

Abstract
A multi-kinase inhibitor, rigosertib (ON 01910.Na) has recently been highlighted as a novel type of anti-cancer agent for the treatment of the myelodysplastic syndromes (MDS), but its action mechanisms remain to be clarified. We investigated the in vitro effects of rigosertib on an MDS-derived cell line MDS-L and a myeloid leukemia cell line HL-60. Rigosertib suppressed the proliferation of both HL-60 and MDS-L cells and induced apoptosis by inhibition of the PI3 kinase/Akt pathway. As the effects on cell cycle, rigosertib treatment promoted the phosphorylation of histone H2AX and led to the DNA damage-induced G2/M arrest. In addition, an immunofluorescence staining study demonstrated the abnormal localization of aurora A kinase, suggesting that rigosertib causes perturbation of spindle assembly and deregulated mitotic patterns towards cell cycle arrest and apoptosis. We also found that rigosertib exerted growth inhibitory effects on two lymphoid cell lines, Jurkat and Ramos. We further examined the molecular pathways influenced by rigosertib from the gene expression profiling data of MDS-L cells and found a possible involvement of rigosertib treatment in the upregulation of the genes related to microtubule kinetics and the downregulation of the mRNA degradation system. The gene set enrichment analysis showed the suppression of “nonsense-mediated mRNA decay (NMD)” as the most significantly affected gene set. These data provide a new aspect and a potential utility of rigosertib for the treatment of refractory hematopoietic malignancies.
Keywords: G2/M arrest, myelodysplastic syndromes, nonsense-mediated mRNA decay, rigosertib, synthetic anticancer agent
The myelodysplastic syndromes (MDS) are a group of acquired hematopoietic disorders characterized by cytopenias and dysplasia, as a result of clonal growth of pathological stem cells and ineffective hematopoiesis, and bear an increased risk of progression to acute myeloid leukemia (AML).1 Patients with MDS are classified from low-risk to high-risk on the basis of the Revised International Prognostic Scoring System (IPSS-R).2,3 Although hematopoietic stem cell transplantation (HSCT) is the only potentially curative treatment for patients with high-risk MDS, it is restricted to a small subset of MDS patients owing to factors such as advanced age, concomitant comorbidities and donor availability.4 DNA methyltransferase inhibitors (DNMT inhibitors), azacitidine (AZA) and decitabine (DAC) have recently been used as chemotherapeutic agents for patients with high-risk MDS who are not candidates for HSCT. Notably, the treatment with AZA significantly increased overall survival in patients with high-risk MDS as compared with conventional therapy (AZA-001), but it has a low potential for a complete cure.5 Hence, novel therapeutic agents are desired for refractory cases after initial therapy with DNMT inhibitors or for those without indication for HSCT.6–8
Rigosertib (ON 01910.Na) is a multi-kinase inhibitor that inhibits cell cycle progression by selectively inducing a mitotic arrest and apoptosis in cancer cells. First, this agent was reported to suppress the activity of polo-like kinase 1 (Plk1),which modulates mitosis, spindle assembly and centrosome maturation in various tumors.9–12 Subsequent reports, however, did not support the relationship between rigosertib and Plk1 inhibition.13–15 In hematological malignancies, Prasad et al.13 demonstrate that rigosertib treatment results in a rapid decrease in cyclin D1 level in mantle cell lymphoma cells, probably due to the inhibition of mRNA translation in conjunction with inhibition of the phosphatidylinositol-3 (PI3) kinase/Akt/mTOR/eIF4E-BP pathway. Chapman et al.14 report that rigosertib is selectively cytotoxic for chronic lymphocytic leukemia cells through a dual mechanism of action involving inhibition of PI3K/Akt pathway and induction of oxidative stress. Oussenko et al.15 demonstrate that the biological activity of rigosertib correlated with prolonged phosphorylation/hyperphosphorylation of RanGAP1SUMO1.
In clinical studies, Steetharam et al.16 show decreased intracellular Akt phosphorylation of bone marrow CD34+ cells of high-risk MDS patients who did not respond to DNMT inhibitor but responded to rigosertib. Olnes et al. demonstrate that rigosertib suppressed cyclin D1 in bone marrow CD34+ cells of MDS patients with trisomy 8 and monosomy 7.17–19 Furthermore, the efficacy of oral rigosertib administration for MDS patients has also been reported.20 However, the action mechanisms of rigosertib remain to be solved.
In this study, we investigated the effects of rigosertib on an MDS cell line established in our laboratory, and demonstrated that rigosertib induces cell death by inhibition of PI3 kinase/Akt pathway and by DNA damage-induced G2/M arrest. We further examined the molecular pathways regulated by rigosertib from the gene expression profiling of MDS-L cells treated with rigosertib. It was also suggested that rigosertib exerts an inhibitory effect on the gene groups involved in the nonsense-mediated mRNA decay system.
Materials and Methods
Reagents
Rigosertib (ON01910.Na, Estybon) was generously provided by Onconova Therapeutics (Newton, PA, USA). It was dissolved in dimethylsulfoxide and stored at −20°C, protected from light. We used rigosertib at concentrations up to 5 μM.
