interatrial septum) and pulmonary veins by straight incisions and stayed quiescent in the absence of electrode pacing. These isolated atrial preparations were studied either by confocal imaging or optical mapping. To maintain resting membrane potential and excitability (Wit & Cranefield, 1977), atrial tissue was continuously paced at 1 Hz. The pacing rate was then switched to 3 Hz for 5–10 stimuli to induce arrhythmia.
Confocal Ca imaging
Isolated atrial myocytes and atrial tissues were stained with intracellular Ca indicators (Fluo4FF for cells, and Rhod2AM for tissue). Rapid confocal scanning was performed using a Nikon A1R confocal microscope. Temporal resolution was 2–4 ms per line for line scanning and 8–16 ms per frame for 2D resonance scanning (256 × 512 pixels). Confocal experiments were conducted at room temperature. For confocal experiments in atrial tissues (n = 5 WT; n = 8 Casq2−/−), 10 μm blebbistatin (Fedorov et al. 2007; Lou et al. 2012) was added to the perfusate to remove motion artefacts. In isolated Casq2-/− atrial myocytes (n = 8), confocal Ca scanning was also performed simultaneously with action potential (AP) recordings by patch clamp using procedures described previously (Belevych et al. 2012).
Optical mapping
Atrial tissues were stained with Rhod2AM and voltage-sensitive dye RH-237 as described previously (Lou et al. 2011; Glukhov et al. 2013). Vm and Ca were simultaneously imaged using a THT microscope and two Ultima-L CMOS cameras (SciMedia, Costa Mesa, CA, USA) at a temporal resolution of 1 ms per frame and a spatial resolution of 0.125 mm per pixel (100 × 100 pixels). Signals were collected from the epicardium of either left or right atrial appendages. Optical mapping experiments (n = 5 WT; n = 5 Casq2−/−) were conducted at both room temperature and at 37 °C with 10 μm blebbistatin in the perfusate.
Data analysis
All data analysis was performed in MATLAB (Mathworks, Inc., Natick, MA, USA). DCR latency, indicative of the recovery of the RyR2s (Belevych et al. 2012), was measured as the average time from the peak of the preceding systolic Ca release to individual Ca releases in a DCR event. The standard deviation of the latencies to individual Ca releases was measured to quantify the synchronicity of a DCR event. The number of wave initiating sites or the wave sources were counted and analysed for their synchronicity for each DCR.
The amplitude of the Ca signals was measured by dividing the total fluorescent signal by background fluorescent signal (F/F0). Frequency (Hz) was calculated by 1/[cycle length]. The decay of each Ca transient was quantified by the time from the peak to 80% recovery. When constructing Ca wave propagation maps from 2D confocal scans, the time of Ca release was measured by the 50% rise of local Ca signal. When the DCRs that triggered an AP and the DCRs that did not trigger an AP were compared (e.g. Fig. 4B), only the portion of DCR from its beginning to its peak was used to calculate the latency and standard deviation. Otherwise, the entire DCR was used for analysis.
Figure 4.
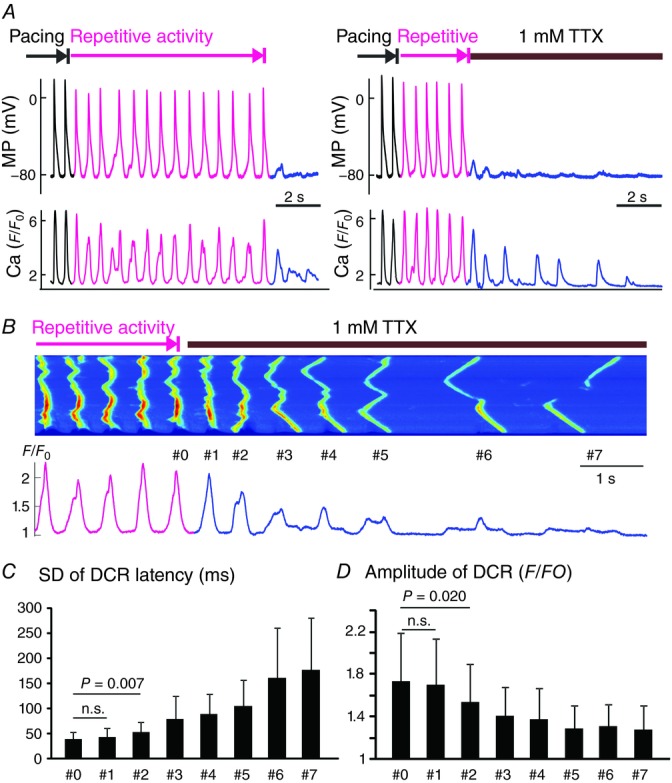
Abolishment of AP desynchronized DCR
A, simultaneous membrane potential (MP) and Ca recordings without (left) and with (right) rapid application of 1 mm TTX during the repetitive activity. B, a representative Ca recording with 1 mm TTX application in Casq2−/− atrial myocytes. C, SD of DCR latency for the last beat associated with a systolic Ca transient (#0) and seven subsequent DCRs (n = 6). D, the amplitude of Ca oscillation for the last beat associated with a systolic Ca transient (#0) and seven subsequent DCRs.
Statistical analysis
Quantitative data are shown as mean ± SD. A paired two-tailed Student's t test was used to compare the data before and after drug or pacing interventions. An unpaired Student's t test was used to determine the difference between WT and Casq2−/− mice. One-way repeated-measures ANOVA with post hoc Tukey's test was used when comparing multiple DCRs from the same recordings. A value of P < 0.05 was considered statistically significant.
Results
Casq2−/− myocytes are prone to self-sustained Ca-dependent rhythmic activity
We investigated the factors underlying potential atrial arrhythmia by monitoring Ca and Vm in atrial myocytes isolated from Casq2−/− and WT mice hearts. To assess the cellular propensity for arrhythmogenic spontaneous/non-driven activity, we applied our pacing protocol in the presence and absence of isoproterenol (ISO, 100 nm). Pacing of Casq2−/− cells in the presence of ISO resulted in runs of self-sustained Ca oscillations (SCOs) (Fig. 1A). These SCOs were characterized by an average frequency of 2.8 ± 0.8 Hz and amplitude of 2.7 ± 0.8 (F/F0) and lasted approximately 31 ± 21 s before dying out spontaneously. Casq2−/− myocytes that were not exposed to ISO, or WT myocytes with ISO exposure showed no SCOs.
Figure 1.

