

Abstract
EML4-ALK lung cancer accounts for approximately 3–7% of non-small-cell lung cancer cases. To investigate the molecular mechanism underlying tumor progression and targeted drug sensitivity/resistance in EML4-ALK lung cancer, clinically relevant animal models are indispensable. In this study, we found that the lung adenocarcinoma cell line A925L expresses an EML4-ALK gene fusion (variant 5a, E2:A20) and is sensitive to the ALK inhibitors crizotinib and alectinib. We further established highly tumorigenic A925LPE3 cells, which also have the EML4-ALK gene fusion (variant 5a) and are sensitive to ALK inhibitors. By using A925LPE3 cells with luciferase gene transfection, we established in vivo imaging models for pleural carcinomatosis, bone metastasis, and brain metastasis, all of which are significant clinical concerns of advanced EML4-ALK lung cancer. Interestingly, crizotinib caused tumors to shrink in the pleural carcinomatosis model, but not in bone and brain metastasis models, whereas alectinib showed remarkable efficacy in all three models, indicative of the clinical efficacy of these ALK inhibitors. Our in vivo imaging models of multiple organ sites may provide useful resources to analyze further the pathogenesis of EML4-ALK lung cancer and its response and resistance to ALK inhibitors in various organ microenvironments.
Keywords: Alectinib, bone metastasis, brain metastasis, crizotinib, pleural effusion
ALK rearrangement, most commonly EML4-ALK, is detected in approximately 3–7% of unselected NSCLC cases.1,2 EML4-ALK NSCLC is more frequently observed in patients with adenocarcinoma than in those with other diseases, in young adults than in older patients, and in non- or light smokers (<15 packs/year) than in heavier smokers.3 EML4-ALK and other driver gene alterations such as EGFR mutations and KRAS mutations are almost mutually exclusive.1 Crizotinib, an ALK TKI, shows dramatic clinical efficacy, with a response rate of approximately 60–80%, and a progression-free survival of approximately 9–10 months in ALK-rearranged NSCLC patients.4,5 However, almost all patients who strongly responded to crizotinib acquired resistance over time.6 Recently, new generation ALK-TKIs such as alectinib7 and ceritinib8 have been approved for treatment of ALK-rearranged NSCLC patients, and it is now clear that resistance may also develop against this class of inhibitors. Thus, further research into the molecular pathogenesis of EML4-ALK lung cancer and the mechanism of ALK inhibitor resistance is necessary to further improve the prognosis of this disease.
For such studies, EML4-ALK lung cancer cell lines are essential resources. However, the number of EML4-ALK lung cancer cell lines is still very limited. In addition, while in vivo imaging is a method for studying mechanisms of cancer progression and the efficacy of targeted drugs,9 clinically relevant in vivo imaging models for EML4-ALK lung cancer have not been established. In this study, we identified a novel human lung adenocarcinoma cell line, A925L, that harbors an EML4-ALK gene fusion (variant 5a, E2:A20, a rare isoform). We established highly tumorigenic A925LPE3 cells from the A925L cells after in vivo selection cycles and further developed in vivo imaging models for pleural carcinomatosis, bone metastasis (bone lesion), and brain metastasis (brain lesion).
Materials and Methods
Cell cultures and reagents
A human lung adenocarcinoma cell line, A925L, established from a surgical specimen obtained from a Japanese male patient (T2N2M0, stage IIIA), was maintained in RPMI-1640 medium, supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (10 μg/mL), in a humidified CO2 incubator at 37°C. The characteristics of this cell line are documented in a previous report.10 The H2228 human lung adenocarcinoma cell line, with the EML4-ALK fusion protein variant 3 (E6;A20) were purchased from the ATCC (Manassas, VA, USA). The H3122 human lung adenocarcinoma cell line, with the EML4-ALK fusion protein variant 1 (E13;A20), was kindly provided by Dr. Jeffrey A. Engelman of the Massachusetts General Hospital Cancer Center (Boston, MA, USA).11 PC-9 cells, an EGFR mutant human lung adenocarcinoma cell line, were obtained from Immunobiological Laboratories Co. (Fujioka, JAPAN), Ltd. All cells were passaged for <3 months before renewal from frozen, early-passage stocks. Cells were regularly screened for mycoplasma by using MycoAlert Mycoplasma Detection Kits (Lonza, Rockland, ME, USA). Crizotinib and alectinib (Fig. S1) were obtained from Selleck Chemicals (Houston, TX, USA).
Tumor cell inoculation in SCID mice
We used 5-week-old female SCID mice (Clea, Tokyo, Japan) for the study. For the pleural carcinomatosis model,12 the skin and subcutaneous tissue on the right side of the chest were cut and the parietal pleura was exposed. Tumor cells (1 × 106/100 μL) were then injected into the right thoracic cavity through the parietal pleura by using a 27-G needle. Subsequently, the incisions were sutured to close the wound.