Cell lines and culture
MDS-L cell line was established as a blastic subline from the parental MDS92 cell line. MDS-L cells were positive for CD34, c-Kit, HLA-DR, CD13 and CD33 and partially positive for CD41. The main karyotype was 49, XY, +1, der(5)(5;19), −7, +8, −12, der(13)t(7,13), der(14)t(12;14), der(15)(15;15), +19, +20, +21, der(22)(11;22) but showed multiple chromosomal abnormalities similar to those of MDS92.21–24 The MDS-L cells were maintained in RPMI1640 medium supplemented with 10% FBS, 50 μM 2-mercaptoethanol, 2.0 mM l-glutamine and 100 U/mL IL-3. A human myeloid leukemia cell line, HL-60, T-acute lymphoblastic leukemia cell line Jurkat and Burkitt lymphoma cell line Ramos were also used in this study. The morphological assessment was performed with May–Gruenwald Giemsa-stained cytospin slides.
Cell growth assay and MTT assay
Cell growth was assessed by counting the number of living cells after trypan blue staining. Cell suspensions were plated into 96-well plates in the presence of the drug or solvent alone, incubated as above at 37°C for 1–4 days, and analyzed using the 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay.
Apoptosis assay
Apoptosis was examined using an AnnexinV Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA) and all samples were analyzed with FACS Calibur flowcytometer and CellQuest software (Becton Dickinson, Franklin Lakes, NJ, USA).25,26
Cell cycle analyses
Cells were fixed with 100% methanol for 30 min and treated with 2 mg/mL ribonuclease A (Nacalai Tesque, Kyoto, Japan) for 30 min at 37°C, then with 50 μg/mL propidium iodide (PI [Sigma, St Louis, MO, USA]) for a further 20 min at room temperature.
Immunoblotting analysis
Cell lysates of MDS-L and HL-60 were prepared in lysis buffer containing 50 mM Tris–HCL, 150 mM NaCl, 5 mM EDTA, 0.5% TritonX-100, 0.05% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 2 mM phenylmethylsulfonyl fluoride and 1 mM Na3VO4. The lysates were separated by SDS-PAGE and immunoblotting analysis was performed as previously described.27 Primary antibodies were obtained from Santa Cruz Biotechnology (p53, bcl2, bcl-XL/Xs; Santa Cruz, CA, USA), Cell Signaling Technology Tokyo, Japan (phospho-aurora A (Thr288), Akt, phospho-Akt (Ser473), phospho-p53 (Ser15), phospho-H2AX (Ser139), cleaved-PARP (cPARP), cyclin A; Danvers, MA, USA), Sigma-Aldrich (α-tubulin; St. Louis, MO, USA), BD biosciences (aurora A, aurora B, cyclin B1, cyclin D; San Jose, CA, USA) and Abcam (Plk-1, phosphor-Plk-1; Cambridge, MA, USA). Horse-radish peroxidase-conjugated mouse and rabbit antibodies were from GE Healthcare Life Sciences (Piscataway, NJ, USA).
Immunofluorescence staining of the cells
Cells were centrifuged onto cytospin slides and fixed for 30 s in 4% formalin/50% acetone, then permeabilized with lysis buffer including 0.2% Triton X-100, 25 mM HEPES, 60 mM PIPES, 10 mM EGTA and 2 mM MgCl2 for 30 s. Immunofluorescence staining was performed using mouse monoclonal anti-aurora kinase A (BD biosciences) to detect centrosomes, and AlexaFluor488-conjugated anti-IgG antibodies (Molecular Probes, Eugene, OR, USA) as secondary antibodies. Spindle assembly was detected with AlexaFluor 555-conjugated rabbit polyclonal anti-β-tubulin antibodies (Sigma-Aldrich). The nucleus was stained with DAPI (Dojindo, Kumamoto, Japan). Cells were observed under an Olympus Tokyo, Japan BX51 fluorescence microscope.
Gene expression profiling and gene set enrichment analysis
Gene expression profiling of MDS-L and HL-60 cells was examined in three independent experiments (rigosertib-treated or untreated cells were harvested after 24 h treatment). Total RNA was extracted with an RNeasy Mini Kit (Qiagen, Germantown, MD, USA), converted to cDNA and amplified with GeneChip WT Terminal Labeling and Controls Kit (Affymetrix, Santa Clara, CA, USA). The fragmentation, the labeling and the hybridization of cDNA were treated with a GeneChip Hybridization, Wash, and Stain Kit (Affymetrix). Chips were scanned with a GeneChip Scanner 3000 7G System (Affymetrix).
The gene set enrichment analysis (GSEA Broad Institute Cambridge, MA, USA) was performed using the gene expression profiling data as obtained above and by handling the GSEA software and the Molecular Signatures Database according to the references.28,29
Statistical analyses
Most data were expressed as mean ± SEM. Comparisons between the groups were done using one-way anova. Differences were considered statistically significant if P-values were <0.05. Data analysis was performed using the Prism software (GraphPad, La Jolla, CA, USA).