Self-sustained repetitive activity and its modulation by ryanodine and caffeine in isolated atrial myocytes from Casq2−/− mice
A, confocal Ca line scan showing self-sustained repetitive activity in a Casq2−/− atrial myocyte. Diastolic Ca releases (DCR), triggered activity (TA) and self-sustained repetitive activity are marked by pink arrows and traces. Pacing is indicated by black line markers. B, simultaneous membrane potential (MP) and intracellular Ca recordings in a Casq2−/− atrial myocyte. A slow phase of Ca release represents DCR and is associated with delayed afterdepolarization (DAD), and a rapid phase of Ca releases (marked by a downward black arrow) represents systolic Ca release and is exclusively linked to action potential (AP). C, the percentage of hearts (WT, n = 10; Casq2−/−, n = 25) that have self-sustained repetitive activity in isolated atrial myocytes indicated as inducibility, together with the duration of repetitive activity. D, representative Ca recordings before and after the application of ryanodine (3 μm) in Casq2−/− atrial myocytes (n = 5). E, the duration of self-sustained repetitive activity before and after the application of ryanodine (Rya). F, representative Ca recordings before and after the application of 0.2–0.5 mm caffeine in WT atrial myocytes (n = 6). G, the duration of self-sustained repetitive activity before and after the application of caffeine (Caf). Isoproterenol (100 nm) was present in all conditions (A–G).
Close examination of Ca changes during SCOs revealed that Ca release during each spontaneous beat comprises a slow ‘foot’ phase and a rapid Ca elevation. Whereas the slow phase reflects diastolic Ca waves arising at several locations, the rapid phase is apparently due to synchronous, cell-wide Ca release secondary to electrical excitation induced by the initial Ca elevation. To further examine the mechanism of SCOs we performed simultaneous Vm (patch clamp) and intracellular Ca recordings (Fig. 1B). These experiments confirmed that spontaneous Ca waves resulted in a slow membrane depolarization (DAD) towards the AP threshold and that the rapid phase of Ca release was closely linked to the AP and was therefore systolic Ca release. Thus, every beat during SCOs was associated with diastolic Ca release triggering an AP and a secondary voltage-dependent Ca release; failure to trigger an AP was immediately followed by dampening Ca oscillations and resulted in termination of spontaneous beating (Fig. 1A).
To further examine the potential role of Ca cycling in spontaneous beating of Casq2−/− cells, we pharmacologically modulated SR Ca release. Direct inhibition of RyR2 with ryanodine (3 μm) completely inhibited SCOs (Fig. 1D and E). Conversely, sensitization of RyR2s by low doses of caffeine (0.2–0.5 mm) resulted in SCOs in WT myocytes that normally lacked the propensity for SCOs (Fig. 1F and G). To examine the potential role of pacemaker current (If) to aberrant depolarization, we inhibited If with 10 μm ivabradine (Bois et al. 1996) and found that SCOs were not affected (data not shown). Collectively, these results demonstrate for the first time that myocytes from AF-susceptible hearts possess the capacity for self-sustained Ca-dependent repetitive activity.
Rhythmic activity in Casq2−/− myocytes is driven by alternating interplay between Vm and Ca, i.e. the ‘leapfrog’ mechanism
Previous studies conducted in ventricular myocytes suggested that DCR needs to be sufficiently synchronized to trigger an extrasystolic AP (Schlotthauer & Bers, 2000). We quantified the synchronicity of DCR in Casq2−/− atrial cells by measuring the standard deviation of latencies (SDL) to individual Ca release in a DCR event from the preceding systolic Ca release. The SDL of the first post-pacing DCR was significantly shorter in Casq2−/− than WT atrial cells (56 ± 28 vs. 217 ± 105 ms, following 2 Hz pacing, P < 0.001). This increased synchronicity of DCR in Casq2−/− cells was caused by Ca release arising simultaneously from several independent foci across the cell. On average, there were 6.3 ± 1.9 DCR sources in Casq2−/− versus 1.7 ± 0.7 in WT cells (P < 0.001). Moreover, within the same Casq2−/− cells the number of DCR sources and their synchrony was significantly higher for DCRs that triggered an aberrant AP than for those that did not (Fig. 2B and C, WT cells showed no DCR that triggered an AP). These data suggested that synchronous DCR involving simultaneous activation of multiple release sites is essential for triggered activity in Casq2−/− atrial cells.
Figure 2.
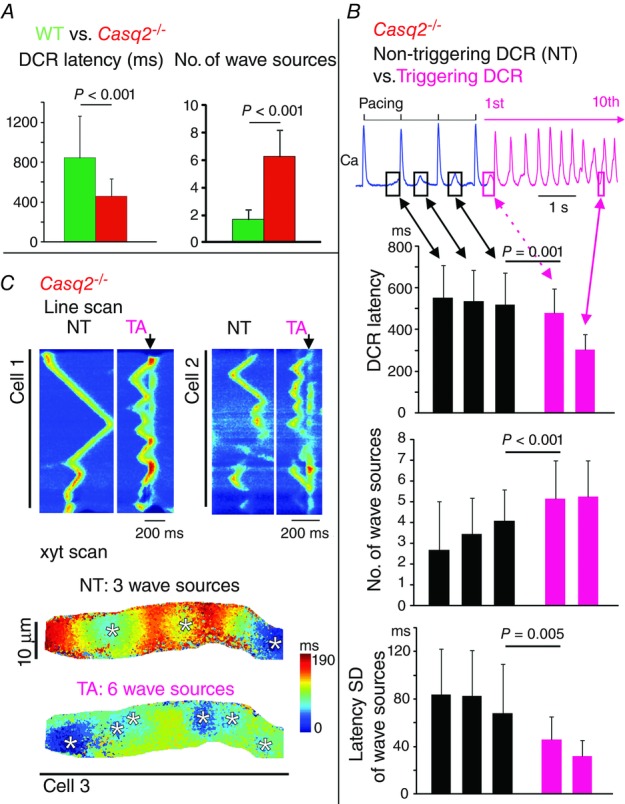
Synchronization of diastolic Ca release (DCR)
A, latency from the preceding systolic Ca release to the DCR (or DCR latency) and number of wave sources of the first DCR following 2 Hz pacing in WT (n = 10) and Casq2−/− (n = 15) atrial myocytes. B, DCR latency, number of wave sources and synchronization of wave sources (quantified by SD of latency from the preceding systolic Ca release to individual wave sources) in Casq2−/− atrial myocytes (n = 33). These parameters were quantified for the last three DCRs that did not trigger an AP (NT) and for the first and 10th DCRs that triggered an AP (TA). The P values are only shown for comparisons between last NT and first TA. C, representative 1D and 2D scans of DCRs associated with NT and TA in Casq2−/− atrial myocytes. The downward black arrows mark the systolic Ca release, indicative of the presence of an AP. The colours in the 2D map indicate the time of Ca release. The transition from blue to red colour indicates the sequence of Ca wave propagation. The spatial origins of Ca waves (or wave sources) are marked by white asterisks.
