

Abstract
Sal-like protein 2 (Sall2), a homeotic transcription factor, is a putative tumor suppressor. We have previously shown that Sall2 activates the transcription of tumor suppressor gene p21 and suppresses tumorigenesis through cell cycle inhibition and induction of apoptosis. To investigate additional Sall2-regulated downstream genes, we analyzed the differences in mRNA expression profiles with and without exogenously expressed Sall2. We identified 1616 Sall2-responsive genes through gene expression arrays. Promoter-reporter assays of p16INK4A and several other tumor-related genes indicated that the Sall2 regulation of these promoters was not significantly different between the two major forms of Sall2 with alternative exon 1 or exon 1A. Additional analysis showed that Sall2-induced p16 promoter activation was Sall2 dose-dependent. Deletion and site-directed mutagenesis of the p16 promoter identified a consensus Sall2 binding site (GGGTGGG) proximal to the p16 transcription start site and was critical for p16 promoter activation. Finally, to confirm the significance of Sall2-activated p16 expression in cell cycle regulation, we co-transfected the SKOV3 cells with a Sall2 expression construct and a p16 minigene and also co-transfected the ES-2 cells with a Sall2 expression construct and the siRNA against p16 for flow cytometry analysis. Our results showed that Sall2 enhanced the p16 minigene blocking of cell cycle progression and p16 knockdown with siRNA abolished most of the Sall2 inhibition of cell cycle progression. These findings indicate that Sall2 targets multiple cell cycle regulators, including p16, through their promoters, adding knowledge to the understanding of Sall2 and p16 gene regulation, and how Sall2 deregulation may promote cancer formation.
Keywords: Cell cycle, p16 promoter, Sall2, transcription factor tumor suppressor
Sal-like protein 2 (Sall2, also known as p150sal2 and hsal2) is a putative tumor suppressor and a polyoma tumor antigen inactivation target.1,2 Sall2 is a zinc-finger transcription factor with a consensus DNA binding sequence, GGG(T/C)GGG, for optimal binding in vitro. Sall2 thus belongs to a group of GC box-binding proteins including the Sp1 family of transcription factors.3 The SALL2 gene generates two similar major protein products through its two distinct promoters, P1 for the distal promoter and P2 for the proximal promoter, with alternatively spliced proteins differing only at their short N-terminus sequences using either exon 1 or exon 1A.4
Sall2 suppresses tumorigenesis in many ways, including the prevention of viral DNA replication and the activation of p21 promoter by means of a consensus-like binding site with growth arrest and pro-apoptotic functions in tumor cells.3,5 Sall2 may be an early tumor marker for gastric carcinoma6 and acts as a suppressor of ovarian cancers.7 Sall2 also plays a significant role in the development and growth of colorectal carcinomas.8 Epigenetic silencing of SALL2 gene expression in different types of cancers is related to tumor formation.7,9 However, the networks of Sall2-regulated target genes that directly act on cell proliferation have not been fully revealed.
p16, also designated MTS1 and p16INK4A, is a cyclin-dependent kinase inhibitor and an important tumor suppressor that plays critical roles in cell cycle progression, cellular senescence and the development of human cancers. p16 expression can be induced by oncogenes, DNA damage response or aging, and triggers cell cycle arrest and accelerates cellular senescence.10–13 Cell cycle arrest and apoptotic induction function of p16 plays a primary role in its multiple anti-tumor functions, including inhibition of tumor proliferation, migration and tumor formation.14,15
Sall2 binds to two natural promoters, p21 (Cip1/Waf1) and BAX, with GC elements GGG(T/C)GGG, placing Sall2 among the Sp1 family of transcription factors.3 Sp1 is known to positively regulate p16 transcription upon binding to the corresponding GC boxes in the p16 promoter.16 Here, we investigated Sall2-regulated downstream target genes through expression arrays and identified p16 gene as a direct downstream target of Sall2 through specific promoter activation, thus adding knowledge to the understanding of the Sall2 gene regulation network and a potential mechanism for Sall2 deregulation in cancer formation.
Materials and Methods
Cell culture
SKOV3 (ATCC HTB-77) and ES-2 (ATCC CRL-1978) were maintained in DMEM (Gibco, Grand Island, NY, USA), supplemented with 10% FBS (Gibco), 1% penicillin and streptomycin (Gibco) at 37°C and 5% CO2.
Expression microarray analysis
E1A version of Sall2 expression construct or a control empty vector is used to transfect SKOV3 cells, and cells were selected in G418 for 14 days to form stable colonies. Over 100 colonies were pooled to isolate total RNA. The RNA is then converted to cDNA through random primer reverse transcription and used in expression array analysis performed using Agilent Human Expression Array (design ID: 014850). Array profiling was carried out by Shanghai Bio Corporation, Shanghai, China. Data was analyzed using Cluster 3 and Treeview software by Michael Eisen, Eisen Lab, QB3 Institute University of California at Berkeley, CA.
Promoter-reporter constructs
Human osteosarcoma cell (U2OS ATCC HTB-96) genomic DNA was used as a template for nested PCR, to amplify gene promoters Ki67, p16, MX1, MX2, hTERT, TK, Sp1 and c-Myc. Amplification primers are shown in Table1; primers for TK,17 Sp118 and c-Myc19 promoters were the same as reported. The fragments containing the promoter transcription start site were sub-cloned into pGL3-Basic luciferase vector (Promega).
Table 1.
