

Abstract
Hypokalaemia is a risk factor for development of ventricular arrhythmias. The aim of this study was to determine the cellular mechanisms leading to triggering of arrhythmias in ventricular myocytes exposed to low Ko. Low Ko, corresponding to moderate hypokalaemia, increased Ca2+ transient amplitude, sarcoplasmic reticulum (SR) Ca2+ load, SR Ca2+ leak and Ca2+ wave probability in field stimulated rat ventricular myocytes. The mechanisms leading to Ca2+ overload were examined. Low Ko reduced Na+,K+-ATPase (NKA) currents, increased cytosolic Na+ concentration and increased the Na+ level sensed by the Na+, Ca2+ exchanger (NCX). Low Ko also hyperpolarized the resting membrane potential (RMP) without significant alterations in action potential duration. Experiments in voltage clamped and field stimulated ventricular myocytes, along with mathematical modelling, suggested that low Ko increases the Ca2+ transient amplitude by reducing NKA activity despite hyperpolarization of the RMP. Selective inhibition of the NKA α2 isoform by low dose ouabain abolished the ability of low Ko to reduce NKA currents, to increase Na+ levels sensed by NCX and to increase the Ca2+ transient amplitude. We conclude that low Ko, within the range of moderate hypokalaemia, increases Ca2+ levels in ventricular myocytes by reducing the pumping rate of the NKA α2 isoform with subsequent Na+ accumulation sensed by the NCX. These data highlight reduced NKA α2-mediated control of NCX activity as a possible mechanism underlying triggered ventricular arrhythmias in patients with hypokalaemia.
Key points
Hypokalaemia is a risk factor for development of ventricular arrhythmias.
In rat ventricular myocytes, low extracellular K+ (corresponding to clinical moderate hypokalaemia) increased Ca2+ wave probability, Ca2+ transient amplitude, sarcoplasmic reticulum (SR) Ca2+ load and induced SR Ca2+ leak.
Low extracellular K+ reduced Na+,K+-ATPase (NKA) activity and hyperpolarized the resting membrane potential in ventricular myocytes. Both experimental data and modelling indicate that reduced NKA activity and subsequent Na+ accumulation sensed by the Na+, Ca2+ exchanger (NCX) lead to increased Ca2+ transient amplitude despite concomitant hyperpolarization of the resting membrane potential.
Low extracellular K+ induced Ca2+ overload by lowering NKA α2 activity. Triggered ventricular arrhythmias in patients with hypokalaemia may therefore be attributed to reduced NCX forward mode activity linked to an effect on the NKA α2 isoform.
Introduction
Hypokalaemia is a common electrolyte disturbance present in over 20% of hospitalized patients (Paice et al. 1986), and increases the risk of ventricular tachycardia and fibrillation more than four-fold (Goyal et al. 2012). Hypokalaemia is suggested to increase the propensity for both early and delayed afterdepolarizations, in addition to promoting re-entry circuits in the myocardium (Osadchii, 2010).
Low extracellular K+ has been reported to result in aftercontractions after a positive inotropic response to K+-free superfusion in guinea pig papillary muscles (Eisner & Lederer, 1979a,b). The inotropic and pro-arrhythmic effect of low Ko was suggested to be caused by reduced Na+,K+-ATPase (NKA) activity, leading to increased intracellular Na+ concentration and subsequent Ca2+ overload through modulation of Na+, Ca2+ exchanger (NCX) activity. Later studies have linked aftercontractions in myocardial samples to induction of Ca2+ waves at the cellular level (Orchard et al. 1983). In contrast to these findings, hyperpolarization of the resting membrane potential (RMP) has been reported to induce lower cellular Ca2+ levels in experiments in isolated ventricular myocytes superfused with 1 mm K+ (Bouchard et al. 1995). Therefore, previous investigations suggest that low Ko may exert two opposite effects on NCX activity, namely increasing cytosolic Na+ levels (increasing cellular Ca2+ levels) and inducing hyperpolarization of the RMP (reducing cellular Ca2+ levels).
The net effect of low Ko on NCX activity depends on the balance between the reduced NKA activity, RMP hyperpolarization and influence of other Ko-sensitive currents. None of the aforementioned studies are directly applicable to hypokalaemic patients, as these studies were performed with either no extracellular K+ or Ko levels lower than observed in hypokalaemic patients. Moderate hypokalaemia is defined as serum K+ levels of 2.5–3.0 mm, at which NKA will pump at reduced rates, in contrast to experiments conducted with 0 Ko, which abolishes NKA activity. Thus, whether low Ko within the clinically relevant range induces Ca2+ overload and subsequent Ca2+ waves in ventricular myocytes is not known.
The first aim of the present study was to determine whether low Ko (corresponding to clinically moderate hypokalaemia) induces Ca2+ overload and Ca2+ waves, which can be a substrate for delayed afterdepolarization in ventricular myocytes. Further, we wanted to determine the relative role of NKA inhibition, hyperpolarization of the RMP and altered action potential duration (APD) in controlling the cellular Ca2+ levels during low Ko. Our second aim was to determine the relative role of NKA α1 and α2 isoforms in mediating the cellular response to low Ko. While NKA α1 is uniformly distributed throughout the sarcolemma, NKA α2 is clustered in the t-tubules and is a potent regulator of NCX activity in ventricular myocytes by a yet undetermined mechanism (Swift et al. 2007; Despa et al. 2012). NKA α1 and NKA α2 also differ in terms of Ko sensitivity; NKA α2 having a lower K+ affinity than NKA α1, with a reported Ko sensitivity in a range corresponding to levels found in hypokalaemic patients (Han et al. 2009). Here we show that reduced NKA α2 activity is the main mechanism leading to Ca2+ overload in response to low Ko in ventricular myocytes, providing a cellular mechanism for triggered arrhythmias in hypokalaemic patients.
