

Abstract
Time-dependent refractoriness of calcium (Ca2+) release in cardiac myocytes is an important factor in determining whether pro-arrhythmic release patterns develop. At the subcellular level of the Ca2+ spark, recent studies have suggested that recovery of spark amplitude is controlled by local sarcoplasmic reticulum (SR) refilling whereas refractoriness of spark triggering depends on both refilling and the sensitivity of the ryanodine receptor (RyR) release channels that produce sparks. Here we studied regulation of Ca2+ spark refractoriness in mouse ventricular myocytes by examining how β-adrenergic stimulation influenced sequences of Ca2+ sparks originating from individual RyR clusters. Our protocol allowed us to separately measure recovery of spark amplitude and delays between successive sparks, and data were interpreted quantitatively through simulations with a stochastic mathematical model. We found that, compared with spark sequences measured under control conditions: (1) β-adrenergic stimulation with isoproterenol (isoprenaline) accelerated spark amplitude recovery and decreased spark-to-spark delays; (2) activating protein kinase A (PKA) with forskolin accelerated amplitude recovery but did not affect spark-to-spark delays; (3) inhibiting PKA with H89 retarded amplitude recovery and increased spark-to-spark delays; (4) preventing phosphorylation of the RyR at serine 2808 with a knock-in mouse prevented the decrease in spark-to-spark delays seen with β-adrenergic stimulation; (5) inhibiting either PKA or Ca2+/calmodulin-dependent protein kinase II (CaMKII) during β-adrenergic stimulation prevented the decrease in spark-to-spark delays seen without inhibition. The results suggest that activation of either PKA or CaMKII is sufficient to speed SR refilling, but activation of both kinases appears necessary to observe increased RyR sensitivity. The data provide novel insight into β-adrenergic regulation of Ca2+ release refractoriness in mouse myocytes.
Key points
Refractoriness of calcium release in heart cells is altered in several disease states, but the physiological mechanisms that regulate this process are incompletely understood.
We examined refractoriness of calcium release in mouse ventricular myocytes and investigated how activation of different intracellular signalling pathways influenced this process.
We found that refractoriness of calcium release is abbreviated by stimulation of the ‘fight-or-flight’ response, and that simultaneous activation of multiple intracellular signalling pathways contributes to this response.
Data obtained under several conditions at the subcellular, microscopic level were consistent with results obtained at the cellular level.
The results provide insight into regulation of cardiac calcium release and how alterations to this process may increase arrhythmia risk under different conditions.
Introduction
In ventricular myocytes, release of calcium (Ca2+) from the sarcoplasmic reticulum (SR) is both critical for contraction and centrally involved in arrhythmia initiation. The SR releases Ca2+ in response to Ca2+ entry through L-type Ca2+ channels each time the heart beats, and this released Ca2+ enables contraction by binding to myofilaments. SR Ca2+ release, however, can occur spontaneously when the SR is overloaded with Ca2+ or the channels responsible for release, ryanodine receptors (RyRs), exhibit abnormal gating. Spontaneous release induces inward membrane current when Ca2+ is extruded from the cell through the electrogenic Na+–Ca2+ exchanger (Pogwizd et al. 2001). When this spontaneous release occurs simultaneously in many cells, it can trigger inappropriate action potentials in ventricular myocytes and initiate potentially lethal arrhythmias (Xie et al. 2010).
After Ca2+ release begins, local depletion of SR Ca2+ stores is the most important factor involved in release termination (DelPrincipe et al. 1999; Sobie et al. 2002; Terentyev et al. 2002; Sobie & Lederer, 2012; Cannell et al. 2013; Stern et al. 2013). This local depletion enables a refractory period, during which it is more difficult (but not impossible) to initiate a second Ca2+ release event (Cheng et al. 1996; Szentesi et al. 2004; Sobie et al. 2005; Ramay et al. 2011). Ca2+ release refractoriness is an important factor in determining whether potentially arrhythmogenic patterns develop. Refractoriness is shortened in disease states that are associated with Ca2+-triggered ventricular arrhythmias, such as catecholaminergic polymorphic ventricular tachycardia (CPVT) (Kornyeyev et al. 2012; Liu et al. 2013; Brunello et al. 2013). Refractoriness is also important in the development of alternating large–small–large patterns of release, so-called Ca2+ transient alternans (Rovetti et al. 2010; Shkryl et al. 2012). These Ca2+ release alternans can produce beat-to-beat alterations in action potential duration, a pattern that predisposes hearts to arrhythmias (Qu et al. 2013). In fact, it has been suggested that refractoriness is governed by the ‘Goldilocks Principle’ such that abnormally short refractoriness and abnormally long refractoriness can both be detrimental, albeit for different reasons (Liu et al. 2012). This fine balance indicates the need to obtain reliable quantitative measurements of changes in release refractoriness under different conditions.
Besides being altered in disease states, Ca2+ release refractoriness can be modified physiologically, specifically after stimulation of β-adrenergic receptors (Szentesi et al. 2004). Our group has previously shown, at the level of the Ca2+ spark, that β-adrenergic stimulation accelerates recovery from refractoriness, and we demonstrated that a combination of experiments and mathematical modelling can provide quantitative insight into the changes that occur after stimulation (Ramay et al. 2011). The previous data (Ramay et al. 2011), however, did not address the particular signalling pathways, or the specific phosphorylation sites, that are responsible for shortening the refractory period. In particular, both protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinase II (CaMKII) are activated when β-adrenergic receptors are stimulated with an agonist such as isoproterenol (Curran et al. 2007, 2014; Gutierrez et al. 2013), even in quiescent cells that are not undergoing Ca2+ transients, and each kinase may contribute to the observed changes.
In this study we examined restitution of Ca2+ sparks in mouse ventricular myocytes under several conditions to delineate the pathways responsible for accelerating the recovery of Ca2+ release after β-adrenergic stimulation. By coupling the experimental results to simulations with a stochastic mathematical model of the Ca2+ spark (Sobie et al. 2002; Ramay et al. 2011; Lee et al. 2013), we developed quantitative predictions about whether particular interventions cause a change in the rate of refilling, altered RyR sensitivity, or both. Our experimental results suggest that activation of either PKA or CaMKII is sufficient to speed the rate of refilling, but that activation of both kinases is required to observe maximal changes in RyR sensitivity during β-adrenergic stimulation. Moreover, the data also suggest that phosphorylation of the RyR at serine 2808 is involved in the increased RyR sensitivity observed during β-adrenergic stimulation, potentially helping to define a physiological role for RyR phosphorylation at this residue.
Methods
Ethical approval
This investigation conforms with the Guide for the Care and Use of Laboratory Animals by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). All experimental protocols were approved by the Institutional Animal Care and Use Committee of Icahn School of Medicine at Mount Sinai and with the permission of the State Veterinary Administration and according to Swiss Federal Animal protection law (permit BE126/12).
