

Abstract
We studied excitation–contraction coupling (ECC) and inositol-1,4,5-triphosphate (IP3)-dependent Ca2+ release in normal and heart failure (HF) rabbit atrial cells. Left ventricular HF was induced by combined volume and pressure overload. In HF atrial myocytes diastolic [Ca2+]i was increased, action potential (AP)-induced Ca2+ transients (CaTs) were larger in amplitude, primarily due to enhanced Ca2+ release from central non-junctional sarcoplasmic reticulum (SR) and centripetal propagation of activation was accelerated, whereas HF ventricular CaTs were depressed. The larger CaTs were due to enhanced IP3 receptor-induced Ca2+ release (IICR) and reduced mitochondrial Ca2+ buffering, consistent with a reduced mitochondrial density and Ca2+ uptake capacity in HF. Elementary IP3 receptor-mediated Ca2+ release events (Ca2+ puffs) were more frequent in HF atrial myoctes and were detected more often in central regions of the non-junctional SR compared to normal cells. HF cells had an overall higher frequency of spontaneous Ca2+ waves and a larger fraction of waves (termed arrhythmogenic Ca2+ waves) triggered APs and global CaTs. The higher propensity of arrhythmogenic Ca2+ waves resulted from the combined action of enhanced IICR and increased activity of sarcolemmal Na+–Ca2+ exchange depolarizing the cell membrane. In conclusion, the data support the hypothesis that in atrial myocytes from hearts with left ventricular failure, enhanced CaTs during ECC exert positive inotropic effects on atrial contractility which facilitates ventricular filling and contributes to maintaining cardiac output. However, HF atrial cells were also more susceptible to developing arrhythmogenic Ca2+ waves which might form the substrate for atrial rhythm disorders frequently encountered in HF.
Key points
Impaired calcium (Ca2+) signalling is the main contributor to depressed ventricular contractile function and occurrence of arrhythmia in heart failure (HF).
Here we report that in atrial cells of a rabbit HF model, Ca2+ signalling is enhanced and we identified the underlying cellular mechanisms. Enhanced Ca2+ transients (CaTs) are due to upregulation of inositol-1,4,5-trisphosphate receptor induced Ca2+ release (IICR) and decreased mitochondrial Ca2+ sequestration.
Enhanced IICR, however, together with an increased activity of the sodium–calcium exchange mechanism, also facilitates spontaneous Ca2+ release in form of arrhythmogenic Ca2+ waves and spontaneous action potentials, thus enhancing the arrhythmogenic potential of atrial cells.
Our data show that enhanced Ca2+ signalling in HF provides atrial cells with a mechanism to improve ventricular filling and to maintain cardiac output, but also increases the susceptibility to develop atrial arrhythmias facilitated by spontaneous Ca2+ release.
Introduction
Excitation–contraction coupling (ECC) refers to the process that links action potential-dependent membrane depolarization, elevation of cytosolic Ca2+ levels ([Ca2+]i) and force development in cardiac myocytes. Disturbances of ECC play a crucial role in cardiac disease resulting in contractile dysfunction and arrhythmogenesis. Atrial contraction is an important determinant of ventricular filling (‘atrial kick’) and it can contribute up to 40% of the end-diastolic ventricular filling during periods of increased haemodynamic demands, e.g. during exercise (Rahimtoola et al. 1975; Nicod et al. 1986). In patients with systolic heart failure (HF), the atrial contribution to the diastolic filling of an impaired ventricle becomes increasingly important to maintain cardiac output (Bonow et al. 1983; Raymond et al. 1998). Furthermore, such patients are especially prone to develop rhythm disorders like atrial fibrillation, the most common form of cardiac arrhythmia, which not only increase the risk of stroke, but also eliminate the atrial kick. The consequence of diminished atrial contractility is a reduced overall blood pumping capacity of the heart by up to a third, which in turn worsens symptoms of HF and facilitates its progression (Alpert et al. 1988). At the single cell level atrial Ca2+ signalling during ECC shows properties distinctly different from ventricular cells. These differences can largely be attributed to structural and functional differences in the organization of the couplon (Bootman et al. 2006) and the Ca2+ release units (CRUs) (Chen-Izu et al. 2006; Li et al. 2012). The couplon is the basic unit of ECC and refers to the ultrastructural arrangement of the sarcoplasmic reticulum (SR) membrane hosting ryanodine receptor (RyR) Ca2+ release channels and the sarcolemmal membrane containing L-type Ca2+ channels (LTCCs), only separated by a narrow gap of a few nanometres. The fact that the LTCCs directly face clusters of RyRs in CRUs assures activation of RyRs despite their rather low cytosolic Ca2+ sensitivity (Cannell & Kong, 2012). These SR regions are referred to as junctional SR (j-SR) where Ca2+ entry through LTCCs allows for an efficient activation of a cluster of RyRs forming a CRU by a process termed Ca2+ induced Ca2+ release (CICR; Cannell et al. 1987; Sheehan & Blatter, 2003). In ventricular cells, invaginations of the sarcolemmal membrane organized in a sarcomeric pattern and known as t-tubules create couplons throughout the entire volume of the cell and allow for rapid synchronous release of Ca2+ from CRUs. Atrial cells of many species, however, are either devoid of or show very sparse t-tubules, resulting in a rather inhomogenous distribution of the structures involved in Ca2+ handling and consequently the spatio-temporal organization of the cytosolic Ca2+ signal during ECC. In the subsarcolemmal space, the sarcolemmal membrane is located in close vicinity to the SR (j-SR). Here LTCCs directly face CRUs, resembling the ultrastructural arrangement found in couplons of ventricular cells (Stern et al. 1999) and are referred to as peripheral couplings (Kockskamper et al. 2001). However in more central regions of the cytosol where t-tubules are absent, abundant RyRs of the non-junctional SR (nj-SR) are found where CRUs are not associated with LTCCs and activation of RyRs relies on centripetal Ca2+ diffusion and propagation (Coombes & Timofeeva, 2003; Thul et al. 2012; Trafford et al. 2013). As a consequence, during ECC striking differences in the shape, magnitude and kinetics of subcellular Ca2+ transients (CaTs) are observed: upon action potential-dependent membrane depolarization fast and high-amplitude CaTs in the subsarcolemmal space are observed first that result from CICR from j-SR elicited by Ca2+ currents through LTCCs. Centripetal Ca2+ diffusion activates CICR from nj-SR CRUs resulting in propagation of CICR towards the cell centre in a Ca2+ wave-like fashion (Huser et al. 1996; Kockskamper et al. 2001; Sheehan & Blatter, 2003). Consequently, central CaTs have a slower rise time, a delayed peak and are smaller in amplitude.
The scarcity of t-tubules in atrial cells also affects beat-to-beat Ca2+ removal that occurs through reuptake into the SR by SERCA and extrusion across the sarcolemma predominantly via Na+–Ca2+ exchange (NCX). In atrial myocytes Ca2+ released from nj-SR CRUs occurs at greater distances from NCX and therefore the contribution of NCX to beat-to-beat Ca2+ removal is likely to differ from ventricular cells. Furthermore, increased NCX activity is a hallmark of myocytes from failing hearts and the electrogenic nature of its activity as cause for arrhythmogenic disturbances in ventricular tissue is well established (Pogwizd et al. 1999); however, the role of NCX in atrial arrythmogenesis still needs to be determined.
Atrial myocytes are also unique compared to ventricular cells in their significantly higher expression levels of a second type of SR Ca2+ release channel, the inositol-1,4,5-trisphosphate receptor (IP3R; Lipp et al. 2000). IP3Rs are part of a Gq-protein coupled receptor (GPCR) signalling pathway and release Ca2+ from the SR upon binding of IP3 in a [Ca2+]i-dependent fashion. IP3R-induced Ca2+ release (IICR) has been hypothesized to lower the threshold for the activation of RyRs by CICR and may directly contribute to Ca2+ transients (Kockskamper et al. 2008). In cardiac disease such as hypertrophy, HF (Wang et al. 2003) and atrial fibrillation (Yamda et al. 2001) IP3R expression levels are further increased. Furthermore, patients with cardiac hypertrophy or HF have elevated levels of Angiotensin II (Ang II; Nattel, 2002), a hormone that binds to GPCRs and exerts its effect, at least in part, by enhancing IICR. Taken together, evidence is growing that IP3R-mediated Ca2+ signalling plays a critical role in cardiac disease, but the specific ramifications for atrial function are poorly understood.
Mitochondria contribute to Ca2+ cycling during ECC at different levels, from providing the fuel (ATP) for ion pumps (including SERCA) and substrate for phosphorylation of Ca2+ handling proteins, to control of the redox environment and to Ca2+ buffering (Dedkova & Blatter, 2012, 2013). While it is generally agreed that mitochondria are capable of sequestering Ca2+, even in large quantities, it has remained controversial whether significant amounts of Ca2+ buffering can occur during ECC on a beat-to-beat basis and thus whether mitochondria can shape the CaT in space and time (Bootman et al. 2006; O'Rourke & Blatter, 2009; Dedkova & Blatter, 2013). Again, the scarcity of t-tubules and the spatially inhomogeneous patterns of Ca2+ release in atrial cells, raise the question of a specific role of mitochondria for the propagation of activation, and specifically, whether the high density of mitochondria (Barth et al. 1992; Mackenzie et al. 2004; Hohendanner et al. 2013) can impede the propagation of Ca2+ release through the network of the nj-SR, and whether this changes in HF.
The goal of this study was a comprehensive characterization of atrial Ca2+ cycling during ECC in single myocytes from normal and from failing hearts. We focused on the interplay between IICR, mitochondrial Ca2+ buffering and altered NCX activity and its consequences for atrial arrhythmogenesis in HF. We found that in HF enhanced IICR and reduced mitochondrial Ca2+ uptake had positive inotropic effects mediated by enhanced CaTs during ECC, but also led to a higher propensity of spontaneous proarrhythmic Ca2+ release mediated by enhanced NCX activity.
Methods
Ethical approval
All protocols were approved by the Institutional Animal Care and Use Committee of Rush University Chicago, and comply with US and UK regulations on animal experimentation (Drummond, 2009).