While synchronous DCR can potentially account for the generation of an aberrant AP, it does not explain the self-sustained rhythmic activity observed in Casq2−/− atrial cells. We hypothesized that rhythmic activity involves a sequential synchronizing, i.e. ‘leapfrog’, interplay between Vm and Ca in the settings of RyR2 dysfunction associated with CPVT. Specifically, while synchronized DCR results in an AP, it is the AP that in turn aligns DCR for the subsequent AP, thereby perpetuating this cyclical phenomenon. To test this aberrant rhythm-generating mechanism we first examined the effect of a single pacing-induced AP in producing a sufficiently synchronous DCR to trigger an aberrant AP. Casq2−/− atrial myocytes exposed to elevated Ca (6 mm) in the bath solution displayed occasional, spontaneous DCRs. Despite the presence of multiple release initiation sites, these sites were out of phase and therefore not sufficiently synchronized to produce a triggered AP (Fig. 3A). In contrast, after a pacing-induced AP, two major DCR sources in the field of view immediately became temporally aligned. The resultant synchrony resulted in an aberrant AP (Fig. 3A and B). AP consistently synchronized DCR by aligning multiple DCR sources (Fig. 3C), which in 75% of cases triggered APs (Fig. 3D). Importantly, when triggered APs occurred they synchronized subsequent DCR usually resulting in another sequential triggered AP (Fig. 3A). By contrast, failure to generate a triggered AP was followed by gradual loss of DCR synchronicity.
Figure 3.

AP synchronized DCR
A, representative Ca recording where one pacing stimulus synchronized the previously out-of-phase Ca waves and induced the self-sustained repetitive activity in Casq2−/− atrial myocytes. B, close-up view of the diastolic Ca releases (DCR) before and after the pacing. C, SD of the DCR latency before and after a single stimulus (n = 10). D, percentage inducibility of DCR-induced triggered activity (TA).
To further confirm the involvement of AP in the maintenance of rhythmic activity, we abolished cellular excitability by inhibiting Na channels with either TTX or by rapid elimination of Na from the extracellular solution. As shown in Fig. 4A, rapid addition of TTX (1 mm) to the superfusate resulted in an immediate termination of AP and dampening of diastolic Ca oscillation. Quantitative analysis of Ca recordings (Fig. 4B–D) indicated that termination of the repetitive activity by TTX was followed by desynchronization of DCRs. Similarly, synchronous rhythmic activity was suppressed when the superfusate was switched to a 0 Na, 0 Ca solution (Fig. 5) that inhibited both the Na and the Na/Ca exchanger (NCX) currents (Schlotthauer & Bers, 2000). Of note, elevation of extracellular Ca to 6 mm in Casq2−/− atria was insufficient to trigger activity. A stimulated AP was necessary to initiate such activity (Fig. 3A). Collectively, these results suggest that AP is necessary for synchronous DCR. This then forms a conceptual framework for a novel mechanism for self-sustained repetitive activity that is based on pathological interplay between Ca and aberrant APs. Importantly, this mechanism could provide the trigger for AF induction in settings of defective RyR2 function.
Figure 5.

Inhibition of Na and NCX currents did not prevent the desynchronization of DCR
A, a representative Ca recording when 0 Ca, 0 Na (substituted by Li) solution was rapidly applied during repetitive activity in Casq2−/− atrial myocytes. B, SD of DCR latency for the last beat associated with a systolic Ca transient (#0) and seven subsequent DCRs (n = 7). C, the amplitude of Ca oscillation for the last beat associated with a systolic Ca transient (#0) and seven subsequent DCRs.
Rhythmic activity is modulated by Ca current, SR Ca uptake and RyR2 function
The rate or frequency of repetitive activity may reflect its pro-arrhythmic potential. To determine how to modulate the frequency, we tested the effects of altered Ca current, SR Ca uptake and RyR2 function on the frequency of repetitive activity.
Application of the Ca channel agonist Bay K8644 (BayK, 50 nm; Schramm et al. 1983) during repetitive activity consistently increased the oscillatory frequency (Fig. 6A and B). Similarly, augmentation of the inward Ca current (Hess & Tsien, 1984) by rapid application of high Ca (6 mm) Tyrode's solution also consistently increased the oscillatory frequency (Fig. 6C and D). Taken together, these results suggest that increased Ca influx through voltage-gated Ca channels accelerates repetitive activity.
Figure 6.
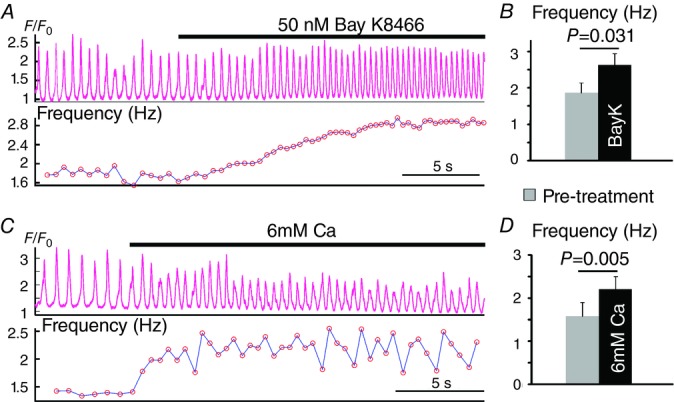
Effects of Ca current enhancement on the frequency of repetitive activity
A, a representative Ca recording of repetitive activity with rapid application of 50 nm Bay K8466 (BayK). B, summarized effects of BayK on the frequency of repetitive activity (n = 5). C, a representative Ca recording of repetitive activity with rapid application of high Ca (6 mm) Tyrode's solution. D, summary of the effects of 6 mm Ca on the frequency of repetitive activity (n = 5).