Methods
Animals and cell isolation
Male Wistar-Hannover rats (Møllegaard, Denmark; ∼300 g) were housed in a temperature-regulated room with 12 h day/12 h night cycling, with ad libitum access to food and water. Rats were sedated with a mixture of 4% isoflurane, 64% N2O and 31% O2, before endotracheal intubation and ventilation (Zoovent, Triumph Technical Services, Milton Keynes, UK) with a mixture of 68% N2O, 29% O2 and 2.5% isoflurane. Then, 200 IU of heparin was injected i.v. prior to excision of the heart in surgical anaesthesia. The heart was rapidly cannulated and retrogradely perfused using the following solution (mm): NaCl 130, Hepes 25, MgCl2 0.5, KCl 5.4, NaH2PO4 0.4, d-glucose 22, pH adjusted to 7.4 with NaOH. Collagenase was added to the perfusion as previously described (Bokenes et al. 2008).
Ca2+ transients
Ca2+ transients were obtained from isolated ventricular myocytes, loaded with 20 μm fluo-4 AM (Molecular Probes, Eugene, OR, USA) and subsequently superfused at 37°C with the following solution (mm): NaCl 140, Hepes 5, KCl 5.0 or 2.7, CaCl2 1.0, MgCl2 0.5, d-glucose 5.5, NaH2PO4 0.4, pH adjusted to 7.4 with NaOH. Cells were excited at 488 nm and emitted light was registered with a photomultiplier (Photon Technology International, Monmouth Junction, NJ, USA) mounted on a Nikon inverted microscope as previously described (Louch et al. 2010). To correct for background fluorescence, cell-free fluorescence was obtained after each experiment and subtracted from the primary recording. The resulting Ca2+ transients were analysed with Clampfit 9.0 (Axon Instruments, Foster City, CA, USA). The following sets of experiments were performed using this setup: (1) Ca2+ transient amplitude, measured as the ratio between maximal (F) and basal (F0) fluorescence during one twitch; (2) Ca2+ wave probability, measured as the probability of evoking a Ca2+ wave within 30 s following field stimulation at 1, 2 and 4 Hz. Ca2+ wave probability was measured at Ko = 5.0 mm, before switching to Ko = 2.7 mm, and back to Ko = 5.0 mm to test reversibility; (3) sarcoplasmic reticulum (SR) Ca2+ load, measured as the ratio between maximal and basal fluorescence after rapid application of 10 mm caffeine; (4) rate constant of Ca2+ extrusion mediated by SERCA2, NCX and other Ca2+ transport mechanisms. The SERCA2 rate constant was calculated as the difference between the rate constant for regular Ca2+ transients and the caffeine-evoked Ca2+ transient. Rate constants were calculated as 1/tau, where tau was obtained by a monoexponential fit of the extrusion phase of the Ca2+ transient (Diaz et al. 1997). The NCX rate constant was measured as the difference between the rate constant after rapid application of caffeine ± 10 mm Ni+ in the superfusate (Bokenes et al. 2008). (5) SR Ca2+ leak, measured as the difference in resting fluorescence ± 1 mm tetracaine (Shannon et al. 2002). Ventricular myocytes were field stimulated in normal Tyrode's solution containing (mm): NaCl 140, KCl 5.0 or 2.7, CaCl2 1.0, MgCl2 1.0, d-glucose 10, Hepes 5, pH adjusted to 7.4 with NaOH. Field stimulation was stopped and leak was measured in a 0 Na+/0 Ca2+ solution (mm): LiCl 140, KCl 5.0 or 2.7, MgCl2 1, d-glucose 10, Hepes 5, EGTA 10, pH adjusted to 7.4 with LiOH, ± 1 mm tetracaine.
NKA and NCX activity
NKA currents and NCX currents were measured using whole-cell voltage clamp as previously described (Swift et al. 2008), using an Axopatch 200B amplifier (Axon Instruments). The pipette solution contained (mm): Hepes 10, tetraethylammonium chloride 20, l-aspartate 42, EGTA 42, CaCl2 29.7, Na2phosphocreatine 5, MgATP 10, NaOH 40, pH adjusted to 7.2 with CsOH. Free Ca2+ concentration was calculated to 300 nm, using WinMAX C 2.01 software (C. Patton, Stanford University, CA, USA). Average pipette resistance was ∼1 MΩ. After gaining whole-cell access, ventricular myocytes were superfused (37°C) with (mm): NaCl 147, MgCl2 2, EGTA 0.1, d-glucose 5.5, Hepes 5, BaCl2 2, nicardipine 0.001, pH adjusted to 7.40 with NaOH. The holding potential was –50 mV, and NKA currents were interpreted as the Ko-sensitive current induced by superfusion with either 2.7 or 5.0 mm KCl. INCX was elicited with 2 mm CaCl2, and was elicited both in the presence and in the absence of Ko to study the effect of NKA-mediated regulation on NCX activity. A low dose of ouabain (0.3 mm) was used to selectively block the NKA α2 isoform (Swift et al. 2007).
NKA I–V relationship was measured as described previously (Swift et al. 2007). Briefly, ventricular myocytes were depolarized (50 ms) to +70 mV from the holding potential of −50 mV, then hyperpolarized to −120 mV (dV/dt = 380 mV s−1), and back to −50 mV. The difference between the recorded current at baseline and during activation of NKA represented the I–V relationship of NKA.
Cytosolic Na+ measurements
To measure the cytosolic Na+ concentration, isolated ventricular myocytes were loaded at room temperature in sodium-binding benzofurzan isophthalate (SBFI) for 120 min, in the presence of 0.15% Pluronic F-127. Isolated ventricular myocytes were superfused with the same solution used for fluo-4 experiments. SBFI ratios were recorded with 5.0 mm KCl in the superfusate during field stimulation (1 Hz), before switching to 2.7 mm KCl. Each cell was calibrated by superfusing the cell with a solution containing gramicidin 2 μg ml−1, ouabain 1 mm, Hepes 5 mm, glucose 5.5 mm, EGTA 2 mm, NaCl 0 or 14 mm, and KCl 140 or 0 mm, pH adjusted to 7.2 with NaOH or KOH. Cytosolic Na+ concentration was calculated as previously described (Swift et al. 2007).