Isolation of ventricular myocytes
C57Bl/6 (Charles River Laboratories; Wilmington, MA, USA) and S2808A knock-in mice (Benkusky et al. 2007), generated in a sv129/C57B1/6 background, were used in this study at 2.5–4 months of age. In total we used N = 25 control mice and N = 5 S2808A mice. Standard enzymatic dissociation techniques (Wolska & Solaro, 1996; Guatimosim et al. 2001) were used to isolate ventricular myocytes. Briefly, mice were given an intraperitoneal injection of a lethal dose of pentobarbital (100 mg kg−1). Hearts were rapidly removed from the chest cavity and retrogradely perfused on a Langendorff apparatus with Ca2+-free-Tyrode solution (NaCl 140 mM, KCl 5.4, MgCl2 1.1, Hepes 5, NaH2PO4 1, glucose 10; pH 7.3, adjusted with NaOH; osmolarity 300 mosmol l–1) for 5 min at 37°C. For tissue digestion, the solution was then switched to Tyrode solution containing 36 μm Ca2+, collagenase (112 U ml−1; Worthington, Lakewood, NJ, USA) and protease (0.16 U ml−1) for approximately 5 min. Ventricles were removed from the heart and cut into small pieces in Tyrode solution containing 200 μm CaCl2, yielding individual cells. [Ca2+] was gradually increased from 200 μm to 1 mm over a period of 30 min.
Solutions
During measurement, cells were perfused with Tyrode solution: NaCl 140, KCl 5.4, MgCl2 1.1, Hepes 5, NaH2PO4 1, glucose 10, CaCl2 1.8, pH 7.4 and osmolarity of 300 mosmol l–1. We studied six experimental groups besides control: (1) H89 (1 μm H89 to block PKA); (2) ISO (100 nm isoproterenol to stimulate β-adrenergic receptors); (3) ISO + KN92 (100 nm isoproterenol plus 1 μm KN92 (EMD chemicals, Darmstadt, Germany), an inactive form of CaMKII blocker); (4) ISO + KN93 (100 nm isoproterenol plus 1 μm KN93, an active CaMKII blocker); (5) ISO + H89 (100 nm isoproterenol plus 1 μm H89, a blocker of PKA); (6) Forskolin (1 μm forskolin (Cayman chemicals, Ann Arbor, MI, USA) to activate adenylyl cyclase). Drugs were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless noted otherwise. Experiments were performed at room temperature (22°C). See Table1 for a summary of the drugs used in the experiments.
Table 1.
Summary of drugs used in the experimental protocol
| Drug | Concentration (μm) | Incubation time (min) | Purpose |
|---|---|---|---|
| Ryanodine | 0.05 | <8 | Activate repetitive sparks |
| Isoproterenol | 0.1 | 30 | β-Adrenergic agonist |
| Forskolin | 1 | 3 | Activate adenylyl cyclase |
| KN92 | 1 | 30 | Inactive CaMKII blocker |
| KN93 | 1 | 30 | CaMKII blocker |
| H89 | 1 | 30 | PKA blocker |
| Caffeine | 10,000 | Instant application | Empty SR |
Confocal recordings
Isolated ventricular myocytes were loaded with the acetoxymethyl ester (AM) form of the Ca2+ indicator fluo-3 (fluo-3-AM; 5 μM, 30 min loading; 30 min deesterification; Invitrogen). During Ca2+ spark restitution measurements, EGTA-AM (5 μM, 30 min loading; 30 min deesterification; Invitrogen, Carlsbad, CA, USA) was also added to suppress intracellular Ca2+ waves (Ramay et al. 2011). Cells were imaged using a confocal microscope (LSM 5 Exciter, Carl Zeiss USA, Thornwood, NY or Fluoview 1000, Olympus USA, Center Valley, PA) operating in line scan mode. To record intracellular [Ca2+], fluo-3 was excited at 488 nm, and fluorescence above 505 nm was acquired.
Ca2+ spark restitution experiments
The ‘ryanodine method’ (Sobie et al. 2005) was used to record repetitive Ca2+ sparks originating from a single cluster of RyRs. This protocol is based on the idea that a ryanodine-bound RyR will open much more frequently and can serve as a potential Ca2+ spark trigger (Buck et al. 1992; Bidasee et al. 2003). Through this approach repetitive sparks can be then recorded from a single cluster of RyRs (Sobie et al. 2005; Ramay et al. 2011).
Before recording, cells were exposed to the drugs in the particular experimental groups (H89, ISO, ISO + KN92; ISO + KN93; ISO + H89) for 30 min except the Forskolin group where exposure lasted 3 min. The Tyrode solution containing drugs and 50 nm ryanodine was then applied to the cell, and active Ca2+ spark sites within the myocyte were identified and scanned at high speed (0.96–3.07 ms line−1) for 10 s. Ryanodine exposure was limited to 8 min to minimize the probability that RyR sub-conductance states could produce extremely long sparks (Ramay et al. 2011).
Ca2+ transient experiments
We followed the same exposure times for different drugs as in spark restitution experiments. Cells were perfused with Tyrode solution containing drugs and field stimulated for 25 s at 1 Hz, at which point steady-state Ca2+ transients were recorded. After stimulation, cells were rapidly exposed to 10 mm caffeine, and resulting changes in intracellular [Ca2+] were recorded to assess the SR Ca2+ load.
Data analysis
Repetitive Ca2+ sparks and Ca2+ transients were analysed using custom programs written in MATLAB (Mathworks, Natick, MA, USA). Briefly, background fluorescence was subtracted from the confocal image. To convert to units of F/F0, fluorescence averaged over either the whole cell (Ca2+ transient experiments) or a 1.8 μm region (Ca2+ spark experiments) was normalized to fluorescence recorded during a quiescent period.
Ca2+ spark restitution analyses consisted of two parameters for each spark pair: (1) amplitude of the second normalized to the first; and (2) the delay between sparks in milliseconds. The same criteria for exclusion were used as in Ramay et al. (2011). Briefly: (1) while all spark-to-spark delays were included in histograms, amplitude ratios were excluded if the initial spark in the pair occurred soon after a previous event (200 ms); (2) active sites were excluded if: (i) there was Ca2+ spark activity recorded from two sites close by, which made it difficult to resolve the position of the sparks; or (ii) very long sparks (>200 ms), indicating RyR sub-conductance states, were observed; (3) confocal images were excluded if the frequency of Ca2+ sparks exceeded 15 sparks (100 μm)–1 s–1.