Myocyte isolation and HF induction
Left atrial and left ventricular myocytes were isolated from male New Zealand White rabbits (Myrtle's Rabbitry, Thompsons Station, TN, USA). Rabbits were killed by intravenous injection of pentobarbital sodium (100 mg kg−1). Hearts were excised, mounted on a Langendorff apparatus, and retrogradely perfused with nominally Ca2+-free Tyrode solution for 5 min followed by minimal essential medium Eagle (MEM) solution containing 20 μm Ca2+ and 22.5 μg ml−1 Liberase Blendzyme TH (Roche Applied Science, Indianapolis, IN, USA) for 20 min at 37°C. The left atrium was removed from the heart and digested for an additional 5 min in the enzyme solution at 37°C. Ventricular or atrial tissue was minced, filtered, and washed in MEM solution containing 50 μm Ca2+ and 10 mg ml−1 BSA. Isolated cells (n = 69 normal hearts) were kept in MEM solution with 50 μm Ca2+ at room temperature (20–24°C) until indicator dye loading and subsequent experimentation or cultured and used for experimentation after 24–48 h.
In addition, left atrial and ventricular myocytes were isolated from male rabbits with non-ischaemic HF (n = 17 hearts) induced by combined volume and pressure overload. Rabbits underwent two survival surgeries. Volume overload was induced by creation of aortic valve insufficiency, followed 2–4 weeks later by induction of pressure overload by abdominal aorta banding. Surgeries were conducted under isoflurane inhalation anaesthesia after ketamine–xylazine–acepromazine premedication. HF developed gradually (over 5–8 months, monitored by serial echocardiography), and was characterized by depressed systolic ventricular function, and arrhythmogenesis in an animal that has human-like Ca2+ handling and elecrophysiological properties (Pogwizd, 1995). Atrial cells were isolated from hearts that showed marked left ventricular dilatation (54 ± 5% increase in left ventricular diastolic internal dimension) and systolic dysfunction (decrease of left ventricular fractional shortening by 37 ± 4%). Ventricular cells from this model have been studied extensively and are well characterized at structural, biochemical, molecular, Ca2+ handling, and electrophysiological levels (Pogwizd, 1995; Pogwizd et al. 2001; Bers et al. 2002; Despa et al. 2002; Shannon et al. 2003; Domeier et al. 2009; Zima et al. 2010).
Cell culture and adenoviral gene transfer
For adenoviral transfections myocytes were kept in short-term culture (≤48 h) using Corning cellgro Medium 199 (VWR, Radnor, PA, USA) supplemented with penicillin and streptomycin (50 μg ml−1). The medium was changed daily. Adenoviral gene transfer (multiplicity of infection of 500) was used to express FIRE1-cyt (fluorescent IP3 responsive element 1 targeted to the cytosol; Remus et al. 2006), an IP3 affinity trap (Domeier et al. 2008), mouse 43 kDa inositol polyphosphate 5′-phosphatase (m43 phosphatase; Kapoor et al. 2014), GFP or Mitycam (Kettlewell et al. 2009). Experiments were performed after 24 h culture for FIRE1-cyt, IP3 affinity trap, m43 phosphatase and GFP, and after 48 h culture for Mitycam.
Solutions and chemicals
All chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) unless noted otherwise. SEA-0400 was obtained from ChemScene (Monmouth Junction, NJ, USA). Fluorescent calcium indicator dyes and MitoTracker Red FM were obtained from Molecular Probes/Life Technologies (Grand Island, NY, USA). Tyrode solution contained (in mm): 130 NaCl, 4 KCl, 2 or 7 CaCl2 (as indicated), 1 MgCl2, 10 d-glucose, 10 Hepes; pH 7.4 (adjusted with NaOH). For experiments on permeabilized myocytes saponin (0.01%) was used for cell membrane permeabilization and cells were kept in an ‘internal solution’ containing (in mm): 100 potassium aspartate, 15 KCl, 5 KH2PO4, 5 MgATP, 0.35 EGTA, 0.067 CaCl2, 0.75 MgCl2, 10 phosphocreatine, and 10 Hepes with 5 U ml−1 creatine phosphokinase and 4% dextran (molecular weight: 40000); pH 7.2 (adjusted with KOH). Free [Ca2+]i and [Mg2+]i of this solution were 150 nm and 1 mm, respectively (calculated using Maxchelator, Stanford University). All cells were plated on laminin-coated glass coverslips and superfused with 2 mm Ca2+ Tyrode solution. To prevent movement of cells during confocal imaging blebbistatin (10 μm; Tocris Bioscience, Bristol, UK) was added to all Tyrode based solutions. ‘Mitochondrial solution’ contained (in mm): 135 KCl, 5 NaCl, 20 Hepes, 5 pyruvate, 2 glutamic acid, 2 malic acid, 0.5 KH2PO4, 1 MgCl2, 5 EGTA, 15 BDM. Free [Ca2+] of this solution was set to 0 or 5 μm by addition of CaCl2 (as determined with Maxchelator).
[Ca2+]i and [Ca2+]SR measurements
Indo-1 AM (5 μm, loading for 30 min at room temperature, followed by 15 min wash) was used to ratiometrically (excitation at 360 nm, emission collected at 405 (F405) and 485 (F485) nm) measure averaged whole-cell [Ca2+]i. Background signals were subtracted and [Ca2+]i changes are expressed as the ratio R = F405/F485. Fluo-4 and Fluo-5N were used for confocal cytosolic and SR [Ca2+] measurements. Changes in [Ca2+] measured with single wavelength dyes are expressed as F/F0 where F0 refers to resting fluorescence in unstimulated cells measured at the begin of an experiment or diastolic fluorescence levels measured under control steady-state conditions during electrical stimulation, respectively. CaT amplitudes were quantified as ΔF/F0 and ΔR, respectively, where ΔF = F − F0 and ΔR = Rpeak − Rdiast. For confocal [Ca2+]i measurements cells were loaded with Fluo-4 AM (5 μm, excitation at 488 nm, emission at 515 nm) for 20 min. For [Ca2+]SR measurements cells were loaded with Fluo-5N AM (10 μm, excitation at 488 nm, emission at 515 nm) for 1 h 45 min. All dye loading procedures were followed by a washing period (>10 min) in Tyrode solution. Confocal line scan images were recorded at 512 lines s–1 using a ×60 oil-immersion objective lens (NA = 1.49) with a Nikon A1R system (Nikon Corporation, Melville, NY, USA). The scan line was placed along the longitudinal or transversal axis (as indicated) of the cell using pixel dimensions of 0.03–0.24 μm. Subcellular CaTs reflecting Ca2+ release from j-SR (SS, subsarcolemmal) and nj-SR (CT, central) were derived from 1 μm wide regions. The centripetal propagation velocity of CICR in atrial myocytes was derived from transverse confocal line scan images and calculated as the distance between two subcellular locations of interest and the interval between the times when the local CaTs reached half-maximal amplitude at these respective locations. The empirically measured point-spread function of the system had full width at half-maximum (FWHM) values of 0.28, 0.29 and 0.7 μm in the x, y and z (axial) dimensions (Maxwell & Blatter, 2012). Action potentials and global CaTs were elicited by electrical field stimulation (0.5 Hz) of intact atrial myocytes using a pair of platinum electrodes (voltage set at 50% above the threshold for contraction). Caffeine (10 mm) was used to deplete SR Ca2+ stores and measure the SR Ca2+ content. Experiments were conducted at room temperature (20–24°C).
IP3 and IICR
A cytosol-targeted FRET-based IP3 biosensor (FIRE1-cyt; for a detailed description see Remus et al. 2006) was used to dynamically study [IP3]i in atrial cells. Briefly, FIRE1-cyt reports fluorescence resonance energy transfer (FRET) between cyan and yellow fluorescent protein (CFP and YFP) upon binding of IP3. FIRE1-cyt is a fusion protein of the IP3 binding domain of the IP3 receptor type 1 with CFP and YFP at the amino and carboxyl termini, respectively. Upon excitation of CFP at 457 nm, an increase in [IP3] leads to increased FRET and a detectable change in the YFP/CFP fluorescence ratio (emissions were measured at >530 and 488 nm).
Inhibition of IICR was achieved by (i) adenoviral expression of FIRE1-cyt lacking CFP and YFP sequences which acts as a buffer of cytosolic IP3 (IP3 affinity trap; Domeier et al. 2008); (ii) adenoviral transfer of mouse inositol polyphosphate 5′ phosphatase (m43; Kapoor et al. 2014) which rapidly and efficiently degrades the biologically active IP3 by removing the 5′ phosphate (Majerus et al. 1999; Vasilevski et al. 2008); and (iii) application (5 min) of the IP3R blocker 2-aminoethoxydiphenyl borate (2-APB, 10 μm).
Elevation of [IP3]i in intact myocytes was achieved by agonist stimulation with angiotensin II (Ang II; 0.25 μm in freshly isolated myocytes; 10 μm in cultured cells) or by photolysis of caged IP3 (cag-IP3 PM; SiChem, Bremen, Germany). Cells were loaded with 100 nm cag-IP3 for 20 min, and liberation of biologically active IP3 was achieved by 405 nm laser illumination in user-defined cellular regions using the Mosaic illumination tool integrated in the Nikon A1R confocal imaging system (Shkryl et al. 2011).
Elementary IP3R-mediated Ca2+ release events (Ca2+ puffs) were recorded in permeabilized atrial cells after stimulation with IP3 (d-myo-inositol 1,4,5-trisphosphate, hexapotassium salt; Tocris Bioscience, Bristol, UK) and inhibition of RyR-mediated Ca2+ release with tetracaine (4 mm; ‘Ca2+ puff protocol’; Zima & Blatter, 2004).
Mitochondria
Cultured (48 h) left atrial myocytes were adenovirally transfected with the mitochondria-targeted fluorescent Ca2+ probe Mitycam (Kettlewell et al. 2009) to measure mitochondrial distribution and density as well as mitochondrial Ca2+ uptake. Mitycam is an inverse pericam that is excited at 488 nm and emission is measured at 530 ± 15 nm. Changes of [Ca2+]mito are expressed as 1 – (F/F0). For structural studies (density and distribution of mitochondria) with Mitycam or MitoTracker Red FM (25 μm, 30 min loading at 37°C, excitation at 543 nm, emission at >600 nm) atrial cardiomycoytes were kept in Tyrode solution and two-dimensional (2D) confocal images were recorded from a central plane. Mitochondrial Ca2+ uptake via mitochondrial Ca2+ uniporter (MCU) was studied in permeabilized cells expressing Mitycam. For the inhibition of the MCU, the ruthenium compound Ru360 (10 μm, EMD Chemicals, Philadelphia, PA, USA) was used.
Data analysis and statistics
The Liscana algorithm (Hohendanner et al. 2013) was used to analyse mitochondrial density and cell area.