Next, we assessed the role of Ca re-sequestration into the SR as a factor influencing rhythmic activity. Studies in ventricular myocytes suggested that synchronization of arrhythmogenic DCR is enhanced by accelerated SR Ca reuptake affecting RyR2 functional recovery (Wasserstrom et al. 2010). We perturbed SR reuptake by rapid application of cyclopiazonic acid (CPA). CPA (25 μm) within seconds prolonged the decay of Ca transient (Fig. 7A). Importantly, this prolongation was accompanied by a deceleration of SCO (Fig. 7A). Of note, a strong linear correlation between the decay of Ca transient and the frequency of repetitive activity was observed (Fig. 7A). As inhibition of SR Ca uptake by CPA in addition to slowing the kinetics of uptake also lowers the SR Ca content (Fig. 7A) (Baudet et al. 1993), both of these factors could contribute to the CPA-induced slowing of repetitive activity.
Figure 7.
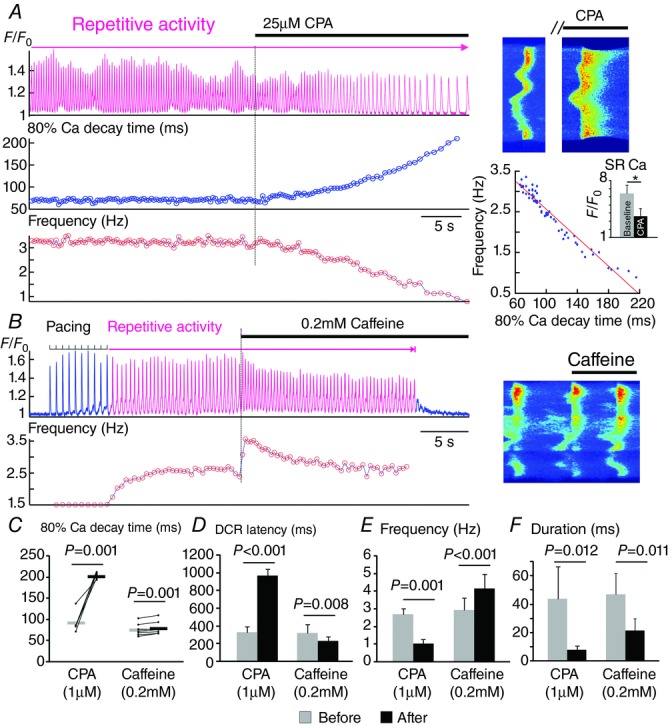
Regulation of repetitive activity by the rate of Ca re-uptake and functional recovery of RyR2s
A, a representative Ca recording showing the immediate effect of 25 μm cyclopiazonic acid (CPA) in the 80% decay time of a Ca transient and the frequency of repetitive activity. Line scan images from the same recording before and after CPA is shown on the right. Frequency vs. 80% Ca decay time is plotted for each individual beat following the application of CPA. The effect of 1 min treatment with 25 μm CPA on the amplitude of 20 mm caffeine-induced Ca transient (indicative of the SR Ca content) is shown in the bar graph. *P < 0.05. B, a representative Ca recording showing the effect of 0.2 mm caffeine on the frequency of repetitive activity. The line scan image at the moment of caffeine application is shown. C–F, effects of CPA (1 μm, n = 5) and caffeine (0.2 mm, n = 7) on 80% Ca decay time (C), DCR latency (D), frequency (E) and duration (F) of the self-sustained repetitive activity are summarized. To better show the direction of change, paired data points are plotted and connected with thin lines in C. The averages are shown as thick horizontal lines in C.
Lastly, we enhanced RyR2 activity by the application of caffeine. Caffeine (0.2 mm) instantly shortened DCR latency and increased the frequency of SCOs (Fig 7B and E). Thus, rhythmic activity appears to be influenced by Ca current, SR Ca reuptake and Ca release functions.
Synchronization of SCOs in atrial tissue
An important unresolved question about the role of DCR in the genesis of atrial arrhythmia is whether and how spontaneous Ca oscillations that arise in individual myocytes synchronize in a cellular agglomeration to lead to triggered activity in the tissue. To directly examine the ability of DCR to synchronize and thereby result in tissue-wide atrial arrhythmia, we carried out confocal Ca imaging in atrial preparations derived from Casq2−/− mice. Consistent with the results of isolated atrial myocyte experiments, when paced in the presence of ISO, Casq2−/− atrial preparations exhibited bursts of spontaneous Ca oscillations that occurred synchronously in neighbouring cells in the field of view (Fig. 8A) at an average frequency of 2.8 ± 1 Hz (n = 5) lasting for 16 ± 8 s. The Ca transients in each cell of the atrial preparation were preceded by slower Ca elevations comprising several independent Ca waves similar to DCRs observed in isolated myocytes (Fig. 8B and D). DCR latencies among different neighbouring cells presented similarly narrow distributions (Fig. 8B), suggesting a high level of DCR synchronicity not only within a given cell but also across different cells of the atrial preparation, thereby overcoming the source–sink mismatch and facilitating the generation of focal activations in atrial tissue.
Figure 8.
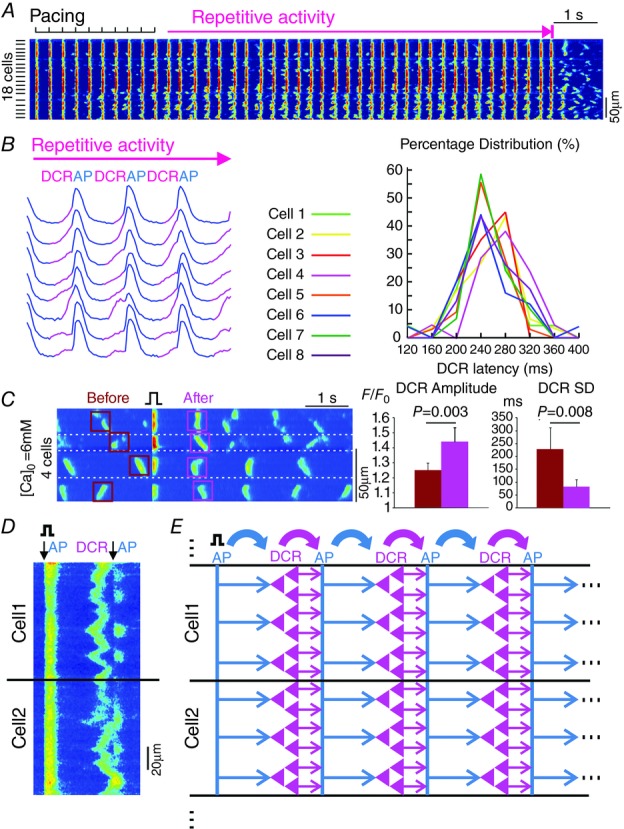
DCR synchronization in Casq2−/− atrial tissue preparations
A, confocal line scanning from 18 atrial myocytes showing repetitive activity following the cessation of pacing. The scanning line was placed perpendicular to the cell axis. B, Ca transients from eight neighbouring atrial myocytes during repetitive activity. DCRs preceding the systolic Ca release are marked in pink. On the right is the DCR latency distributions of neighbouring atrial myocytes. C, on the left is a representative Ca recording where one pacing stimulus synchronized the previously out-of-phase Ca waves in neighbouring myocytes in atrial tissue. Cell boundaries are marked by white dashed lines. On the right are amplitude and SD of DCRs across atrial myocytes before and after the single stimulus. D, confocal line scanning from two neighbouring atrial myocytes. The scanning line was placed along the cell axis. The downward arrow indicates the systolic Ca release reflecting the occurrence of an action potential (AP). E, schematic illustration of the alternating ‘leapfrog’ interplay between AP and synchronized DCR during self-sustained repetitive activity. That is, AP first synchronizes tissue-wide DCR, which results in an ectopic AP; the AP, in turn, temporally aligns the next DCR. As such, this process repeats itself and maintains the self-sustained repetitive activity.