Ca2+ transient analyses included two parameters for each experimental group: (1) transient decay; and (2) fractional release (Fig.1B). Transient decay is represented by time constant (τ, in s), extracted from an exponential function fit to the decaying phase of the Ca2+ transient (amplitude × e−time/τ). Fractional release, calculated as field-stimulated Ca2+ transient amplitude divided by caffeine-induced Ca2+ transient amplitude, describes the fraction of the SR depleted in response to the field stimulus.
Figure 1.
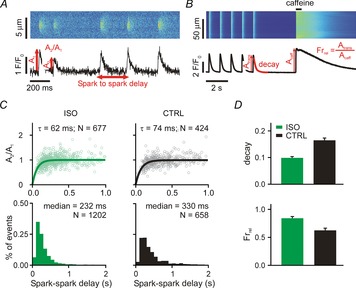
Ca2+ spark restitution and cellular Ca2+ transients after β-adrenergic stimulation
A, line scan and time courses of repetitive Ca2+ sparks induced by 50 nm ryanodine. Spark-to-spark delay and the amplitude of the second spark relative to the first were calculated and analysed (see annotations). B, line scan and time course of whole cell Ca2+ transient (1 Hz for 25 s) and subsequent caffeine application (10 mm). Ca2+ transient decay and fractional release (Frrel = Atrans/Acaff) were measured for analyses (see annotations). C, each column shows spark amplitude restitution (top; normalized amplitude of the second spark vs. the delay between sparks) and spark-to-spark delay histogram (bottom) for different conditions: (1) ISO, ryanodine plus isoproterenol (100 nm); (2) CTRL, ryanodine only. Bold lines show fits to the data of exponential recovery curves with indicated time constant and number of spark pairs. Histogram binning is 100 ms; N refers to number of spark pairs in each group. D, Ca2+ transient decay and fractional release in (1) ISO (recorded from 25 and 17 cells, respectively) and (2) CTRL (recorded from 20 and 15 cells, respectively) conditions. If not indicated otherwise, P < 0.01.
To statistically compare fractional release and Ca2+ transient decay rates between groups, we used ANOVA, followed by Student's t test for two-group comparisons. To compute confidence intervals (10–90%) for spark restitution time constants and spark-to-spark delay medians, we implemented a bootstrapping approach (Calmettes et al. 2012). This entailed repeatedly sampling 600 random pairs from each group, then calculating the resulting summary statistics (recovery τ and delay median) only from that subset of data. This step was repeated 300 times to produce a distribution for each summary statistic from the randomly sampled data sets. We then calculated P values and confidence intervals from these distributions.
Mathematical modelling
Simulations were performed with a modified version of the sticky cluster model (Sobie et al. 2002; Ramay et al. 2011; Lee et al. 2013). The model contains four compartments: (1) subspace, (2) cytosol, (3) junctional SR (JSR) and (4) network SR. Ca2+ buffering within each compartment, Ca2+ diffusion between compartments, and stochastic gating of RyRs are calculated. Sparks occur when Ca2+ flux through a single RyR stochastically causes most of the 28 RyRs to open. Each RyR has two states (open and closed), with no explicit inactivation process (Liu et al. 2012), and gating depends on subspace [Ca2+], JSR [Ca2+] and allosteric coupling between RyRs within the cluster.
To mimic the ability of a ryanodine molecule to induce repetitive Ca2+ sparks, we make a single RyR within the cluster hyperactive by reducing this channel's mean closed time from 28,175 s to 55 ms. All other RyR properties were unchanged. Frequent opening of the hyperactive channel provided random triggers for Ca2+ sparks, but, due to stochastic gating of the cluster, not every opening was sufficient to activate a spark. We generated sequences of random, repetitive Ca2+ sparks by simulating the autonomous stochastic gating of an RyR cluster for 1250 s (time step of 10−6 s). This model sufficiently simulated each experimental group by modifying just two parameters: (1) the maximum opening rate of the RyRs and (2) the time constant of refilling (see Table2).
Table 2.
Model parameters that matched experimental data
| Experimental group | τrefill (ms) | kopen (ms−1) |
|---|---|---|
| CTRL | 6.55 | 42 |
| ISO | 4.9 | 60 |
| Forskolin | 4.6 | 30 |
| H89 | 8.8 | 35 |
| S2808A | 5.5 | 42 |
| S2808A + ISO | 4.9 | 42 |
| ISO + KN92 | 4.5 | 90 |
| ISO + KN93 | 5.3 | 42 |
| ISO + H89 | 5.7 | 40 |
τrefill, time constant of refilling; kopen, maximum opening rate of the RyRs.
The number of open RyRs and the current through the cluster of RyRs (IRyR) were recorded during simulations. The threshold for Ca2+ spark triggering was a peak of at least five open RyRs, as simulations have shown that such an event should be detectable (Williams et al. 2011). For each simulated Ca2+ spark pair, two variables were calculated: (1) the delay between sparks; and (2) the amplitude of the second spark relative to the first (A2/A1) (Fig.1A). The integral of IRyR during the spark was used instead of spark amplitude because simulations demonstrated that these two variables were strongly correlated (results not shown).
Simulation execution
To speed up the simulations we performed the calculations on a Graphical Processing Unit (GPU) and used MATLAB's parallel computing toolkit. A file created in the parallel computing platform CUDA that described the sticky cluster model was created, using the curand_kernel.h. This file was compiled to a parallel thread execution (PTX) file using nvcc compiler in the NVIDIA CUDA Toolkit. Both files .CU and .PTX were used to create an executable kernel in MATLAB (CUDAKernel object). A computer containing an NVIDIA QUADRO 4000 GPU unit was used for all simulations.
Results
β-Adrenergic stimulation accelerates Ca2+ spark restitution in mouse ventricular myocytes
We have previously shown, in rat ventricular myocytes, that stimulation of β-adrenergic receptors (βARs) leads to accelerated recovery of Ca2+ spark amplitude and dramatically shorter spark-to-spark delays when repetitive Ca2+ sparks are induced with low-dose ryanodine (Ramay et al. 2011). Mathematical modelling of this phenomenon supported the conclusion that βAR stimulation led to both faster refilling of local SR Ca2+ stores and to increased sensitivity of RyRs. Our initial goal was to confirm these results in mouse ventricular myocytes. Treatment of cells with 50 nm ryanodine led to repeated Ca2+ sparks from the same cluster of RyRs (Fig.1A), as previously seen (Sobie et al. 2005; Ramay et al. 2011). These repeated sparks were analysed by computing Ca2+ spark amplitude recovery as a function of time and by generating histograms of spark-to-spark delays (Fig.1C). Consistent with previous results in rat myocytes (Ramay et al. 2011), βAR stimulation with 100 nm isoproterenol (ISO) led to accelerated recovery of Ca2+ spark amplitude (time constant τ = 74 ms in control; τ = 62 ms with ISO) and a leftward shift in the delay histograms (median = 330 ms in control, 232 ms with ISO).