Results
Rigosertib inhibits the proliferation of myeloid and lymphoid cell lines and causes apoptosis
We added rigosertib into the culture of HL-60, MDS-L, Jurkat and Ramos cells and found that all four cell lines were susceptible to rigosertib and each cell viability was reduced at concentrations <200 nM (Fig.1a and data not shown). We determined dose-response curves and the half-maximal inhibitory concentration (IC50) of rigosertib in HL-60 cells (24 and 48 h), MDS-L cells (24, 48, 72 and 96 h), Jurkat cells (24 and 48 h) and Ramos cells (24 and 48 h) with MTT assay, respectively. The IC50 value of rigosertib for HL-60 cells, Jurkat cells and Ramos cells at 48 h was 58.5 ± 1.8, 95.4 ± 0.6 and 193.1 ± 2.5 nM, whereas that of rigosertib for MDS-L cells at 96 h was 52.1 ± 0.8 nM. Cell counting by trypan blue staining indicated a similar tendency to the result of MTT assay (data not shown).
Figure 1.
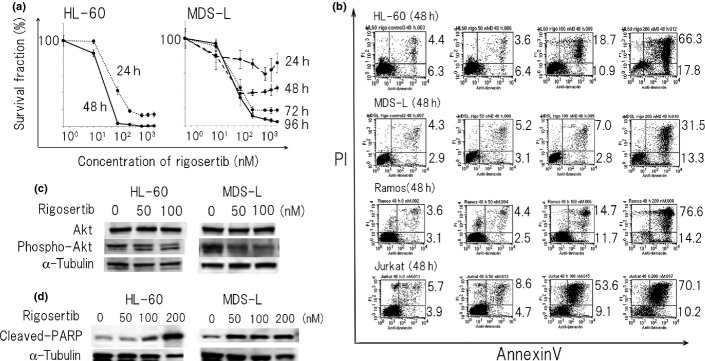
Rigosertib suppresses the growth of myeloid and lymphoid cells mainly by apoptosis. (a) HL-60, MDS-L, Jurkat and Ramos cells were treated with rigosertib (0–5000 nM) for indicated times and cell count was assessed by MTT assay. The value without rigosertib was adjusted to 100%. The data represent the mean values with SD from five independent experiments. (b) HL-60, MDS-L, Jurkat and Ramos cells were treated with different concentrations of rigosertib (0, 50, 100 and 200 nM) for 48 h and apoptosis was assessed by flow cytometry using annevin V/propiodium iodide (PI) staining. The single-positive fraction for annexin V implies early apoptosis, and the double-positive fraction for annexin V/PI implies late apoptosis. The values of the lower right area and the upper right area indicate the percentage of the cells in early apoptosis and late apoptosis, respectively. (c) HL-60 and MDS-L cells were treated with indicated concentrations of rigosertib for 24 and 48 h, respectively, and protein lysates were analyzed by immunoblotting analysis for the detection of Akt and phospho-Akt with each antibody. The amount of alpha-tubulin was shown as a loading control. (d) HL-60 and MDS-L cells were treated with indicated concentrations of rigosertib for 24 and 48 h, respectively, and protein lysates were analyzed by immunoblotting analysis for the detection of cleaved PARP. The amount of alpha-tubulin was shown as a loading control.
Next, we examined the presence and the degree of apoptosis of regosertib-treated HL-60, MDS-L, Jurkat and Ramos cells by dual staining of annexinV and PI. Rigosertib induced apoptosis in these cells in a dose-dependent (0, 50, 100 and 200 nM) and time-dependent manner (24 and 48 h), respectively (Fig.1b). Because rigosertib was previously reported to inhibit PI3K/Akt pathway, we examined the status of phospho-Akt by immunoblotting analysis. The phospholylation of Akt in HL-60 and MDS-L cells was suppressed by rigosertib in a dose-dependent manner (0, 50, 100 nM at 24 h) (Fig.1c). The amount of cPARP, a marker of undergoing apoptosis in HL-60 and MDS-L cells, was increased in a dose-dependent manner (0, 50, 100 and 200 nM at 48 h) by rigosertib treatment (Fig.1d). In contrast, the expression of bcl-2 family members (Bcl-2, Bax, Bcl-XL, Bcl-Xs) did not change in spite of massive cell death after rigosertib treatment (data not shown).
Rigosertib has inhibitory effects on the cell cycle in myeloid and lymphoid cell lines
Because rigosertib has been reported to affect the cell cycle, we checked the morphology of rigosertib-treated HL-60, MDS-L, Jurkat and Ramos cells by May–Gruenwald–Giemsa staining and calculated the percentage of mitotic cells (mitotic index) (Fig.2a). The mitotic index of HL60 cells after rigosertib treatment (at 50 nM for 24 and 48 h) was increased as compared with that of the control cells (7.9% at 50 nM for 24 h and 16.2% at 50 nM for 48 h vs 2.0% in the control). Likewise, the mitotic index of MDS-L cells after rigosertib treatment was increased (9.0% at 100 nM and 15.5% at 200 nM for 48 h vs 1.5% in the control). In addition, lymphoid cells also had a similar and marked tendency of mitotic arrest (Jurkat: 7.4% at 50 nM, 27.5% at 100 nM and 34.9% at 200 nM for 24 h vs 2.4% in the control; Ramos: 5.3% at 50 nM, 15% at 100 nM and 43.7% at 200 nM for 24 h vs 1.9% in the control).