Voltage clamp protocols
For Ca2+ transient measurements, ventricular myocytes were voltage clamped using discontinuous mode (switching rate 9 kHz) on an Axoclamp 2B amplifier and pCLAMP software (Axon Instruments). Patch pipettes (1.5-2.5 MΩ) were filled with (in mm): CsCl 120, TEACl 20, Hepes 10, Na2ATP 5, CsEGTA 0.02, and fluo-5F pentapotassium salt 0.1, pH adjusted to 7.2 with CsOH. Cells were patched in a solution containing (in mm): NaCl 140, Hepes 5, KCl 5.0, CaCl2 1.0, MgCl2 0.5, d-glucose 5.5, NaH2PO4 0.4, pH adjusted to 7.4 with NaOH. After gaining whole cell access, cells were superfused with the following solution (in mm): NaCl 134, glucose 10, Hepes 10, MgCl2 1, KCl 5, 4-aminopyridine 5, BaCl2 0.1 and probenecid 2, pH adjusted to 7.4 with NaOH. The holding potential was −45 mV, and Ca2+ transients were triggered at 1 Hz by a 100 ms square voltage step from −45 to 0 mV. After stable Ca2+ transients were obtained, the superfusate was switched to a comparable solution, except KCl was reduced to 2.7 mm. Ca2+ transients were obtained with Cairn Research Optoscan Monochromator (excitation 485 nm, emission 515 nm long pass) (Cairn Research Ltd., Faverham, UK).
Background K+ current was measured in ventricular myocytes by continuous mode voltage clamp using patch pipettes (2.0–2.5 MΩ) filled with the following solution (in mm): KCl 130, NaCl 10, Hepes 10, MgATP 5, MgCl2 1, EGTA 0.5, pH adjusted to 7.2 with KOH. The myocytes were superfused with a solution containing (in mm): NaCl 135, Glucose 10, Hepes 10, KCl 5.4, MgCl2 1, CaCl2 1.8, CdCl2 0.2, pH adjusted to 7.4 with NaOH. CdCl2 was used to inhibit Ca2+ currents. The voltage clamped myocytes were held at -80 mV, and currents were elicited by 500 ms voltage steps to various test potentials in the range −170 mV to 50 mV. In order to study the effect of Ba2+ on background K+ currents, 2 mm BaCl2 (same concentration as used to study NKA and NCX currents) was added to the superfusate. The stable K+ currents were measured in the same myocytes both with and without Ba2+ present.
Current clamp protocols
RMP and action potentials (APs) were recorded as previously described (Mork et al. 2009). APs were triggered by a 3 ms suprathreshold current injection. The pipette solution contained (in mm) potassium aspartate 120, MgCl2 0.5, NaCl 6, EGTA 0.06, Hepes 10, glucose 10, KCl 25 and K2-ATP 4, pH adjusted to 7.2 with KOH.
Mathematical modelling
A computational simulation of the rat ventricular myocytes was performed using a whole-cell model (Niederer & Smith, 2007). This model integrates the electrophysiological components of the endocardial model of Pandit et al. (2009), the Ca2+ handling model of Hinch et al. (2004) and the contraction model of Niederer et al. (2006). The model features a single cytoplasmic pool of Ca2+ and Na+, i.e. without involving a specific subspace whose specific concentration is sensed by NKA. The mathematical code was implemented in the CellML language (www.cellml.org; Terkildsen et al. 2008) and the differential equations integrated with the COR software (Cellular Open Resource, cor.physiol.ox.ac.uk), using the CVODE solver. Further analysis was performed in Matlab (MathWorks, Natick, MA, USA). The hypokalaemia experiments were simulated by instantaneously switching the value of the Ko exposed to each K+-transporting channel separately, to allow the evaluation of the contribution of each channel in isolation.
Statistics
All data are presented as means ± SEM. Numbers of observations are presented as n. Statistical significance was calculated by Student's t test using either paired or unpaired tests. P < 0.05 was considered significant.
Results
Low Ko increased propensity for Ca2+ waves in isolated ventricular myocytes
Superfusion with Ko = 2.7 mm increased the Ca2+ wave probability at all frequencies tested (Fig.1A–C, P < 0.05, n = 7). None of the included ventricular myocytes showed Ca2+ waves at 1 Hz with Ko = 5.0 mm, while 8 and 17% of the same cells showed Ca2+ waves at 2 and 4 Hz, respectively. Ca2+ wave probability was 63%, 92% and 92% in ventricular myocytes superfused with Ko = 2.7 mm and field stimulated at 1, 2 and 4 Hz, respectively. After again superfusing the cells with Ko = 5.0 mm, the Ca2+ wave probability was largely reversed, as 25%, 0% and 20% of the ventricular myocytes showed Ca2+ waves at 1, 2 and 4 Hz, respectively.
Figure 1.
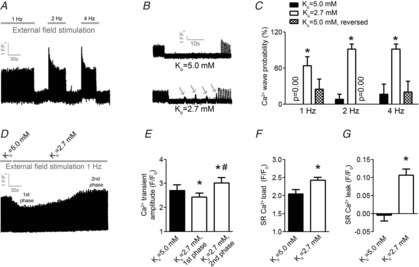
Low extracellular K+ increases probability for Ca2+ waves, Ca2+ transient amplitude, SR Ca2+ load and SR Ca2+ leak in ventricular myocytes
A, protocol for evaluation of Ca2+ wave frequency in field-stimulated ventricular myocytes. Stimulation frequencies were as indicated, with 30 s pause after each stimulation frequency. B, representative tracings of cellular fluorescence after superfusion with Ko = 5.0 mm (top) and Ko = 2.7 mm (bottom) at 1 Hz stimulation frequency. Grey arrows indicate Ca2+ waves. C, number of cells exerting Ca2+ waves at various stimulation frequencies. D, representative tracing of Ca2+ transient amplitude in 1 Hz field-stimulated ventricular myocytes after switch from Ko = 5.0 to 2.7 mm. E, Ca2+ transient amplitude at baseline, and 1st and 2nd phase as indicated in D. F, SR Ca2+ load evoked by rapid caffeine application after reaching stable (2nd) phase at 1 Hz field stimulation. G, SR Ca2+ leak. *P < 0.05 vs. Ko = 5.0 mm, #P < 0.05 vs. Ko = 2.7 mm.