The time constants and medians reported above were computed from complete data sets: hundreds of Ca2+ spark pairs obtained at numerous repetitive spark sites from several cells. To compute confidence intervals of these estimates and statistically compare differences between groups, we employed a bootstrapping approach (Calmettes et al. 2012). As described in more detail in Methods, this involved repeatedly estimating time constants and medians from samples of the complete datasets. Using this procedure, we determined 10–90% τ confidence intervals (CIs) of 68.7–80.4 ms for control (CTRL) and 57.3–67.4 ms for ISO, and median CIs of 316.1–349.3 ms for CTRL, 221.4–242.7 ms for ISO. We further calculated P values of 0.02 for τ, CTRL vs. ISO, and <0.001 for median, CTRL vs. ISO.
We complemented these Ca2+ spark recordings with measurements at the cellular level of steady-state Ca2+ transients and the response of myocytes to rapid application of 10 mm caffeine (Fig.1B). The two primary metrics we extracted from these whole cell recordings were the time constant of Ca2+ transient decay and the ‘fractional release,’ defined as the ratio of Ca2+ transient amplitude to the amplitude of the caffeine response (Atrans/Acaff). Consistent with previous results (Ginsburg & Bers, 2005), ISO led to a faster decay of Ca2+ transients (smaller time constant) due to stimulation of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), and to increased fractional release.
Activation of PKA explains some but not all effects of β-adrenergic stimulation
Stimulation of βARs leads to activation of protein kinase A (PKA), but this may induce additional downstream changes as well. To begin to delineate the specific role of PKA activation in Ca2+ spark restitution, we analysed repeated sparks after application of either forskolin, an activator of adenylyl cyclase, or H89, a PKA inhibitor (Fig.2A). Forskolin led to acceleration of Ca2+ spark amplitude recovery (τ = 74 ms in control; τ = 54 ms with forskolin; 10–90% CIs: 68.7–80.4 ms CTRL, 49.17–59.52 ms forskolin). This faster restitution would be expected to induce a leftward shift in the spark-to-spark delay histogram, and indeed a minor shift is seen with forskolin (median = 330 ms in control, 304 ms with forskolin; 10–90% CIs: 316.1–349.3 ms CTRL, 288.2–319.9 forskolin). Together these results suggest that selective activation of PKA leads to faster local SR refilling but does not increase sensitivity of RyRs to Ca2+. It also implies that additional signalling pathways must be activated to explain the full range of effects seen with βAR stimulation. In contrast, PKA inhibition by H89 caused slower restitution of Ca2+ spark amplitude (τ = 74 ms in control; τ = 92 ms with H89; 10–90% CIs: 68.7–80.4 ms CTRL, 85.2–99.0 ms H89) and a rightward shift in the delay histogram (median = 330 ms in control, 442 ms with H89; 10–90% CIs: 316.1–349.3 ms CTRL, 421.5–464.4 H89). This result implies that mouse ventricular myocytes have substantial PKA activation at baseline. Cellular-level measurements were in agreement with the Ca2+ spark restitution recordings. Compared with control conditions, forskolin decreased the Ca2+ transient time constant and increased fractional release, whereas H89 caused the opposite effects.
Figure 2.
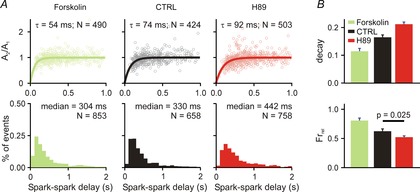
Ca2+ spark restitution and cellular Ca2+ transients after PKA activation/inhibition
A, each column shows spark amplitude restitution (top) and spark-to-spark delay histogram (bottom) for different conditions: (1) forskolin (1 μm), an activator of PKA; (2) CTRL (replotted from Fig.1 for comparison); (3) H89 (1 μm), an inhibitor of PKA. Bold lines show fits to the data of exponential recovery curves with indicated time constant and number of spark pairs. Histogram binning is 100 ms; N refers to number of spark pairs in each group. Spark restitution was performed in presence of 50 nm ryanodine to induce repetitive Ca2+ sparks. B, Ca2+ transient decay and fractional release in (1) forskolin (recorded from 8 cells); (2) CTRL (recorded from 20 and 15 cells, respectively) and (3) H89 (recorded from 23 and 20 cells, respectively) group. If not stated otherwise, P < 0.01.
Phosphorylation of RyRs at S2808 contributes to the response to βAR stimulation
Next we wished to gain insight into which phosphorylation sites might contribute to the increased RyR sensitivity seen with βAR stimulation. A serine residue located at position 2808 in the mouse RyR (2809 in human RyR) has been hypothesized to be critical for both physiological changes to RyR gating and to the development of heart failure (Marx et al. 2000; Marx & Marks, 2013), although this idea has been challenged (Bers, 2012; Houser, 2014). We examined Ca2+ spark restitution in myocytes from S2808A knock-in mice (Benkusky et al. 2007), in which phosphorylation at this particular residue is impossible. In these cells (Fig.3A), βAR stimulation with ISO led to accelerated restitution of Ca2+ spark amplitude, but only a minor shift in the median of the delay histogram (294 ms to 261 ms; 10–90% CIs: 281.9–305.9 S2808A, 247.9–274.4 S2808A ISO). This contrasts with the much larger shift in median observed in control mice (330 ms CTRL to 232 ms with ISO). This lack of a robust shift in spark-to-spark delays implies that phosphorylation of the RyR at S2808 does indeed contribute to the complete physiological response induced by βAR stimulation. Consistent with the Ca2+ spark restitution experiments, cellular-level recordings (Fig.3B) showed that ISO caused a decrease in Ca2+ transient decay time constant and an increase in fractional release in cells from S2008A mice.
Figure 3.
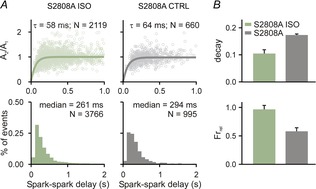
Spark restitution in S2808 mice
A, Ca2+ spark amplitude restitution (top row) and spark-to-spark delay histogram (bottom row) for different conditions (columns): (1) ISO, ryanodine plus isoproterenol; and (2) CTRL, only ryanodine exposure. Bold lines show fits to the data of exponential recovery curves with indicated time constant and number of spark pairs. Histogram binning is 100 ms; N refers to number of spark pairs in each group. B, whole cell Ca2+ transient statistics for S2808 ISO and S2808A groups, recorded from 8 cells in each group. If not indicated otherwise, P < 0.01.