For the bone metastasis model,13 the knee joint was sterilized with 70% ethanol, and a percutaneous intraosseal injection was carried out by drilling a 27-G needle into the tibia, immediately proximal to the tuberositas tibiae. After penetration of the cortical bone, the needle was further inserted into the shaft of the tibia and was used to deposit 4 μL tumor cell suspension (4 × 105/4 μL) in the cortex.
For the brain metastasis model,14 the scalp was sterilized with 70% ethanol, and a small hole was bored into the skull, 0.5 mm anterior and 3.0 mm lateral to the bregma, using a dental drill. Cell suspensions (1.5 × 105/1.5 μL) were injected into the right striatum, 3 mm below the surface of the brain, using a 10-μL Hamilton syringe with a 26-G needle. The scalp was closed using an Autoclip Applier, which were purchased from the BD (Franklin Lakes, NJ, USA).
After 7–11 days, the mice were randomized and treated orally with or without crizotinib or alectinib. The size of s.c. tumors and mouse body weights were measured twice per week, and tumor volume was calculated in mm3, as width2 × length/2. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Education, Culture, Sports, Science and Technology, Japan. The protocol was approved by the Committee on the Ethics of Experimental Animals, and the Advanced Science Research Center, Kanazawa University, Kanazawa, Japan (approval no. AP-132618). All surgeries were carried out under sodium pentobarbital anesthesia, and efforts were made to minimize the suffering of the animals.
Multiplex RT-PCR analysis of EML4-ALK
RNA was extracted using an automated BioRobot EZ1 workstation (Qiagen: Tokyo, JAPAN). The isolated RNA was subjected to RT with a ReverTra Ace qPCR RT kit (Toyobo: Osaka, JAPAN), and the resulting cDNA was subjected to PCR for 50 cycles of 94°C for 15 s, 60°C for 30 s, and 72°C for 1 min with AmpliTaq Gold DNA polymerase (Applied Biosystems: Carlsbad, CA, USA) and 2 μmol/L each primer listed in Table S1.15 The PCR products were subjected to Sanger sequencing to confirm the presence of EML4-ALK.
Fluorescence in situ hybridization
The FISH technique, used to detect ALK fusion genes, was carried out as reported previously16 using the Vysis ALK break-apart FISH probe, according to the hybridization and scoring protocol in the package insert (Abbott Molecular, Des Plaines, IL, USA).
Cell viability assay
Cell viability was measured by the MTT dye reduction method. Tumor cells, plated at a density of 2 × 103/100 μL RPMI-1640 plus 10% FBS per well in 96-well plates, were incubated for 24 h. Crizotinib or alectinib were then added to each well, and incubation was continued for another 72 h. Cell growth was measured with MTT solution (2 mg/mL; Sigma, St. Louis, MO, USA), as described.17
Antibodies and Western blot analysis
Protein aliquots of 25 μg each were used for Western blotting. The primary antibodies used were anti-ALK (C26G7), anti-phospho-ALK (Tyr1604), anti-STAT3 (79D7), antiphospho-STAT3 (Y705), Akt, and p-Akt (S473) (all Cell Signaling Technology, Beverly, MA, USA), as well as human/mouse/rat ERK1/ERK2 and p-ERK1/ERK2 (T202/Y204) (R&D Systems, Minneapolis, MN, USA). The membranes treated with the primary antibodies were incubated for 1 h at room temperature with species-specific HRP-conjugated secondary antibodies. Immunoreactive bands were visualized with SuperSignal West Dura Extended Duration Substrate, an enhanced chemiluminescent substrate (Pierce Biotechnology, Rockford, IL, USA). The data presented are representative of three independent experiments.
Histological analyses of tumors
Proliferating cells were detected by incubating tissue sections with Ki-67 antibody (Clone MIB-1; Dako, Glostrup, Denmark). Antigen was retrieved by microwaving tissue sections in 10 mM citrate buffer (pH 6.0). After incubation with secondary antibody and treatment with the Vectastain ABC Kit (Vector Laboratories, Burlingame, CA, USA), peroxidase activity was visualized using the DAB reaction. The sections were counterstained with hematoxylin.
EGFP-Eluc gene transfection
The cDNA of EGFP from pIRES-EGFP Vector (Invitrogen, Carlsbad, CA, USA) and Emerald Luc (Eluc) from Emerald Luc Vector (ELV-101; Toyobo) were cloned into MaRXIVf Puro retroviral vector (Fig. S2). All cDNA were sequenced. Transfection was carried out using Lipofectamine LTX and PLUS Reagent (Invitrogen) and retroviral infection was carried out using Retrovirus Packaging Kit Ampho (Takara: Otsu, JAPAN) according to the manufacturer's instructions. The cells were then selected in puromycin.