Low Ko increased Ca2+ transient amplitude, SR Ca2+ load and SR Ca2+ leak
Switching from Ko = 5.0 to 2.7 mm induced a biphasic response in Ca2+ transient amplitude in ventricular myocytes field stimulated at 1 Hz (Fig.1D and E). Initially, within 30–60 s after the switch, the Ca2+ transient amplitude decreased in amplitude by 13 ± 2% (P < 0.05, n = 9)(1st phase in Fig.1D). Subsequently, the Ca2+ transient amplitude gradually increased and reached a steady state 25 ± 7% above baseline within 4–6 min (2nd phase in Fig.1D).
The increase in Ca2+ transient amplitude by low Ko was associated with a 36% increase in SR Ca2+ load compared to Ko = 5.0 mm (Fig.1F, P < 0.05, n = 6–9). No SR Ca2+ leak was detected in cells superfused with Ko = 2.7 mm, while there was significant SR Ca2+ leak at Ko = 2.7 mm (Fig.1G, P < 0.05, n = 9). Hence, increased frequency of Ca2+ waves in intact cells was associated with Ca2+ overload after exposure to low Ko.
Low Ko hyperpolarized RMP without altering AP duration
To determine the underlying cellular mechanism leading to Ca2+ overload by low Ko in field-stimulated ventricular myocytes, we investigated the relative contribution of RMP hyperpolarization and APD. In voltage-clamped ventricular myocytes stimulated at 1 Hz, switching Ko from 5.0 to 2.7 mm induced a significant hyperpolarization of the RMP (Fig.2A and B, P < 0.05, n = 6). None of the analysed APD parameters were significantly altered by low Ko (Fig.2C).
Figure 2.
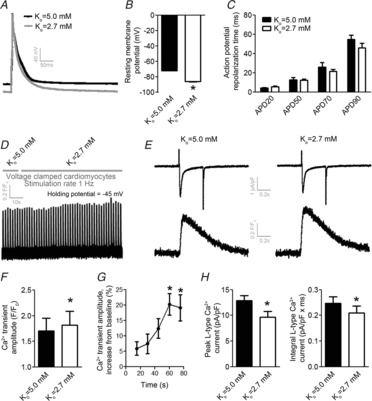
Low extracellular K+ increases Ca2+ transient amplitude in ventricular myocytes with clamped membrane potential
A, representative tracings of RMPs and APs at 1 Hz stimulation. B, mean RMPs. C, mean AP repolarization time. D, representative tracing of Ca2+ transients obtained from voltage-clamped cells and stimulated at 1 Hz (holding potential –45 mV). E, representative Ca2+ transients and L-type Ca2+ currents during a single twitch in the same cells at Ko = 5.0 mm and after switch to Ko = 2.7 mm. F, mean Ca2+ transient amplitude in voltage-clamped ventricular myocytes. G, time development of Ca2+ transient amplitude after switch from Ko = 5.0 to 2.7 mm. H, peak and integral L-type Ca2+ current before and after switch from Ko = 5.0 to 2.7 mm. *P < 0.05 vs. baseline.
Voltage-clamped ventricular myocytes did not exhibit the 1st negative phase induced by low Ko
To test whether the increase in Ca2+ transient amplitude was dependent upon RMP hyperpolarization, we next recorded Ca2+ transients in cells with clamped RMP to prevent the hyperpolarization by low Ko at 1 Hz stimulation. As shown in Fig.2D, the Ca2+ transient amplitude gradually increased after switching Ko from 5.0 to 2.7 mm, reaching a stable state in ∼60 s. Notably, in contrast to the intact field-stimulated ventricular myocytes, Ca2+ transient amplitude increased in all cells compared to baseline at all analysed time points after switching Ko from 5.0 to 2.7 mm (Fig.2D–G). In the same ventricular myocytes, both peak and the integral of L-type Ca2+ currents were decreased by switching Ko from 5.0 to 2.7 mm (Fig.2H and I). Decreased L-type Ca2+ currents concomitant with increased Ca2+ transient amplitude indicate low Ko that to increase Ca2+ levels by enhancing SR fluxes relative to trans-sarcolemmal Ca2+ cycling, possibly by reducing forward mode NCX activity.
Low Ko decreased NKA activity and increased Na+ concentration in ventricular myocytes
To test whether reduced NKA activity mediated the increased Ca2+ levels observed by low Ko, we next examined NKA and NCX currents in a series of experiments shown in Fig.3. The NKA current was 25±1% lower by adding 2.7 mm K+ to the superfusate compared to 5.0 mm K+ (Fig.3H, P < 0.05, n = 7), and NKA currents were lower by switching Ko from 5.0 to 2.7 mm at all membrane potentials (Fig.3A and B). Based on the NKA I-V plot shown in Fig.3B and the detected hyperpolarization of the RMP by Ko = 2.7 mm (Fig.2B), switching Ko from 5.0 mm to 2.7 mm was calculated to reduce NKA currents by ∼48%. Corresponding to the reduced NKA activity, we detected an increase in cytosolic Na+ in intact cells field stimulated at 1 Hz (Fig.3C and D; P < 0.05, n = 8).
Figure 3.
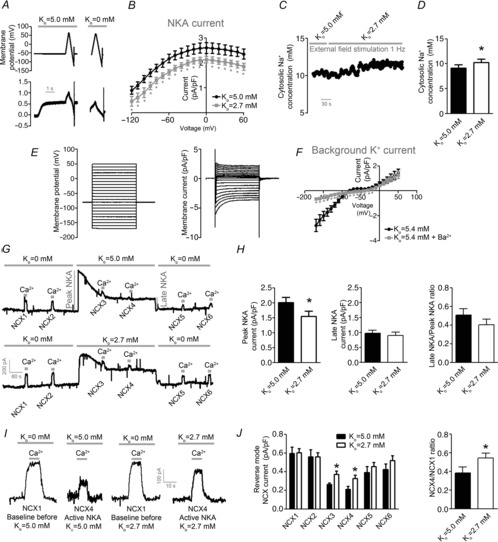
Low extracellular K+ reduces NKA activity and increases Na+ level sensed by NCX
A, protocol and representative tracing used to obtain I–V plots for NKA currents. B, I–V plots for NKA currents. C, representative tracing of cytosolic Na+ level converted from SBFI fluorescence in 1 Hz field-stimulated ventricular myocytes. D, cytosolic Na+ concentration. E, protocol and representative tracing used to obtain I–V plots of background K+ current. F, I–V plot for background K+ currents with and without Ba2+ present in the superfusate. G, representative tracing of protocol used to obtain NKA-dependent regulation of NCX currents. H, NKA currents in whole cell voltage-clamped ventricular myocytes. I, representative tracings of reverse mode NCX currents evoked by activation of Ca2+ before and after activation of NKA with either Ko = 5.0 mm (left) or Ko = 2.7 mm (right). J, mean NCX currents (left) and NCX4/NCX1 ratio (right). *P < 0.05.