Nested PCR primers for cloning promoters
| Gene | Primer 5′—3′ |
|---|---|
| p16 | Forward1 CATATTGCCAATCCTACAATGCC |
| Reverse1 CTCCCCGCCGCCCGCTGCCTGCTC | |
| Forward2 GCTACACAGCTAATTGAGAGGTACC | |
| Reverse2 CGTCCGTAACTATTCGGTGC | |
| hTERT | Forward1 GCTGCTCTCCGCATGTCGCTGGTTCC |
| Reverse1 GCACGCTCGATGCAGCGACTCTGTTG | |
| Forward2 GACGCAGCGCTGCCTGAAACTC | |
| Reverse2 AGGTGTGAGCGCGGGACTAAGATGTA | |
| MX1 | Forward1 GATCATGTGCTGAATGCCTAGCAC |
| Reverse1 GTGGAGCCTGACCTTGTGGCACTG | |
| Forward2 CTGGACTTCCAGCTTCTGGAATGAGC | |
| Reverse2 GGCACAGCGTGGTTCGGTGCTTCTG | |
| MX2 | Forward1 GTGGAGTTCCACTCCAAGGTGC |
| Reverse1 CATTCACCAAGCAGATGTCACG | |
| Forward2 CCATTGCCTTTGTCCTGTTAATTCAG | |
| Reverse2 GCTGCACGTTCAGGATGGAGAAATC | |
| Ki67 | Forward1 CGCCTGGCCTAGCACCTTCTAGTTC |
| Reverse1 CGGTGGCCCTACAGGCTACGTC | |
| Forward2 CTGCATCCATGAGGGTGGAAGAAC | |
| Reverse2 GCTCTAGCGGCTCCCACCGA |
Data show the sequence of the primer.
p16 promoter-reporter constructs
The p16 (1318 bp) promoter was cloned into the pGL3-Basic luciferase vector. The deletion panel was constructed using a KOD-plus-Mutagenesis Kit (Toyobo, Osaka, Japan). The sequences of mutagenesis primers are shown in Table2.
Table 2.
The sequences of mutagenesis primers
| Mutation | Primer 5′—3′ |
|---|---|
| Δ983 | Forward GAAGCTGGTCTTTGGATCACTG |
| Reverse CTTGAGCTCGGTACCTATCG | |
| Δ571 | Forward CTCACAACTAGGAAAGAATAGT |
| Reverse CTTGAGCTCGGTACCTATCG | |
| Δ417 | Forward AGTGAACGCACTCAAACACG |
| Reverse CTTGAGCTCGGTACCTATCG | |
| Δ160 | Forward GTCCCTCCAGAGGATTTGAG |
| Reverse CTTGAGCTCGGTACCTATCG | |
| Δ97 | Forward AGGAGGGGCTGGCTGGTCACC |
| Reverse CTTGAGCTCGGTACCTATCG | |
| Δ52 | Forward CTCGGCGGCTGCGGAGAGG |
| Reverse CTTGAGCTCGGTACCTATCG | |
| Δ160-del-GG | Forward CTCGGCGGCTGCGGAGAGG |
| Reverse CTCTTTCTTCCTCCGGTGCTGG | |
| Δ160-del-ETS2 | Forward AGGAGGGGCTGGCTGGTCACC |
| Reverse GACCCTGTCCCTCAAATCCTC | |
| Δ160-del-ETS2&GG | Forward CTCGGCGGCTGCGGAGAGG |
| Reverse GACCCTGTCCCTCAAATCCTC |
Data show the sequence of the primer.
Construction of p16 minigene
The p16 promoter fragment was obtained by digestion of p16-Luc with ScaI and XhoI and ligated to a SpeI fragment of p16 cDNA in a promoter-less pcDNA3 based expression plasmid to generate a circular plasmid designated p16 minigene.
siRNA targeting p16
siRNA against p16 and a scrambled control were purchased from Invitrogen, Grand Island, NY. The three independent siRNA used for p16 knockdown are shown as follows. sip16-1: antisense 5′-AACUAUUCGGUGCGUUGGGTT-3′, sense 5′-CCCAACGCACCGAAUAGUUTT-3′; sip16-2: antisense 5′-AUGGUUACUGCCUCUGGUGTT-3′, sense 5′-CACCAGAGGCAGUAA CCAUTT-3′; sip16-3: antisense, 5′-CACCAGAGGCAGUAACC AUTT-3′, sense 5′-AUGGU UACUGCCUCUGGUGTT-3′. siRNA was labeled with FAM at 5′ terminal to track transfection.
Transfection
Transfection of p16 minigene and siRNA was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's suggestions. All other transfections were carried out using PEI (Polysciences, Warrington, PA) according to Cellntec's protocol (Cedarlane Laboratories, Burlington, Ontario, Canada). All follow-up assays were performed 48 h after transfection.
Luciferase assay
Firefly and Renilla luciferase activities present in cellular lysates were assayed using the dual-luciferase assay system (Promega) 48 h after transfection.
Western blot analysis
Whole cell lysate was used for the assay. Antibody against Sall2 was a gift from Dr Thomas Benjamin, Harvard Medical School, Boston, MA, USA. Antibodies against p16 and beta-actin (#3700) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell cycle analysis
Flow cytometry analysis was performed using a BD LSRFortessa flow cytometer (BD Biosciences, San Jose, CA) according to manufacturer's suggestions 48 s after transfection. Flow cytometry analysis was performed with at least 10 000 cells from each sample and cell cycle profiles were evaluated using Modifit 3.0 (Verity Software House, Topsham, ME).