Next, to test the role of an AP in synchronization of subsequent diastolic release in atrial tissue, we examined the effects of an isolated electrical stimulus on the properties of subsequent diastolic release in atrial preparations bathed in a solution with elevated Ca (6 mm). At rest, occasional DCRs occurred randomly in various individual cells within the tissue. Application of a single electrical stimulus resulted in synchronization of DCRs across the tissue, as evidenced by both increased amplitude of tissue-averaged DCR and decreased standard deviation of latencies to DCR (Fig. 8C). These results suggest that an AP is able to synchronize DCR both on the cellular level as well as in tissue, giving rise to tissue-wide self-sustained repetitive activity.
DCR-dependent repetitive focal activity in atrial appendages from Casq2−/− mice
To determine the contribution of a Ca-dependent triggered mechanism to atrial arrhythmia, we conducted macroscopic optical mapping of both Vm and Ca in atrial tissue preparations derived from WT mice and Casq2−/− mice (Glukhov et al. 2013; Faggioni et al. 2014) prone to AF (Fig. 9). SAN pacemaker complex was removed so that the native pacemaking mechanism would not potentially contribute to such aberrant events. Vulnerability to atrial arrhythmia in atrial appendages was assessed by applying an electrical pacing protocol (3 Hz) in the presence of ISO (100 nm). Pacing consistently induced runs of repetitive self-sustained oscillations in Vm and intracellular Ca in atrial preparations from Casq2−/− (in 5 out of 5) but not WT mice (Fig. 9A and B). Runs of rhythmic activity consisted of reoccurring APs and corresponding Ca transients occurring at a frequency of 7.0 ± 0.3 Hz at 37 °C (2.5 ± 0.6 Hz at room temperature) lasting 19 ± 11 s. As demonstrated in the activation maps in Fig. 9E, rhythmic activity consistently originated from the same focal area, thus pointing to focal firing as basis of the activity. Importantly, at the site of origin, the upstrokes of AP during the runs of rhythmic activity were always preceded by intracellular Ca elevations (Fig. 9C), thereby further suggesting that focal firing was mediated by a Ca-dependent leapfrog mechanism rather than Vm-driven automaticity.
Figure 9.
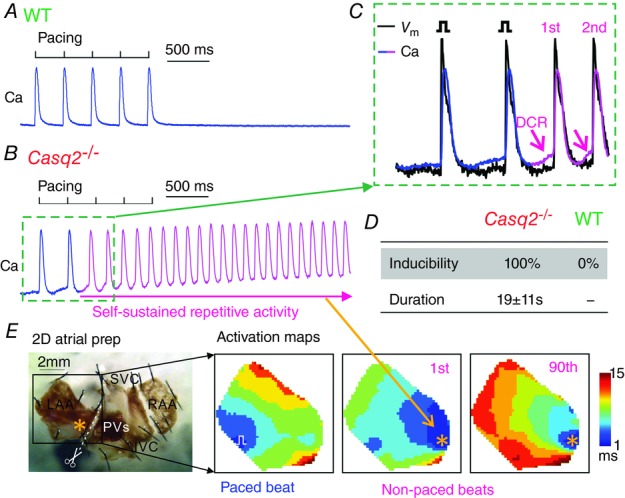
Optical mapping demonstrating repetitive focal activity in Casq2−/− atrial tissue preparations
A, representative Ca transient during and after pacing in WT atrial preparation. B, representative Ca transient during and after pacing in Casq2−/− atrial preparations. Pink colour indicates the presence of self-sustained repetitive activity. C, simultaneous optical membrane potential (Vm) and Ca signals during the initiation of self-sustained repetitive activity (marked by green dotted rectangles in B). Diastolic Ca releases (DCRs) are marked by the pink arrows. D, inducibility and duration of the repetitive activity under 100 nm isoproterenol in WT (n = 5) and Casq2−/− atrial tissue (n = 5). E, activation maps showing the sequence of electrical conduction from blue to red colours during pacing, 1st non-paced beat, and 90th non-paced beat during the recording in B. Bottom left is a representative unwrapped 2D atrial tissue preparation, where the left atrial appendage (LAA) is separated from the pacemaker complex and studied. RAA, right atrial appendage; SVC, superior vena cava; IVC, inferior vena cava; PVs, pulmonary veins.
Discussion
We used a multi-scale approach, including atrial optical Vm/Ca mapping and confocal Ca imaging, in atrial tissue and myocyte preparations to examine the mechanisms of focal atrial arrhythmia in the setting of familial Ca-dependent arrhythmia (i.e. CPVT) caused by deletion of Casq2. Our major findings are: (1) atrial arrhythmia could be generated through ectopic self-sustained repetitive activity that manifests itself in both myocytes and intact atrial tissue in the form of spatiotemporally synchronized, rapid SCOs; and (2) SCOs arise via a novel rhythm-generating mechanism that involves a sequential synchronizing interplay between Vm and SR Ca in the settings of dysregulated RyR2 function. These findings suggest a link between abnormal Ca handling and triggering mechanisms for AF and provide a mechanistic basis for using RyR2 modulators to treat AF.