Mathematical modelling suggests that ISO does not alter RyR gating in S2808A mice
To interpret the experimental results obtained so far, we performed simulations with a mathematical model of the cardiac Ca2+ spark (Sobie et al. 2002; Ramay et al. 2011; Lee et al. 2013). The basic model structure and parameters were as previously described (Ramay et al. 2011), but the model was improved to include a single hyperactive RyR that opens stochastically and frequently (mean closed time = 55 ms) and can potentially trigger repeated Ca2+ sparks from the remaining 27 RyRs in the cluster. These sequences of events were analysed to generate plots of simulated spark amplitude restitution and delay histograms, analogous to the experimental data. To reproduce the results seen in control myocytes (Fig.4), we assumed a refilling time constant of 6.55 ms and a maximum opening rate of 42 ms−1.
Figure 4.
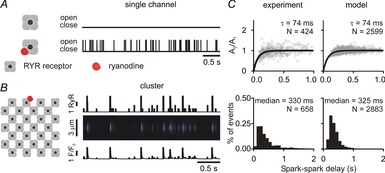
Mathematical modelling results
A, simulations of 4 s of activity of isolated RyR either without (top) or with (bottom) a single ryanodine molecule bound. Ryanodine was assumed to cause repetitive openings due to a dramatic decrease in mean closed time (28,175 s to 55 ms). Mean open time was 2 ms in either condition. B, simulations of a single ryanodine-bound RyR within a cluster of channels resulted in repetitive Ca2+ sparks, as seen in either the number of open channels (top), simulated line scan (middle) or simulated Ca2+ spark time course (bottom). We set a threshold of 5 open channels for an event to be defined as an identifiable spark (Williams et al. 2011). C, Ca2+ spark amplitude restitution and spark-to-spark delays as observed experimentally (left) and as simulated numerically (right).
Each experimental condition was simulated, and two parameters were adjusted to recapitulate the experimental data: (1) the time constant of JSR refilling was modified to match spark amplitude restitution; and (2) the maximum opening rate of the RyR was adjusted, if necessary, to match the median of the delay histogram. This allowed us to make inferences of the most likely changes occurring under each experimental condition. For instance, recapitulating the effects of ISO in control cells required decreasing the refilling time constant (from 6.55 to 4.9 ms) and increasing the maximal RyR opening rate (from 42 to 60 ms−1). In contrast, the effects of ISO in cells from S2808A mice could be reproduced by altering the refilling time constant (5.5 to 4.9 ms) without assuming any changes in RyR gating. The simulation results therefore support the suggestion that phosphorylation at S2808 contributes to the alterations observed with full stimulation of βARs.
Activation of both PKA and CaMKII is required for full effects of βAR stimulation
The next set of experiments aimed to delineate the contributions of PKA and CaMKII to the overall response to βAR stimulation. To do this we measured Ca2+ spark restitution and whole-cell Ca2+ transients after application of ISO plus drugs to block either PKA or CaMKII activity. As a control, and as used in previous studies (Sossalla et al. 2010), we incubated cells with ISO plus KN92, the inactive form of the CaMKII blocker KN93, and this led to effects nearly identical to those observed with ISO alone – an acceleration of both Ca2+ spark amplitude restitution and a dramatic leftward shift in the delay histogram (Fig.5A, first column). When ISO was added along with either the PKA inhibitor H89 or the CaMKII inhibitor KN93, we observed acceleration of Ca2+ spark amplitude restitution but only a much smaller leftward shift in the delay histogram (Fig.5A, second and third columns). For instance, the median of the delay histogram was 330 ms in control, 232 ms with ISO only, and 286 ms with ISO plus KN93 (10–90% CIs: 183.1–193.6 ISO + KN92, 270.0–295.4 ISO + KN93, 283.0–310.2 ISO + H89). Measurements made at the cellular level were consistent with the spark-level results. All three conditions (ISO + KN92, ISO + KN93, ISO + H89) caused a decrease in Ca2+ transient decay time constant and an increased fractional release, but less dramatic effects were seen with the inhibitors than with ISO alone or ISO plus the inactive inhibitor. These results suggest that both PKA and CaMKII must be activated to achieve the full effects of βAR stimulation.
Figure 5.
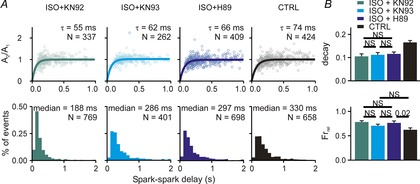
Ca2+ spark restitution and cellular Ca2+ transients after β-adrenergic stimulation combined with PKA or CAMKII inhibition
A, Ca2+ spark amplitude restitution (top) and spark-to-spark delay histogram (bottom) for different conditions (columns): (1) ISO (100 nm) + KN92 (1 μm), an inactive CaMKII inhibitor; (2) ISO + KN93 (1 μm), a CaMKII inhibitor; (3) ISO + H89 (1 μm), a PKA inhibitor; (4) CTRL (replotted from Fig.1 for comparison). Bold lines show fit to the data of exponential recovery curves with indicated time constant and number of spark pairs. Histogram binning is 100 ms; N refers to number of spark pairs in each group. Experiments performed in presence of 50 nm ryanodine to induce repetitive Ca2+ sparks. B, whole cell Ca2+ transient statistics in (1) ISO + KN92 (recorded from 15 and 13 cells, respectively); (2) ISO + KN93 (recorded from 17 and 12 cells, respectively); (3) ISO + H89 (recorded from 21 and 19 cells, respectively); (4) CTRL (recorded from 20 and 15 cells, respectively). If not indicated otherwise, P < 0.01 (NS, not significant).
Figure6 illustrates the consistency of the spark restitution and whole-cell measurements over the different experimental conditions examined. In Fig.6A we plot the time constant of Ca2+ transient decay at the cellular level versus the time constant of restitution at the Ca2+ spark level. The two variables are positively correlated, which is expected since both should depend on the activity of the SERCA pump. In Fig.6B we plot fractional release assessed at the cellular level versus the median of the delay histogram assessed at the spark level. A smaller median, implying increased RyR sensitivity, is associated with larger fractional release. Although fractional release will also depend on the magnitude of L-type Ca2+ current, which was not measured in these experiments but which should increase with β-adrenergic stimulation, the strong correlation between the two variables supports the idea that alterations in RyR Ca2+ sensitivity should affect both quantities.
Figure 6.
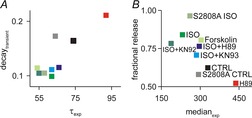
Relationship between single spark and whole cell transient measurements to summarize experimental results
A, median of spark-to-spark delays vs. fractional release. B, time constant of calcium spark restitution vs. whole cell transient decay. Each symbol corresponds to a different experimental condition as labelled.