Luciferase expression analysis and radiographic analysis by IVIS imaging system
Two to three weeks after inoculation, the quantity of tumors and the quality of bones were tracked in live mice by repeated non-invasive optical imaging of tumor-specific luciferase activity using the IVIS Lumina XR Imaging System (PerkinElmer, Alameda, CA, USA). Twenty minutes after luciferase substrate luciferin (150 mg/kg) i.p. injection, mice were photographed under bright-field illumination and images were overlaid with luminescence data gathered over the maximum exposure period (5–30 s) while anesthetized by 2% isoflurane. The intensity of the bioluminescence signal was analyzed using Living Image 4.0 (PerkinElmer) by serially quantifying peak photon flux at the selected region of interest covering the tumor and corrected for total area of region of interest and time lapse, during which the bioluminescence signals were picked by the charge-coupled device camera and expressed as photons/s/cm2/sr (sr:steradian). At the same time, the X-ray was converted to visible light by a scintillator, and the image captured on a charge-coupled device by the IVIS Lumina XR Imaging System.
Results
A925LPE3 cells have high potential to produce thoracic tumors and pleural effusion in SCID mice
Almost all EML4-ALK lung cancers are adenocarcinomas, which are often associated with pleural carcinomatosis in the advanced stages. Pleural carcinomatosis is, sometimes, the first sign of progression observed during the onset of resistance to crizotinib treatment.4 In fact, Choi et al.18 discovered a resistant ALK mutation by using pleural effusion cell DNA obtained from an EML4-ALK lung cancer patient who acquired crizotinib resistance. We inoculated A925L cells into the thoracic cavity of SCID mice and found that two out of eight mice developed pleural effusion-containing tumor cells (Fig.1). We harvested the pleural effusion-containing tumor cells and cultured them in vitro. These cells were then inoculated into the thoracic cavity of SCID mice. After three cycles, we established A925LPE3 cells. Compared to the parental A925L cells, A925LPE3 cells display a higher potential to produce pleural effusion in SCID mice (Fig.1).
Figure 1.
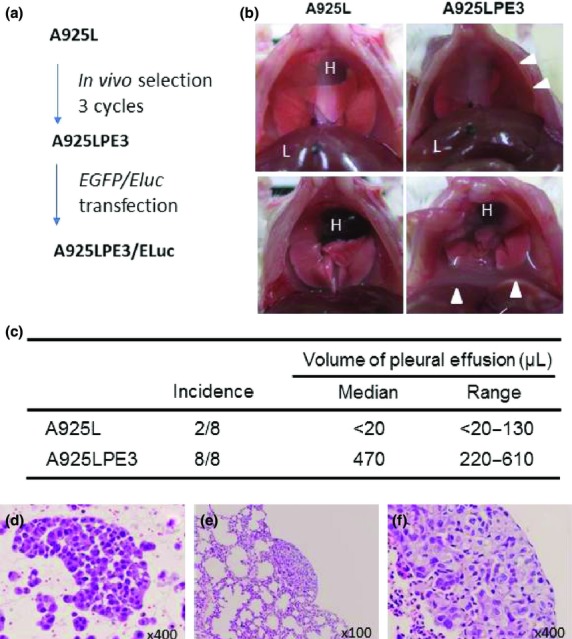
Establishment of highly tumorigenic EML4-ALK non-small-cell lung cancer cell line. (a) A925L is the parental cell line. A925LPE3 cells were established after three cycles of in vivo selection. A925LPE3/Eluc cells were established after transfection of Eluc-EGFP gene into A925LPE3 cells. (b, c) Tumor cells were inoculated into the right thoracic cavity of SCID mice. A925L cells inoculated into the thoracic cavity rarely produced pleural effusion or thoracic tumors, whereas A925LPE3 cells developed pleural effusion in all recipient mice. Arrow heads, pleural effusion; H, heart; L, liver. (d) Tumor cells were detected in the pleural effusion produced by A925LPE3 cells. (e, f) A925LPE3 cells produced disseminated small lesions on the lung.
A925L and A925LPE3 harbor an EML4-ALK gene fusion and are sensitive to ALK-TKIs
The RT-PCR analysis revealed that both the parental A925L cells and the derivative A925LPE3 cells expressed an EML4-ALK gene fusion product (variant 5a, E2:A20) (Fig.2a), as confirmed against the sequence of EML4-ALK (Fig. S3). Break-apart FISH also indicated the presence of ALK fusion genes in both A925L and A925LPE3 cells (Fig.2b). In addition, both the parental and derivative cell lines displayed a similar sensitivity to ALK-TKIs such as crizotinib and alectinib. A925L and A925LPE3 cells showed an intermediate sensitivity to crizotinib and alectinib compared with that shown by other EML4-ALK lung cancer cell lines such as H3122 (variant 1, E13:A20) and H2228 (variant 3a+3b, E6:A20), which are highly and slightly sensitive to ALK-TKIs, respectively (Fig.2c, Table S2). We verified that the observed sensitivity of A925L and A925LPE3 cells to crizotinib or alectinib treatment correlated with inhibition of both phosphorylation of ALK and phosphorylation of downstream molecules such as STAT3, AKT, and ERK (Fig.2d).
Figure 2.