The frequency of Ca2+ puffs and the frequency, amplitude, spatial width (FWHM), full duration (full duration at half-maximum, FDHM) and time to peak (TTP) of Ca2+ sparks were analysed with SparkMaster (Picht et al. 2007). Because of the small amplitude of the signal, Ca2+ puff amplitude, FWHM, FDHM and TTP were further verified by manual analysis.
The relative contribution of NCX to Ca2+ removal during CaTs (referred to as ‘NCX activity’) was calculated as:
| 1 |
where the time constants τCaT and τCaffeine were obtained by mono-exponential fits to the decay of electrically and caffeine-evoked CaTs in the same cell. The assay is based on the notion that the decline of an AP-induced CaT results from Ca2+ extrusion via NCX and reuptake into the SR via SERCA pump, whereas during caffeine (10 mm) exposure [Ca2+]i declines solely by NCX extrusion. By considering the time constants τCaT and τCaffeine (cf. Fig. 7Ea) according to eqn 1 a measure of NCX activity can be derived that estimates the percentage contribution of NCX to the decline of an electrically evoked CaT.
Figure 7.
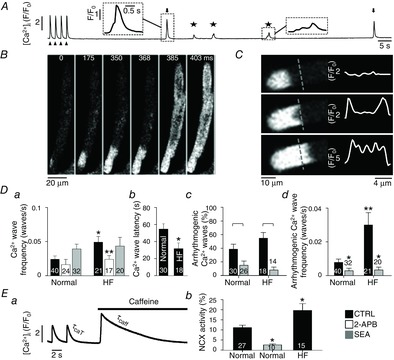
Arrhythmogenic Ca2+ waves and NCX activity in normal and HF atrial myocytes
A, protocol to monitor spontaneous Ca2+ wave activity. Cells were electrically paced (arrowheads) at 0.5 Hz in 7 mm extracellular [Ca2+] to enhance SR Ca2+ loading. Ca2+ waves were quantified during a 2-min rest period after cessation of pacing. The stars mark spontaneous Ca2+ waves without global cell-wide Ca2+ release and arrows mark arrhythmogenic Ca2+ waves. B, spontaneous Ca2+ wave in a single atrial myocyte observed during a rest period after pacing. The Ca2+ wave led to subsequent global Ca2+ release by triggering an AP and SR Ca2+ release typical for atrial ECC (‘arrhythmogenic Ca2+ wave’). C, confocal 2D image from a central focal plane revealing the ‘U-shaped’ wave front (left) and normalized fluorescence profiles (F/F0) recorded along the dashed line (right). Da, frequency of spontaneous Ca2+ waves in normal and HF cells under control (CTRL) conditions (black), after the application of 2-APB (white) and SEA (grey). *P < 0.05 versus normal control; **P < 0.05 versus HF control, Student's t test. Db, latency of spontaneous Ca2+ waves. *P < 0.05, Student's t test. Dc, fraction of Ca2+ waves that induce a spontaneous AP and global CaTs (‘arrhythmogenic Ca2+ waves’) in HF atrial cells before (black) and after application of SEA (grey). In panel Dc square brackets indicate significant differences at P < 0.05, one-way ANOVA. Dd, absolute frequency of arrhythmogenic Ca2+ waves in normal and HF atrial myocytes in the absence and presence of SEA. To calculate absolute wave frequencies (panels a and d), also cells showing no waves were included. **P < 0.05 versus normal cells in the absence of SEA, Student's t test. *P < 0.05 versus control, Student's t test. Ea, protocol used to determine τCaT and τCaffeine for quantification of NCX activity. Eb, NCX activity in normal atrial cells (left), after inhibition of NCX in normal atrial cells with SEA, and in HF atrial cells. *P < 0.05 against normal cells, Student's t test.
Statistical comparisons were made using Student's t test or one-way ANOVA for paired or unpaired data, with statistical significance set at P < 0.05. Data are presented as individual observations or as means ± SEM of n measurements, where n is the number of cells.
Results
Ca2+ transients and SR Ca2+ depletion during ECC in normal and HF myocytes
We compared cytosolic CaTs between left atrial myocytes from normal rabbits and rabbits with systolic heart failure (Maxwell et al. 2012). Cytosolic CaTs were elicited by electrical field stimulation and recorded with Fluo-4 by line scan confocal imaging (Fig.1Aa and d). As expected for a cardiac cell type lacking transverse tubules (Huser et al. 1996; Kockskamper et al. 2001) [Ca2+]i rose first and to the highest level in the cell periphery (SS in Fig. 1Aa, corresponding to Ca2+ release from j-SR), followed by propagation of Ca2+ release in centripetal direction where [Ca2+]i peaked with a delay at significantly (Fig. 1Ab and C) lower levels (CT in Fig. 1Aa, corresponding to Ca2+ release from nj-SR). In atrial cells from failing hearts, CaTs were prolonged and significantly higher in amplitude in central regions of the cell (Fig. 1Ae and C). This is in stark contrast to ventricular myocytes where CaTs were significantly depressed in HF (Fig. 1Af and E) compared to normal myocytes (Fig. 1Ac and E). Furthermore, the centripetal propagation velocity of Ca2+ release was significantly accelerated in atrial HF myocytes (Fig. 1D; HF: 205 ± 19 μm s−1; normal: 147 ± 13 μm s−1). Consistent with the larger amplitude of cytosolic CaTs in HF atrial myocytes, the beat-to-beat SR Ca2+ depletion transients (Fig. 1B) were significantly larger in HF atrial myocytes (Fig. 1F).
Figure 1.
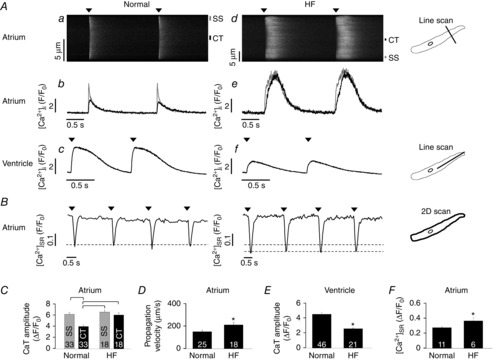
Ca2+ cycling during ECC in normal and HF left atrial myocytes
A, control line scan images (a, d) and CaTs (b, e) from central (CT) and subsarcolemmal (SS) cytosolic regions of normal and HF atrial myocytes and from control (c) and HF (f) ventricular cells. Arrowheads indicate electrical stimulation. B, [Ca2+]SR traces obtained from whole-cell imaging of Fluo-5 N fluorescence in normal (left) and HF (right) atrial myocytes. Arrowheads indicate electrical stimulation. C, summary data of local CaT amplitudes in CT and SS regions of normal and HF atrial myocytes obtained from transversal line scans. Square brackets indicate significant differences at P < 0.05, one-way ANOVA. D, summary data for the centripetal Ca2+ propagation velocity in normal and HF atrial cells derived from transversal line scans. E, summary data for the CaT amplitude in normal and HF ventricular myocytes obtained from longitudinal line scans. F, summary data for the [Ca2+]SR depletion amplitude in normal and HF atrial myocytes as obtained from 2D confocal measurements of Fluo-5 N fluorescence. Panels D–F: *P < 0.05, Student's t test. Here and subsequent figures: numbers in bar graphs indicate n = number of cells.
IICR induced by angiotensin II
Ang II is known to increase [IP3]i and to exert a positive inotropic effect in cardiac mycoytes (Meissner et al. 1998; Remus et al. 2006). Ang II levels as well as Ang II receptor expression on cell surfaces are increased in vivo in different models of cardiac disease (e.g. atrial fibrillation; Nattel, 2002); however, Ang II has also been shown to depress left ventricular contractile performance in HF (Cheng et al. 1996). We compared the effects of Ang II stimulation on diastolic [Ca2+]i, CaTs and SR Ca2+ load in normal and HF atrial myocytes. CaTs were elicited by field stimulation and in each experiment SR Ca2+ load was estimated from the amplitude of the [Ca2+]i signal elicited by rapid application of caffeine (10 mm; Fig. 2A). We confirmed the Ang II-dependent intracellular IP3 production using the FRET-based IP3 biosensor FIRE1-cyt (Remus et al. 2006). As shown in Fig. 2B, Ang II stimulation increased [IP3]i significantly in normal and HF atrial myocytes, but there was no difference in magnitude of this increase between normal and HF cells, although the sensitivity and dynamic range of the IP3 biosensor may not be sufficient to detect these potentially small changes (Fig. 2F). In atrial myocytes from failing hearts, diastolic [Ca2+]i levels, measured ratiometrically with Indo-1, were significantly increased compared to normal cells (Fig. 2C), and treatment with Ang II increased diastolic [Ca2+]i in normal and HF myocytes even further. The whole-cell CaT amplitudes measured with Indo-1, were significantly increased in HF cells (+32%; Fig. 2A and D), thus confirming the results obtained with Fluo-4 (Fig. 1C). However, Ang II significantly decreased the CaT amplitude in HF cells by 36% (Fig. 2D, P < 0.05). Additionally, the SR Ca2+ content was unchanged in HF atrial cells compared to normal cells. However, exposure of cells to Ang II for >5 min decreased the SR Ca2+ load significantly in HF, whereas it had no effect in normal atrial cells (Fig. 2E).
Figure 2.
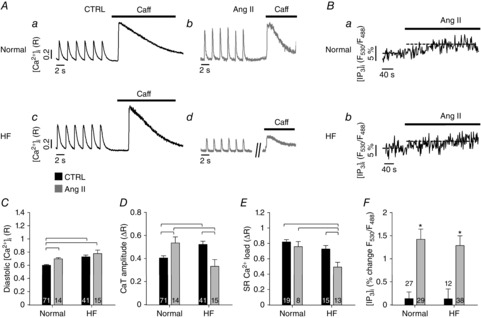
Effect of Ang II on Ca2+ signalling during ECC in atrial cells
A, electrically and caffeine-evoked CaTs in normal (top) and HF (bottom) atrial cells before (a, c; CTRL) and after (b, d) application of Ang II. B, changes of [IP3]i in normal (a) and HF (b) atrial myocytes revealed by measurement of FRET between CFP and YFP in cells expressing the IP3 biosensor FIRE1-cyt. C, diastolic [Ca2+]i during field stimulation in normal and HF myocytes under control conditions (black) or after treatment with Ang II (grey). D, CaT amplitudes during field stimulation in normal and HF myocytes under control conditions (black) or after treatment with Ang II (grey). E, SR Ca2+ load assessed from changes of [Ca2+]i induced by rapid application of 10 mm caffeine, in normal and HF myocytes under control conditions (black) or after treatment with Ang II (grey). In panels C–E square brackets indicate significant differences at P < 0.05, one-way ANOVA. F, grey bars show average increase of FIRE1-cyt FRET signal (F530/F488) after application of AngII in normal and HF myocytes. Black bars represent changes of control FRET signal over the same time interval in the absence of AngII. *P < 0.05, Student's t test.