Simulations provide quantitative insight into changes in SR refilling and RyR gating
Finally, we performed additional numerical simulations to interpret the Ca2+ spark restitution data obtained under different experimental conditions. For each condition, we simulated the effects of low-dose ryanodine by dramatically shortening the mean closed time of one RyR within a cluster of 28 channels, thereby allowing that single RyR to potentially initiate repetitive Ca2+ sparks (Fig.4). We adjusted the time constant of local SR refilling in the model and the maximal opening rate of the other RyRs within the cluster to simulate the changes that occurred under different experimental conditions. Changes to these parameters that were required to reproduce experimental results are shown in Table2. From these model-derived parameter values we can make the following experimental inferences: (1) activation of either PKA or CaMKII is sufficient to accelerate Ca2+ spark restitution through faster SR refilling; (2) simultaneous activation of both PKA and CaMKII is necessary for the increase in RyR sensitivity seen with βAR stimulation; (3) forskolin causes faster SR refilling and may in fact lead to a decrease in RyR sensitivity, although the evidence is not strong enough to conclude this with certainty.
Discussion
The results presented in this study provide new insight into changes in SR Ca2+ release that occur in heart cells after β-adrenergic stimulation. Consistent with our previous results (Ramay et al. 2011), we show that β-adrenergic stimulation accelerates the recoveries of both Ca2+ spark amplitude and Ca2+ spark triggering probability. The acceleration of amplitude recovery occurs because of faster SR refilling whereas the changes to spark triggering probability depend on both refilling and increased RyR Ca2+ sensitivity. These changes, however, are not mediated solely through activation of PKA, since more modest changes to spark triggering probability are observed when either CaMKII activity is blocked (Fig.5), or when activation of PKA occurs independently of β-adrenergic receptors (Fig.2). Moreover, the results suggest that mouse ventricular myocytes, unlike rat myocytes, exhibit substantial PKA activity at baseline, since inhibition of PKA with H89 slows recovery from refractoriness (Fig.3). When the experimental data are interpreted using simulations with our mathematical model, the results suggest the scheme illustrated in Fig.7. Block of basal PKA activity can slow local SR refilling, with little effect on RyR sensitivity, whereas activation of PKA or CaMKII alone will accelerate SR refilling, again with little effect on RyR sensitivity. It is only when PKA and CaMKII are activated in concert that refilling will be faster and RyRs will become more sensitive, leading to the greatest changes in refractoriness. The implications of these different effects and the possible clinical consequences are discussed below.
Figure 7.
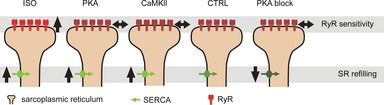
Schematic summary of changes seen under different experimental conditions
β-Adrenergic stimulation led to increases in both SR refilling and apparent RyR sensitivity. Activation of either PKA alone or CaMKII alone led to faster SR refilling but no apparent change in RyR sensitivity. Block of PKA activity led to slower SR refilling.
One surprising finding in the present results was that blockade of PKA activity with H89 slowed the recovery from refractoriness, even in control myocytes in which βARs had not been stimulated. H89 applied to unstimulated myocytes caused a slower recovery of Ca2+ spark amplitude and a pronounced rightward shift in the spark-to-spark delay histogram (Fig.2). Importantly, this intervention also increased the time constant of decay of the cellular Ca2+ transient and decreased fractional release (Fig.2), providing a critical confirmation of the Ca2+ spark data. These results imply that mouse ventricular myocytes exhibit substantial PKA activity at baseline, and they confirm previous findings suggesting that basal phosphorylation of key PKA targets may be higher in mouse than in other species (Huke & Bers, 2008; Parks & Howlett, 2012).
Two lines of evidence indicate that both PKA and CaMKII are activated when β-adrenergic receptors are stimulated with isoproterenol, and that activity of both kinases is required for maximal acceleration of recovery from refractoriness. First, direct activation of adenylyl cyclase with forskolin, an intervention that presumably activates PKA without activating CaMKII in resting cells, does not lead to increased sensitivity of RyRs (Fig.2 and Table2), and may in fact cause decreased RyR sensitivity. Second, when βARs are activated in the presence of KN93, a CaMKII inhibitor, RyR sensitivity is similarly not increased. These results are consistent with previous studies indicating that both PKA and CaMKII are activated when βARs are stimulated with isoproterenol (Curran et al. 2007, 2014; Gutierrez et al. 2013). At present, however, it remains unclear how β-adrenergic stimulation, in the absence of Ca2+ transients, may lead to activation of CaMKII. One possibility is that signalling downstream of Epac (exchange protein directly activated by cAMP) leads to an increase in CaMKII activity, although forskolin would presumably activate such a mechanism. Therefore a more likely possibility, supported by recent results (Gutierrez et al. 2013; Curran et al. 2014), is that CaMKII becomes activated through a nitric oxide-dependent pathway.
Our results also suggest that phosphorylation of the RyR at serine 2808 is essential for the changes in refractoriness seen during β-adrenergic stimulation. In the S2808A knock-in mouse, where phosphorylation of this residue is impossible, β-adrenergic stimulation caused faster recovery of Ca2+ spark amplitude, consistent with increased SERCA activity, but only a minimal leftward shift in the spark-to-spark delay histogram, implying that RyR function was not altered. This contrasted with the much larger leftward shift seen in myocytes from control mice, confirming that S2808 was necessary for the full effects of β-adrenergic stimulation on refractoriness. This is consistent with recent results showing that S2808A mice, while exhibiting a subtle phenotype, do indeed show altered responsiveness to β-adrenergic stimulation (Ullrich et al. 2012). Phosphorylation at S2808 has been hypothesized to play a central role in the development of heart failure (Marx & Marks, 2013), although this hypothesis remains extremely controversial (Bers, 2012; Dobrev & Wehrens, 2014; Houser, 2014). Although our data do not address the controversy relating to heart failure development, the results do suggest that phosphorylation at S2808 has functional consequences, in particular a shortening of the Ca2+ release refractory period. It seems fairly clear, however, that the positive inotropic response seen after β-adrenergic stimulation results primarily from phosphorylation of the L-type Ca2+ channel and phospholamban rather than from altered RyR function (Houser, 2014). It is intriguing that the relatively mild alteration in refractoriness seen in S2808A mice was similar to the effect seen after ISO application when CaMKII activity was inhibited by KN93. This could indicate that phosphorylation at S2808 occurs primarily through CaMKII rather than PKA. Although this residue is commonly considered a PKA site rather than a CaMKII site, it should be noted that early publications describing this site noted the ability of both CaMKII and PKA to phosphorylate this particular serine (Witcher et al. 1991; Rodriguez et al. 2003).