A925L and 925LPE3 cells harbor the EML4-ALK fusion gene and are sensitive to anaplastic lymphoma kinase (ALK)–tyrosine kinase inhibitors. (a) Multiplex RT-PCR for EML4-ALK was carried out. V, variant. (b) Break-apart FISH revealed that A925L and A925LPE3 cells have an ALK fusion gene. (c) Tumor cells were treated with crizotinib or alectinib for 72 h. The cell viability was determined by MTT assay. (d) Tumor cells were treated with crizotinib (1 μM) or alectinib (1 μM) for 1 h. The cell lysates were harvested and proteins and phosphorylation (P-) statuses were examined by Western blot analysis. STAT3, signal transducer and activator of transcription 3.
Subcutaneously inoculated A925L cells are sensitive to ALK-TKIs
We next examined the effect of ALK-TKIs on s.c. tumors produced by A925L cells. Compared to treatment with control, treatment with 25 mg/kg crizotinib retarded tumor growth; however, 50 mg/kg crizotinib was required to induce tumor shrinkage. Under the same experimental conditions, treatment with 25 mg/kg alectinib caused remarkable tumor shrinkage, and the tumors disappear after only 7 days of treatment (Fig.3a).
Figure 3.
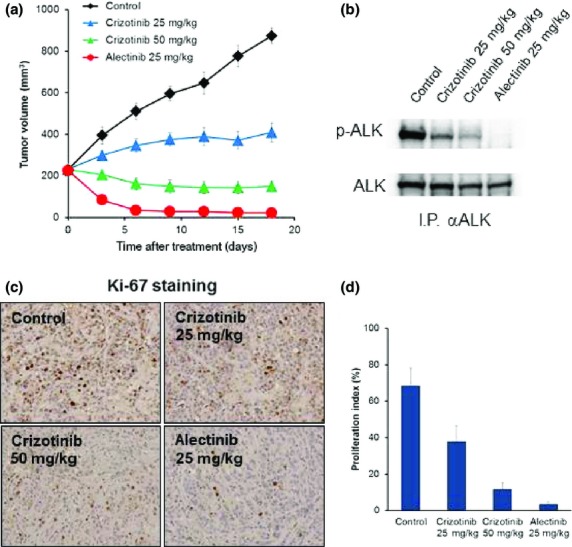
Crizotinib and alectinib show antitumor activity on s.c. tumors produced by A925L cells. (a) A925L (1 × 106) cells were inoculated s.c. into SCID mice. After tumor volumes reached 200 mm3, daily oral treatment with crizotinib or alectinib was initiated. (b) Tumors were harvested after 18 days from treatment initiation, and the lysates were evaluated for the expression and phosphorylation (p-) of anaplastic lymphoma kinase (ALK) by Western blot analysis after immunoprecipitation for ALK protein. (c) Tumors harvested were evaluated for proliferating tumor cells by immunohistochemical staining for Ki-67. (d) Proliferating tumors cells were quantified.
Western blot analysis with in vivo treated tumors showed that crizotinib treatment suppressed ALK phosphorylation and that alectinib treatment, more impressively, inhibited it (Fig.3b). In association with the degree of ALK phosphorylation inhibition, proliferation of tumors cells was also inhibited by treatment with either crizotinib or alectinib (Fig.3c,d). These results indicate that parental A925L cells were sensitive to ALK-TKIs when evaluated in a s.c. tumor model.
Establishment of A925LPE3/Eluc cells for in vivo imaging
In vivo imaging is a powerful tool for evaluating the degree of tumor progression and the efficacy of treatment. To this end, we transfected the Eluc/EGFP gene (Fig. S2) into highly tumorigenic A925LPE3 cells and established A925LPE3/Eluc cells. A925LPE3/Eluc cells constitutively expressed green fluorescence signal (Fig. S4a); luminescence signal was detected after the addition of luciferin (Fig. S4b). In the s.c. model, tumor volume and the luminescence shown by A925LPE3/Eluc cells increased in a time-dependent manner, indicating that luminescence was proportional to the tumor volume produced by A925LPE3/Eluc cells (Fig. S5).
In vivo imaging model for pleural carcinomatosis
To create a pleural carcinomatosis model, A925LPE3/Eluc cells were inoculated into the thoracic cavities of SCID mice. We were able to detect luminescence in the chest 12 days after tumor cell inoculation, and the luminescence increased in a time-dependent manner, reflecting tumor progression (Fig.4). The mice became moribund between days 28 and 35. Daily oral treatment with 50 mg/kg crizotinib or 25 mg/kg alectinib, starting on day 12, decreased luminescence until it almost disappeared on day 28, indicating the high efficacies of crizotinib and alectinib in the pleural carcinomatosis model developed using A925LPE3/Eluc cells.
Figure 4.