The composite Vm/Ca oscillator
Despite early efforts, the molecular to macroscopic mechanisms of AF remain poorly understood. Recently, abnormal Ca handling has been recognized as a major factor in AF (Nattel & Dobrev, 2012). In particular, several studies have suggested that aberrant DCR due to leaky RyR2s may contribute to AF by causing ectopic activity that can serve as a trigger for either re-entry (King et al. 2013) or focal arrhythmia (Faggioni et al. 2014). However, it is unclear how triggered activity can lead to the repetitive characteristic of focal atrial arrhythmia. Similarly it remains elusive how aberrant Ca signals in individual cells are synchronized across the atrial tissue to overcome the current source–sink mismatch thus culminating in ectopic activity. Here we demonstrate a new mechanism of abnormal cardiac rhythm generation that involves a sequential synchronizing, i.e. ‘leapfrog’, interplay between Vm and SR Ca in the settings of dysregulated RyR2 function. Specifically, in a setting of abbreviated RyR2 refractoriness as is evidenced in CPVT, temporal alignment of DCR in neighbouring atrial myocytes by preceding AP results in synchronized DCR sufficient to trigger an aberrant AP. This aberrant AP, in turn, injects Ca and aligns refractory periods of RyR2s to result in a subsequent synchronized DCR. The process repeats itself, resulting in self-sustained rhythmic activity in Casq2−/− atrial cells (Fig. 8E).
In addition to accounting for repetitive firing in myocytes, this mechanism also explains how DCR becomes synchronized across atrial tissue, thus resolving a principal question in the generation of focal atrial arrhythmia. Note that the ability of an AP to serve as a tissue-wide synchronizing event depends on its rapid propagation across the tissue. It could be inferred that a slower propagation of an AP may result in a desynchronized DCR in tissue, which would decrease the potential of an ectopic focal arrhythmia. On the other hand, factors that slow AP propagation (such as fibrosis and reduced gap junctional coupling) also reduce the source–sink mismatch and thereby may promote ectopic focal arrhythmia (Myles et al. 2012). In addition, slow propagation is a key facilitator of re-entrant arrhythmia (Kleber & Rudy, 2004). It therefore remains to be determined whether slowing AP propagation could desynchronize the DCR, and whether the resultant desynchronized DCR would outweigh the pro-arrhythmic effects of slowed conduction.
Previously it has been demonstrated (Maltsev & Lakatta, 2009; Lakatta et al. 2010) that oscillations in intracellular Ca can contribute to normal heart rhythm by influencing the pacemaking automaticity (i.e. two separate interacting oscillators: Vm clock and Ca clock). The mechanism of self-sustained repetitive activity described here is different in the sense that neither the Ca nor the Vm component is individually capable of generating rhythmic activity but instead these components come together to form a composite Vm/Ca oscillator. Moreover, this mechanism does not involve the pacemaker current. While it remains to be determined whether the mechanism described here contributes to normal pacemaking, the main implication of our study is that the composite Vm/Ca oscillator forms a pathological rhythm generator that can potentially precipitate AF.
Subcellular and molecular mechanisms of the Vm/Ca oscillator
Recently we showed (Brunello et al. 2013) that aberrant DCR in the setting of CPVT is associated with impaired Ca signalling refractoriness. These events are caused by the failure of RyR2s to deactivate, thereby predisposing Ca release units to spontaneous activation. Here we demonstrate that loss of Casq2 affects atrial myocyte Ca cycling in a similar way, resulting in DCR with decreased latency. We also demonstrate that an AP is required for synchronous DCR both within and across the cell boundary, thus serving as a synchronizing event for DCR (Belevych et al. 2012; Brunello et al. 2013; Izu et al. 2013). These results are consistent with the concept of store-dependent deactivation previously developed for ventricular myocytes (Terentyev et al. 2002; Zima et al. 2008) and thereby synchronization of DCR is a result of a combination of abbreviated RyR2 refractoriness and the preceding synchronous Ca release/reuptake dynamics resulting from an AP (Brunello et al. 2013). Furthermore we demonstrate that besides leading to isolated trigger events, shortened RyR2 refractoriness gives rise to self-sustained repetitive activity due to sequential synchronizing interplay between synchronized DCR and AP (Fig. 8E). It remains to be determined whether such pro-arrhythmic repetitive activity occurs in ventricular myocytes/tissue.
Our findings also provide insights into the molecular and subcellular factors affecting the properties of repetitive triggered activity including their frequency and duration. In particular our results demonstrate that the cycle length of repetitive triggered activity is influenced by DCR latency, which is in turn dependent on RyR2 refractoriness (Belevych et al. 2012; Radwanski et al. 2013). Indeed, further shortening RyR2 refractoriness by low concentrations of caffeine (Venetucci et al. 2007; Belevych et al. 2012) shortened cycle length and increased frequency of repetitive triggered activity, whereas increased DCR latency by CPA prolonged cycle length and decreased the frequency of repetitive activity in Casq2−/− myocytes (Fig. 7D and E). On the other hand, our study suggests that the duration of the oscillation is determined by intracellular Ca reserve and, in particular, by the levels of Ca that the myocyte is able to recycle back to SR during SCOs. Indeed, SCOs evidently die out when intra-SR Ca falls below a certain level, thus resulting in reduced release that fails to trigger an extrasystolic AP. For example, both increasing leak by caffeine and slowing SR Ca uptake rate, interventions that would inhibit SR Ca re-accumulation, shortened the duration of SCOs (Fig. 7F). This proposed view also accounts for the pro-arrhythmic effects of β-adrenergic stimulation in this and other studies of AF mechanisms in CPVT (Chelu et al. 2009; Shan et al. 2012; Faggioni et al. 2014). Indeed, accelerating SR Ca uptake should increase both the frequency and the duration of SCOs by increasing SR refilling and abbreviating Ca release restitution.
Limitations
One potential limitation of our study is the difference in atrial Ca handling between mice and humans (Bers, 2008; Rudy et al. 2008). It remains to be determined whether the specific phenomena and mechanisms observed herein apply to the human variant of AF (Wit & Boyden, 2007). Recent studies have functionally demonstrated dysregulated RyR2s in isolated atrial myocytes from AF patients (Hove-Madsen et al. 2004; Neef et al. 2010; Voigt et al. 2012, 2014). Future studies are warranted to evaluate whether the RyR2 dysfunction due to Casq2 deletion could functionally recapitulate the RyR2 dysregulation and associated atrial arrhythmias in AF patients.
To avoid the involvement of pacemaker current in the role of observed atrial arrhythmias, we only studied atrial appendages, which is also a limitation of this study. We have previously observed focal atrial arrhythmias in the intercaval region and pulmonary veins from Casq2−/− mice (Glukhov et al. 2013), which were not studied here. It remains to be determined whether the same Vm/Ca oscillator underlies repetitive focal activity in those regions, and whether these regions are associated with shortened RyR2 refractoriness or accelerated Ca uptake dynamics, both of which are shown to enhance the rate of repetitive triggered activity in this study.
