

Abstract
Background and Purpose
The non-nucleoside analogue reverse transcriptase inhibitor efavirenz is associated with hepatic toxicity and metabolic disturbances. Although the mechanisms involved are not clear, recent evidence has pinpointed a specific mitochondrial action of efavirenz accompanied by the induction of an endoplasmic reticulum (ER) stress/unfolded protein response in human hepatic cells. The aim of this study was to further investigate the involvement of this organelle by evaluating efavirenz's effects in cells lacking functional mitochondria (rho°) and comparing them with those of the typical mitotoxic agent rotenone, a standard complex I inhibitor, and the ER stress inducer thapsigargin.
Experimental Approach
Hep3B rho+ and rho° cells were treated with clinically relevant concentrations of efavirenz, then mitochondrial function and cytotoxicity were studied using standard cell biology techniques.
Key Results
Efavirenz-treated rho° cells exhibited a substantial reduction in parameters indicative of mitochondrial interference, such as increased superoxide production, mitochondrial mass/morphology alterations and enhanced expression of LONP, a highly conserved mitochondrial protease. In line with these results, the cytotoxic effect (cell number, chromatin condensation, cell cycle alterations and induction of apoptosis) of efavirenz was less pronounced in Hep3B respiration-depleted cells than in wild-type cells. The effect of efavirenz was both similar and different from those of two distinct mitochondrial stressors, thapsigargin and rotenone.
Conclusions and Implications
Cells lacking normal mitochondria (rho°) are less vulnerable to efavirenz. Our results provide further evidence that the hepatic damage induced by efavirenz involves acute interference with mitochondria and extend our knowledge of the response of mitochondria/ER to a stress stimulus.
Tables of Links
| TARGETS |
|---|
| ATP synthase |
| LIGANDS | |
|---|---|
| 2-deoxyglucose (2-DG) | Staurosporine |
| ATP | Thapsigargin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
The current pharmacological approach for treatment of human immunodeficiency virus (HIV) infection, known as combined antiretroviral therapy (cART), has transformed acquired immune deficiency syndrome (AIDS) into a chronic illness. Efavirenz, a non-nucleoside analogue reverse transcriptase inhibitor, is among the most widely used drugs in this therapeutic combination. Although generally considered safe, there is a concern about the side effects induced by efavirenz-containing therapies (Tashima et al., 2003; Gutiérrez et al., 2005; Maggiolo, 2009; Loko et al., 2011). In particular, there is growing concern whether lifelong treatment with this and other drugs used in cART leads to accumulative hepatotoxicity as liver-related complications are the second cause of mortality among AIDS patients (Jones and Núñez, 2012). The mechanisms behind this liver damage are unclear, but recent evidence has revealed the presence of acute mitochondrial dysfunction in cultured human hepatocytes treated with clinically relevant concentrations of this drug. The mechanism of such interference is specific and, unlike that exhibited by other anti-HIV drugs (namely nucleoside analogue reverse transcriptase inhibitors, NRTI), is not related to chronic inhibition of the mitochondrial DNA polymerase (pol-γ) (Apostolova et al., 2011a). Studies with the human hepatoma cell line Hep3B and primary cultures of human hepatocytes have revealed that the mitochondrial effect of efavirenz involves the specific inhibition of complex I of the electron transport chain (ETC), which leads to reduced O2 consumption, decreased mitochondrial membrane potential (ΔΨm), bioenergetic changes and elevated reactive oxygen species (ROS) generation (Apostolova et al., 2010, 2011b; Blas-García et al., 2010). Parallel to these manifestations, efavirenz triggers endoplasmic reticulum (ER) stress and activates the unfolded protein response (UPR) observed as altered ER morphology and increased expression of several UPR genes (Apostolova et al., 2013).