Statistical analysis
Excluding cell cycle analysis, all the data has been expressed as mean ± SD. GraphPad Prism (version 5; GraphPad Software, La Jolla, CA) was used to analyze and illustrate the results. The statistical significance is described in the respective figure legends (*P < 0.05, Student t-test).
Results
Hierarchical cluster analysis of differentially expressed genes in SKOV3 cells impacted by Sall2
Sall2 is an Sp1-like transcription factor with a wide range of functions from tumorigenesis to stem cell marker. To identify the global influence on gene expression of Sall2, we prepared cDNA from SKOV3 cells transfected with either an E1A version of Sall2 expression construct or an empty vector with pooled stable G418-resistant colonies. Using two-color microarray-based gene expression analysis (Agilent, Santa Clara, CA) with Cluster 3.0 and Treeview expression microarray analysis software, we identified approximately 1616 genes with altered expression level of more than 1.5-fold after Sall2 transfection compared with the vector control group among 44 000 probes (Fig.1). There are 739 upregulated genes and 877 downregulated genes, including tumor suppressor p16 and oncogenes hTERT and c-Myc (Fig.1a). The expressions of several genes that inhibit tumor and cell growth, such as p16,14 were upregulated while the expressions of genes known to promote tumor formation, such as hTERT and c-Myc,20,21 were often inhibited (Fig.1b).
Figure 1.
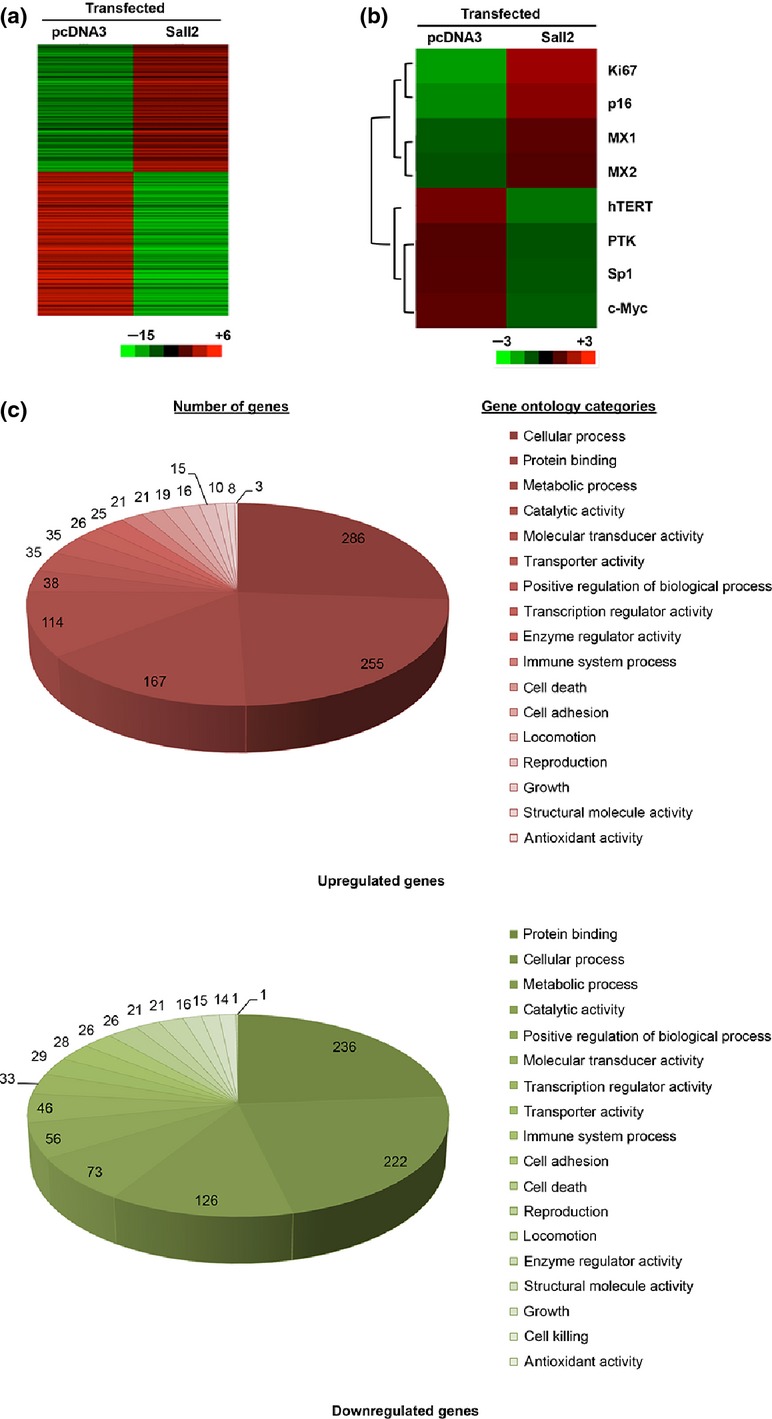
Hierarchical clustering of differentially expressed genes in SKOV3 cell transfected with Sall2. The microarray data for 44 000 gene probes were filtered by applying two criteria for significance, P < 0.05 and fold change (FC) >1.5, between the cells transfected with Sall2 and pcDNA3. (a) The selected data is represented by hierarchical cluster analysis of the normalized expression values of 1616 genes of SKOV3 cell using Sall2 transfected versus control samples. Each row represents a single gene, while each column represents the gene expression levels for cells transfected with Sall2 expression or control plasmid. In the color-coded gene expression heat map, red represents the highest level of expression and green the lowest. (b) Eight selected Sall2-target genes. Ki67, p16, MX1 and MX2 are upregulated by Sall2; hTERT, TK, Sp1 and c-Myc are downregulated by sal2. (c) Ontology list of functions of differentially expressed genes in SKOV3 cells transfected with Sall2. Upper panel: upregulated genes. Lower panel: downregulated genes. The number of genes in each category is shown.