IICR induced by cytosolic uncaging of IP3
To further elucidate the role of IP3R-mediated Ca2+ release during ECC in normal and HF atrial myocytes, we loaded atrial myocytes with the caged IP3 compound cag-IP3. Upon UV-light exposure, cag-IP3 instantaneously released biologically active IP3 which led to an increase of [IP3]i levels as confirmed by FIRE1-cyt measurements (Fig. 3A and B). Global IP3 uncaging significantly increased the CaT amplitude in regions of the nj-SR (CT in Fig. 3C) in normal atrial cells with little effect on SS CaTs. In HF atrial myocytes, however, IP3 uncaging led to a similar decrease of Ca release from j-SR and nj-SR (Fig. 3E). This decrease in CaT amplitude in HF myocytes was due to a combined effect of reduced peak CaT (by 17 ± 2 and 19 ± 3% in CT and SS regions) and an increase in diastolic [Ca2+]i (by 57 ± 7 and 70 ± 8% in CT and SS regions). Averaged over the entire cells the increase in diastolic [Ca2+]i 20 s after IP3 uncaging was almost twice as large in HF as in normal cells ([Ca2+]diast (F/F0): 1.36 ± 0.06 in normal cells (n = 12); 1.67 ± 0.13 in HF cells (n = 6); P < 0.05). The IP3R blocker 2-APB (10 μm) abolished the IP3 effects (Fig. 3D), confirming that the observed effects on CaT amplitude were mediated by IICR, and actually decreased CaT amplitudes below control levels, suggesting that basal IP3 activity made a contribution towards the magnitude of CaTs under control conditions.
Figure 3.
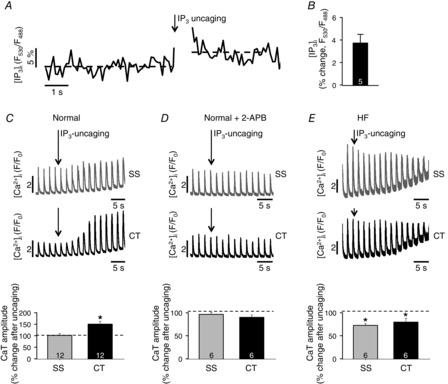
Effect of IP3 uncaging on Ca2+ signalling during ECC in atrial cells
A, change of [IP3]i in an atrial myocyte expressing FIRE1-cyt before and after IP3 uncaging. B, average percentage change of the [IP3]i-dependent FIRE1-cyt FRET signal (F530/F488) averaged over 3 s after uncaging. C and E, effect of global whole-cell IP3 uncaging on local CaTs in the subsarcolemmal (SS) and central (CT) cytosol in normal (C) and HF (E) atrial myocytes. D, same experiment as in panel C but in the presence of the IP3R blocker 2-APB. Panels C–E, bottom: summary data of IP3 uncaging effects on CaT amplitudes. *P < 0.05, Student's t test. 100% corresponds to the average [Ca2+]i levels before IP3 uncaging.
Effect of basal [IP3]i on CaTs
We investigated the effect of basal IP3 activity on CaTs further, using three different methodological approaches. First, we used the pharmacological IP3R blocker 2-APB. In the presence of 2-APB the CaT amplitude was reduced in atrial myocytes; however, this basal IP3 effect was significantly more pronounced in HF (Fig. 4E; cf. also Fig. 3D). In two additional series of experiments we adenovirally expressed the IP3 binding domain of the IP3R type 1 (IP3 affinity trap) or the m43 phosphatase in the cytosol. Both strategies aimed to reduce [IP3]i levels. As a control we transfected cells with an adenovirus encoding GFP and determined in control measurements that CaT amplitudes were identical in the presence or absence of the virus (data not shown). Figure 4Aa and Ba shows CaT in virus infected normal and HF atrial cells. As in uninfected cells (Figs 1 and 2) the CaT amplitude was increased in HF atrial myocytes. In atrial cells expressing the IP3 affinity trap (Fig. 4Ab and Bb) or m43 phosphatase (Fig. 4Ac and Bc) the CaT amplitude was reduced in normal cells (albeit statistically not significant) and to a significantly larger degree in HF atrial myocytes. For comparison, m43 phosphatase expression in normal ventricular myocytes had no effect on CaT amplitudes, suggesting a lower basal IP3 activity in ventricular cells. The effect of inhibition of IP3 signalling with three distinctly different methods yielded extremely consistent results (Fig. 4C–E) and validated the use of 2-APB as a valuable tool to inhibit IP3Rs in cardiac cells. As shown in Fig. 4F the effect of basal [IP3]i levels on CaT amplitude affected predominantly release of Ca2+ from j-SR in normal cells, whereas in HF atrial myocytes the effect was overall more pronounced but affected j-SR and nj-SR to a similar degree.
Figure 4.
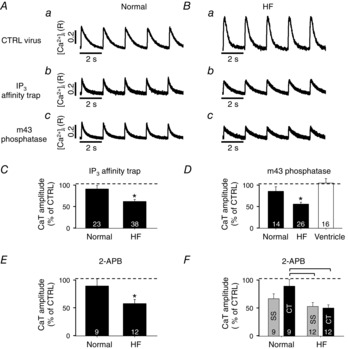
Effect of basal IICR on CaT amplitude
A and B, CaTs in field stimulated normal (A) and HF (B) atrial myocytes after 24 h in culture, expressing a control (CTRL) virus (a), IP3 affinity trap (b) or m43 phosphatase (c). C and D, summary data of the percentage change of CaT amplitudes in normal and HF atrial myocytes expressing an IP3 affinity trap (C) or m43 phosphatase (D). *P < 0.05 against normal cells, Student's t test. In D, comparison of m43 phosphatase effect in normal ventricular cells. E, summary data of the effect of 2-APB on CaT amplitudes in normal and HF cells. Panels C–E: *P < 0.05, Student's t test. F, same as panel E but spatially resolved for the subsarcolemmal (SS) and the central (CT) cytosol. Square brackets indicate significant differences at P < 0.05, one-way ANOVA. In panels C–F, 100% corresponds to the average CaT amplitudes before inhibition of IP3 signalling.
Elementary IP3R-dependent Ca2+ release events
Elementary IP3R-dependent Ca2+ release events from clusters of IP3Rs (termed ‘Ca2+ puffs’, cf. Tovey et al. 2001) have been observed in a variety of cell types including atrial myocytes. In cat atrial myocytes their amplitude has been reported to be 70–80% smaller, the duration three to four times longer and the rise time approximately twofold longer than RyR-mediated Ca2+ release events (Ca2+ sparks; Zima & Blatter, 2004). In order to quantify Ca2+ puffs from rabbit atrial myocytes, localized non-propagating elementary Ca2+ release events were recorded in permeabilized left atrial myocytes from normal (Fig. 5A) and HF hearts (Fig. 5B). To abolish RyR-mediated Ca2+ release from the SR, the RyR blocker tetracaine (4 mm) was added which resulted in an almost complete cessation of local Ca2+ release event activity (Fig. 5Ab and Bb). For further analysis cells were only included if they displayed no more than 1 event s−1 and 100 μm of confocally scanned distance (s−1 (100 μm)−1) in the presence of tetracaine. Cells were subsequently exposed to tetracaine + IP3 (5 μm) to record elementary Ca2+ release events that originated essentially exclusively from IP3Rs (Fig. 5Ac and Bc), thus representing Ca2+ puffs. In comparison to RyR-mediated Ca2+ sparks (Fig. 5C), Ca2+ puffs recorded in normal (Fig. 5D) and HF atrial rabbit myocytes were >50% smaller in amplitude, their duration was more than twofold longer and the rise time of [Ca2+]i was about 40% longer. The IP3R origin of Ca2+ puffs was confirmed with the cessation of Ca2+ release after the addition of 2-APB (Fig. 5Ad and Bd). The spatio-temporal properties of Ca2+ puffs were strikingly similar between normal and HF atrial myocytes: amplitude (ΔF/F0: 0.38 ± 0.04 in normal cells vs. 0.32 ± 0.03 in HF cells), FDHM (62 ± 7 ms normal vs. 60 ± 7 ms HF), FWHM (2.7 ± 0.1 μm normal vs. 2.7 ± 0.2 HF) and rise time (29 ± 2 ms normal vs. 28 ± 2 ms HF). The overall frequency of Ca2+ puffs was significantly increased in HF (Fig. 5E; Ca2+ puffs: 0.8 ± 0.3 s−1 (100 μm)−1 vs. 1.8 ± 0.3 s−1 (100 μm)−1). The subcellular distribution of Ca2+ puffs revealed interesting inhomogeneities (Fig. 5F). The highest Ca2+ puff frequencies were found within a distance of ≤2 μm from the sarcolemma with no obvious difference between normal and HF myocytes. In contrast, a robust increase in Ca2+ puff frequency in HF was found in the central cytosol (Fig. 5F; defined as regions with a minimal distance of 2 μm to the sarcolemmal or nuclear membrane) which increased from 0.4 ± 0.2 in normal cells to 1.9 ± 0.3 Ca2+ puffs s−1 (100 μm)−1. Interestingly, in normal cells Ca2+ puffs were more frequent in the perinuclear region (i.e. at distances <2 μm from the nuclear membrane) compared to the central cytosol; however, in HF myocytes central and perinuclear frequencies were similar. In summary, these results indicate that overall in HF IICR became more prevalent and shifted spatially towards more central regions of the cell, indicative of substantial structural and/or functional remodelling of IP3-dependent Ca2+ signalling in HF.
Figure 5.

Ca2+ puff activity in normal and HF atrial myocytes
A and B, examples of longitudinal line scan images from permeabilized normal (A) and HF (B) myocytes under control (CTRL) conditions (a), after application of tetracaine (4 mm) (b), tetracaine + IP3 (5 μm) (c), and tetracaine + IP3 + 2-APB (10 μm) (d). nuc, nucleus; c, cytosol; e, extracellular space. C, line scan image of a Ca2+ spark (top) and Ca2+ spark profile (bottom) under control conditions in a normal atrial cell. D, line scan image of a Ca2+ puff (top) and Ca2+ puff profile (bottom) under control conditions in a normal atrial cell. E, overall frequencies of Ca2+ release events under the four conditions of panels A and B in normal (black) and HF (grey) cells. F, Ca2+ puff frequencies in the subsarcolemmal, central cytosolic and perinuclear compartments of normal (black) and HF (grey) atrial cells. Panels E and F: *P < 0.05, Student's t test.