In order to better understand the role of mitochondria in these effects, we assessed the cellular actions of efavirenz in a model of hepatic cells that lack functional mitochondria (rho° cells generated in Hep3B background). The aim was to analyse the possibility of the existence of a differential mitochondrial effect of efavirenz resulting from differences in the mitochondrial background present in the cells treated with this drug. The model of rho° cells was employed as a robust approach to mimic a scenario of non-fully functional mitochondria in which we wished to evaluate if the toxic effects of efavirenz were potentiated or not. Moreover, the action of efavirenz was compared with that of two different mitotoxic stimuli: a direct one (rotenone) and an indirect one (thapsigargin) both at sub-lethal concentrations. Rotenone is a widely used pharmacological inhibitor of mitochondrial complex I and, like efavirenz, enhances mitochondrial superoxide production and induces apoptosis (Ishiguro et al., 2001; Li et al., 2003). Thapsigargin is a bona fide ER stressor that provokes a depletion of Ca2+ in the ER lumen (Thastrup et al., 1990). Several studies have reported a mitotoxic effect of thapsigargin that may occur independently of or secondary to its actions on the ER. Studies in hepatic models have shown specific mitochondrial alterations induced by thapsigargin, such as the biphasic fragmentation of mitochondria through Ca2+-mediated mitochondrial fission and apoptosis in rat liver cells (Hom et al., 2007) and triggering of mitochondrial permeability transition in isolated rat liver mitochondria (Korge and Weiss, 1999).
The results of the present study demonstrate that the stress response triggered by clinical concentrations of efavirenz in hepatic cells is diminished in those lacking functional mitochondria. These findings (i) highlight the participation of this organelle in the effects induced by efavirenz in hepatic cells and (ii) reveal both similarities and differences when compared with the responses invoked by two other distinct mitochondrial stressors, pointing out the specificity and complexity of efavirenz's actions. The effects of efavirenz described here may throw light on the hepatic stress induced by the clinical use of this drug.
Methods
Cell culture
Experiments were performed with the human hepatoblastoma cell line Hep3B (ATCC HB-8064), which displays certain degree of cytochrome P450 activity (specifically CYP2B6) capable of metabolizing efavirenz (Zhu et al., 2007; Lin et al., 2012). Some confirmatory experiments were performed in HepaRG cells, terminally differentiated hepatic cells derived from a human hepatic progenitor cell line that retains many characteristics of primary human hepatocytes. Unless otherwise stated, all the reagents employed in the cell culture were from GIBCO (Invitrogen, Eugene, OR, USA). Hep3B cells were cultured in minimal essential medium supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 1 mM non-essential amino acids, 1 mM sodium pyruvate, penicillin (50 u·mL−1) and streptomycin (50 μg·mL−1). Undifferentiated HepaRG cells (HPRGC10) purchased from GIBCO were initially grown at a density of 2.7 × 104 cells·cm−2 in William's medium E (Sigma-Aldrich) supplemented with 10% FBS, 2 mM L-glutamine, 5 μg·mL−1 bovine insulin and 50 μM hydrocortisone hemisuccinate (Sigma-Aldrich), 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. After 2 weeks, cell differentiation was induced by adding 2% dimethyl sulfoxide (DMSO) to the medium and cells were further cultured in its presence for a 2 week period (Anthérieu et al., 2010). Thereafter, differentiated hepatocyte-like cells were selectively collected through mild trypsinization (trypsin EDTA 0.125%) and reseeded at a density of 7–9 × 104 cells·cm−2 in medium containing 2% DMSO. Treatments were performed in DMSO-free medium. All cell cultures were maintained in an incubator (IGO 150, Jouan, Saint-Herblain Cedes, France) at 37°C, with a humidified atmosphere of 5% CO2/95% air (AirLiquide Medicinal, Valencia, Spain). Treatments were always performed in complete cell culture medium. In some experiments, treatments were performed on cells exposed to 10 mM 2-deoxyglucose (2-DG), a glucose analogue that inhibits glycolysis via its action on hexokinase (Pelicano et al., 2006).