To characterize the Sall2 impact on the overall cell transcription profile changes, we analyzed these mRNA expression changes by Gene Ontology (GO) classification organized by gene function (Fig.1c). According to the hit-ratio, without considering the single digit number of gene changes in antioxidant activity with only four genes, the top Sall2-impacted GO categories are locomotion (7% hit ratio), cell adhesion (5.6%), transporter activity (5.5%) and growth (5.5%) (Table3). Within the functional categories, the ratio of upregulated or downregulated genes is mostly very similar, with the upregulated or downregulated gene ratio varying around 1 (±0.3). However, Sall2-regulated genes in the categories of catalytic and enzyme regulator activities are exceptionally unbalanced, with 1.6-fold more of the upregulated genes than the downregulated genes. The GO catalytic activity category includes hTERT, MX1 and MX2, as well as the enzyme regulator category includes p21 and p16 cyclin kinase inhibitors and tumor suppressors, suggesting that Sall2 may also regulate p16, which is defective in many cancer cells. In SKOV3 cells, the p16 gene has large deletions but leaves a wild type p16 promoter and 405 bp normal exon 1 intact (Table S1, Fig. S1 and Data S1). Therefore, p16 promoter is regulated normally and will produce a 405 bp partial p16 transcript that reflects p16 promoter activity in SKOV3 cells and in our expression array analysis.
Table 3.
Differentially expressed genes in Gene Ontology (GO) categories
| GO name | Downregulated genes | Upregulated genes | Upregulated gene/Downregulated gene | Total | Hits ratio % |
|---|---|---|---|---|---|
| Cell death | 26 | 21 | 0.8 | 1167 | 4 |
| Growth | 14 | 10 | 0.7 | 440 | 5.5 |
| Locomotion | 21 | 16 | 0.8 | 525 | 7 |
| Positive regulation of biological process | 56 | 35 | 0.6 | 1859 | 4.9 |
| Cell adhesion | 26 | 19 | 0.7 | 808 | 5.6 |
| Catalytic activity | 73 | 114 | 1.6 | 5095 | 3.7 |
| Structural molecule activity | 15 | 8 | 0.5 | 603 | 3.8 |
| Transporter activity | 29 | 35 | 1.2 | 1160 | 5.5 |
| Protein binding | 236 | 255 | 1.1 | 12467 | 3.9 |
| Antioxidant activity | 1 | 3 | 3.0 | 46 | 8.7 |
| Enzyme regulator activity | 16 | 25 | 1.6 | 836 | 4.9 |
| Transcription regulator activity | 33 | 26 | 0.8 | 1479 | 2.2 |
| Molecular transducer activity | 46 | 38 | 0.8 | 2123 | 4 |
| Reproduction | 21 | 15 | 0.7 | 743 | 4.8 |
| Cell killing | 1 | / | 0.0 | 46 | 2.2 |
| Immune system process | 28 | 21 | 0.8 | 1085 | 4.5 |
| Metabolic process | 126 | 167 | 1.3 | 8264 | 3.5 |
| Cellular process | 222 | 286 | 1.3 | 12 605 | 4 |
Data show the number of genes and ratio.
Sall2 regulates selected target genes though the regulation of promoter activity independent of the alternative Sall2 exons
To confirm the results from expression array, we selected four upregulated genes and four downregulated genes closely related to tumor formation, including p16, for promoter-reporter analysis. These selected genes are spread in six categories, including tumor-suppressor gene p16, oncogenes hTERT and c-Myc, transcription factor Sp1, cell cycle regulated gene TK, immune response related genes MX1 and MX2, and DNA replication related gene Ki67 (Table4). We also investigated the difference in Sall2 regulatory effects using Sall2 constructs with alternative exon 1 or exon 1A (Sall2E1 or Sall2E1A) and the effect of high or low levels of serum on the Sall2-induced downstream promoter activity changes. Our results showed that the promoter activities of tumor suppressor p16, DNA replication marker Ki67, immune response gene MX1 and MX2 were stimulated by the co-transfected Sall2 expression construct (Fig.2a,b) while promoters of oncogene hTERT and c-Myc, cell cycle-induced gene TK and transcription factor Sp1 were repressed by Sall2 (Fig.2c,d). At the same time, serum concentration did not have a significant effect on either Sall2-induced activation or repression when we compared the luciferase activities of 0.1% serum incubation (Fig.2a,c) with that of 10% serum incubation (Fig.2b,d). The promoter activity changes were not significantly different whether the co-transfected Sall2 constructs contained E1 or E1A alternative exons, except Sp1. Therefore, unless specified otherwise, the EIA version of Sall2 is discussed in the rest of this paper.
Table 4.