Effects of mitochondrial Ca2+ uptake on Ca2+ cycling during ECC
Mitochondria can take up significant amounts of Ca2+ and have been shown to influence the spread of activation of Ca2+ release during ECC in rat atrial myocytes (Mackenzie et al. 2004). This was confirmed here for rabbit atrial myocytes. In cells exposed to the MCU blocker Ru360 CaT amplitudes were increased, but no significant effects on diastolic [Ca2+]i and SR Ca2+ load were observed (Fig. 6A). These results suggest that mitochondrial Ca2+ uptake can contribute to [Ca2+]i regulation on a beat-to-beat basis.
Figure 6.
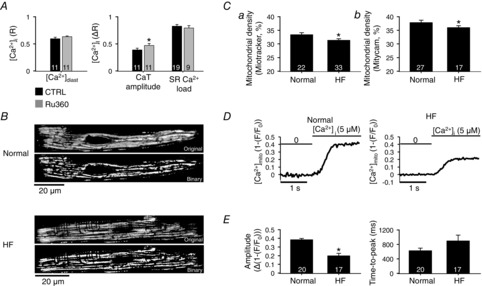
Mitochondrial density and mitochondrial Ca2+ uptake
A, average effects of Ru360 on diastolic [Ca2+]i, amplitude of AP-induced CaTs and SR Ca2+ load (CaT evoked by 10 mm caffeine). B, normal (top) and HF (bottom) atrial cell stained with Mitotracker Red FM. Raw fluorescence images and binary images used to calculate mitochondrial density. C, estimated cell volume occupied by mitochondria using Mitotracker (a) or Mitycam (b). D, effect of rapidly increasing [Ca2+]i from 0 to 5 μm on mitochondrial Ca2+ uptake in permeabilized normal and HF atrial cells. E, average effect of increasing [Ca2+]i to 5 μm on amplitude and the time to peak of [Ca2+]mito. *P < 0.05, Student's t test.
Since changes in mitochondrial density in ventricular cells associate with cardiac disease – whereby both, increases and decreases have been reported (Coleman et al. 1987; Barth et al. 1992; Hohendanner et al. 2013) – we measured mitochondrial density in rabbit atrial myocytes. For this purpose we used two different mitochondria-targeted fluorescent probes and measured mitochondrial density in normal and HF atrial myocytes. From 2D confocal images of dye-loaded myocytes, binary images were constructed (Fig. 6B) that then allowed us to determine the fraction of a cellular cross-section occupied by mitochondria (for details and quantitative evaluation of the algorithm used see Hohendanner et al. 2013). Using the probe Mitotracker (Fig. 6Ca), HF atrial myocytes revealed a significantly reduced mitochondrial content (estimated cell volume occupied by mitochondria: 33 ± 0.5% in normal cells vs. 31 ± 0.7% in HF; P < 0.05). Because accumulation of Mitotracker is membrane potential dependent and disease-related potential differences between normal and HF cannot be excluded, we used the fluorescent Ca2+-sensitive mitochondria targeted protein Mitycam (Kettlewell et al. 2009) as a second approach to quantify mitochondrial density (Fig. 6Cb). Using Mitycam the estimated mitochondrial volume was 38 ± 0.8% in normal and 36 ± 0.7% in HF myocytes (P < 0.05). Although the values obtained with Mitycam tended to be higher, both probes reported independently a significantly decreased density of mitochondria in HF. The consequences of reduced mitochondrial density for mitochondrial Ca2+ uptake were measured directly using the Mitycam probe. As shown in Fig. 6D, exposing Mitycam-loaded mitochondria in permeabilized normal atrial cells to 5 μm extramitochondrial Ca2+ resulted in a fast (<1 s) increase of mitochondrial [Ca2+] ([Ca2+]mito) that rapidly levelled off; 5 μm is approximately the [Ca2+] for half-maximal activation of the MCU (Sedova et al. 2006). In HF atrial myocytes the Ca2+ uptake response was blunted. As summarized in Fig. 6E, the [Ca2+]mito levels reached in HF cells amounted to only about half of that observed in normal cells, and the uptake kinetics were slowed in HF myocytes. These data indicate that mitochondrial Ca2+ uptake occurs on a time scale similar to CaTs and therefore has the potential to buffer Ca2+ released during ECC. The reduced mitochondrial density and Ca2+ uptake in HF may therefore contribute to the robust increase of the CaT, particularly in the centre of atrial cells (Fig. 1).
Arrhythmogenic spontaneous Ca2+ release in HF atrial myocytes
Heart failure patients are significantly more prone to develop atrial fibrillation (Cohn et al. 1991; Johnstone et al. 1992; Tsang et al. 2002). We therefore tested the hypothesis that in HF atrial myocytes spontaneous pro-arrhythmic Ca2+ release events in the form of Ca2+ waves occur more frequently, and we explored the effects of spontaneous Ca2+ waves on the electrical stability of these cells. Spontaneous Ca2+ waves and spontaneous (not electrically triggered) APs were quantified during a 2-min period of rest after >1 min of electrical pacing (arrowheads in Fig. 7A) in elevated extracellular Ca2+ (7 mm) to facilitate Ca2+ loading of the SR (Domeier et al. 2012). Spontaneous Ca2+ waves were observed in both ventricular (after stimulation at 1 Hz) and atrial (after stimulation at 0.5 Hz) myocytes at frequencies of 0.017 ± 0.003 (n = 11) and 0.025 ± 0.005 waves s–1, respectively. (No waves were observed in ventricular cells stimulated at 0.5 Hz). However, the propensity to produce spontaneous waves was significantly increased in HF atrial myocytes. Not only was the fraction of cells revealing waves increased (normal: 75% (30/40 cells); HF: 86% (18/21 cells)), but wave frequency also increased (Fig. 7Da: increase to 0.050 ± 0.010 waves s–1 in HF) and the latency (the time interval between cessation of pacing and the onset of the first spontaneous Ca2+ wave) was reduced from 56 ± 6 s to 32 ± 7 s (Fig. 7Db). The spontaneous Ca2+ waves in atrial cells revealed a unique spatio-temporal organization. As illustrated in Fig. 7B, typically the waves propagated from the site of origin (t = 0) along the longitudinal axis of the cell; however, upon closer examination it became evident that in atrial cells the leading wave front propagated through the subsarcolemmal space (thus presumably via release of Ca2+ from the j-SR) giving rise to a ‘U’-shaped wave front (t = 368 ms), illustrated in more detail in Fig. 7C. Furthermore, 39 ± 7% of all Ca2+ waves observed in normal atrial cells and 55 ± 8% of all Ca2+ waves in HF atrial cells at some point during propagation induced global Ca2+ release, presumably as a consequence of inducing an AP (Fig. 7Dc). These events showed the typical [Ca2+]i pattern of an atrial CaT with an initial ring of elevated [Ca2+]i in the cell periphery (t = 385 ms) that subsequently propagated towards the cell centre (t = 403 ms) (Huser et al. 1996; Kockskamper et al. 2001; Sheehan et al. 2006). We will refer to this type of Ca2+ wave here as an ‘arrhythmogenic Ca2+ wave’ (arrows in Fig. 7A). The overall absolute frequency of arrhythmogenic Ca2+ waves was significantly higher in HF atrial myocytes (Fig. 7Dd; 0.030 ± 0.007 vs. 0.008 ± 0.002 in normal). In contrast to atrial cells, no arrhythmogenic Ca2+ waves were observed in ventricular myocytes.
In the next set of experiments we investigated the mechanisms underlying spontaneous and arrhythmogenic Ca2+ waves. As shown in Fig. 7Da in the presence of the IP3R blocker 2-APB the frequency of spontaneous Ca2+ waves was reduced in atrial myocytes with a more pronounced effect in HF myocytes. In normal cells 2-APB reduced the wave frequency by nearly a third (statistically not significant), whereas in HF the reduction amounted to over half and became statistically significant. Since 2-APB has been reported to potentially also affect Ca2+ pumps (Peppiatt et al. 2003), we checked for this possibility by assessing the SR Ca2+ content with caffeine (10 mm) in normal cells and found no significant difference (amplitude of the caffeine-induced CaT (ΔR): 0.81 ± 0.03 (n = 19) control vs. 0.75 ± 0.07 (n = 10) in 2-APB). The data indicate that basal IP3-dependent Ca2+ signalling facilitated spontaneous SR Ca2+ release (Ca2+ waves; see also Li et al. 2005; Domeier et al. 2008), a phenomenon that was more pronounced in HF atrial cells, consistent with enhanced IP3 activity in HF atrial cells (Figs 2–5).
We further investigated the involvement of NCX in the generation of spontaneous Ca2+ waves. The electrogenic nature of NCX action has the potential to depolarize the membrane potential and to initiate Ca2+ release. Addition of the NCX inhibitor SEA-0400 (SEA) had no significant effect on the overall Ca2+ wave frequency in normal and HF atrial cells. However, NCX inhibition had a very pronounced effect when considering exclusively arrhythmogenic Ca2+ waves (Fig. 7Dc and Dd). SEA significantly reduced the fraction of cells revealing arrhythmogenic Ca2+ waves (by 62%) as well as their absolute frequency in normal atrial myocytes, and almost completely abrogated this wave type in HF atrial myocytes, suggesting an enhanced role of NCX for the occurrence of arrhythmogenic Ca2+ waves, especially in HF. We quantified NCX activity from the time constants of the decline of electrically (τCaT) and caffeine (τCaffeine)-evoked CaTs (Fig. 7Ea) as outlined in the Methods section (eqn 1). The experiments revealed two important results. First, as shown in Fig. 7Eb, SEA efficiently reduced NCX activity. Second, in HF atrial cells NCX activity was significantly increased (by 76 ± 30%; P < 0.05). The latter results are similar to the increase of NCX activity found in rabbit HF ventricular cells (Bassani et al. 1994; Pogwizd et al. 1999). Together these data indicate that HF atrial cells have a lower Ca2+ wave threshold, are more likely to show spontaneous Ca2+ waves and are more prone to develop Ca2+ waves with subsequent cell-wide depolarization due to an increased NCX activity compared to normal atrial myocytes.