Rho° cells in Hep3B background were obtained after 8 weeks of treatment of the wild-type cells with the chemical ethidium bromide (EtBr, 500 ng·mL−1) to deplete mtDNA in medium supplemented with 50 μg·mL−1 uridine. These mitochondrial ‘ghosts’ without DNA, which are known as mitoids, do not produce mitochondrial ATP, and cells harbouring them survive and proliferate only in medium supplemented with pyruvate and uridine (King and Attardi, 1989). As expected, rho° cells have a slower proliferation rate compared with the parental cells. This fact was taken into account and the two cell types were seeded at different cell density (in the case of 48-well plates, 50 000 cells per well for rho° cells and 30 000 cells per well for WT) in order to ensure the same cell density when the drug was added.
Analysis of cellular morphology through live-cell imaging
Bright field individual images (40×) were captured with IX81 Olympus microscope (Olympus, Hamburg, Germany) using ‘CellR’ software v.2.8.
Western blotting (WB)
WB was performed using total cell protein extracts as described elsewhere (Blas-García et al., 2010). Primary antibodies: anti-CV subunit β and anti-CIV subunit II were used at 1:1000 (both mouse monoclonal antibodies from Molecular Probes, Invitrogen); anti-porin at 1:1000, anti-LONP at 1:600 and anti-CI subunit ND1 at 1:600 (all three rabbit polyclonal antibodies from Abcam, Cambridge, UK) and anti-actin rabbit polyclonal at 1:1000 (Sigma-Aldrich). Secondary antibodies: peroxidase-labelled anti-mouse (Thermo Scientific, Rockford, IL, USA; 1:2000) and anti-rabbit IgG (Vector laboratories, Burlingame, CA, USA; 1:5000).
Determination of mtDNA copy number
The mtDNA copy number was determined by using total cellular DNA as described elsewhere (Apostolova et al., 2010). PCR reactions of mtDNA and nDNA were performed using 100 ng of total DNA mixed with LightCycler® FastStart DNA MasterPLUS SYBR Green I master mix (Roche Applied Science, Penzberg, Germany) following the instructions in the manual. Forward and reverse primers (0.2 μM) were added in a final reaction volume of 10 μL. To quantify the amounts of the template, a standard curve for the analysed gene was included in each run. The specificity of the amplified products was verified by melting curve analysis. Data were calculated as number of DNA copies. The results shown are a summary of four independent experiments (in duplicate) and are expressed as a ratio of mtDNA/nDNA copy.
Fluorescence microscopy and static cytometry
In order to avoid trypsinization and further manipulation of the cells, which often provokes artefacts in flow cytometry, we employed a life cell imaging method to detect fluorescent markers in which cells remain adherent and vital during the whole procedure. Fluorescence was detected with an IX81 Olympus fluorescence microscope and CellR software v.2.8 (Olympus) was employed to capture individual images. The fluorescent signal was quantified individually (per cell) by static cytometry software ‘ScanR’ version 2.03.2 (Olympus). All treatments – efavirenz, vehicle, thapsigargin and rotenone (24 h) – were performed in duplicate in 48-well plates. Treated cells were then washed in HBSS and 16–30 images per well were immediately recorded. Nuclei were stained with the fluorochrome Hoechst 33342 (2.5 μM) for the last 30 min of the treatment.
Mitochondrial function assessment: membrane potential, superoxide production and mitochondrial mass
Fluorochromes: 5 μM TMRM, 2.5 μM MitoSOX (both from Molecular Probes, Invitrogen) and 1 μM NAO (Sigma-Aldrich) were added for the last 30 min of treatment to assess ΔΨm, superoxide production and mitochondrial mass respectively. Carbonyl cyanide m-chloro phenyl hydrazone (CCCP 10 μM; uncoupler of oxidative phosphorylation) was used as a control for assessment of ΔΨm (Lou et al., 2007).
Cell proliferation/survival and cell cycle analysis
Cells were treated, allowed to proliferate exponentially (24 h) and then counted according to mean Hoechst fluorescence (2.5 μM Hoechst 33342, 25 images per well). Hoechst area and total intensity of its fluorescence were also recorded for analysis of nuclear area (nuclear size) and cell cycle.