Conclusions
This study presents a novel mechanistic insight into repetitive triggered atrial activity. Specifically, abbreviated RyR2 refractoriness sets the stage for aberrant oscillations of both intracellular Ca and membrane potential. AP and synchronized DCR regenerate each other and thereby maintain a ‘leapfrog’-like oscillation. This repetitive activity may serve as an important mechanism for initiation and perpetuation of AF. Besides revealing a new mechanism for ectopic rhythm generation, our study shows that desynchronizing aberrant DCR and prolonging DCR latency by modulating RyR2 refractoriness may be effective means of treating atrial arrhythmias.
Acknowledgments
We thank Dr Lai-hua Xie for his valuable technical suggestions. We thank Dr Lucia Brunello for helpful comments on the manuscript.
Glossary
- AF
atrial fibrillation
- AP
action potential
- Casq2
calsequestrin 2
- CPA
cyclopiazonic acid
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- DAD
delayed afterdepolarization
- DCR
diastolic Ca release
- ISO
isoproterenol
- NCX
Na/Ca exchanger
- RyR
ryanodine receptor
- SCO
self-sustained Ca oscillation
- SDL
standard deviation of latency
- SR
sarcoplasmic reticulum
- Vm
membrane potential
- WT
wild-type
Additional information
Competing interests
None.
Author contributions
Q.L., V.V.F. and S.G. contributed to the conception and design of the experiments. Q.L., A.E.B., P.B.R., B.L. and A.K. contributed to the data collection and analysis. Q.L., A.E.B., P.B.R., V.V.F. and S.G. contributed to the data interpretation. Q.L., A.E.B., P.B.R., B.L., A.K., B.C.K, V.V.F. and S.G. contributed to the drafting and critical revision of the article. All authors have read and approved the final submission.
Funding
This work was supported primarily by an Ohio State University, Department of Physiology & Cell Biology pilot grant (to Q.L.) and by National Institutes of Health (NIH) grants HL074045 and HL063043 (to S.G.), and partially by NIH HL115580 (to V.V.F.).
References
- Baudet S, Shaoulian R. Bers DM. Effects of thapsigargin and cyclopiazonic acid on twitch force and sarcoplasmic reticulum Ca2+ content of rabbit ventricular muscle. Circ Res. 1993;73:813–819. doi: 10.1161/01.res.73.5.813. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE. Gyorke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin JR, Cannell MB. Lederer WJ. Cellular origins of the transient inward current in cardiac myocytes. Role of fluctuations and waves of elevated intracellular calcium. Circ Res. 1989;65:115–126. doi: 10.1161/01.res.65.1.115. [DOI] [PubMed] [Google Scholar]
- Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- Bhuiyan ZA, van den Berg MP, van Tintelen JP, Bink-Boelkens MT, Wiesfeld AC, Alders M, Postma AV, van Langen I, Mannens MM. Wilde AA. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation. 2007;116:1569–1576. doi: 10.1161/CIRCULATIONAHA.107.711606. [DOI] [PubMed] [Google Scholar]
- Bois P, Bescond J, Renaudon B. Lenfant J. Mode of action of bradycardic agent, S 16257, on ionic currents of rabbit sinoatrial node cells. Br J Pharmacol. 1996;118:1051–1057. doi: 10.1111/j.1476-5381.1996.tb15505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunello L, Slabaugh JL, Radwanski PB, Ho HT, Belevych AE, Lou Q, Chen H, Napolitano C, Lodola F, Priori SG, Fedorov VV, Volpe P, Fill M, Janssen PM. Gyorke S. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc Natl Acad Sci U S A. 2013;110:10312–10317. doi: 10.1073/pnas.1300052110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capes EM, Loaiza R. Valdivia HH. Ryanodine receptors. Skelet Muscle. 2011;1:18. doi: 10.1186/2044-5040-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D. Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D. Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet. 2010;375:1212–1223. doi: 10.1016/S0140-6736(10)60096-7. [DOI] [PubMed] [Google Scholar]
- Faggioni M, Savio-Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D. Knollmann BC. Suppression of spontaneous Ca elevations prevents atrial fibrillation in calsequestrin 2-null hearts. Circ Arrhythm Electrophysiol. 2014;7:313–320. doi: 10.1161/CIRCEP.113.000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov VV, Lozinsky IT, Sosunov EA, Anyukhovsky EP, Rosen MR, Balke CW. Efimov IR. Application of blebbistatin as an excitation–contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4:619–626. doi: 10.1016/j.hrthm.2006.12.047. [DOI] [PubMed] [Google Scholar]
- Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, Belevych AE, Mohler PJ, Knollmann BC, Periasamy M, Gyorke S. Fedorov VV. Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex. Eur Heart J. 2013 doi: 10.1093/eurheartj/eht452. doi: http://dx.doi.org/10.1093/eurheartj/eht452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV. Singer DE. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–2375. doi: 10.1001/jama.285.18.2370. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D. Turner MB. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P. Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- Hess P. Tsien RW. Mechanism of ion permeation through calcium channels. Nature. 1984;309:453–456. doi: 10.1038/309453a0. [DOI] [PubMed] [Google Scholar]
- Ho HT, Liu B, Snyder JS, Lou Q, Brundage EA, Velez-Cortes F, Wang H, Ziolo MT, Anderson ME, Sen CK, Wehrens XH, Fedorov VV, Biesiadecki BJ, Hund TJ. Gyorke S. Ryanodine receptor phosphorylation by oxidized CaMKII contributes to the cardiotoxic effects of cardiac glycosides. Cardiovasc Res. 2014;101:165–174. doi: 10.1093/cvr/cvt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser SR. When does spontaneous sarcoplasmic reticulum Ca2+ release cause a triggered arrythmia? Cellular versus tissue requirements. Circ Res. 2000;87:725–727. doi: 10.1161/01.res.87.9.725. [DOI] [PubMed] [Google Scholar]
- Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A. Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- Izu LT, Xie Y, Sato D, Banyasz T. Chen-Izu Y. Ca2+ waves in the heart. J Mol Cell Cardiol. 2013;58:118–124. doi: 10.1016/j.yjmcc.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jais P, Haissaguerre M, Shah DC, Chouairi S, Gencel L, Hocini M. Clementy J. A focal source of atrial fibrillation treated by discrete radiofrequency ablation. Circulation. 1997;95:572–576. doi: 10.1161/01.cir.95.3.572. [DOI] [PubMed] [Google Scholar]
- January CT. Fozzard HA. Delayed afterdepolarizations in heart muscle: mechanisms and relevance. Pharmacol Rev. 1988;40:219–227. [PubMed] [Google Scholar]