The data also provide new insight into factors that may increase the risk of arrhythmias during β-adrenergic stimulation. Just as the heart's electrical refractory period protects against propagation of improper electrical signals, the Ca2+ release refractory period helps to minimize the possibility that spontaneous release events will occur soon after triggered release. An abbreviated refractory period is associated with conditions that increase the risk of Ca2+-triggered arrhythmias, such as CPVT (Kornyeyev et al. 2012; Liu et al. 2013; Brunello et al. 2013) and heart failure (Belevych et al. 2012). The results presented here provide additional evidence that a shortened refractory period may be an important factor in the increased risk of Ca2+-triggered arrhythmias seen during β-adrenergic stimulation. Moreover, since our results suggest that both PKA and CaMKII are activated in response to isoproterenol, and since both kinases contribute to the effects that are observed, these results support the idea that CaMKII inhibition is a viable therapeutic strategy (Swaminathan et al. 2012). Targeting this kinase may be a way to prevent some of the negative consequences of β-adrenergic stimulation without fully inhibiting all of the positive consequences.
We should note some limitations of the study. One is that, besides the insight gained from the experiments with S2808A mice, we do not know the precise residues involved in the acceleration of refractoriness seen with β-adrenergic stimulation. The increase in RyR sensitivity seen with concurrent activation of PKA and CaMKII may involve phosphorylation at S2814 (Wehrens et al. 2004) and/or S2030 (Xiao et al. 2005) in addition to phosphorylation at S2808. A second caveat is that, while we can conclude that RyR sensitivity is increased in the presence of ISO, we cannot conclusively determine whether RyR sensitivity is unchanged, or perhaps slightly decreased, under other experimental conditions (Forskolin, ISO plus KN93, ISO plus H89). This issue is important and challenging in light of results suggesting that phosphorylation may either increase or decrease RyR Ca2+ sensitivity, depending on the baseline phosphorylation state (Lokuta et al. 1995; Liu et al. 2014). Careful additional experiments will be required to fully determine the relationship between phosphorylation and RyR sensitivity.
In summary, we have shown that both PKA and CaMKII contribute to the abbreviation of the Ca2+ release refractory period that is observed during β-adrenergic stimulation. By combining the experimental results with numerical simulations, we developed quantitative predictions about changes in SERCA activity or RyR sensitivity that occur under different conditions. The simulations suggest that activation of either PKA or CaMKII can increase the rate of local SR refilling, but both kinases must be active to observe increased RyR sensitivity. The data provide new insight into the regulation of SR Ca2+ release, and in particular the factors that control the release refractory period, a critical factor in determining the risk of Ca2+-triggered arrhythmias.
Acknowledgments
The authors thank Dr Hector Valdivia of the University of Michigan for the generous gift of the S2808A mice. We also thank Frank Fabris, Christine Falce and Ghislaine Rigoli for expert technical assistance.
Glossary
- βAR
β-adrenergic receptor
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CI
confidence interval
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- GPU
graphical processing unit
- ISO
isoproterenol (isoprenaline)
- JSR
junctional sarcoplasmic reticulum
- PKA
protein kinase A
- RyR
ryanodine receptor
- SERCA
sarco/endoplasmic reticulum Ca2+ ATPase
- SR
sarcoplasmic reticulum
Additional information
Competing interests
The authors declare that no potential competing interests exist.
Author contributions
E.P., A.I., E.N. and E.A.S. conceived the study. E.P. and A.I. performed the experiments and analysed the data. E.P. and E.A.S. performed the simulations and wrote the manuscript. All authors approved the final version of the manuscript.
Funding
This work is supported by the National Institutes of Health (HL076230, GM071558 to E.A.S.) and the Swiss National Science Foundation (31-132689 and 31-109693 to E.N.). A.I. was a recipient of a SciEx fellowship.
Translational perspective.
Triggered release of calcium from the sarcoplasmic reticulum in heart cells is the critical physiological process that links electrical excitation to contraction. Improper regulation of this process, however, increases the risk of spontaneous calcium release, which may initiate life-threatening ventricular arrhythmias. The refractory period after release is an important factor in determining whether harmful spontaneous calcium release occurs. The refractory period can be abbreviated in disease states and is regulated by β-adrenergic stimulation. We examined the calcium release refractory period at the microscopic level in mouse ventricular myocytes, and we tested the hypothesis that multiple downstream pathways activated during β-adrenergic stimulation work together to alter the release refractory period. Consistent with previous studies, we found that β-adrenergic stimulation accelerates recovery from refractoriness. We further determined that both protein kinase A and Ca2+/calmodulin-dependent protein kinase II contribute to this accelerated recovery. Using a genetically modified mouse, we also found that phosphorylation of the release channels, ryanodine receptors, at a particular residue (serine 2808), plays a role in abbreviated release refractoriness during β-adrenergic stimulation. The results provide new insight into the mechanisms by which β-adrenergic stimulation may increase the risk of calcium-triggered ventricular arrhythmias, and the data provide support for particular therapeutic interventions that may help to reduce the risk of these arrhythmias.