Apoptosis
Apoptosis was studied as bivariate Annexin V/Propidium iodide (PI) analysis (apoptosis detection kit, Abcam). After 24 h of treatment, the medium was replaced with HBSS containing 0.9 μL per well of Annexin V fluorescein (to detect phosphatidyl serine exteriorization) and 2.5 μM Hoechst 33342 (to mark nuclei). Following incubation (20 min), 0.3 μL per well of the chromatin-detecting dye PI was added (10 min) to label dead or damaged cells. Staurosporine (STS, 1 μM), a widely used protein kinase inhibitor, was employed as a positive pro-apoptotic control (Lakhani et al., 2006).
Analysis of mitochondrial morphology through confocal fluorescence microscopy
Cells were stained with 2.5 μM Hoechst 33342 (to mark nuclei) and 1 μM NAO (to mark mitochondria) for the last 30 min of treatment. After washing with HBSS, life cell images were acquired with a Leica TCS-SP2 confocal laser scanning unit (Leica Microsystems, Wetzlar, Germany) with argon and helium-neon laser beams and attached to a Leica DMIRBE inverted microscope. Images were captured at (63×) magnification with HCX PL APO 40.0 (Leica Microsystems) × 1.32 oil UV objective.
Determination of intracellular ATP concentration
ATP concentration (pmol·μg−1 protein) inside cells incubated for 24 h with efavirenz, vehicle, thapsigargin or rotenone (or a combination of these with 10 mM 2-DG) was determined using an ATP Bioluminescence Assay Kit HSII (Roche Applied Science) and a Fluoroskan microplate reader (Thermo Labsystems, Thermo Scientific). Protein concentrations were determined with the BCA protein assay kit (Pierce Chemicals, Thermo Scientific).
Electrochemical measurement of oxygen (O2) consumption
O2 consumption was evaluated with a Clark-type O2 electrode (Oxytherm, Hansatech Instruments, Norfolk, UK) and Custom Windows® software was used for instrument control and data analysis. Cells (4 × 106) were placed in gas-tight chambers containing 1 mL respiration buffer (HBSS) and agitated at 37°C.
Presentation of data and statistical analysis
Data were analysed using GraphPad Prism v.3 software (GraphPad Software, La Jolla, CA, USA) with Student's t-test. In most cases, data are presented as % of control, the negative control (untreated cells) being considered to be 100%. All values are expressed as a mean ± SEM (statistical significance in the case of efavirenz treatment was analysed vs. vehicle and represented as *P < 0.05, **P < 0.01 and ***P < 0.001, whereas thapsigargin, rotenone, CCCP and STS were analysed separately and their significance was shown as: #P < 0.05, ##P < 0.01 and ###P < 0.001.
Reagents and drugs
Unless otherwise stated, chemical reagents were purchased from Sigma-Aldrich (Steinheim, Germany). Efavirenz (Sequoia Research Products, Pangbourne, UK) was dissolved in methanol (3 mg·mL−1). The efavirenz concentrations employed (10, 25 and 50 μM) are clinically relevant and were chosen by considering the important interindividual variability in its pharmacokinetics (Burger et al., 2006; Carr et al., 2010; Gounden et al., 2010).
Results
Mitochondrial function in Hep3B rho° cells and the effects of efavirenz
After generation of rho° cells, we assessed several markers of this phenotype, which is characterized by the presence of not fully functional mitochondria. Electrochemical measurements of O2 consumption in intact cells using a Clark-type oxygen electrode revealed a major drop in the respiration of rho° cells, which maintained only 15% of the basal levels recorded in WT cells (Figure 1A). Depletion of mtDNA and hence the rho° state of cells was controlled by quantitative genomic PCR analysis of mtDNA versus the amount of nuclear DNA. This experiment revealed that rho° cells possess roughly 30% of the mtDNA compared with WT cells (Figure 1B), indicating a major depletion but meaning that they are not completely devoid of mtDNA or not fully ‘rho°’ cells. WB analysis using whole-cell protein extracts demonstrated the complete absence of subunit II of the mitochondrial complex IV and a major reduction (80%) in expression of ND1, a complex I subunit, both mtDNA-encoded proteins, while no alterations were observed in the expression of porin and CV-β, mitochondrial proteins encoded by nuclear genes (Figure 1C).