Discussion
In this study, we investigated IP3-dependent Ca2+ release and its role for ECC in normal atrial cells and atrial myocytes from a left ventricular heart failure model. The key findings of our investigation were as follows: first, in HF atrial myocytes, AP-dependent CaTs were enhanced in amplitude and duration, whereas ventricular CaTs were depressed. Primarily more robust Ca2+ release from nj-SR and faster propagation of activation were responsible for this positive inotropic response. Second, HF atrial myocytes revealed enhanced IICR that preferentially facilitated Ca2+ release from central nj-SR and was, at least in part, the cause of enhanced inotropy in HF atrial myocytes. Third, HF atrial myocytes revealed a reduced mitochondrial density that resulted in reduced Ca2+ sequestration, with the consequence of larger amplitude CaTs. Finally, HF atrial myocytes revealed a higher propensity of proarrhythmic Ca2+ waves that resulted from the combined effect of enhanced IICR and increased NCX activity.
Enhanced IICR in HF: consequences for atrial ECC
Heart failure is characterized by depressed ventricular contractility and arrhythmogenesis. At the cellular level the combination of increased RyR-mediated SR Ca2+ leak (Maxwell et al. 2012), lower SERCA activity and higher NCX activity (Pogwizd et al. 1999), resulting in diminished CaTs and contractility, are hallmarks of systolic HF. The observation of depressed CaTs in single ventricular myocytes presented here (Fig. 1Af) confirmed the impaired Ca2+ cycling in ventricular myocytes in our rabbit HF model, induced by volume and pressure overload. Patients with systolic heart failure increasingly rely on the atrial contribution to diastolic ventricular filling to maintain cardiac output, but are also more prone to develop atrial fibrillation (AF; Bonow et al. 1983; Raymond et al. 1998). The irregular rhythm of AF resulting in the loss of the atrial systole and the neurohumoral activation of the renin–angiotensin–aldosterone system due to depressed ventricular contractility are contributing factors to adverse haemodynamic changes (Rahimtoola et al. 1975; Linderer et al. 1983; Daoud et al. 1996; Wijffels et al. 1997). Furthermore, atrial (Cao et al. 2002; Zhao et al. 2007) and ventricular (Go et al. 1995; Ai et al. 2005) cells show an increase of cytosolic IP3R expression under conditions of HF and AF. Although IP3Rs are outnumbered by the much more abundant RyRs by one to two orders of magnitude, they can provide an inotropic reserve by increasing diastolic [Ca2+]i in the vicinity of the Ca2+-sensitive RyR. This is particularly the case in atrial myocytes that have been shown to express up to six times as many IP3Rs as ventricular cells (Lipp et al. 2000). IICR has been postulated to have positive inotropic effects by sensitizing RyRs and bringing them closer to threshold for activation by Ca2+ and to facilitate CICR (Domeier et al. 2008; Kockskamper et al. 2008; Harzheim et al. 2009). Furthermore, Ca2+ released through IP3Rs may contribute directly to systolic CaTs (Foskett et al. 2007). A role for IICR directly contributing to systolic CaTs could be explained by the need of IP3Rs for binding of both Ca2+ and IP3 to release Ca2+ from the SR. For ‘sensitized’ IP3Rs (with pre-bound IP3) Ca2+ release is expected to be augmented during systolic CaTs, when Ca2+ fluxes through LTCC or RyRs are maximal. In our study, photorelease of caged IP3 or neurohumoral stimulation with Ang II to activate the endogenous IP3 production confirmed that increased IICR enhanced CaT amplitudes in field-stimulated normal rabbit atrial myocytes. The previously reported upregulation of IP3Rs in HF and AF (Go et al. 1995; Yamda et al. 2001; Ai et al. 2005) is consistent with our functional results. In HF atrial cells the CaT amplitude was significantly increased, predominantly resulting from enhanced Ca2+ release from the central nj-SR. This is consistent with the observed overall increased frequency of elementary Ca2+ release events originating from clusters of IP3Rs (Ca2+ puffs) in HF atrial myocytes that was particularly pronounced in areas of the nj-SR. The latter may suggest that the reported increased IP3R expression in HF is preferentially located to the central cytosolic regions in atrial cells. Interestingly, the spatial and temporal properties of Ca2+ puffs were unchanged in HF, only the frequency was increased. This would suggest that IP3R upregulation in HF may not occur through changes in IP3R cluster size, but through formation of new clusters that are preferentially located in the nj-SR. This is consistent with the finding that pharmacological IP3R inhibition (2-APB) predominantly affected j-SR Ca2+ release in normal cells whereas j-SR and nj-SR were equally affected in HF atrial cells (Fig. 4F).
The notion that IICR significantly contributed to SR Ca2+ release during ECC is underscored by the fact that IP3R inhibition (2-APB), enhanced IP3 buffering (IP3 affinity trap) or IP3 degradation (m43 phosphatase) all decreased the amplitude of CaTs in normal atrial cells and did so even more in HF atrial cells, providing additional experimental support for IICR becoming increasingly more important in HF. The fact that enhancing cytosolic IP3 degradation by expressing m43 phosphatase had no effect on the CaTs in ventricular cells further underscored the relevance of IP3 signalling for atrial ECC.
A common finding in patients with developing cardiac hypertrophy and HF is an increased level of Ang II, a hormone that exerts its effects in part by increasing IICR (Li et al. 2005; Bootman et al. 2007). In our experiments, in normal atrial cells Ang II enhanced Ca2+ signalling during ECC, similar to IP3 uncaging. However, both Ang II and IP3 uncaging had the opposite effect in HF atrial myocytes. Under steady-state pacing conditions, elevating [IP3]i levels decreased the CaT amplitude, an observation that was paralleled by a significant decrease in the SR Ca2+ content. Our results, together with the previous observation of enhanced SR Ca2+ leak in HF and the sensitivity of Ca2+ leak to stimulation of IICR (Zima et al. 2010), suggest, that in HF higher IP3R activity and enhanced IICR ultimately result in Ca2+ depletion of the SR. The data suggest that in HF atrial myocytes basal IICR is activated to the extent that it boosts atrial contractility to optimize ventricular filling. However, further stimulation of IP3 production via humoral pathways (Ang II) or by directly elevating [IP3]i (IP3 uncaging) converts this compensatory positive inotropic effect of IICR into adverse consequences for contractility by depleting the SR of Ca2+ and therefore depressing atrial CaTs and subsequently ventricular filling.
Mitochondrial Ca2+ uptake
Mitochondria occupy approximately 30% of the cell volume of a cardiac myocyte and play a crucial role in energy metabolism, redox regulation and Ca2+ homeostasis under normal and pathophysiological conditions. Although the question of whether they contribute to Ca2+ signalling during ECC on a beat-to-beat basis (O'Rourke & Blatter, 2009) is still a matter of controversy, the organelle is equipped for efficient and rapid Ca2+ uptake and release (Wendt-Gallitelli & Isenberg, 1989; Miyata et al. 1991; Sedova et al. 2006) and is able to buffer significant amounts of Ca2+ (Solaro & Briggs, 1974; Dedkova & Blatter, 2012, 2013). Mitochondria further contribute (indirectly) to shaping CaTs by providing ATP to SERCA, influencing RyR activity via ATP-dependent regulation, and generally by setting the redox environment that in turn regulates the activity of Ca2+ handling proteins (Zima et al. 2013). Mitochondria have been suggested to attenuate the propagation of release through the nj-SR by sequestering cytosolic Ca2+ to the extent that mitochondria can act as a ‘firewall’ against activation of Ca2+ release in central regions of atrial myocytes (Mackenzie et al. 2004; Bootman et al. 2011). When mitochondrial Ca2+ uptake was blocked with Ru360 (Matlib et al. 1998) in our experiments, CaT amplitudes during ECC were increased without affecting SR Ca2+ content. Changes in mitochondrial function and/or structure are of great interest in cardiac disease (Dominic et al. 2014). In several disease models increases as well as decreases in mitochondrial density have been reported, suggesting that mitochondrial remodelling in cardiac disease is complex (Coleman et al. 1987; Barth et al. 1992; Hohendanner et al. 2013). We observed a decreased mitochondrial density in HF atrial cells, suggesting a smaller capacity to buffer [Ca2+]i while the cell size was not significantly increased (1017 ± 50 vs. 1255 ± 118 μm2 in normal (n = 29) vs. HF (n = 36) cells; P = 0.09). Even though the difference in mitochondrial density between normal and HF atrial myocytes appeared rather small at first glance, the consequences for mitochondrial Ca2+ uptake were significant. When testing mitochondrial Ca2+ uptake directly, not only was the rate of uptake slowed, the maximum uptake was significantly reduced to approximately half (Fig. 6). Our data suggest that atrial remodelling during HF involves a reduction in mitochondrial density that also turns out to have beneficial effects for ECC. By attenuating mitochondrial Ca2+ uptake during ECC, SR Ca2+ release became more robust and efficient which ultimately resulted in larger and prolonged CaTs and a positive inotropic effect that would facilitate ventricular filling.
Arrhythmogenic SR Ca2+ release in HF
Our data show that HF atrial cells reveal a higher frequency of spontaneous Ca2+ waves (Fig. 7). Our experiments indicate that increased IICR plays a critical role. While IP3R inhibition decreased the Ca2+ wave frequency in both normal and HF atrial cells, the effect was significantly more pronounced in HF cells. This is consistent with the upregulation of IP3 signalling in HF and the more pronounced effect of IICR on electrically evoked CaTs (Fig. 3). In addition, atrial cells in general were prone to develop arrhythmogenic Ca2+ waves (around 40% of all Ca2+ waves in normal atrial cells), i.e. waves that led to subsequent global Ca2+ release by triggering an AP and SR Ca2+ release typical for atrial ECC. Arrhythmogenic Ca2+ waves were not observed in ventricular cells, thus appear to be a unique feature of atrial cells and occurred more often in HF. This is consistent with findings in atrial cells from hearts with myocardial infarction, where the frequency of spontaneous depolarizations was significantly increased (Kettlewell et al. 2013). The spatial propagation pattern of Ca2+ waves in atrial cells revealed a unique feature where the front of the propagating wave advanced through the subsarcolemmal space via j-SR Ca2+ release, thus preceding the centripetal propagation of CICR from nj-SR. In rabbit atrial cells NCX is exclusively located in the cell periphery due to the lack of transverse tubules. Thus, the subsarcolemmal propagation of the Ca2+ wave front exposed NCX for a prolonged period of time to high [Ca2+]i, which might play a critical role in the generation of arrhythmogenic Ca2+ waves. Because of the electrogenic nature of NCX, extrusion of elevated [Ca2+]i will lead to membrane depolarization to the extent that it can trigger an AP. We tested this hypothesis by inhibiting NCX pharmacologically (SEA). NCX inhibition decreased the frequency of arrhythmogenic Ca2+ waves significantly in normal atrial cells and did so even more in HF atrial cells, indicating that NCX in forward mode can trigger a depolarization of the cell during spontaneous Ca2+ waves. Consistent with our finding of an increased propensity of arrhythmogenic Ca2+ waves in HF, NCX activity was indeed enhanced in HF atrial myocytes (Fig. 7E).