Category of genes regulated by Sall2
| Tumor suppressor gene | Oncogene | Transcription factor | Cell cycle regulatory gene | Immune response | DNA replication gene | |
|---|---|---|---|---|---|---|
| Upregulated genes | p16 | / | / | / | MX1 MX2 | Ki67 |
| Downregulated genes | / | c-Myc | Sp1 | TK | / | / |
| hTRET |
Data show the genes.
Figure 2.
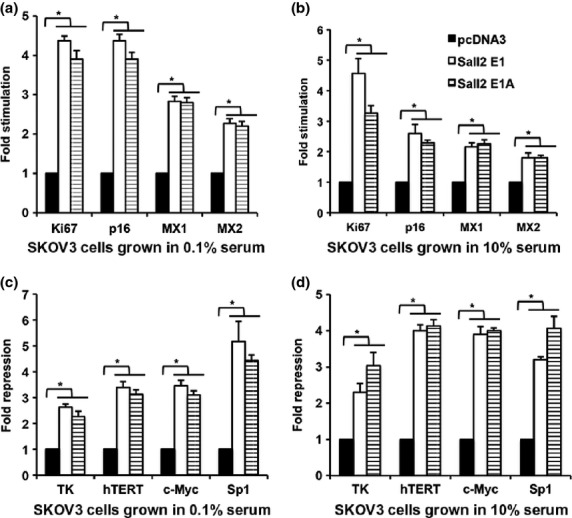
Two forms of Sall2, Sall2E1 and Sall2E1A, affect the activity of the promoter in a similar manner. Co-transfected Sall2 either stimulates or inhibits the activity of some tumor-related promoters in different growth conditions. Sall2E1, Sall2E1A and control vector were co-transfected with selected gene promoter-reporter constructs into SKOV3 cells. Cells were incubated in DMEM and 0.1% serum for 48 h and then in 0.1% serum (a,c) or 10% serum (b,d) for an additional 20 h. Firefly luciferase activities were normalized by Renilla luciferase activity. (a,b) Sall2 induces the expression of p16, MX1, MX2, Ki67 promoters and (c,d) Sall2 inhibits the expression of hTERT, c-Myc, Sp1, TK in SKOV3 cells. Data is shown as mean ± SD (n = 3; *P < 0.05, t-test).
Sall2 upregulates p16 in a dose-dependent manner
One of the most significant Sall2-induced global gene expression changes is the 1.6-fold more up than downregulation of enzyme-regulator genes including the upregulation of tumor suppressors p21 and p16, which both regulate CDK activities. We investigated whether Sall2 regulates p16 expression by western blot and promoter-reporter analysis. Because SKOV3 cells are defective in the p16 gene, Sall2 expression construct was transiently transfected to ES-2 cells that have minimal expression of Sall2 and a normal p16 gene. The cell extracts were assayed for the expression of endogenous p16 48 h after transfection with Sall2. Western blot results showed an increase in p16 expression when normalized with β-actin as a loading control (Fig.3a,b). We further confirmed the induction of p16 expression by the co-transfection of various amount of Sall2 with p16 promoter-luciferase constructs. Our data suggest that upregulation of p16 promoter is Sall2 dose-dependent (Fig.3c).
Figure 3.
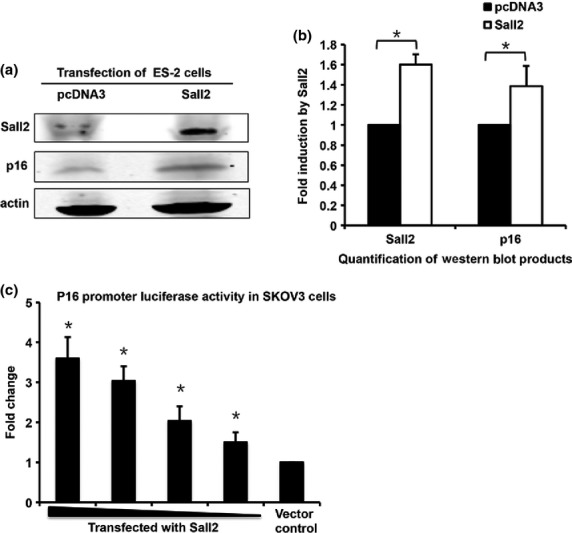
Sall2 upregulates p16 in a dose-dependent manner. (a) p16 is induced by exogenously expressed Sall2 and quantified (b) according to actin level in western blot assay. (c) SKOV3 cells were co-transfected with p16 promoter luciferase construct (0.2 μg/well) and a reducing amount of Sall2 expression plasmid (0.8, 0.6, 0.4 and 0.2 μg/well) with TK-Renilla as an internal control and normalized by Renilla luciferase activity. Data is shown as mean ± SD (n = 3; *P < 0.05, t-test).
A Sall2 binding site located at the proximal of p16 promoter is responsible for Sall2-induced p16 promoter activation
In order to find the Sall2 responsive binding site on p16 promoter, we constructed a series of p16 promoter mutants combining large segment deletions with specific deletions of some of the known transcription factor binding sites and analyzed the reporter luciferase activities in SKOV3 cells (Fig.4).
Figure 4.

Identification of a novel Sall2-responsive element on p16 proximal promoter. (a) Luciferase assay with various deletions of p16-luc that covers −1318 to −52 bp promoter region in SKOV3 cells. Sall2 was transiently co-transfected with p16 promoter deletion luciferase constructs into SKOV3 cells with TK-Renilla as an internal control. Luciferase activity was normalized and expressed as fold differences compared with p16-luc. (b) Luciferase assay with various deletions of p16-luc that covers −160 to −52 bp promoter region in SKOV3 cells. A potential Sall2 transcription factor-binding site of p16 promoter sequence is GGGTGGG (−72 to −66 bp). Data is shown as mean ± SD (n = 4; *P < 0.05, t-test).