Figure 1.

Determination of the Hep3B rho° phenotype. (A) Electrochemical measurement of O2 consumption. Representative trace showing O2 consumption in rho+ and rho° cells detected in intact cells using Clark-type O2 electrode (4 × 106 cells were employed in each run). (B) Quantitative genomic PCR used to quantify the relative ratio of mtDNA/nDNA in rho+ and rho° cells. (C) Representative WB image and histogram expressing quantification of complex IV subunit II, complex I subunit ND1, complex V subunit β and porin expression in total cell extracts of rho+ and rho° cells; β-actin was used as a housekeeping control. Data represent mean ± SEM, n = 4, and were analysed by Student's t-test (**P < 0.01, ***P < 0.001 vs. WT cells).
The effect of efavirenz (24 h treatment) on mitochondria in WT and mtDNA-depleted cells was evaluated through three parameters indicative of mitochondrial function: superoxide production, ΔΨm and mitochondrial morphology/mass. Rho° cells under basal conditions display a slightly higher mitochondrial superoxide production (MitoSOX fluorescence) than WT cells. All three stimuli – efavirenz, thapsigargin and rotenone – induced an increase in mitochondrial superoxide production in Hep3B WT cells that was significantly lower (with thapsigargin or rotenone) or even absent (with efavirenz) in cells lacking normal mitochondria (Figure 2A). Assessment of ΔΨm revealed a decrease in this parameter in untreated rho° cells compared with rho+ (Figure 2B) under basal conditions. Cells exposed to efavirenz or rotenone exhibited a similar drop in ΔΨm to that observed with 10 μM of the uncoupler CCCP, which was employed as a control. Importantly, this effect was present in rho° cells and was even more pronounced with efavirenz 50 μM and rotenone. Unlike efavirenz and rotenone, thapsigargin provoked an increase in TMRM fluorescence in WT cells, an effect that was absent in rho° cells.
Figure 2.

Mitochondrial effect of efavirenz (EVF) on respiration-deficient hepatic cells. (A) Superoxide production (MitoSOX fluorescence), (B) mitochondrial membrane potential (TMRM fluorescence) with or without 2-DG 10 mM, (C) intracellular ATP levels with or without 2-DG 10 mM and (D) mitochondrial mass (10-N-nonyl-acridine orange chloride, NAO, fluorescence) in WT and rho° cells treated for 24 h with increasing concentrations of efavirenz, vehicle, rotenone (Rot)10 or 25 μM or thapsigargin (TG) 2 μM. CCCP 10 μM was used as a positive control. Data (mean ± SEM, n = 4–6) were calculated as % of control WT value (untreated cells) and analysed by Student's t-test (*P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle). Data for rotenone, thapsigargin and CCCP were analysed separately (#P < 0.05, ##P < 0.01, ###P < 0.001 vs. untreated cells). (E) Representative confocal microscopy images (63×) of WT and rho° cells treated for 24 h with efavirenz 25 μM, thapsigargin 2 μM or rotenone 25 μM and stained with Hoechst 33342 and NAO.