Low Ko increased Na+ levels sensed by NCX in voltage-clamped ventricular myocytes
Whether low Ko increases Na+ concentration sensed by NCX was next tested in a voltage clamp experiment designed to measure NKA-dependent control of NCX activity (Fig.3G). To control for background K+ currents, we first obtained I–V plots for the background K+ current and tested the effect of adding Ba2+ to the superfusate. As shown in Fig.3E and F, Ba2+ blocked the background K+ currents at −60 to −30 mV.
Based on the obtained I–V plots for NKA and background K+ currents, we examined NKA-dependent regulation of NCX at a holding potential of −45 mV and in the presence of Ba2+. Stepping Ko from 0 to 5.0 mm induced a larger peak NKA current than stepping Ko from 0 to 2.7 mm (Fig.3H, left panel). The NKA current gradually declined in all cells examined, and stabilized at a lower level than the initial peak current (late NKA current in Fig.3G, H). The NCX current during activation of NKA was ∼30% higher in cells superfused with Ko = 2.7 mm compared to Ko = 5.0 mm (NCX3–4) (Fig.3I, J, P < 0.05, n = 6–8). Because we measured reverse mode NCX current in this protocol, these data suggest that NKA activation by 2.7 mm K+ increases the Na+ level sensed by the NCX compared to 5.0 mm K+.
Low Ko reduced NCX-mediated Ca2+ extrusion
The results described above suggest low Ko both reduces NKA activity and leads to RMP hyperpolarization, which would have opposite effects on NCX driving forces in intact cells. As shown in Fig.4, the rate constant for NCX-mediated Ca2+ extrusion (forward mode) was lower in cells superfused with 2.7 mm K+ compared to 5.0 mm K+ (P < 0.05, n = 6–9) at 1 Hz field stimulation. The removal of cytosolic Ca2+ by SERCA2 and additional, slow Ca2+ extrusion mechanisms were not significantly different between cells superfused with 2.7 and 5.0 mm K+. These results support a model where reduced NKA activity exerts a more pronounced effect on NCX activity than hyperpolarization of RMP.
Figure 4.
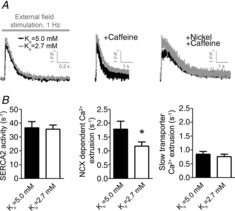
Low extracellular K+ decreases NCX-mediated Ca2+ extrusion in ventricular myocytes
A, representative tracings of Ca2+ transients in field-stimulated (1 Hz) ventricular myocytes (left), after rapid application of caffeine (middle) and after rapid application of caffeine in the presence of nickel (right). B, rate constant of SERCA2 activity (left), NCX-mediated Ca2+ extrusion (middle) and Ca2+ extrusion by other Ca2+ transporters (right). *P < 0.05.
Modelling supported a role for NKA in mediating increased Ca2+ levels during low Ko
To further investigate the role of NKA activity in the Ca2+ response induced by low Ko, we utilized a previously published mathematical model of rat ventricular myocyte electro-mechanical function (Niederer & Smith, 2007). In line with the experimental results, the model predicted that switching Ko from 5.0 to 2.7 mm would induce a comparable biphasic response in Ca2+ transient amplitude (Fig.5A, B) at 1 Hz contraction frequency. The 2nd phase with increased Ca2+ transient amplitude was further characterized by increased SR Ca2+ load, reduced NKA currents, increased cytosolic Na+ concentration and reduced NCX currents (Fig.5C).
Figure 5.
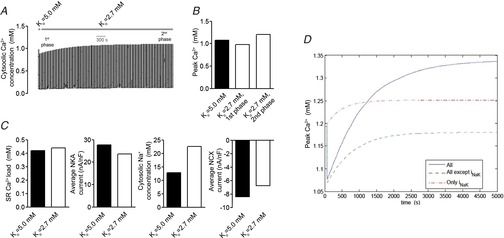
Modelling predicts NKA inhibition as a mechanism for increased Ca2+ transients by low extracellular K+
A, cytosolic Ca2+ concentration at 1 Hz contraction rate in whole cell model after switch from Ko = 5.0 to 2.7 mm. B, peak Ca2+ levels in 1st and 2nd phase after switch from Ko = 5.0 to 2.7 mm. C, SR Ca2+ load, mean NKA current, cytosolic Na+ concentration and mean NCX current after switch from Ko = 5.0 to 2.7 mm. D, factorial analysis of peak Ca2+ levels after switch from Ko = 5.0 to 2.7 mm without Ko dependency of NKA or Ko dependency of all Ko-sensitive ion transporters other than NKA.
A factorial analysis was performed (Fig.5D) to test the relative role of NKA versus other ion transporters sensitive to Ko in mediating the increased Ca2+ levels by low Ko. In this analysis, the 2nd positive phase observed experimentally was not present when holding NKA pump activity at the rate obtained with Ko at 5.0 mm while all other ion transporters were exposed to Ko at 2.7 mm. Thus, the factorial analysis suggests that reduced NKA activity is the main mechanism leading to the increase in Ca2+ transient amplitude by low Ko.
Inhibition of NKA α2 abolished the increase in Ca2+ transient amplitude by low Ko
We next investigated the hypothesis that reduction of NKA α2 currents mediates the increase in Ca2+ transient amplitude by low Ko. NKA I–V relationships at Ko at 5.0 and 2.7 mm in voltage clamp cardiomyocytes exposed to a low dose of ouabain that selectively inhibits NKA α2 (Swift et al. 2007) are shown in Fig.6A and B.
Figure 6.