Figure 2.
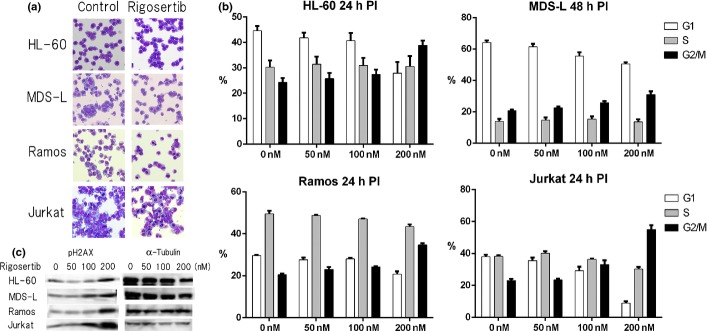
Effects of rigosertib on the morphology and cell cycle of myeloid and lymphoid cells. (a) Morphological changes of rigosertib-treated HL-60, MDS-L, Jurkat and Ramos cells. Four cell lines were cultured without treatment (control) or treated with 50, 100 and 200 nM rigosertib for 24 and 48 h, respectively, and cytospin slides are indicated (May–Gruenwald–Giemsa stain, original magnification ×400). (b) The cell cycle analyses by propiodium iodide (PI) staining are shown. HL-60, MDS-L, Jurkat and Ramos cells were treated with indicated concentrations of rigosertib for 24 and 48 h, respectively, and the cells were stained with PI and analyzed by flow cytometry. The cell fractions at G1, S and G2/M phase are presented by white, gray and black bars, respectively. (c) HL-60, MDS-L, Jurkat and Ramos cells were treated with indicated concentrations of rigosertib for 24 and 48 h, respectively, and protein lysates were analyzed by immunoblotting analysis for the detection of phospho-H2AX. The amount of alpha-tubulin is shown as a loading control.
Next, we performed the cell cycle analysis of rigosertib-treated HL-60, MDS-L, Jurkat and Ramos cells by flow cytometry and demonstrated that exposure of each cell line to 50–200 nM rigosertib for 24 or 48 h led to the increase in the number of the cells at G2/M phase in a dose-dependent manner (Fig.2b). Phosphorylation of histone H2AX (phospho-H2AX) is the most reliable marker for activated DNA damage response (DDR), and we actually observed that the amount of phospho-H2AX was increased by rigosertib treatment in a dose-dependent manner (Fig.2c). In addition, we also evaluated the status of TP53, p21, cyclin A, cyclin B1 and cyclin D1, which were associated with cell cycle arrest, but the expression of them was apparently unchanged (data not shown). Although rigosertib is considered as a multi-kinase inhibitor including Plk-1 inhibition, the present study could not demonstrate the in vitro data indicating Plk-1 inhibition clearly (data not shown).
Rigosertib causes profound spindle abnormalities and abnormal centrosome localization in HL-60 and MDS-L cells
Since we demonstrated that rigosertib exerts inhibitory effects on the cell cycle, particularly on the progress of M phase, we attempted to explore whether rigosertib affects aurora A and B kinases, which are involved in mitosis and cell division, and examined their expression and localization in HL-60 and MDS-L cells. Although the protein amount of both kinases after rigosertib treatment appeared unchanged, the phosphorylation of aurora A kinase was found to be increased (Fig.3a).
Figure 3.
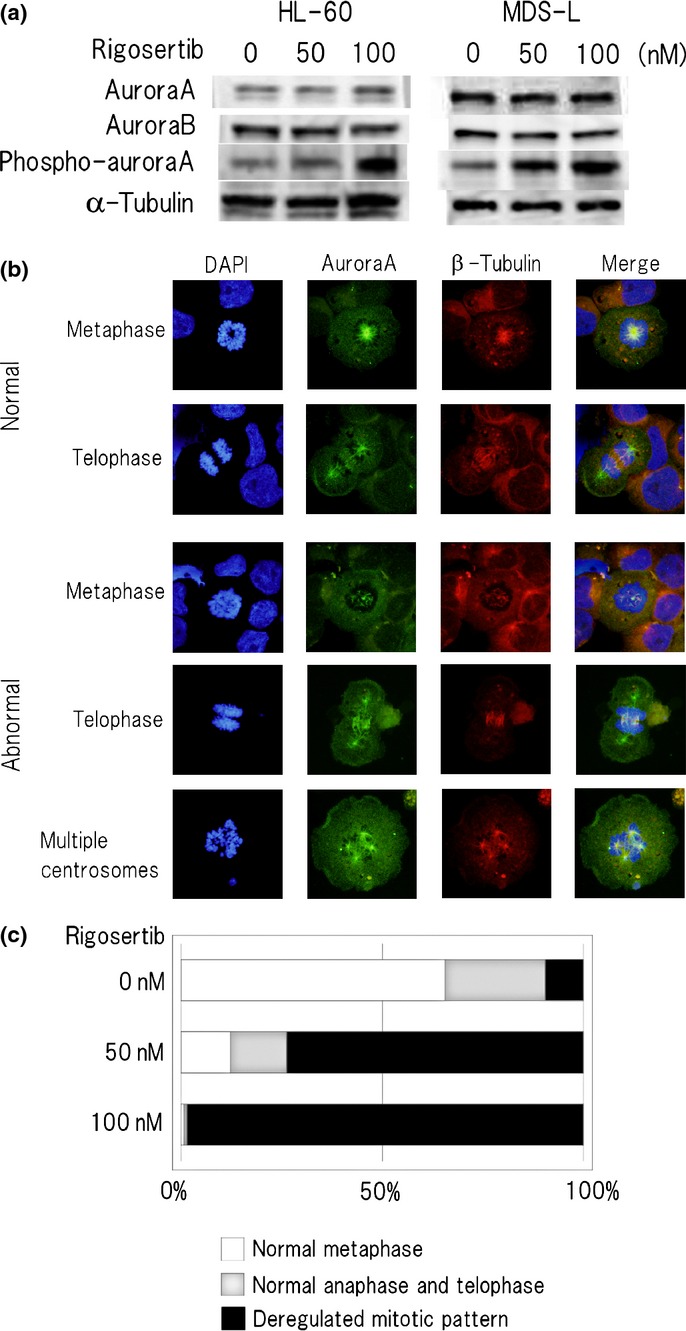
Rigosertib causes aberrant multiplication of centrosomes and abnormal spindle assembly in MDS-L cells. (a) HL-60 and MDS-L cells were treated with indicated concentrations of rigosertib for 24 h, and protein lysates were analyzed by immunoblotting analysis for the detection of aurora A kinase, aurora B kinase and phospho-aurora A kinase with each antibody. The amount of alpha-tubulin is shown as a loading control. (b) Immunofluorescence staining of rigosertib-treated MDS-L cells. Cells were treated with or without 50 nM rigosertib for 24 h and immunostained by anti-aurora A kinase followed by