Many cell types have the ability to maintain ΔΨm under conditions of diminished mitochondrial respiration or OXPHOS uncoupling through the reverse (ATP spending) activity of ATP synthase (complex V of ETC) (Faccenda and Campanella, 2012). Taking this into account, we also assessed the effect of efavirenz on cells where glycolysis has been inhibited (by addition of 10 mM 2-DG). All three stimuli – efavirenz, thapsigargin and rotenone – provoked a similar response although slightly more pronounced in cells treated with 2-DG compared with WT. Moreover, while rho° cells under basal conditions displayed only a slightly lower ΔΨm in comparison with WT cells, this difference was greater (about 50% reduction) when both cell populations cells were exposed to 2-DG. Both efavirenz and rotenone, as well as the uncoupler CCCP employed as a positive control, were able to induce a similar ΔΨm decrease in 2-DG-exposed rho° cells compared with the WT counterpart, whereas interestingly, in 2-DG-exposed rho° cells the increase triggered by thapsigargin was absent (Figure 2B). In parallel to these experiments, we evaluated the intracellular ATP levels after 24 h of treatment and this assessment revealed several interesting results. Firstly, in rho+ all three stimuli led to an increase in the ATP level and clearly this effect was due to activation of glycolysis as it was abolished when cells were co-treated with 2-DG (Figure 2C). In the case of efavirenz, we observed a concentration-dependent decrease in ATP in 2-DG-exposed WT cells. Rotenone provoked an even greater drop in the ATP level in these cells, whereas ATP levels were preserved in thapsigargin-treated cells. Secondly, the changes in intracellular ATP were generally less pronounced in rho° cells compared with WT. Both efavirenz and rotenone did not seem to induce an increase in ATP in cells with active glycolysis and the reduction in ATP levels previously described in 2-DG-exposed cells was much smaller (or even absent). Thapsigargin-treated cells seem to be less affected by the rho° phenotype in accordance to the action of this drug being primarily on ER and not mitochondria.
In accordance with its potential to produce lactic acidosis in human toxicology, methanol (the vehicle of efavirenz) by itself led to an increase in intracellular ATP, an effect that seem to be due to glycolysis as it was absent when cells were co-treated with 2-DG.
In order to evaluate mitochondrial mass and morphology, cells were stained with the mitochondria-specific fluorochrome NAO. An increase in NAO fluorescence has been associated with mitochondrial damage in this model (Apostolova et al., 2010), and in the present study, we observed an increase in NAO fluorescence in WT cells exposed for 24 h to efavirenz (25 and 50 μM, but not 10 μM), thapsigargin and rotenone (Figure 2D). All three stimuli induced clear changes in the mitochondrial network, as shown in Figure 2E. Although mitochondria in untreated cells were dispersed, efavirenz- and thapsigargin-treated cells emitted a more compact mitochondrial signal. Rotenone-exposed cells exhibited a granulated mitochondrial network with an intense signal around the nucleus. Importantly, the alterations in NAO fluorescence were less prominent in rho° cells. In sharp contrast to WT cells, Hep3B cells lacking normal mitochondria showed either no increase or a lower increase in mean NAO fluorescence intensity when exposed to rotenone and thapsigargin respectively. In the case of efavirenz, NAO fluorescence revealed a major variance between the different concentrations of the drug evaluated. Both efavirenz 25 and 50 μM increased NAO fluorescence in WT cells, whereas such an increase was detected in rho° cells only with efavirenz 50 μM, which points to a more severe level of mitochondrial damage that exceeded a certain threshold.
In order to confirm that despite its cancerous nature, the Hep3B model was appropriate, several experiments were also performed in WT HepaRG, a terminally differentiated hepatic cell line derived from human hepatic progenitor cells. Importantly, the responses observed in parental Hep3B and WT HepaRG cells were similar in the parameters studied. As shown in Figure 3A and B, 24 h of treatment with efavirenz led to a concentration-dependent increase in mitochondrial ROS production (mitoSOX fluorescence) paralleled by a decrease in cell number (nuclei stained with Hoechst 33342), although the effect was less pronounced in HepaRG than in Hep3B cells. Also, HepaRG cells seem to be particularly susceptible to rotenone and less susceptible to thapsigargin in comparison with Hep3B.
Figure 3.