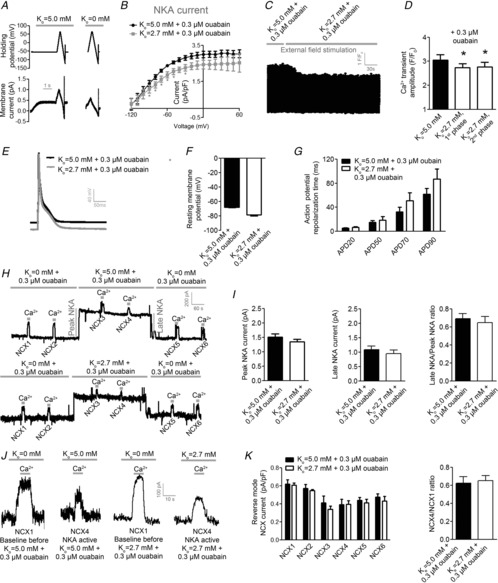
NKA α2 inhibition abolishes the increase in Ca2+ transient by low extracellular K+
A, protocol and representative tracing used to obtain I–V plots for NKA currents. B, I–V plots for NKA currents in the presence of 0.3 μm ouabain. C, representative tracing of Ca2+ transients in ventricular myocytes after switch from Ko = 5.0 to 2.7 mm in the presence of 0.3 μm ouabain and field stimulated at 1 Hz. D, mean Ca2+ transient amplitude. E, representative tracings of RMPs and APs. F, mean RMP. G, mean AP repolarization time. H, representative tracings of experiments used to obtain NKA-dependent regulation of NCX currents in the presence of 0.3 μm ouabain. I, NKA currents in whole cell voltage-clamped ventricular myocytes in the presence of 0.3 μm ouabain. J, reverse mode NCX currents evoked by Ca2+ in whole cell voltage-clamped ventricular myocytes. K, mean NCX currents (left) and NCX4/NCX1 ratio. *P < 0.05.
To test whether NKA α2 mediates the increase in cellular Ca2+ levels by low Ko, we obtained Ca2+ transients in ventricular myocytes field stimulated at 1 Hz in the presence of 0.3 μm ouabain. At baseline, pretreatment with ouabain increased Ca2+ transient amplitude (data not shown). Switching Ko from 5.0 to 2.7 mm induced the 1st negative phase (Fig.6C, D, P < 0.05, n = 9) within a comparable time frame as in cells without ouabain present. Importantly, the 2nd positive phase was abolished in cells pretreated with ouabain (Fig.6C, D), indicating that reduced NKA α2 current is a key mediator of the increase in Ca2+ transient amplitude seen with low Ko.
Selective NKA α2 inhibition blunted the ability of low Ko to increase Na+ levels sensed by NCX
As low Ko was not able to increase the amplitude of Ca2+ transients in ventricular myocytes pretreated with ouabain, we investigated whether Na+ levels sensed by NCX was increased by low Ko in the presence of ouabain (Fig.6H–K). Both peak and late-stage NKA currents were comparable at Ko at 5.0 and 2.7 mm in ventricular myocytes exposed to ouabain. Importantly, neither of the measured NCX currents (NCX3–4) or the NCX4/NCX1 ratio were different between cells perfused with 5.0 and 2.7 mm K+ in the presence of ouabain. These results are consistent with a model where low Ko increases Na+ levels sensed by NCX by preferentially lowering NKA α2 activity in ventricular myocytes.
Selective NKA α2 inhibition did not alter RMP or AP response to low Ko
To exclude that 0.3 μm ouabain altered the RMP or AP response to low Ko, we obtained RMP and AP in ventricular myocytes superfused with ouabain before and after switching Ko from 5.0 to 2.7 mm. As for experiments without ouabain, switching Ko from 5.0 to 2.7 mm hyperpolarized the RMP in the presence of ouabain (Fig.6E, F, P < 0.05, n = 6). None of the analysed APD times were significantly altered by switching Ko from 5.0 to 2.7 mm in the presence of ouabain (Fig.6G).
Discussion
In this study, we show that low Ko, corresponding to the serum K+ levels in patients with moderate hypokalaemia, leads to increased Ca2+ transient amplitude, SR Ca2+ load and Ca2+ wave probability in ventricular myocytes. As we found that reduced NKA activity exerts a more pronounced effect on NCX activity than the concomitant RMP hyperpolarization, we suggest that reduced NKA activity is the main modulator of Ca2+ levels in ventricular myocytes in response to low Ko. Ca2+ overload induced by low Ko was not exacerbated by the presence of an NKA α2 selective dose of ouabain, suggesting that triggered arrhythmias in hypokalaemic patients are linked to reduced NKA α2-mediated control of NCX activity.
Previous studies have reported both negative (Bouchard et al. 2004) and positive inotropic effects (Eisner & Lederer, 1979a,b; Christe, 1983; White & Terrar, 1991) in response to low Ko. Hyperpolarization of the RMP, altered APD and NKA inhibition have all been suggested to contribute to the inotropic response to low Ko in various cardiac preparations. In the present study, we detected a biphasic response eventually leading to an increase in Ca2+ transient amplitude by switching Ko from 5.0 to 2.7 mm in field-stimulated ventricular myocytes, corresponding to the ‘early’ and ‘late’ effects previously reported in guinea-pig papillary muscles (Eisner & Lederer, 1979a). Several investigators have linked the positive inotropic response to low Ko to reduced NKA activity (Eisner & Lederer, 1979b; Godfraind & Ghysel-Burton, 1980), although it has been reported that alterations in NKA activity could depress contractility through excessive Ca2+ loading (Schouten et al. 1990).
We found that NKA currents were sensitive to switching Ko from 5.0 to 2.7 mm (Fig.2B), in accordance with earlier reports (Nakao & Gadsby, 1986; Han et al. 2009). The decline of NKA currents observed over minutes in the protocol used to study NKA-dependent control of NCX activity (Fig.3G) may be caused by a gradual cellular Na+ depletion as a result of NKA activation. However, it might also involve post-translational modifications of NKA (Aronsen et al. 2013). In the present study, Ko = 5.0 mm induced a larger peak NKA current than Ko = 2.7 mm, while the late phase NKA currents were similar. To explain these findings, we suggest that low Ko initially decreases NKA currents due to the Ko dependency of NKA, and subsequently leads to increased cellular Na+ levels. In line with this idea, reverse mode NCX currents during stable state NKA currents were higher at Ko = 2.7 mm than at 5.0 mm (Fig.3I, J). As NCX currents in this protocol are expected to be mostly regulated by the intracellular Na+ levels, these data suggest that low Ko leads to cytosolic Na+ accumulation sensed by the NCX.