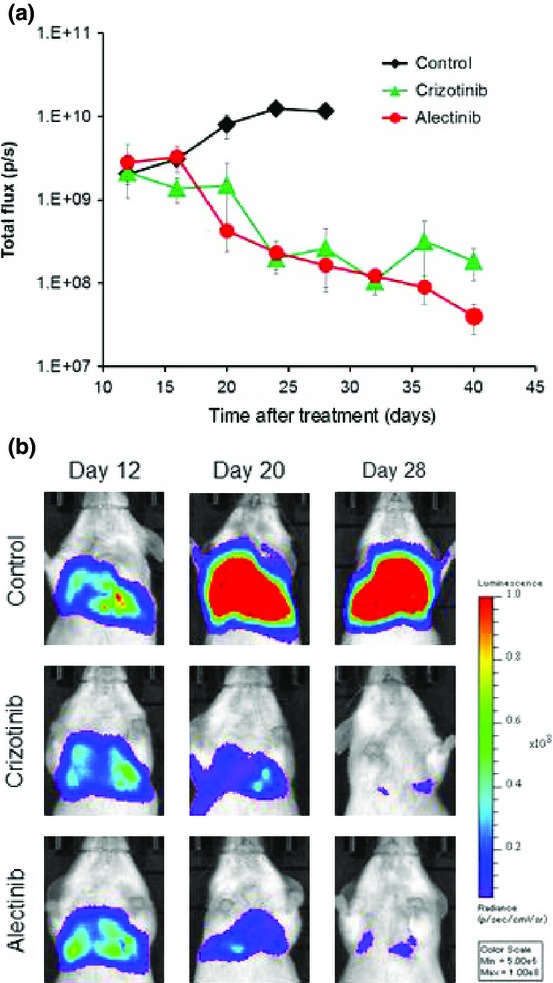
Pleural carcinomatosis model of A925LPE3/Eluc cells and efficacy of anaplastic lymphoma kinase–tyrosine kinase inhibitors. (a) A925LPE3/Eluc cells were inoculated into the thoracic cavity of SCID mice. Mice were treated with control (n = 5), 50 mg/kg crizotinib (n = 5), or 25 mg/kg alectinib (n = 5). Treatment was given daily on days 12–28. Luminescence was evaluated twice per week. Bars indicate standard error (p/s: photons/second). (b) Appearance of representative mice is shown.
In vivo imaging model for bone metastasis
To create a bone metastasis model, A925LPE3/Eluc cells were inoculated into the tibias of SCID mice. We were able to detect luminescence in tibias 10 days after tumor cell inoculation, and the luminescence increased in a time-dependent manner, reflecting tumor progression in the bone (Fig.5). X-ray photographs revealed that A925LPE3/Eluc cells produced osteolytic lesion by day 28, and histological examinations confirmed osteolytic changes in tibias harboring tumor cells (Fig.5a). Daily oral treatment with 50 mg/kg crizotinib, starting on day 10, prevented a further increase in luminescence (Fig.5b) as well as osteolytic changes (Fig.5c), but did not reduce the luminescence below the starting point. However, daily oral treatment with 25 mg/kg alectinib, starting on day 10, significantly decreased luminescence, indicating that alectinib was highly efficacious against bone lesions produced by A925LPE3/Eluc cells.
Figure 5.

Bone metastasis model of A925LPE3/Eluc cells and efficacy of anaplastic lymphoma kinase–tyrosine kinase inhibitors. (a) H&E staining of bone lesions. Arrow head indicates osteolytic change. (b) A925LPE3/Eluc cells were inoculated into the tibia of SCID mice. Mice were treated orally with control (n = 5), 50 mg/kg crizotinib (n = 5), or 25 mg/kg alectinib (n = 5). Treatment was given daily on days 10–28. Luminescence was evaluated twice per week. Bars indicate standard error. (c) Appearance of representative mice is shown.
In vivo imaging model for brain metastasis
To create a brain metastasis model, A925LPE3/Eluc cells were inoculated into the cerebra of SCID mice, and histological examinations confirmed the presence of tumors in the cerebra (Fig.6a,b). Furthermore, luminescence was detected in the brain 11 days after tumor cell inoculation, and the luminescence increased in a time-dependent manner, reflecting tumor progression in the cranial space (Fig.6c). The mice became moribund between days 26 and 30. Daily oral treatment with 50 mg/kg crizotinib, starting on day 11, delayed the increase of luminescence, but did not decrease it, whereas daily oral treatment with 25 mg/kg alectinib, starting on day 11, significantly decreased luminescence (Fig6c,d), indicating that alectinib was highly efficacious against brain lesions produced by A925LPE3/Eluc cells.
Figure 6.