AlexaFluor488(green) -conjugated secondary antibody and AlexaFluor555 (red)-conjugated rabbit polyclonal anti-β-tubulin antibody. The chromosome area was stained with DAPI (blue) (original magnification ×1000). Representative images of normal and abnormal mitotic patterns are shown. (c) The location of aurora A kinase in mitotic cells was classified into normal metaphase, normal anaphase and telophase, and deregulated mitotic pattern, and each rate (percentage) is indicated below from counting more than 500 cells at M phase in each concentration. Rigosertib 0 nM: normal metaphase 66%, normal anaphase and telophase 25%, and deregulated mitotic pattern 9%. Rigosertib 50 nM: normal metaphase 12%, normal anaphase and telophase 14%, and deregulated mitotic pattern 74%. Rigosertib 100 nM: normal metaphase 1%, normal anaphase and telophase 1%, and deregulated mitotic pattern 98%.
The behavior of aurora A and B kinases in mitosis is well-known. In G1 phase the activity of both kinases is markedly reduced; in prophase, aurora A kinase is located around the centrosome; in metaphase, aurora A kinase is on the microtutbules whereas aurora B kinase is concentrated in the spindle midzone; in cytokinesis, both kinases are concentrated in the midbody.9,30 After rigosertib treatment, the mitotic cells with abnormal localization of aurora A kinase around the centrosome and deregulated spindle assembly were observed frequently in both cell lines (Fig.3b). We defined them as deregulated mitotic patterns and evaluated them by counting more than 500 cells. The percentage of deregulated mitotic patterns following rigosertib treatment in MDS-L cells was significantly increased as compared with that of control cells (Fig.3c). These data suggest that the suppression of mitosis due to deregulated spindle formation as above described resulted in massive cell death.
Rigosertib is possibly implicated in the kinetics of microtubules and inhibition of the quality control of mRNA biogenesis in MDS-L cells
To further investigate the action mechanisms of rigosertib, the molecular pathways influenced by rigosertib were explored using the gene expression profiling of MDS-L cells treated with or without 50 nM rigosertib for 24 h. Genes whose expression changed by more than 1.5 fold or <0.66 following the treatment were defined as regulated genes. In fact, 54 genes were upregulated and 44 genes downregulated in rigosertib-treated MDS-L cells, and gene expression profiling suggested that rigosertib significantly affected various gene biogroups. To better understand the action mechanisms of rigosertib, we examined the Gene Ontology (GO) categories. Table1 shows the list of nine top-ranked biogroups that were upregulated after rigosertib treatment, including microtuble organizing center, DNA-dependent DNA replication, mitotic spindle elongation and microtubule binding, and nine top-ranked biogroups whose expression was decreased after the treatment, including deoxynucleotide transport, nuclear-transcribed mRNA catabolic process and deadenylation-dependent decay. To further investigate the gene groups related with the targets of rigosertib, we performed the gene set enrichment analysis (GSEA) and found that the most significantly suppressed GSEA set was “nonsense-mediated mRNA decay (NMD)” (Fig.4).
Table 1.