- Kass RS, Lederer WJ, Tsien RW. Weingart R. Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibres. J Physiol. 1978;281:187–208. doi: 10.1113/jphysiol.1978.sp012416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian P, Gollob MH, Pantano A. Oudit GY. A novel mutation in the RYR2 gene leading to catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation: dose-dependent arrhythmia-event suppression by β-blocker therapy. Can J Cardiol. 2011;27:870.e7–e10. doi: 10.1016/j.cjca.2011.02.003. [DOI] [PubMed] [Google Scholar]
- King JH, Zhang Y, Lei M, Grace AA, Huang CL. Fraser JA. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol (Oxf) 2013;207:308–323. doi: 10.1111/apha.12006. [DOI] [PubMed] [Google Scholar]
- Kleber AG. Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431–488. doi: 10.1152/physrev.00025.2003. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C. Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, Maltsev VA. Vinogradova TM. A coupled system of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res. 2010;106:659–673. doi: 10.1161/CIRCRESAHA.109.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Ho HT, Velez-Cortes F, Lou Q, Valdivia C, Knollmann B, Valdivia H. Gyorke S. Genetic ablation of ryanodine receptor 2 phosphorylation at Ser-2808 aggravates Ca2+-dependent cardiomyopathy by exacerbating diastolic Ca2+ release. J Physiol. 2014;592:1957–1973. doi: 10.1113/jphysiol.2013.264689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Q, Fedorov VV, Glukhov AV, Moazami N, Fast VG. Efimov IR. Transmural heterogeneity and remodeling of ventricular excitation–contraction coupling in human heart failure. Circulation. 2011;123:1881–1890. doi: 10.1161/CIRCULATIONAHA.110.989707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Q, Glukhov AV, Kalyanasundaram A, Hanson B, Hage L, Belevych AE, Gyorke S, Mohler PJ, Knollmann BC, Periasamy M. Fedorov VV. Calsequestrin 2 deletion causes abnormal atrial automaticity and conduction associated with degenerative fibrosis of the sinoatrial node pacemaker complex. Circulation. 2012;126:A10945. doi: 10.1093/eurheartj/eht452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA. Lakatta EG. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am J Physiol Heart Circ Physiol. 2009;296:H594–615. doi: 10.1152/ajpheart.01118.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka Y, Barnes ME, Gersh BJ, Cha SS, Bailey KR, Abhayaratna WP, Seward JB. Tsang TS. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation. 2006;114:119–125. doi: 10.1161/CIRCULATIONAHA.105.595140. [DOI] [PubMed] [Google Scholar]
- Myles RC, Wang L, Bers DM. Ripplinger CM. Decreased IK1 and increased ryanodine receptor sensitivity synergistically contribute to sustained focal arrhythmia in the intact rabbit heart. J Physiol. 2014;593:1479–1493. doi: 10.1113/jphysiol.2014.279638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles RC, Wang L, Kang C, Bers DM. Ripplinger CM. Local β-adrenergic stimulation overcomes source–sink mismatch to generate focal arrhythmia. Circ Res. 2012;110:1454–1464. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattel S. Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J. 2012;33:1870–1877. doi: 10.1093/eurheartj/ehs079. [DOI] [PubMed] [Google Scholar]
- Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G. Maier LS. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106:1134–1144. doi: 10.1161/CIRCRESAHA.109.203836. [DOI] [PubMed] [Google Scholar]
- Pizzale S, Gollob MH, Gow R. Birnie DH. Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J Cardiovasc Electrophysiol. 2008;19:1319–1321. doi: 10.1111/j.1540-8167.2008.01211.x. [DOI] [PubMed] [Google Scholar]
- Radwanski PB, Belevych AE, Brunello L, Carnes CA. Gyorke S. Store-dependent deactivation: cooling the chain-reaction of myocardial calcium signaling. J Mol Cell Cardiol. 2013;58:77–83. doi: 10.1016/j.yjmcc.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy Y, Ackerman MJ, Bers DM, Clancy CE, Houser SR, London B, McCulloch AD, Przywara DA, Rasmusson RL, Solaro RJ, Trayanova NA, Van Wagoner DR, Varro A, Weiss JN. Lathrop DA. Systems approach to understanding electromechanical activity in the human heart: a national heart, lung, and blood institute workshop summary. Circulation. 2008;118:1202–1211. doi: 10.1161/CIRCULATIONAHA.108.772715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotthauer K. Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- Schramm M, Thomas G, Towart R. Franckowiak G. Novel dihydropyridines with positive inotropic action through activation of Ca2+ channels. Nature. 1983;303:535–537. doi: 10.1038/303535a0. [DOI] [PubMed] [Google Scholar]
- Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A. Marks AR. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2012;111:708–717. doi: 10.1161/CIRCRESAHA.112.273342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumitomo N, Sakurada H, Taniguchi K, Matsumura M, Abe O, Miyashita M, Kanamaru H, Karasawa K, Ayusawa M, Fukamizu S, Nagaoka I, Horie M, Harada K. Hiraoka M. Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ J. 2007;71:1606–1609. doi: 10.1253/circj.71.1606. [DOI] [PubMed] [Google Scholar]
- Ter Keurs HE. Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007;87:457–506. doi: 10.1152/physrev.00011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL. Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW. Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, O'Neill SC. Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285–292. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XH, Nattel S. Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–156. doi: 10.1161/CIRCULATIONAHA.113.006641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH. Dobrev D. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+–Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakili R, Voigt N, Kaab S, Dobrev D. Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011;121:2955–2968. doi: 10.1172/JCI46315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstrom JA, Shiferaw Y, Chen W, Ramakrishna S, Patel H, Kelly JE, O'Toole MJ, Pappas A, Chirayil N, Bassi N, Akintilo L, Wu M, Arora R. Aistrup GL. Variability in timing of spontaneous calcium release in the intact rat heart is determined by the time course of sarcoplasmic reticulum calcium load. Circ Res. 2010;107:1117–1126. doi: 10.1161/CIRCRESAHA.110.229294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wit AL. Boyden PA. Triggered activity and atrial fibrillation. Heart Rhythm. 2007;4:S17–23. doi: 10.1016/j.hrthm.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wit AL. Cranefield PF. Triggered and automatic activity in the canine coronary sinus. Circ Res. 1977;41:434–445. doi: 10.1161/01.res.41.4.434. [DOI] [PubMed] [Google Scholar]
- Xie Y, Sato D, Garfinkel A, Qu Z. Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Picht E, Bers DM. Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]