Conclusions
In hearts with ventricular failure and decreased cardiac output, atrial cells might compensate to maintain ventricular filling despite increased end-diastolic pressure, with a positive inotropic response that results from enhanced atrial CaTs due to increased IICR. The positive inotropic response of atrial cells is aided by reduced Ca2+ buffering by mitochondria during ECC. Upregulation of IICR, on the other hand, also has undesirable consequences in form of a significantly higher propensity for arrhythmogenic Ca2+ release. The combined action of enhanced IICR together with a higher NCX activity are likely contributors to the arrhythmogenicity found in HF and may form the substrate for the frequently encountered AF in this disease state.
Glossary
- AF
atrial fibrillation
- Ang II
angiotensin II
- AP
action potential
- 2-APB
2-aminoethoxydiphenyl borate
- BSA
bovine serum albumin
- [Ca2+]i
cytosolic free Ca2+ concentration
- [Ca2+]mito
mitochondrial free Ca2+ concentration
- [Ca2+]SR
sarcoplasmic reticulum free Ca2+ concentration
- CaT
Ca2+ transient
- CICR
Ca2+-induced Ca2+release
- CRU
Ca2+ release unit
- ECC
excitation–contraction coupling
- FDHM
full duration at half-maximum
- FIRE
fluorescent IP3-responsive element
- FRET
florescence resonance energy transfer
- FWHM
full width at half-maximum
- GFP
green fluorescent protein
- GPCR
Gq-protein coupled receptor
- HF
heart failure
- IICR
IP3-induced Ca2+release
- IP3
inositol-1,4,5-trisphosphate
- IP3R
IP3 receptor
- j-SR
junctional sarcoplasmic reticulum
- LTCC
L-type Ca2+ channel
- MCU
mitochondrial calcium uniporter
- NCX
Na+–Ca2+ exchange
- nj-SR
non-junctional sarcoplasmic reticulum
- RyR
ryanodine receptor
- SEA
SEA-0400, 2-[4-[(2,5-diuorophenyl) methoxy]phenoxy]-5-ethoxyaniline
- SERCA
sarcoplasmic–endoplasmic reticulum Ca2+ ATPase
- SR
sarcoplasmic reticulum
- TTP
time to peak
Additional information
Competing interests
None declared.
Author contributions
Conception and design of the experiments: F.H, L.A.B. Collection, analysis and interpretation of data: F.H., S.W., J.T.M. S.K., S.A., G.L.S., V.A.L., L.A.B. Drafting the article: F.H., L.A.B. All authors have approved the final version of the manuscript.
Funding
This work was supported by National Institutes of Health grants HL62231, HL80101 and HL101235 and the Leducq Foundation (to L.A.B.).
References
- Ai X, Curran JW, Shannon TR, Bers DM. Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- Alpert JS, Petersen P. Godtfredsen J. Atrial fibrillation: natural history, complications, and management. Annu Rev Med. 1988;39:41–52. doi: 10.1146/annurev.me.39.020188.000353. [DOI] [PubMed] [Google Scholar]
- Barth E, Stammler G, Speiser B. Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24:669–681. doi: 10.1016/0022-2828(92)93381-s. [DOI] [PubMed] [Google Scholar]
- Bassani JW, Bassani RA. Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Pogwizd SM. Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97(Suppl. 1):I36–42. doi: 10.1007/s003950200027. [DOI] [PubMed] [Google Scholar]
- Bonow RO, Frederick TM, Bacharach SL, Green MV, Goose PW, Maron BJ. Rosing DR. Atrial systole and left ventricular filling in hypertrophic cardiomyopathy: effect of verapamil. Am J Cardiol. 1983;51:1386–1391. doi: 10.1016/0002-9149(83)90317-x. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Harzheim D, Smyrnias I, Conway SJ. Roderick HL. Temporal changes in atrial EC-coupling during prolonged stimulation with endothelin-1. Cell Calcium. 2007;42:489–501. doi: 10.1016/j.ceca.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Higazi DR, Coombes S. Roderick HL. Calcium signalling during excitation–contraction coupling in mammalian atrial myocytes. J Cell Sci. 2006;119:3915–3925. doi: 10.1242/jcs.03223. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Smyrnias I, Thul R, Coombes S. Roderick HL. Atrial cardiomyocyte calcium signalling. Biochim Biophys Acta. 2011;1813:922–934. doi: 10.1016/j.bbamcr.2011.01.030. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Berlin JR. Lederer WJ. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;238:1419–1423. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- Cannell MB. Kong CH. Local control in cardiac E–C coupling. J Mol Cell Cardiol. 2012;52:298–303. doi: 10.1016/j.yjmcc.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Cao K, Xia X, Shan Q, Chen Z, Chen X. Huang Y. Changes of sarcoplamic reticular Ca2+-ATPase and IP3-I receptor mRNA expression in patients with atrial fibrillation. Chin Med J (Engl) 2002;115:664–667. [PubMed] [Google Scholar]
- Chen-Izu Y, McCulle SL, Ward CW, Soeller C, Allen BM, Rabang C, Cannell MB, Balke CW. Izu LT. Three-dimensional distribution of ryanodine receptor clusters in cardiac myocytes. Biophys J. 2006;91:1–13. doi: 10.1529/biophysj.105.077180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CP, Suzuki M, Ohte N, Ohno M, Wang ZM. Little WC. Altered ventricular and myocyte response to angiotensin II in pacing-induced heart failure. Circ Res. 1996;78:880–892. doi: 10.1161/01.res.78.5.880. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Johnson G, Ziesche S, Cobb F, Francis G, Tristani F, Smith R, Dunkman WB, Loeb H, Wong M, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med. 1991;325:303–310. doi: 10.1056/NEJM199108013250502. [DOI] [PubMed] [Google Scholar]
- Coleman R, Silbermann M, Gershon D. Reznick AZ. Giant mitochondria in the myocardium of aging and endurance-trained mice. Gerontology. 1987;33:34–39. doi: 10.1159/000212851. [DOI] [PubMed] [Google Scholar]
- Coombes S. Timofeeva Y. Sparks and waves in a stochastic fire–diffuse–fire model of Ca2+ release. Phys Rev E Stat Nonlin Soft Matter Phys. 2003;68:021915. doi: 10.1103/PhysRevE.68.021915. [DOI] [PubMed] [Google Scholar]
- Daoud EG, Weiss R, Bahu M, Knight BP, Bogun F, Goyal R, Harvey M, Strickberger SA, Man KC. Morady F. Effect of an irregular ventricular rhythm on cardiac output. Am J Cardiol. 1996;78:1433–1436. doi: 10.1016/s0002-9149(97)89297-1. [DOI] [PubMed] [Google Scholar]
- Dedkova EN. Blatter LA. Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol. 2012;52:48–61. doi: 10.1016/j.yjmcc.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedkova EN. Blatter LA. Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol. 2013;58:125–133. doi: 10.1016/j.yjmcc.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Islam MA, Pogwizd SM. Bers DM. Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J Physiol. 2002;539:133–143. doi: 10.1113/jphysiol.2001.012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier TL, Blatter LA. Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier TL, Maxwell JT. Blatter LA. β-Adrenergic stimulation increases the intra-sarcoplasmic reticulum Ca2+ threshold for Ca2+ wave generation. J Physiol. 2012;590:6093–6108. doi: 10.1113/jphysiol.2012.236117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA. Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation–contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H596–H604. doi: 10.1152/ajpheart.01155.2007. [DOI] [PubMed] [Google Scholar]
- Dominic EA, Ramezani A, Anker SD, Verma M, Mehta N. Rao M. Mitochondrial cytopathies and cardiovascular disease. Heart. 2014;100:611–618. doi: 10.1136/heartjnl-2013-304657. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH. Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS. Marks AR. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest. 1995;95:888–894. doi: 10.1172/JCI117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harzheim D, Movassagh M, Foo RS, Ritter O, Tashfeen A, Conway SJ, Bootman MD. Roderick HL. Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:11406–11411. doi: 10.1073/pnas.0905485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohendanner F, Ljubojevic S, Macquaide N, Sacherer M, Sedej S, Biesmans L, Wakula P, Platzer D, Sokolow S, Herchuelz A, et al. Intracellular dyssynchrony of diastolic cytosolic [Ca2+] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ Res. 2013;113:527–538. doi: 10.1161/CIRCRESAHA.113.300895. [DOI] [PubMed] [Google Scholar]
- Huser J, Lipsius SL. Blatter LA. Calcium gradients during excitation–contraction coupling in cat atrial myocytes. J Physiol. 1996;494:641–651. doi: 10.1113/jphysiol.1996.sp021521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone D, Limacher M, Rousseau M, Liang CS, Ekelund L, Herman M, Stewart D, Guillotte M, Bjerken G, Gaasch W, et al. Clinical characteristics of patients in studies of left ventricular dysfunction (SOLVD) Am J Cardiol. 1992;70:894–900. doi: 10.1016/0002-9149(92)90734-g. [DOI] [PubMed] [Google Scholar]
- Kapoor N, Maxwell JT, Mignery GA, Will D, Blatter LA. Banach K. Spatially defined InsP3-mediated signaling in embryonic stem cell-derived cardiomyocytes. PLoS One. 2014;9:e83715. doi: 10.1371/journal.pone.0083715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettlewell S, Burton FL, Smith GL. Workman AJ. Chronic myocardial infarction promotes atrial action potential alternans, afterdepolarizations, and fibrillation. Cardiovasc Res. 2013;99:215–224. doi: 10.1093/cvr/cvt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettlewell S, Cabrero P, Nicklin SA, Dow JA, Davies S. Smith GL. Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. J Mol Cell Cardiol. 2009;46:891–901. doi: 10.1016/j.yjmcc.2009.02.016. [DOI] [PubMed] [Google Scholar]
- Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA. Blatter LA. Activation and propagation of Ca2+ release during excitation–contraction coupling in atrial myocytes. Biophys J. 2001;81:2590–2605. doi: 10.1016/S0006-3495(01)75903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockskamper J, Zima AV, Roderick HL, Pieske B, Blatter LA. Bootman MD. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol. 2008;45:128–147. doi: 10.1016/j.yjmcc.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, O'Neill SC, Tao T, Li Y, Eisner D. Zhang H. Mechanisms by which cytoplasmic calcium wave propagation and alternans are generated in cardiac atrial myocytes lacking T-tubules-insights from a simulation study. Biophys J. 2012;102:1471–1482. doi: 10.1016/j.bpj.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zima AV, Sheikh F, Blatter LA. Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate (IP3)-receptor type 2-deficient mice. Circ Res. 2005;96:1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- Linderer T, Chatterjee K, Parmley WW, Sievers RE, Glantz SA. Tyberg JV. Influence of atrial systole on the Frank–Starling relation and the end-diastolic pressure–diameter relation of the left ventricle. Circulation. 1983;67:1045–1053. doi: 10.1161/01.cir.67.5.1045. [DOI] [PubMed] [Google Scholar]
- Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W. Bootman MD. Functional InsP3 receptors that may modulate excitation–contraction coupling in the heart. Curr Biol. 2000;10:939–942. doi: 10.1016/s0960-9822(00)00624-2. [DOI] [PubMed] [Google Scholar]
- Mackenzie L, Roderick HL, Berridge MJ, Conway SJ. Bootman MD. The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J Cell Sci. 2004;117:6327–6337. doi: 10.1242/jcs.01559. [DOI] [PubMed] [Google Scholar]
- Majerus PW, Kisseleva MV. Norris FA. The role of phosphatases in inositol signaling reactions. J Biol Chem. 1999;274:10669–10672. doi: 10.1074/jbc.274.16.10669. [DOI] [PubMed] [Google Scholar]
- Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N. Bers DM. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem. 1998;273:10223–10231. doi: 10.1074/jbc.273.17.10223. [DOI] [PubMed] [Google Scholar]
- Maxwell JT. Blatter LA. Facilitation of cytosolic calcium wave propagation by local calcium uptake into the sarcoplasmic reticulum in cardiac myocytes. J Physiol. 2012;590:6037–6045. doi: 10.1113/jphysiol.2012.239434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell JT, Domeier TL. Blatter LA. Dantrolene prevents arrhythmogenic Ca release in heart failure. Am J Physiol Heart Circ Physiol. 2012;302:H953–H963. doi: 10.1152/ajpheart.00936.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Min JY. Simon R. Effects of angiotensin II on inotropy and intracellular Ca2+ handling in normal and hypertrophied rat myocardium. J Mol Cell Cardiol. 1998;30:2507–2518. doi: 10.1006/jmcc.1998.0813. [DOI] [PubMed] [Google Scholar]
- Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD. Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 1991;261:H1123–H1134. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- Nicod P, Hillis LD, Winniford MD. Firth BG. Importance of the "atrial kick" in determining the effective mitral valve orifice area in mitral stenosis. Am J Cardiol. 1986;57:403–407. doi: 10.1016/0002-9149(86)90761-7. [DOI] [PubMed] [Google Scholar]
- O'Rourke B. Blatter LA. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol. 2009;46:767–774. doi: 10.1016/j.yjmcc.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT. Roderick HL. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium. 2003;34:97–108. doi: 10.1016/s0143-4160(03)00026-5. [DOI] [PubMed] [Google Scholar]
- Picht E, Zima AV, Blatter LA. Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM. Nonreentrant mechanisms underlying spontaneous ventricular arrhythmias in a model of nonischemic heart failure in rabbits. Circulation. 1995;92:1034–1048. doi: 10.1161/01.cir.92.4.1034. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Qi M, Yuan W, Samarel AM. Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W. Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium–calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Rahimtoola SH, Ehsani A, Sinno MZ, Loeb HS, Rosen KM. Gunnar RM. Left atrial transport function in myocardial infarction. Importance of its booster pump function. Am J Med. 1975;59:686–694. doi: 10.1016/0002-9343(75)90229-6. [DOI] [PubMed] [Google Scholar]
- Raymond RJ, Lee AJ, Messineo FC, Manning WJ. Silverman DI. Cardiac performance early after cardioversion from atrial fibrillation. Am Heart J. 1998;136:435–442. doi: 10.1016/s0002-8703(98)70217-0. [DOI] [PubMed] [Google Scholar]
- Remus TP, Zima AV, Bossuyt J, Bare DJ, Martin JL, Blatter LA, Bers DM. Mignery GA. Biosensors to measure inositol 1,4,5-trisphosphate concentration in living cells with spatiotemporal resolution. J Biol Chem. 2006;281:608–616. doi: 10.1074/jbc.M509645200. [DOI] [PubMed] [Google Scholar]
- Sedova M, Dedkova EN. Blatter LA. Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am J Physiol Cell Physiol. 2006;291:C840–C850. doi: 10.1152/ajpcell.00619.2005. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Pogwizd SM. Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- Sheehan KA. Blatter LA. Regulation of junctional and non-junctional sarcoplasmic reticulum calcium release in excitation–contraction coupling in cat atrial myocytes. J Physiol. 2003;546:119–135. doi: 10.1113/jphysiol.2002.026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan KA, Zima AV. Blatter LA. Regional differences in spontaneous Ca2+ spark activity and regulation in cat atrial myocytes. J Physiol. 2006;572:799–809. doi: 10.1113/jphysiol.2005.103267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkryl VM, Maxwell JT. Blatter LA. A novel method for spatially complex diffraction-limited photoactivation and photobleaching in living cells. J Physiol. 2011;590:1093–1100. doi: 10.1113/jphysiol.2011.223446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ. Briggs FN. Estimating the functional capabilities of sarcoplasmic reticulum in cardiac muscle. Calcium binding. Circ Res. 1974;34:531–540. doi: 10.1161/01.res.34.4.531. [DOI] [PubMed] [Google Scholar]
- Stern MD, Song LS, Cheng H, Sham JS, Yang HT, Boheler KR. Rios E. Local control models of cardiac excitation–contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol. 1999;113:469–489. doi: 10.1085/jgp.113.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul R, Coombes S, Roderick HL. Bootman MD. Subcellular calcium dynamics in a whole-cell model of an atrial myocyte. Proc Natl Acad Sci USA. 2012;109:2150–2155. doi: 10.1073/pnas.1115855109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovey SC, de Smet P, Lipp P, Thomas D, Young KW, Missiaen L, De Smedt H, Parys JB, Berridge MJ, Thuring J, Holmes A. Bootman MD. Calcium puffs are generic InsP3-activated elementary calcium signals and are downregulated by prolonged hormonal stimulation to inhibit cellular calcium responses. J Cell Sci. 2001;114:3979–3989. doi: 10.1242/jcs.114.22.3979. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Clarke JD, Richards MA, Eisner DA. Dibb KM. Calcium signalling microdomains and the t-tubular system in atrial mycoytes: potential roles in cardiac disease and arrhythmias. Cardiovasc Res. 2013;98:192–203. doi: 10.1093/cvr/cvt018. [DOI] [PubMed] [Google Scholar]
- Tsang TS, Gersh BJ, Appleton CP, Tajik AJ, Barnes ME, Bailey KR, Oh JK, Leibson C, Montgomery SC. Seward JB. Left ventricular diastolic dysfunction as a predictor of the first diagnosed nonvalvular atrial fibrillation in 840 elderly men and women. J Am Coll Cardiol. 2002;40:1636–1644. doi: 10.1016/s0735-1097(02)02373-2. [DOI] [PubMed] [Google Scholar]
- Vasilevski O, Grubb DR, Filtz TM, Yang S, McLeod-Dryden TJ, Luo J, Karna D, Chen J. Woodcock EA. Ins(1,4,5)P3 regulates phospholipase Cβ1 expression in cardiomyocytes. J Mol Cell Cardiol. 2008;45:679–684. doi: 10.1016/j.yjmcc.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Levy D, Vasan RS, Leip EP, Wolf PA, D'Agostino RB, Murabito JM, Kannel WB. Benjamin EJ. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation. 2003;107:2920–2925. doi: 10.1161/01.CIR.0000072767.89944.6E. [DOI] [PubMed] [Google Scholar]
- Wendt-Gallitelli MF. Isenberg G. X-ray microanalysis of single cardiac myocytes frozen under voltage-clamp conditions. Am J Physiol Heart Circ Physiol. 1989;256:H574–H583. doi: 10.1152/ajpheart.1989.256.2.H574. [DOI] [PubMed] [Google Scholar]
- Wijffels MC, Kirchhof CJ, Dorland R, Power J. Allessie MA. Electrical remodeling due to atrial fibrillation in chronically instrumented conscious goats: roles of neurohumoral changes, ischemia, atrial stretch, and high rate of electrical activation. Circulation. 1997;96:3710–3720. doi: 10.1161/01.cir.96.10.3710. [DOI] [PubMed] [Google Scholar]
- Yamda J, Ohkusa T, Nao T, Ueyama T, Yano M, Kobayashi S, Hamano K, Esato K. Matsuzaki M. Up-regulation of inositol 1,4,5 trisphosphate receptor expression in atrial tissue in patients with chronic atrial fibrillation. J Am Coll Cardiol. 2001;37:1111–1119. doi: 10.1016/s0735-1097(01)01144-5. [DOI] [PubMed] [Google Scholar]
- Zhao ZH, Zhang HC, Xu Y, Zhang P, Li XB, Liu YS. Guo JH. Inositol-1,4,5-trisphosphate and ryanodine-dependent Ca2+ signaling in a chronic dog model of atrial fibrillation. Cardiology. 2007;107:269–276. doi: 10.1159/000095517. [DOI] [PubMed] [Google Scholar]
- Zima AV. Blatter LA. Inositol-1,4,5-trisphosphate-dependent Ca2+ signalling in cat atrial excitation–contraction coupling and arrhythmias. J Physiol. 2004;555:607–615. doi: 10.1113/jphysiol.2003.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Bers DM. Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Pabbidi MR, Lipsius SL. Blatter LA. Effects of mitochondrial uncoupling on Ca2+ signaling during excitation–contraction coupling in atrial myocytes. Am J Physiol Heart Circ Physiol. 2013;304:H983–H993. doi: 10.1152/ajpheart.00932.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]