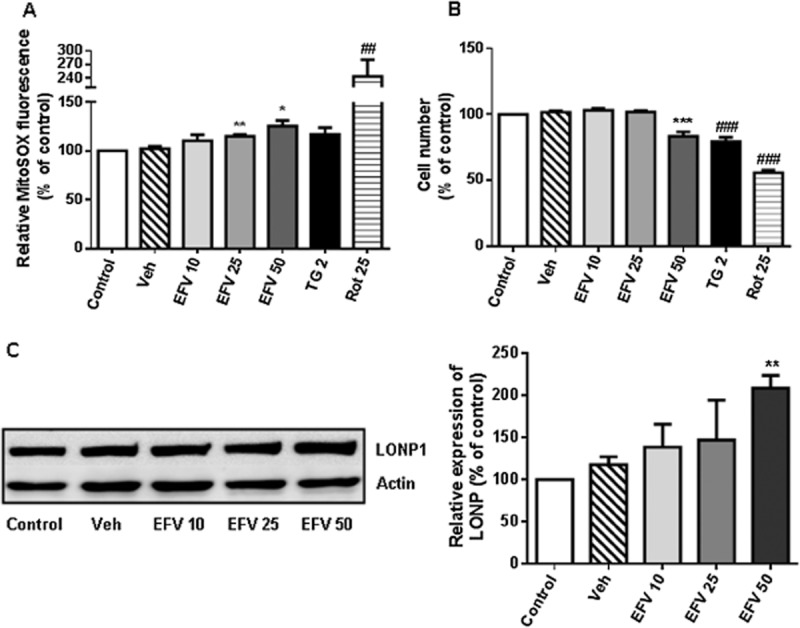
Effect of efavirenz (EVF) treatment on HepaRG cells. (A) Mitochondrial superoxide production (MitoSOX fluorescence) in cells treated for 24 h with increasing concentrations of efavirenz, rotenone (Rot) 25 μM and thapsigargin (TG) 2 μM. (B) Cell viability expressed as cell number (nuclei according to Hoechst fluorescence) after treatment for 24 h with increasing concentrations of efavirenz, rotenone 25 μM and thapsigargin 2 μM. Data (mean ± SEM, n = 4–6) were calculated as % of control value (untreated cells) and analysed by Student's t-test (*P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle). Data for rotenone and thapsigargin were analysed separately (##P < 0.01, ###P < 0.001 vs. untreated cells). (C) Representative WB image and histogram showing quantification of LONP expression in total cell extracts after 24 h of treatment with increasing concentrations of efavirenz. Quantification was performed by densitometry and the results are expressed as relative protein expression in relation to expression in untreated cells, which was considered to be 100%. Data (mean ± SEM, n = 4) were analysed by Student's t-test, **P < 0.01.
Activation of efavirenz-induced adaptive mechanisms linking mitochondria and ER in rho° cells
Previous studies have shown that efavirenz not only disrupts mitochondrial function in hepatic cells but also induces ER stress with the consequent activation of UPR. Notably, the effect of efavirenz on ER has been shown to involve mitochondria as it is radically reduced in cells with respiration-deficient mitochondria (Apostolova et al., 2013). In the present work, we have analysed the expression of Lon protease (LONP), a highly conserved mitochondrial ATP-dependent protease demonstrated to protect cells during both oxidative and ER stress (Hori et al., 2002). As shown in Figure 4, in Hep3B WT cells, 24 h of treatment with efavirenz led to a modest but consistent and concentration-dependent increase in LONP expression. The ER stressor thapsigargin and rotenone also increased LONP expression but to a much lesser extent than efavirenz. The basal expression of LONP in rho° cells was slightly higher than in rho+ cells. Importantly, in cells lacking functional mitochondria the level of LONP was not enhanced by any of the stressors –efavirenz, thapsigargin or rotenone. The efavirenz-induced concentration-dependent increase in LONP protein levels detected in wild-type Hep3B cells was confirmed in HepaRG cells (Figure 3C).
Figure 4.