SKOV3 cells were first co-transfected with p16 promoter large deletions and Sall2 and assayed for luciferase activities. Several significant deletions near the proximal region were noticed for their dramatic reduction in the promoter activities including Δ983, Δ571, Δ417, Δ160, Δ97, Δ52 (Fig.4a). The reduction in p16 promoter activity of Δ417 and activity recovery for Δ160 indicating a negative regulating DNA element may exist between −417 and −160 region. Between −160 and −52, there is a further significant reduction in p16 activity. We further concentrated our study on the Sall2 regulation in this region of the p16 promoter (Fig.4b).
Compared with mutant Δ160, additional deletion Δ157-del-ETS2 between −160 and −97 with a ETS2 binding region, did not reduce the p16 promoter activity but a deletion mutant Δ160-del-GG of the −97 to −52 with a GC region containing GGGTGGG site reduced the p16 promoter response to Sall2 stimulation by nearly a third (Fig.4b). These results suggested that the major Sall2 stimulation effect on the p16 promoter might act through a Sall2 responsive element with Sall2 binding consensus sequence GGGTGGG.
Sall2 activation of p16 activity contributes to Sall2-induced reduction of cell cycle progression
To confirm that the cell cycle regulation functions of Sall2 also include the promotion of p16 expression, we studied the effect of Sall2 on the cell cycle progression by Sall2-induced p16 expression (Fig.5). To take advantage of the defective p16 background in SKOV3 cells for comparison, we transfected a p16 expression plasmid under p16 promoter control, named p16 minigene (Fig.5a) and a Sall2 expression construct, either separately or in combination, to SKOV3 cells co-transfected with EGFP to mark the transfected cells in flow cytometry assays (Fig.5b). Cell population at G0/G1, G2/M or S phase were analyzed (Fig.5c) and showed that the percentage of cell population in G2/M phase changed significantly with different transfections. Compared with pcDNA3 empty vector control, the transfection of p16 minigene alone reduced the G2/M population from 6.97% to 4.06%, a 0.58 fold of the control cells (Fig.5d); similarly, Sall2 transfection alone reduced the G2/M population to 4.43%, a 0.64 fold of the control level. However, the co-transfection of both Sall2 and p16 minigene reduced G2/M level to 1.02% or 0.15 fold of the control level, less than half of the expected 0.37 fold if no interaction between p16 minigene (0.58 fold of control) and Sall2 (0.64 fold of control). The additional G2/M reduction caused by co-transfection of Sall2 and p16 minigene is consistent with the upregulation of p16 minigene expression by the co-transfected Sall2.
Figure 5.

Sall2 inhibition of cell cycle progression through the activation of p16 transcription. p16 minigene and Sall2 were co-transfected into p16-defective SKOV3 cells with EGFP. p16 siRNA labeled with FAM and Sall2 expression or control plasmid were transiently transfected into p16-wild-type ES-2 cells. EGFP co-transfection and FAM fluorescent labeling were used to trace the transfected cells in flow cytometry analysis. (a) Schematic presentation of p16 minigene construct. (b,f) 48 h after transfection, the transfected cells (marked with co-transfected green fluorescence protein or FAM labeled siRNA) were gated for green fluorescence in flow cytometry analysis. (P2, boxed area). (c) Flow cytometry cell cycle analysis of SKOV3 cells co-transfected with p16 minigene and Sall2 expression construct or control plasmid. (d,h) The percentage of cells in the G2/M phase of the cell cycle in each transfection is depicted as bar graphs. (e) Western blot analysis of siRNA knockdown of p16 expression in ES-2 cells. (g) Flow cytometry cell cycle analysis of ES-2 cells co-transfected with p16 siRNA and Sall2 expression or control plasmid. Data is shown as mean ± SD (n = 3; **<0.01, *<0.05, t-test).
To study the p16 knockdown effect in the p16-wild-type ES2 cells, we tested three p16 siRNA labeled with fluorescent FAM by western blot analysis (Fig.5e). We selected sip16-2 to knockdown p16 expression in ES-2 flow cytometry analysis (Fig.5f) and analyzed cell cycle populations (Fig.5g). When compared with the control, the transfection of sip16 alone increased the G2/M population from 7.58 (control) to 11.34%, a 1.5 fold increase (Fig.5h). The transfection of Sall2 alone reduced the G2/M population to 4.35%, or 0.57-fold the control level. However, co-transfection with both sip16 and Sall2 generated a G2/M population of 9.35%, or 1.2-fold control level, a significant decrease from the 1.5-fold sip16 alone caused by co-transfected Sall2, but p16 knockdown dramatically abolished most Sall2-induced G2/M reduction (from 0.57-fold of control to 1.2-fold of control) indicating that p16 is critical in Sall2-induced cell cycle inhibition.
Discussion
As a Sp1-like zinc finger transcription factor, Sall2 upregulates the transcription of p21 and inhibits tumor growth.5 We investigated Sall2-induced changes in the gene expression profile by expression array analysis to identify Sall2 downstream target genes. We confirmed that the p16 tumor suppressor gene is one of the Sall2 target genes and our results suggest that Sall2 upregulates p16 transcription through a Sall2 responsive element with a Sall2 binding site near the proximal region of p16 promoter.