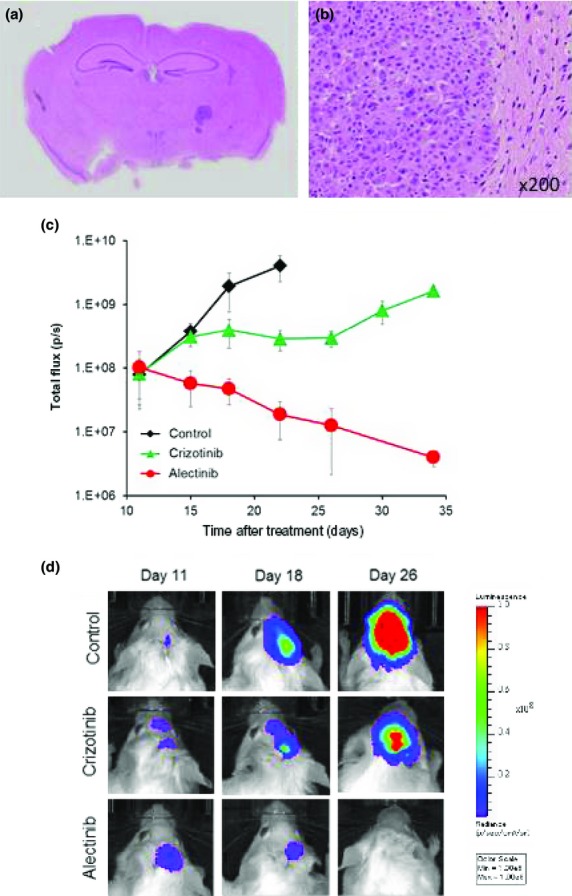
Brain metastasis model of A925LPE3/Eluc cells and efficacy of anaplastic lymphoma kinase–tyrosine kinase inhibitors. A925LPE3/Eluc cells were inoculated into the cerebrum of SCID mice. (a) Macroscopic appearance of brain tumors. (b) Microscopic appearance of brain lesions (×200). (c) Mice were treated orally with control (n = 5), 50 mg/kg crizotinib (n = 5), or 25 mg/kg alectinib (n = 5). Treatment was given daily on days 11–40. Luminescence was evaluated twice per week. Bars indicate standard error. (d) Appearance of representative mice is shown.
Discussion
The EML4-ALK fusion gene accounts for more than 90% of ALK fusion genes. However, only a limited number of NSCLC cell lines that contain EML4-ALK fusion, such as H3122 and H2228, are available.19,20 In the present study, we identified that human lung adenocarcinoma cell line A925L had EML4-ALK variant 5 (E2:A20). At least 14 isoforms of EML4-ALK have been reported so far,15 and variants 1 and 3 account for 90% of EML4-ALK variants.6 Ethnicity is partly responsible for these high incidence rates, with EML4-ALK variants 1 and 3 being dominant in Caucasians21 and East Asians,22 respectively. It was also reported that different EML4-ALK variants show differential sensitivity to ALK-TKIs;23 variant 2 is the most sensitive to ALK-TKIs, followed by variants 1 and 3b, whereas variant 3a is the most resistant. Variant 5 is a rare isoform (accounting for only 1.5%),21 and the sensitivity of variant 5 to ALK-TKIs was largely unknown. We observed that A925L and A925LPE3 cells with EML4-ALK variant 5a showed intermediate sensitivity to crizotinib and alectinib compared to that observed with H3122 (variant 1) and H2228 (variant 3a+3b) cells, thereby proving the usefulness of A925L and A925LPE3 cells as tools for the molecular and biological analysis of EML4-ALK variant 5.
Several xenograft models of human lung cancer cell lines and immunodeficient mice have been reported, including the s.c. inoculation model,24 the orthotopic implantation model,25 and the experimental metastasis model.26 Each model has advantages and disadvantages. The s.c. inoculation model is technically easy to perform and allows for a wide variety of human lung cancer cell lines; however, tumors grow in an ectopic, clinically less relevant, microenvironment. The orthotopic implantation model allows tumors to grow in an orthotopic, clinically relevant microenvironment.27 However, human lung cancer cell lines adaptive to this model are limited, and clinically relevant metastasis to distant organs such as the brain and bone almost never occurs. Finally, the experimental metastasis model with a tail vein injection is technically easy to carry out, and lung metastasis occurs frequently, if adequate cell lines are used. In fact, A925LPE3 cells constantly produced lung metastasis after i.v. injection. But they rarely produce metastasis into the bone or brain after i.v. or intracardiac injection (Table S3). Once again, however, metastases to the brain or bone, which are serious clinical concerns of advanced lung cancer patients, are rarely witnessed. When we used the well-characterized EML4-ALK lung cancer cell lines H3122 and H2228, both of H3122 and H2228 cell lines produced tumors after inoculation into the skin, but not thoracic cavity, bone, or brain of SCID mice (Table S4). In addition, compared with H3122 and A925L cells, H2228 cells are much less sensitive to ALK-TKIs, rendering these cells inadequate for investigation of acquired ALK-TKI resistance.
As documented in this study, A925LPE3 cells are more tumorigenic than A925L, H3122, or H2228 cells, and they constantly produce tumor lesions after inoculation into the thoracic cavity, brain, and bone. Although A925LPE3 cells in these models skipped the invasion of the pleura and early metastatic cascades (from the steps of growth in the primary lesion to extravasation into the metastatic lesion), these models can be powerful tools for elucidating the mechanisms underlying tumor progression and the response and resistance to ALK-TKIs in the thoracic cavity, brain, and bone.