References
- Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE. Gyorke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA. Valdivia HH. Intact β-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- Bers DM. Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ Res. 2012;110:796–799. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- Bidasee KR, Xu L, Meissner G. Besch HR., Jr Diketopyridylryanodine has three concentration-dependent effects on the cardiac calcium-release channelryanodine receptor. J Biol Chem. 2003;278:14237–14248. doi: 10.1074/jbc.M208372200. [DOI] [PubMed] [Google Scholar]
- Brunello L, Slabaugh JL, Radwanski PB, Ho HT, Belevych AE, Lou Q, Chen H, Napolitano C, Lodola F, Priori SG, Fedorov VV, Volpe P, Fill M, Janssen PM. Györke S. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc Natl Acad Sci U S A. 2013;110:10312–10317. doi: 10.1073/pnas.1300052110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck E, Zimanyi I, Abramson JJ. Pessah IN. Ryanodine stabilizes multiple conformational states of the skeletal muscle calcium release channel. J Biol Chem. 1992;267:23560–23567. [PubMed] [Google Scholar]
- Calmettes G, Drummond GB. Vowler SL. Making do with what we have: use your bootstraps. J Physiol. 2012;590:3403–3406. doi: 10.1113/jphysiol.2012.239376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Kong CH, Imtiaz MS. Laver DR. Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys J. 2013;104:2149–2159. doi: 10.1016/j.bpj.2013.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Lederer WJ. Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol Cell Physiol. 1996;270:C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- Curran J, Hinton MJ, Rios E, Bers DM. Shannon TR. β-Adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calciumcalmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, Bers DM, Mohler PJ, Ziolo MT. Shannon TR. Nitric oxide-dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS One. 2014;9:e87495. doi: 10.1371/journal.pone.0087495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelPrincipe F, Egger M. Niggli E. Calcium signalling in cardiac muscle: refractoriness revealed by coherent activation. Nat Cell Biol. 1999;1:323–329. doi: 10.1038/14013. [DOI] [PubMed] [Google Scholar]
- Dobrev D. Wehrens XH. Role of RyR2 phosphorylation in heart failure and arrhythmias: controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. 2014;114:1311–1319. doi: 10.1161/CIRCRESAHA.114.300568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg KS. Bers DM. Isoproterenol does not enhance Ca-dependent NaCa exchange current in intact rabbit ventricular myocytes. J Mol Cell Cardiol. 2005;39:972–981. doi: 10.1016/j.yjmcc.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Guatimosim S, Sobie EA, Dos Santos CJ, Martin LA. Lederer WJ. Molecular identification of a TTX-sensitive Ca2+ current. Am J Physiol Cell Physiol. 2001;280:C1327–C1339. doi: 10.1152/ajpcell.2001.280.5.C1327. [DOI] [PubMed] [Google Scholar]
- Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J. Niggli E. NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc Res. 2013;100:392–401. doi: 10.1093/cvr/cvt201. [DOI] [PubMed] [Google Scholar]
- Houser SR. Role of RyR2 phosphorylation in heart failure and arrhythmias: protein kinase A-mediated hyperphosphorylation of the ryanodine receptor at serine 2808 does not alter cardiac contractility or cause heart failure and arrhythmias. Circ Res. 2014;114:1320–1327. doi: 10.1161/CIRCRESAHA.114.300569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huke S. Bers DM. Ryanodine receptor phosphorylation at serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B. Escobar AL. Calsequestrin 2 deletion shortens the refractoriness of Ca2+ release and reduces rate-dependent Ca2+-alternans in intact mouse hearts. J Mol Cell Cardiol. 2012;52:21–31. doi: 10.1016/j.yjmcc.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Liu OZ, Hwang HS, Knollmann BC. Sobie EA. Parameter sensitivity analysis of stochastic models provides insights into cardiac calcium sparks. Biophys J. 2013;104:1142–1150. doi: 10.1016/j.bpj.2012.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Ho HT, Velez-Cortes F, Lou Q, Valdivia C, Knollmann B, Valdivia H. Gyorke S. Genetic ablation of ryanodine receptor 2 phosphorylation at Ser-2808 aggravates Ca2+-dependent cardiomyopathy by exacerbating diastolic Ca2+ release. J Physiol. 2014;592:1957–1973. doi: 10.1113/jphysiol.2013.264689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Denegri M, Dun W, Boncompagni S, Lodola F, Protasi F, Napolitano C, Boyden PA. Priori SG. Abnormal propagation of calcium waves and ultrastructural remodeling in recessive catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2013;113:142–152. doi: 10.1161/CIRCRESAHA.113.301783. [DOI] [PubMed] [Google Scholar]
- Liu OZ, Lederer WJ. Sobie EA. Does the Goldilocks Principle apply to calcium release restitution in heart cells? J Mol Cell Cardiol. 2012;52:3–6. doi: 10.1016/j.yjmcc.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokuta AJ, Rogers TB, Lederer WJ. Valdivia HH. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J Physiol. 1995;487:609–622. doi: 10.1113/jphysiol.1995.sp020904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO. Marks AR. Dysfunctional ryanodine receptors in the heart: new insights into complex cardiovascular diseases. J Mol Cell Cardiol. 2013;58:225–231. doi: 10.1016/j.yjmcc.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N. Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Parks RJ. Howlett SE. H-89 decreases the gain of excitation-contraction coupling and attenuates calcium sparks in the absence of beta-adrenergic stimulation. Eur J Pharmacol. 2012;691:163–172. doi: 10.1016/j.ejphar.2012.07.012. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W. Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Qu Z, Nivala M. Weiss JN. Calcium alternans in cardiac myocytes: order from disorder. J Mol Cell Cardiol. 2013;58:100–109. doi: 10.1016/j.yjmcc.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramay HR, Liu OZ. Sobie EA. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res. 2011;91:598–605. doi: 10.1093/cvr/cvr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez P, Bhogal MS. Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278:38593–38600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- Rovetti R, Cui X, Garfinkel A, Weiss JN. Qu Z. Spark-induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circ Res. 2010;106:1582–1591. doi: 10.1161/CIRCRESAHA.109.213975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkryl VM, Maxwell JT, Domeier TL. Blatter LA. Refractoriness of sarcoplasmic reticulum Ca2+ release determines Ca2+ alternans in atrial myocytes. Am J Physiol Heart Circ Physiol. 2012;302:H2310–H2320. doi: 10.1152/ajpheart.00079.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobie EA, Dilly KW, Dos Santos CJ, Lederer WJ. Jafri MS. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobie EA. Lederer WJ. Dynamic local changes in sarcoplasmic reticulum calcium: physiological and pathophysiological roles. J Mol Cell Cardiol. 2012;52:304–311. doi: 10.1016/j.yjmcc.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobie EA, Song LS. Lederer WJ. Local recovery of Ca2+ release in rat ventricular myocytes. J Physiol. 2005;565:441–447. doi: 10.1113/jphysiol.2005.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G. Maier LS. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107:1150–1161. doi: 10.1161/CIRCRESAHA.110.220418. [DOI] [PubMed] [Google Scholar]
- Stern MD, Ríos E. Maltsev VA. Life and death of a cardiac calcium spark. J Gen Physiol. 2013;142:257–274. doi: 10.1085/jgp.201311034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan PD, Purohit A, Hund TJ. Anderson ME. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–1677. doi: 10.1161/CIRCRESAHA.111.243956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentesi P, Pignier C, Egger M, Kranias EG. Niggli E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ Res. 2004;95:807–813. doi: 10.1161/01.RES.0000146029.80463.7d. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL. Györke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- Ullrich ND, Valdivia HH. Niggli E. PKA phosphorylation of cardiac ryanodine receptor modulates SR luminal Ca2+ sensitivity. J Mol Cell Cardiol. 2012;53:33–42. doi: 10.1016/j.yjmcc.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR. Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- Williams GS, Chikando AC, Tuan HT, Sobie EA, Lederer WJ. Jafri MS. Dynamics of calcium sparks and calcium leak in the heart. Biophys J. 2011;101:1287–1296. doi: 10.1016/j.bpj.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher DR, Kovacs RJ, Schulman H, Cefali DC. Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- Wolska BM. Solaro RJ. Method for isolation of adult mouse cardiac myocytes for studies of contraction and microfluorimetry. Am J Physiol Heart Circ Physiol. 1996;271:H1250–H1255. doi: 10.1152/ajpheart.1996.271.3.H1250. [DOI] [PubMed] [Google Scholar]
- Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, Walsh MP, Warltier DC, Cheng H. Chen SR. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res. 2005;96:847–855. doi: 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]
- Xie Y, Sato D, Garfinkel A, Qu Z. Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]