Low Ko has previously been reported to decrease cellular Ca2+ levels by increasing forward mode NCX due to hyperpolarization of the RMP (Bouchard et al. 2004). In line with several earlier studies (Eisner & Lederer, 1979a; Christe 1983; Bouchard et al. 2004), we observed a marked hyperpolarization of the RMP by low Ko. In a protocol designed to study Ca2+ transients with a fixed membrane potential, low Ko was still able to increase Ca2+ levels in ventricular myocytes without the initial decline in Ca2+ transient amplitude (1st phase). Based on this observation, we suggest that the 1st and negative phase observed in field-stimulated ventricular myocytes is due to hyperpolarization of the RMP by low Ko, while the stable 2nd phase is due to reduced NKA activity.
Mathematical modelling showed that the Ko dependency of NKA is necessary for low Ko to increase Ca2+ transient amplitude, and thus supports the experimental data. Two notable discrepancies between the model and the experiments were observed. First, the time from switching to low Ko until the Ca2+ transient amplitude was increased compared to baseline was longer in the model simulation compared to the experiments (∼15 vs. ∼5 min). The model is parameterized at room temperature, which may significantly prolong the time until equilibrium. Furthermore, the model predicted an increase in cytosolic Na+ that was ∼10 times larger than what was observed experimentally. The model does not take into account any of the suggested subcellular Na+ gradients that could regulate Ca2+ levels in ventricular myocytes (Aronsen et al. 2013). Absence of such microdomains in the model could explain the slow development of the cellular response to low Ko and the higher predicted Na+ concentration. Future experimental and mathematical studies will be needed to shed more light on the NKA-dependent control of NCX activity within possible cellular microdomains.
We and others have previously reported that the NKA α2 isoform is a key determinant of cardiac contractility and excitation–contraction coupling (James et al. 1999; Swift et al. 2007, 2008; Despa et al. 2012). Selective reduction of NKA α2 currents have been shown to increase Ca2+ transient amplitude independent of the global cytosolic Na+ level (Swift et al. 2007, Despa et al. 2012). Based on these reports, we proposed that low Ko induces Ca2+ overload by reducing NKA α2 activity and thereby increases Na+ concentrations sensed by NCX. In line with this idea, in contrast to experiments without ouabain, low Ko was not able to increase NCX currents in the presence of ouabain. Furthermore, we did not observe any increase in Ca2+ transient amplitude by switching Ko from 5.0 to 2.7 mm in cells pretreated with ouabain. Based on the NKA currents reported in Figs 3 and 6, NKA α2 was calculated to contribute by 24% and 12% to the total NKA current at Ko = 5.0 and 2.7 mm, respectively, which is in line with previous reports that NKA α2 is more sensitive to low Ko than NKA α1 in the range of clinical hypokalaemia (Han et al. 2009). We did not detect any significant alterations in AP duration by low Ko, indicating that increased intracellular Ca2+ levels by low Ko is not caused by AP alterations as previously reported (White & Terrar, 1991). Altogether, we suggest a model where low Ko induces Ca2+ overload by lowering NKA α2 activity in ventricular myocytes, thereby increasing Na+ levels sensed by NCX (Fig.7).
Figure 7.
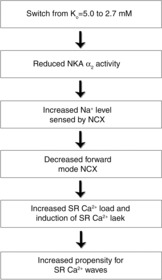
Proposed mechanism for increased propensity for Ca2+ waves in ventricular myocytes by low extracellular K+
In conclusion, we demonstrate that low Ko, within the range of moderate hypokalaemia, leads to increased Ca2+ wave probability in isolated ventricular myocytes. This effect was linked to a reduction in NKA α2 activity and a subsequent increase of Na+ levels that is sensed by the NCX. This provides a possible mechanism underlying triggered ventricular arrhythmias in patients with hypokalaemia.
Glossary
- AP
action potential
- APD
action potential duration
- Ko
extracellular potassium concentration
- NCX
Na+, Ca2+ exchanger
- NKA
Na+,K+-ATPase
- RMP
resting membrane potential
- SR
sarcoplasmic reticulum
Additional information
Competing interests
None of the authors has any conflict of interest.
Author contributions
J.M.A., J.S., W.E.L., K.H., M.K.S., F.S., O.M.S., I.S.: conception and design of the cellular experiments, and collection, analysis and interpretation of data from the cell experiments. A.L., S.A.N., N.P.S.: conception and design of the mathematical models, computational simulations and analysis of simulation results. All authors approved the final version of the manuscript.
Funding
This work was supported by the Research Council of Norway, The South-Eastern Norway Regional Health Authority, The Norwegian Health Association, Anders Jahre's Fund for the Promotion of Science, and Rakel and Otto Kr. Bruun's legacy. A.L., S.N. and N.S. are supported by the United Kingdom Engineering and Physical Sciences Research Council (grants EP/F043929/1, EP/F059361/1 and EP/ G007527/2) and the Virtual Physiological Rat Project (NIH 1 P50 GM094503-01).