Gene expression profiling in MDS-L cells treated with 50 nM rigosertib for 24 h
| Ontology | Term | P-value |
|---|---|---|
| Category (upregulation) | ||
| Cellular component | MCM complex | 3.04E-04 |
| Microtuble organizing center | 1.35E-03 | |
| Cytoplasmic microtubule | 4.03E-03 | |
| Biological process | DNA-dependent DNA replication | 9.43E-04 |
| Vitellogenesis | 2.54E-03 | |
| Hydroxylysine biosynthetic process | 2.54E-03 | |
| Positive regulation of ion transport | 3.27E-03 | |
| Mitotic spindle elongation | 5.07E-03 | |
| Molecular function | Microtubule binding | 3.05E-03 |
| Category (downregulation) | ||
| Biological process | Deoxynucleotide transport | 1.92E-03 |
| Maintenance of ER location | 1.92E-03 | |
| Endoplasmic reticulum localization | 3.85E-03 | |
| Nuclear-transcribed mRNA catabolic process, deadenylation-dependent decay | 3.92E-03 | |
| Positive regulation of anoikis | 6.72E-03 | |
| Molecular function | 3′-5′ exonuclease activity | 1.15E-03 |
| Deoxynucleotide transmembrane transporter activity | 2.05E-03 | |
| Glycogen binding | 3.07E-03 | |
| Exonuclease activity | 3.99E-03 | |
Figure 4.
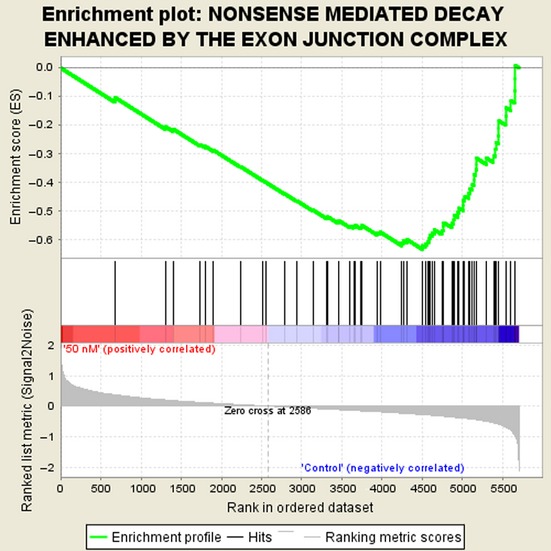
Suppression of the gene set “nonsense-mediated mRNA decay (NMD)” in the gene set enrichment analysis (GSEA). Gene expression profiling of MDS-L cells was examined in three independent experiments (rigosertib-treated or untreated cells were harvested after 24 h treatment) and obtained data were used for GSEA by handling the GSEA software and the Molecular Signatures Database according to the references.28,29 Nominal P-value: 0.000, FDR false discovery rate q-value: 0.126, FWER P-value: 0.000.
Taken together, the gene expression profiling suggested that rigosertib suppresses the genetic pathway involved in the quality control of mRNA biogenesis in MDS-L cells.
We further examined the gene expression profiling and the molecular pathways affected by rigosertib using HL-60 cells treated with rigosertib. However, the gene expression profiles of rigosertib-treated HL-60 showed minimal changes including upregulation of USP17L9P, SNORA11C and downregulation of MIR4720, and GSEA did not show any significant deviation of the gene sets such as NMD, which was significantly affected in MDS-L cells (data not shown).
Discussion
Rigosertib is expected to be a novel therapeutic drug for acute leukemia and MDS. In the present study, we investigated the effects of rigosertib on leukemia cell lines, and demonstrated that rigosertib induces cell death by DNA damage-induced G2/M arrest and that the suppression of mitosis due to deregulated spindle formation may contribute partly to massive cell death. Gumireddy et al.10 examine the effects of rigosertib on several tumor cell lines and report that rigosertib induces apoptosis of all cell lines with IC50, which ranged from 50 to 250 nM. In our in vitro study, IC50 of the four cell lines varied from 50 to 200 nM (Fig.1a), and the time point of IC50 evaluation was different, probably due to the difference of the doubling time of the cell lines (e.g. the doubling time of HL-60 and MDS-L is around 24 and 48 h, respectively).
We confirmed that rigosertib shows a remarkable cytocidal effect on all cell lines studied in the present study, but we also found a difference in the degree of the influence on apoptosis and the cell cycle, a difference in the sensitivity to rigosertib among cell lines. Prasad et al. report that rigosertib rapidly suppresses the level of cyclin D1 expression through the inhibition of PI3 kinase/Akt pathway in mantle cell lymphoma cells. We confirmed that rigosertib inhibits the phosphorylation of Akt (Fig.1c) but it did not accompany the suppression of cyclin D1 expression in our study (data not shown). This discrepancy might be explained by the difference of the cyclin D1-dependence for cell growth between mantle cell lymphoma cells which bear cyclin D1 gene activation and myeloid cells which do not. The phosphorylation of H2AX, a marker of DDR, increased after rigosertib treatment (Fig.2c). We assessed the expression of TP53 and its phosphorylation (Serine 15) in our model, but the involvement of TP53 seemed unlikely because the amount of TP53 was very low in MDS-L and HL-60 was lacking in TP53 molecule itself.