Molecular targeted drugs sometimes show heterogeneous efficacy among lesions in different organs. In the case of ALK-TKIs, crizotinib is less effective on brain lesions compared with lesions in other locations. In fact, CNS lesions, including brain metastases, frequently progress during crizotinib treatment,4 leading some to refer to the CNS as a sanctuary site for crizotinib treatment.28 On the contrary, a recent study reported that alectinib has high efficacy on CNS lesions that became refractory to crizotinib.29 Our data are consistent with these clinical observations; crizotinib was not as effective in our brain metastasis model as alectinib, which was highly effective in the same model. A transgenic mouse model of EML4-ALK is a powerful tool for the analysis of NSCLC tumorigenesis and tumor progression, as well as the efficacy of ALK-TKIs in the lung.30 However, as tumors are selectively developed in the lung, it is impossible to assess the efficacy of ALK-TKIs against lesions in locations other than lung in the transgenic mouse model. Therefore, our model is complementary to other models, such as the s.c. model and transgenic mouse model, in that it will allow researchers to evaluate the heterogeneous efficacy of ALK-TKIs on multiple organ lesions and contribute to the development of novel therapies for EML4-ALK lung cancer. We are now intensively undertaking such experiments by using these models.
In summary, we identified A925L cells as an EML4-ALK variant 5 lung cancer cell line. Moreover, we established A925LPE3 cells with high tumorigenicity in SCID mice after in vivo selection cycles and further developed in vivo imaging models for pleural carcinomatosis, brain metastasis (brain lesion), and bone metastasis (bone lesion). These models may be useful resources for analyzing the pathogenesis of EML4-ALK variant 5, elucidating the molecular mechanisms underlying response and resistance to ALK-TKIs, and developing novel therapies against EML4-ALK lung cancer.
Acknowledgments
We thank Ms. Yuki Togashi, Ms. Satoko Baba, and Dr. Kengo Takeuchi (Japanese Foundation for Cancer Research) for valuable discussions and confirming EML4-ALK variant 5a in A925L and A925LPE3 cells by PCR and FISH.
Glossary
- ALK
anaplastic lymphoma kinase
- CNS
central nervous system
- EGFP
enhanced green fluorescent protein
- EGFR
epidermal growth factor receptor
- Eluc
Emerald luciferase
- EML4
echinoderm microtubule-associated protein-like 4
- NSCLC
non-small-cell lung cancer
- STAT
signal transducer and activator of transcription
- TKI
tyrosine kinase inhibitor
Disclosure Statement
Seiji Yano obtained honoraria from Chugai Pharmaceutical Co. Ltd. Fumihiro Tanaka obtained grants and honoraria from Chugai Pharmaceutical Co. Ltd. The other authors have no conflict of interest.
Supporting Information
Fig. S1. Structures of crizotinib and alectinib.
Fig. S2. Schematics of Eluc/EGFP gene expression vector and MaRX IVf puro-EGFP-Eluc
Fig. S3. DNA sequencing confirmed the presence of EML4-ALK variant 5a in A925LPE3 cells
Fig. S4. Fluorescence and luminescence of A925LPE3/Eluc. (a) Fluorescence of GFP was evaluated by flow cytometry. (b) Luciferase activity was determined after addition of luciferin to the tumor cells.
Fig. S5. Luminescence represents the tumor volume produced by A925LPE3/Eluc cells. To assess the correlation between tumor volume and luminescence, A925LPE3/Eluc (3 × 106) cells were s.c. inoculated into SCID mice. Tumor volume and luminescence were evaluated twice per week.