References
- Aronsen JM, Swift F. Sejersted OHM. Cardiac sodium transport and excitation–contraction coupling. J Mol Cell Cardiol. 2013;61:11–19. doi: 10.1016/j.yjmcc.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Bokenes J, Aronsen JM, Birkeland JA, Henriksen UL, Louch WE, Sjaastad I. Sejersted OM. Slow contractions characterize failing rat hearts. Basic Res Cardiol. 2008;103:328–344. doi: 10.1007/s00395-008-0719-y. [DOI] [PubMed] [Google Scholar]
- Bouchard R, Clark RB, Juhasz AE. Giles WR. Changes in extracellular K+ concentration modulate contractility of rat and rabbit cardiac myocytes via the inward rectifier K+ current IK1. J Physiol. 2004;556:773–790. doi: 10.1113/jphysiol.2003.058248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard RA, Clark RB. Giles WR. Effects of action potential duration on excitation–contraction coupling in rat ventricular myocytes. Action potential voltage-clamp measurements. Circ Res. 1995;76:790–801. doi: 10.1161/01.res.76.5.790. [DOI] [PubMed] [Google Scholar]
- Christe G. Effects of low [K+]o on the electrical activity of human cardiac ventricular and Purkinje cells. Cardiovasc Res. 1983;17:243–250. doi: 10.1093/cvr/17.4.243. [DOI] [PubMed] [Google Scholar]
- Despa S, Lingrel JB. Bers DM. Na+/K+-ATPase 1537;2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc Res. 2012;95:480–486. doi: 10.1093/cvr/cvs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz ME, Trafford AW, O'Neill SC. Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997;501:3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA. Lederer WJ. Inotropic and arrhythmogenic effects of potassium-depleted solutions on mammalian cardiac muscle. J Physiol. 1979a;294:255–277. doi: 10.1113/jphysiol.1979.sp012929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA. Lederer WJ. The role of the sodium pump in the effects of potassium-depleted solutions on mammalian cardiac muscle. J Physiol. 1979b;294:279–301. doi: 10.1113/jphysiol.1979.sp012930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfraind T. Ghysel-Burton J. Independence of the positive inotropic effect of ouabain from the inhibition of the heart Na+/K+ pump. Proc Natl Acad Sci U S A. 1980;77:3067–3069. doi: 10.1073/pnas.77.5.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A, Spertus JA, Gosch K, Venkitachalam L, Jones PG, Van den Berghe G. Kosiborod M. Serum potassium levels and mortality in acute myocardial infarction. JAMA. 2012;307:157–164. doi: 10.1001/jama.2011.1967. [DOI] [PubMed] [Google Scholar]
- Han F, Tucker AL, Lingrel JB, Despa S. Bers DM. Extracellular potassium dependence of the Na+-K+-ATPase in cardiac myocytes: isoform specificity and effect of phospholemman. Am J Physiol Cell Physiol. 2009;297:C699–C705. doi: 10.1152/ajpcell.00063.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinch R, Greenstein JL, Tanskanen AJ, Xu L. Winslow RL. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys J. 2004;87:3723–3736. doi: 10.1529/biophysj.104.049973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA. Lingrel JB. Identification of a specific role for the Na,K-ATPase α2 isoform as a regulator of calcium in the heart. Mol Cell. 1999;3:555–563. doi: 10.1016/s1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- Louch WE, Hougen K, Mork HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G. Sejersted OM. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J Physiol. 2010;588:465–478. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mork HK, Sjaastad I, Sejersted OM. Louch WE. Slowing of cardiomyocyte Ca2+ release and contraction during heart failure progression in postinfarction mice. Am J Physiol Heart Circ Physiol. 2009;296:H1069–H1079. doi: 10.1152/ajpheart.01009.2008. [DOI] [PubMed] [Google Scholar]
- Nakao M. Gadsby DC. Voltage dependence of Na translocation by the Na/K pump. Nature. 1986;323:628–630. doi: 10.1038/323628a0. [DOI] [PubMed] [Google Scholar]
- Niederer SA, Hunter PJ. Smith NP. A quantitative analysis of cardiac myocyte relaxation: a simulation study. Biophys J. 2006;90:1697–1722. doi: 10.1529/biophysj.105.069534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederer SA. Smith NP. A mathematical model of the slow force response to stretch in rat ventricular myocytes. Biophys J. 2007;92:4030–4044. doi: 10.1529/biophysj.106.095463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard CH, Eisner DA. Allen DG. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. 1983;304:735–738. doi: 10.1038/304735a0. [DOI] [PubMed] [Google Scholar]
- Osadchii OE. Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam Clin Pharmacol. 2010;24:547–559. doi: 10.1111/j.1472-8206.2010.00835.x. [DOI] [PubMed] [Google Scholar]
- Paice BJ, Paterson KR, Onyanga-Omara F, Donnelly T, Gray JM. Lawson DH. Record linkage study of hypokalaemia in hospitalized patients. Postgrad Med J. 1986;62:187–191. doi: 10.1136/pgmj.62.725.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit SV, Clark RB, Giles WR. Demir SS. A mathematical model of action potential heterogeneity in adult rat left ventricular myocytes. Biophysical J. 2009;81:3029–3051. doi: 10.1016/S0006-3495(01)75943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten VJ, Bucx JJ, de Tombe PP. ter Keurs HE. Sarcolemma, sarcoplasmic reticulum, and sarcomeres as limiting factors in force production in rat heart. Circ Res. 1990;67:913–922. doi: 10.1161/01.res.67.4.913. [DOI] [PubMed] [Google Scholar]
- Schouten VJ. ter Keurs HE. The slow repolarization phase of the action potential in rat heart. J Physiol. 1985;360:13–25. doi: 10.1113/jphysiol.1985.sp015601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS. Bers DM. Quantitative assessment of the SR Ca2+ leak–load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- Swift F, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, Louch WE, Sjaastad I. Sejersted OM. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase α2-isoform in heart failure. Cardiovasc Res. 2008;78:71–78. doi: 10.1093/cvr/cvn013. [DOI] [PubMed] [Google Scholar]
- Swift F, Tovsrud N, Enger UH, Sjaastad I. Sejersted OM. The Na+/K+-ATPase α2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc Res. 2007;75:109–117. doi: 10.1016/j.cardiores.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Terkildsen JR, Niederer S, Crampin EJ, Hunter P. Smith NP. Using Physiome standards to couple cellular functions for rat cardiac excitation-contraction. Exp Physiol. 2008;93:919–929. doi: 10.1113/expphysiol.2007.041871. [DOI] [PubMed] [Google Scholar]
- White E. Terrar DA. Action potential duration and the inotropic response to reduced extracellular potassium in guinea-pig ventricular myocytes. Exp Physiol. 1991;76:705–716. doi: 10.1113/expphysiol.1991.sp003537. [DOI] [PubMed] [Google Scholar]