For the general understanding of the Sall2 effect on downstream gene expression, we analyzed 44,000 gene probes by double fluorescent gene expression microarray and found a global change in the gene expression profile. We noted that genes with catalytic activity and enzyme regulator activity were more likely to be upregulated than downregulated, while more genes for structural molecules were downregulated than upregulated in response to Sall2 overexpression. We selected eight genes for further analysis, including four upregulated genes (p16, Ki67, MX1 and MX2) and four downregulated genes (TK, hTERT, c-Myc and Sp1) that are closely related to tumor formation and cell cycle control.14,20,21 Our findings suggest that both the E1 and E1A forms of Sall2 regulate these genes similarly and consistent with the expression array results (Fig.2).
p16 or p16INK4A, also known as cyclin-dependent kinase inhibitor 2A (CDKN2A), are very similar to p21, cyclin-dependent kinase inhibitor 1A (CDKN1A), in that they both inhibit cyclin-dependent kinases to block cell cycle progression.22,23 Here, we confirmed that p16 is also regulated by Sall2 binding to its promoter in a similar manner as the Sall2 regulation of p21.
Our results suggest that the major Sall2 responsive element is located near the p16 transcription start region with a consensus Sall2 binding site GGG(T/C)GGG. Because the p16 proximal promoter region contains multiple GC boxes, which are potentially both Sp1 and Sall2 binding sites, the possibility that Sall2 competes with Sp1 for these sites to alter the p16 expression level remains to be confirmed but it is consistent with our finding that both Sall2E1 and E1A forms repress Sp1 promoter expression (Fig.2). Compared with upstream deletions, the deletion between −160 and −52 totally abolished the Sall2 stimulating effect on p16 promoter (Fig.4a). This region contains an Ets2 binding site and a Sall2 binding site. Deletion of the potential Sall2 binding site, but not the Ets2 site, abolished the Sall2 stimulation effect on the p16 promoter, suggesting that the Sall2 upregulation of the p16 promoter is independent of Ets2 binding (Fig.4b).
We showed that Sall2 enhanced the p16 inhibitory effect on cell cycle progression though the co-transfection of a p16 minigene together with a Sall2 expression construct in SKOV3 cells and analyzed the effect of Sall2 and p16 by flow cytometry (Fig.5). Cell cycle analysis data revealed that co-transfection of Sall2 and p16 minigene resulted in an S-phase block different from a G2M phase arrest caused by Sall2 induction of p213,5 and p16 arrest at G0/G1 phase.24 Considering that Sall2 is a multi-gene regulator in cell growth (Figs1 and 2), we suspect this may be a mixed result of Sall2, Sall2-induced p21 and Sall2-induced p16.
To evaluate the contribution of the upregulation of p16 by Sall2 expression in p16 wild-type ES-2 cells, we performed the p16 knockdown analysis by flow cytometry. In addition, we re-evaluated the effect of p16 minigene in SKOV3 cells with or without Sal2 to collaborate the p16 siRNA knockdown results. By analyzing the cell cycle populations with transfected Sall2, p16 and co-transfection of Sall2 and p16, we found that knockdown of p16 expression abolished over half of the Sall2-induced cell cycle G2/M phase reduction; therefore, Sall2 regulation of p16 plays a major role in Sall2-induced blocking of cell cycle progression in addition to other Sall2 effects.
We also analyzed the p16 promoter for conserved regions using Dcode ECR Brower (Fig.6).25 Surprisingly, the DNA sequence of the human p16 promoter region had little homology with other mammals, other than dogs and monkeys around the two potential ID1-associated HLH transcription factor binding sites. In the deletion analysis, only the second ID1-associated HLH transcription factor binding site is required for high-level p16 promoter activity (Fig.4a).
Figure 6.
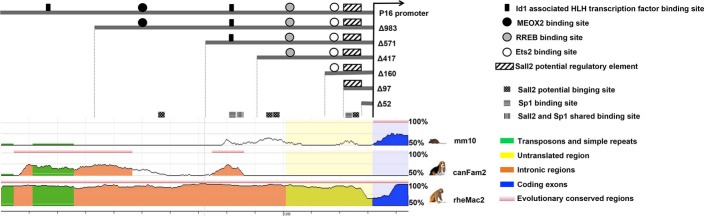
Comparative genomic analysis of p16 promoter region. Schematic diagram shows comparative genomic analysis of p16 promoter regions using ECR browser program (http://ecrbrowser.dcode.org). The DNA sequence of humans is compared with corresponding sequences of mice, dogs and monkeys. Areas with >50% sequence similarities are depicted above the black line.
Taken together, we suggest that Sall2 regulates multiple downstream targets, including upregulation of p16 through promoter binding to a GGGTGGG consensus binding site and causing cell cycle arrest. Further investigation is essential to clarify whether Sall2 binds GC boxes and competes with the Sp1 family of zinc finger proteins to regulate promoter activities. We hope that this study will add to the knowledge on the multifunctional zinc finger transcription factors Sall2 function and p16 cell cycle regulation in cancer cells.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (30771212, 81201769 and 81373319). The authors are indebted to Dr Thomas Benjamin for providing Sall2 plasmid and a monoclonal anti-Sall2 antibody.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting Information
Data S1. Comparison of p16 sequences by Clustalw2.