Based on the observation that rigosertib exerts inhibitory effects on the cell cycle, particularly on the progress of M phase (Fig.2a,b), we explored whether rigosertib affects aurora A and B kinases. The expression of both kinases was not significantly changed following rigosertib treatment by immunoblotting analysis, whereas the phosphorylation of aurora A kinase was enhanced after rigosertib treatment (50–100 nM) in both cell lines (Fig.3a). It is known that the activity of aurora A kinase is markedly reduced in G1 phase and it increases with progression from S phase to M phase with a shift of intracellular localization. As it is unlikely that rigosertib promotes the phosphorylation of aurora A kinase, this treatment is supposed to have induced cell cycle arrest at G2/M phase and resulted in the relative increase of the cell population whose aurora A kinase phophorylation was upregulated. We next examined the effect of rigosertib on the intracellular location of aurora A kinase by immunofluorescence staining of the treated cells and found that rigosertib causes multipolar centrosomes and deregulated spindle assembly (Fig.3b).
It might not be surprising that rigosertib exerts multiple functions and affects plenty of genes or gene sets related to cell survival or death. Hence, we compared the gene expression profile of MDS-L cells treated with or without 50 nM rigosertib for 24 h. Under this treatment condition most cells were alive as shown in Figure1(a) but such treated cells finally underwent cell death. Therefore, serious changes in gene expression would occur under this condition. The GO term analysis suggested rigosertib-induced upregulation of the genes related to microtubule kinetics and DNA replication system, and such-induced downregulation of deoxynucleotide transport and mRNA degradation system (Table1). To further investigate the gene groups related with the targets of rigosertib, we performed the gene set enrichment analysis (GSEA) and found that the most significantly suppressed GSEA set was “nonsense-mediated mRNA decay (NMD)” (Fig.4) and it coincided with the part of the results of GO term analysis (Table1).
Nonsense-mediated mRNA decay is an essential system for the quality control of mRNA, which eliminates abnormal mRNA containing premature stop codons and reduces errors in gene expression. The disturbance of NMD might proceed toward tumorigenesis.31 However, the defect in this system might be toxic to the cells and our study raised the possibility that rigosertib suppresses NMD and potentially causes cell death. Ishigaki et al. report that depletion of NMD-associated molecules causes cell cycle arrest at G2/M phase and subsequent apoptosis.32 Their report provides the data supporting our experimental results and could explain a possible relation of cell cycle inhibition with reduction of NMD. Inhibition of NMD brings autophagy,33 or causes upregulation of TP53 and finally leads to cell death.34 Therefore, NMD might be a potential target of a novel treatment strategy of MDS.
On the contrary to the above result of MDS-L cells, the gene expression changes of rigosertib-treated HL-60 were minimal and GSEA did not show any significant deviation of the gene sets including NMD, although both cell lines are of the similar myeloid lineage. It is uncertain and should be clarified whether the effect of rigosertib on NMD is specific to MDS-L cells. These data suggest that different tumor cells reveal different cell death patterns or different sensitivity to rigosertib probably due to different cell features including genetic/epigenetic changes or altered gene expression profiles. Further investigation regarding the effects of rigosertib on malignant diseases including MDS is needed.
Acknowledgments
Rigosertib was generously provided by Onconova Therapeutics Inc. The authors greatly thank Ms Aki Kuyama and Ms Asami Matsuo for their technical assistance. This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science, by a grant from the Japanese Cooperative Study Group for Intractable Bone Marrow Diseases, Ministry of Health, Labor and Welfare of Japan, and by a Kawasaki Medical School Project Grant.
Disclosure Statement
Tohyama K is a member of the data and safety monitoring committee of Japanese clinical study of rigosertib on MDS (SymBio Pharmaceuticals, Tokyo, Japan). The other authors have no competing financial interests to declare.
References
- Brunning RD, Orazi A, Germing U. “Myelodysplastic Syndrome/Neoplasms, Overview”. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 6th edn. Lyon: IARC; 2008. pp. 87–107. [Google Scholar]
- Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–99. [PubMed] [Google Scholar]
- Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintás-Cardama A, Santos FP, Garcia-Manero G. Therapy with azanucleosides for myelodysplastic syndromes. Nat Rev Clin Oncol. 2010;7:433–44. doi: 10.1038/nrclinonc.2010.87. [DOI] [PubMed] [Google Scholar]
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. International Vidaza High-Risk MDS Survival Study Group Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadia TM, Jabbour E, Kantarjian H. Failure of hypomethylating agent-based therapy in myelodysplastic syndromes. Semin Oncol. 2011;38:682–92. doi: 10.1053/j.seminoncol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List AF. New therapeutics for myelodysplastic syndromes. Leuk Res. 2012;36:1470–4. doi: 10.1016/j.leukres.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Bachegowda L, Gligich O, Mantzaris I, et al. Signal transduction inhibitors in treatment of myelodysplastic syndromes. J Hematol Oncol. 2013;10:6–50. doi: 10.1186/1756-8722-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöffski P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist. 2009;14:559–70. doi: 10.1634/theoncologist.2009-0010. [DOI] [PubMed] [Google Scholar]