Table S1. Primers to detect EML4-ALK variants
Table S2. Inhibitory concentration, 50% (IC50) of EML4-ALK lung cancer cell lines to crizotinib and alectinib
Table S3. Tumorigenesis rate of A925LPE3 injection
Table S4.Tumorigenesis rate of EML4-ALK lung cancer cell line inoculation
References
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Camidge DR, Doebele RC. Treating ALK-positive lung cancer–early successes and future challenges. Nat Rev Clin Oncol. 2012;9:268–77. doi: 10.1038/nrclinonc.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–53. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011–9. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- Mano H. ALKoma: a cancer subtype with a shared target. Cancer Discov. 2012;2:495–502. doi: 10.1158/2159-8290.CD-12-0009. [DOI] [PubMed] [Google Scholar]
- Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013;14:590–8. doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–97. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Carragher NO, Timpson P. Developments in preclinical cancer imaging: innovating the discovery of therapeutics. Nat Rev Cancer. 2014;14:314–28. doi: 10.1038/nrc3724. [DOI] [PubMed] [Google Scholar]
- Sugaya M, Takenoyama M, Osaki T, et al. Establishment of 15 cancer cell lines from patients with lung cancer and the potential tools for immunotherapy. Chest. 2002;122:282–8. doi: 10.1378/chest.122.1.282. [DOI] [PubMed] [Google Scholar]
- Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–83. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakataki E, Yano S, Matsumori Y, et al. Novel orthotopic implantation model of human malignant pleural mesothelioma (EHMES-10 cells) highly expressing vascular endothelial growth factor and its receptor. Cancer Sci. 2006;97:183–91. doi: 10.1111/j.1349-7006.2006.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara H, Kim SJ, Karashima T, et al. Effects of blocking platelet-derived growth factor-receptor signaling in a mouse model of experimental prostate cancer bone metastases. J Natl Cancer Inst. 2003;95:458–70. doi: 10.1093/jnci/95.6.458. [DOI] [PubMed] [Google Scholar]
- Muraguchi T, Tanaka S, Yamada D, et al. NKX2.2 suppresses self-renewal of glioma-initiating cells. Cancer Res. 2011;71:1135–45. doi: 10.1158/0008-5472.CAN-10-2304. [DOI] [PubMed] [Google Scholar]
- Soda M, Isobe K, Inoue A, et al. A prospective PCR-based screening for the EML4-ALK oncogene in non-small cell lung cancer. Clin Cancer Res. 2012;18:5682–9. doi: 10.1158/1078-0432.CCR-11-2947. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Choi YL, Soda M, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14:6618–24. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- Green LM, Reade JL, Ware CF. Rapid colorimetric assay for cell viability: application to the quantitation of cytotoxic and growth inhibitory lymphokines. J Immunol Methods. 1984;70:257–68. doi: 10.1016/0022-1759(84)90190-x. [DOI] [PubMed] [Google Scholar]
- Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanizaki J, Okamoto I, Takezawa K, et al. Evaluation of EML4-ALK fusion proteins in non-small cell lung cancer using small molecule inhibitors. Br J Cancer. 2012;106:763–7. doi: 10.1038/bjc.2011.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Maus MK, Desai SJ, et al. Large-scale screening and molecular characterization of EML4-ALK fusion variants in archival non-small-cell lung cancer tumor specimens using quantitative reverse transcription polymerase chain reaction assays. J Thorac Oncol. 2014;9:18–25. doi: 10.1097/JTO.0000000000000030. [DOI] [PubMed] [Google Scholar]
- Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115:1723–33. doi: 10.1002/cncr.24181. [DOI] [PubMed] [Google Scholar]
- Heuckmann JM, Balke-Want H, Malchers F, et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res. 2012;18:4682–90. doi: 10.1158/1078-0432.CCR-11-3260. [DOI] [PubMed] [Google Scholar]
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014;351:215–21. doi: 10.1016/j.canlet.2014.05.020. [DOI] [PubMed] [Google Scholar]
- Miyoshi T, Kondo K, Ishikura H, et al. SCID mouse lymphogenous metastatic model of human lung cancer constructed using orthotopic inoculation of cancer cells. Anticancer Res. 2000;20:161–3. [PubMed] [Google Scholar]
- Yano S, Nishioka Y, Izumi K, et al. Novel metastasis model of human lung cancer in SCID mice depleted of NK cells. Int J Cancer. 1996;67:211–7. doi: 10.1002/(SICI)1097-0215(19960717)67:2<211::AID-IJC11>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Orthotopic implantation of human colon carcinomas into nude mice provides a valuable model for the biology and therapy of metastasis. Cancer Metastasis Rev. 1991;10:229–43. doi: 10.1007/BF00050794. [DOI] [PubMed] [Google Scholar]
- Gainor JF, Ou SH, Logan J, et al. The central nervous system as a sanctuary site in ALK-positive non-small-cell lung cancer. J Thorac Oncol. 2013;8:1570–3. doi: 10.1097/JTO.0000000000000029. [DOI] [PubMed] [Google Scholar]
- Gadgeel SM, Gandhi L, Riely GJ, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15:1119–28. doi: 10.1016/S1470-2045(14)70362-6. [DOI] [PubMed] [Google Scholar]
- Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci USA. 2008;105:19893–7. doi: 10.1073/pnas.0805381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Structures of crizotinib and alectinib.
Fig. S2. Schematics of Eluc/EGFP gene expression vector and MaRX IVf puro-EGFP-Eluc
Fig. S3. DNA sequencing confirmed the presence of EML4-ALK variant 5a in A925LPE3 cells
Fig. S4. Fluorescence and luminescence of A925LPE3/Eluc. (a) Fluorescence of GFP was evaluated by flow cytometry. (b) Luciferase activity was determined after addition of luciferin to the tumor cells.
Fig. S5. Luminescence represents the tumor volume produced by A925LPE3/Eluc cells. To assess the correlation between tumor volume and luminescence, A925LPE3/Eluc (3 × 106) cells were s.c. inoculated into SCID mice. Tumor volume and luminescence were evaluated twice per week.
Table S1. Primers to detect EML4-ALK variants
Table S2. Inhibitory concentration, 50% (IC50) of EML4-ALK lung cancer cell lines to crizotinib and alectinib
Table S3. Tumorigenesis rate of A925LPE3 injection
Table S4.Tumorigenesis rate of EML4-ALK lung cancer cell line inoculation