Fig. S1. Wild-type promoter and most of the exon 1 is preserved in SKOV3 cells.
Table S1. PCR primers for p16 gene region, exon 1, promoter region.
References
- Sung CK, Yim H, Gu H, et al. The polyoma virus large T binding protein p150 is a transcriptional repressor of c-MYC. PLoS One. 2012;7:e46486. doi: 10.1371/journal.pone.0046486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CK, Dahl J, Yim H, Rodig S, Benjamin TL. Transcriptional and post-translational regulation of the quiescence factor and putative tumor suppressor p150(Sal2) FASEB J. 2011;25:1275–83. doi: 10.1096/fj.10-173674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Li D, Sung CK, Yim H, Troke P, Benjamin T. DNA-binding and regulatory properties of the transcription factor and putative tumor suppressor p150(Sal2) Biochim Biophys Acta. 2011;1809:276–83. doi: 10.1016/j.bbagrm.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Li D, Chai L, et al. Cloning and characterization of two promoters for the human HSAL2 gene and their transcriptional repression by the Wilms tumor suppressor gene product. J Biol Chem. 2001;276:48223–30. doi: 10.1074/jbc.M106468200. [DOI] [PubMed] [Google Scholar]
- Li D, Tian Y, Ma Y, Benjamin T. p150(Sal2) is a p53-independent regulator of p21(WAF1/CIP) Mol Cell Biol. 2004;24:3885–93. doi: 10.1128/MCB.24.9.3885-3893.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Burger MM. p150 overexpression in gastric carcinoma: the association with p53, apoptosis and cell proliferation. Int J Cancer. 2004;112:393–8. doi: 10.1002/ijc.20443. [DOI] [PubMed] [Google Scholar]
- Sung CK, Li D, Andrews E, Drapkin R, Benjamin T. Promoter methylation of the SALL2 tumor suppressor gene in ovarian cancers. Mol Oncol. 2013;7:419–27. doi: 10.1016/j.molonc.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haybaeck J, O'Connor T, Spilka R, et al. Overexpression of p150, a part of the large subunit of the eukaryotic translation initiation factor 3, in colon cancer. Anticancer Res. 2010;30:1047–55. [PubMed] [Google Scholar]
- Farkas C, Martins CP, Escobar D, et al. Wild type p53 transcriptionally represses the SALL2 transcription factor under genotoxic stress. PLoS One. 2013;8:e73817. doi: 10.1371/journal.pone.0073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Fang X, Jiang Z, Peng J, Yao N, Fang X, Zheng S. [Proliferative and invasive effects of inhibitors of kinase 4(P15(ink4b) and P16(ink4a)/CDKN2) gene activation on RKO human colorectal cancer cells] Zhonghua Wei Chang Wai Ke Za Zhi. 2014;17:31–5. [PubMed] [Google Scholar]
- Lu Y, Zhang X, Zhang J. Inhibition of breast tumor cell growth by ectopic expression of p16/INK4A via combined effects of cell cycle arrest, senescence and apoptotic induction, and angiogenesis inhibition. J Cancer. 2012;3:333–44. doi: 10.7150/jca.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Xue L, Weng M, et al. Sp1 is essential for p16 expression in human diploid fibroblasts during senescence. PLoS One. 2007;2:e164. doi: 10.1371/journal.pone.0000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basundra R, Kumar A, Amrane S, Verma A, Phan AT, Chowdhury S. A novel G-quadruplex motif modulates promoter activity of human thymidine kinase 1. FEBS J. 2010;277:4254–64. doi: 10.1111/j.1742-4658.2010.07814.x. [DOI] [PubMed] [Google Scholar]
- Grande L, Bretones G, Rosa-Garrido M, et al. Transcription factors Sp1 and p73 control the expression of the proapoptotic protein NOXA in the response of testicular embryonal carcinoma cells to cisplatin. J Biol Chem. 2012;287:26495–505. doi: 10.1074/jbc.M112.376319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Tsai CF, Hsu CY, et al. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J. 2012;26:778–87. doi: 10.1096/fj.11-191742. [DOI] [PubMed] [Google Scholar]
- Gartner S, Gunesch A, Knyazeva T, et al. PTK 7 is a transforming gene and prognostic marker for breast cancer and nodal metastasis involvement. PLoS One. 2014;9:e84472. doi: 10.1371/journal.pone.0084472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jiao S. Increased lung cancer risk associated with the TERT rs2736100 polymorphism: an updated meta-analysis. Tumour Biol. 2014;35:5763–9. doi: 10.1007/s13277-014-1765-8. [DOI] [PubMed] [Google Scholar]
- Santamarina M, Hernandez G, Zalvide J. CDK redundancy guarantees cell cycle progression in Rb-negative tumor cells independently of their p16 status. Cell Cycle. 2008;7:1962–72. doi: 10.4161/cc.7.13.6071. [DOI] [PubMed] [Google Scholar]
- Nishiwaki E, Turner SL, Harju S, et al. Regulation of CDK7-carboxyl-terminal domain kinase activity by the tumor suppressor p16(INK4A) contributes to cell cycle regulation. Mol Cell Biol. 2000;20:7726–34. doi: 10.1128/mcb.20.20.7726-7734.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–67. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32:W280–6. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Comparison of p16 sequences by Clustalw2.
Fig. S1. Wild-type promoter and most of the exon 1 is preserved in SKOV3 cells.
Table S1. PCR primers for p16 gene region, exon 1, promoter region.