- Gumireddy K, Reddy MV, Cosenza SC, et al. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. 2005;7:275–86. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Jimeno A, Li J, Messersmith WA, et al. Phase I study of ON 01910.Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J Clin Oncol. 2008;26:5504–10. doi: 10.1200/JCO.2008.17.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24:267–76. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- Prasad A, Park IW, Allen H, et al. Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells. Oncogene. 2009;28:1518–28. doi: 10.1038/onc.2008.502. [DOI] [PubMed] [Google Scholar]
- Chapman CM, Sun X, Roschewski M, et al. ON 01910.Na is selectively cytotoxic for chronic lymphocytic leukemia cells through a dual mechanism of action involving PI3K/AKT inhibition and induction of oxidative stress. Clin Cancer Res. 2012;18:1979–91. doi: 10.1158/1078-0432.CCR-11-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oussenko IA, Holland JF, Reddy EP, Ohnuma T. Effect of ON 01910.Na, an anticancer mitotic inhibitor, on cell-cycle progression correlates with RanGAP1 hyperphosphorylation. Cancer Res. 2011;71:4968–76. doi: 10.1158/0008-5472.CAN-10-1603. [DOI] [PubMed] [Google Scholar]
- Seetharam M1, Fan AC, Tran M, et al. Treatment of higher risk myelodysplastic syndrome patients unresponsive to hypomethylating agents with ON 01910.Na. Leuk Res. 2012;36:98–103. doi: 10.1016/j.leukres.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olnes MJ, Shenoy A, Weinstein B, et al. Directed therapy for patients with myelodysplastic syndromes (MDS) by suppression of cyclin D1 with ON 01910.Na. Leuk Res. 2012;36:982–9. doi: 10.1016/j.leukres.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundle SD. Targeting cyclin D1 for high risk myelodysplastic syndromes. Leuk Res. 2012;36:964–5. doi: 10.1016/j.leukres.2012.04.023. [DOI] [PubMed] [Google Scholar]
- Silverman LR, Greenberg P, Raza A, et al. Clinical activity and safety of the dual pathway inhibitor rigosertib for higher risk myelodysplastic syndromes following DNA methyltransferase inhibitor therapy. Hematol Oncol. 2014 doi: 10.1002/hon.2137. ; in press. [DOI] [PubMed] [Google Scholar]
- Komrokji RS, Raza A, Lancet JE, et al. Phase I clinical trial of oral rigosertib in patients with myelodysplastic syndromes. Br J Haematol. 2013;162:517–24. doi: 10.1111/bjh.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama K, Tsutani H, Ueda T, Nakamura T, Yoshida Y. Establishment and characterization of a novel myeloid cell line from the bone marrow of a patient with the myelodysplastic syndrome. Br J Haematol. 1994;87:235–42. doi: 10.1111/j.1365-2141.1994.tb04904.x. [DOI] [PubMed] [Google Scholar]
- Tsujioka T, Yokoi A, Uesugi A, et al. Effects of DNA methyltransferase inhibitors (DNMTIs) on MDS-derived cell lines. Exp Hematol. 2013;41:189–97. doi: 10.1016/j.exphem.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Matsuoka A, Tochigi A, Kishimoto M, et al. Lenalidomide induces cell death in an MDS-derived cell line with deletion of chromosome 5q by inhibition of cytokinesis. Leukemia. 2010;24:748–55. doi: 10.1038/leu.2009.296. [DOI] [PubMed] [Google Scholar]
- Drexler HG, Dirks WG, Macleod RA. Many are called MDS cell lines: one is chosen. Leuk Res. 2009;33:1011–6. doi: 10.1016/j.leukres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Ormerod MG, Collins MKL, Rodriguez-Tarduchy G, Robertson D. Apoptosis in Interleukin-3 dependent haemopoietic cells: quantification by two flow cytometric methods. J Immunol Methods. 1992;153:57–65. doi: 10.1016/0022-1759(92)90305-d. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Shi Y, Tohyama Y, Kadono T, et al. Protein tyrosine kinase Syk is required for pathogen engulfment in complement-mediated phagocytosis. Blood. 2006;107:4554–62. doi: 10.1182/blood-2005-09-3616. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GSEA Home page in Broad Institute . Available from URL: http://www.broadinstitute.org/gsea/index.jsp.
- Farag SS. The potential role of Aurora kinase inhibitors in haematological malignancies. Br J Haematol. 2011;155:561–79. doi: 10.1111/j.1365-2141.2011.08898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LB. Nonsense-mediated RNA decay regulation by cellular stress: implication for tumorigenesis. Mol Cancer Res. 2010;8:295–308. doi: 10.1158/1541-7786.MCR-09-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki Y, Nakamura Y, Tatsuno T, et al. Depletion of RNA-binding protein RBM8A (Y14) causes cell cycle deficiency and apoptosis in human cells. Exp Biol Med. 2013;238:889–97. doi: 10.1177/1535370213494646. [DOI] [PubMed] [Google Scholar]
- Wengrod J, Martin L, Wang D, Frischmeyer-Guerrerio P, Dietz HC, Gardner LB. Inhibition of nonsense-mediated RNA decay activates autophagy. Mol Cell Biol. 2013;33:2128–35. doi: 10.1128/MCB.00174-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Grigoryan A, Wang D, et al. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014;74:1–10. doi: 10.1158/0008-5472.CAN-13-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]