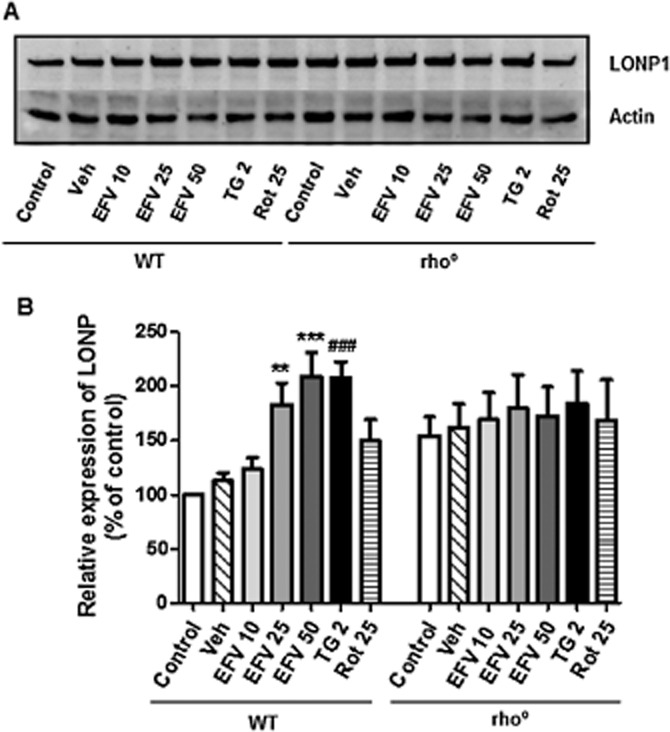
Western blot analysis of LONP expression. (A) Representative WB image and (B) histogram expressing quantification of protein expression in total cell extracts in rho+ and rho° cells treated for 24 h with increasing concentrations of efavirenz (EVF), vehicle, rotenone (Rot) 25 μM and thapsigargin (TG) 2 μM. Quantification was performed by densitometry and the results (mean ± SEM, n = 8, except for rotenone n = 4) are expressed as protein expression in relation to untreated WT cells, which was considered to be 100%. Statistical analysis was performed by Student's t-test (*P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle). Thapsigargin and rotenone were analysed separately (#P < 0.05, ##P < 0.01, ###P < 0.001 vs. untreated cells).
The cytotoxic effect of efavirenz is ameliorated in rho° cells
Light microscopy images of WT cells after 24 h of treatment with efavirenz 25 μM, efavirenz 50 μM, thapsigargin 2 μM or rotenone 25 μM revealed alterations in cell size and morphology with all three stressors, with major differences between the treatments (Figure 5A). Efavirenz 25 μM-treated cells were thinner than controls; this ‘spider net’ appearance was particularly evident in cells treated with thapsigargin, whereas rotenone-treated cells were bigger, had a rounder shape and exhibited a high amount of aggregates (lipofuscin golden brown finely granular pigment granules). Moreover, cell numbers were markedly reduced in rotenone- and thapsigargin-treated samples.
Figure 5.

Effect of efavirenz treatment on the viability of rho° cells. (A) Representative inverted light microscopy images (40×) of cell culture treated for 24 h with efavirenz (EVF) 25 μM, efavirenz 50 μM, thapsigargin (TG) 2 μM or rotenone (Rot) 25 μM. Hoechst fluorescence data – (B) cell number, (C) nuclear area and (D) nuclear signal – in Hep3B WT and rho° cells treated for 24 h with increasing concentrations of efavirenz, rotenone 25 μM and thapsigargin 2 μM. Data (mean ± SEM, n = 3–6) were calculated as % of control WT fluorescence value (untreated cells) and analysed by Student's t-test (*P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle). Data for rotenone and thapsigargin were analysed separately (#P < 0.05, ##P < 0.01, ###P < 0.001 vs. untreated cells). The cell number of untreated cells in either cellular background (rho° and rho+) was considered to be 100%. (E) Cell cycle analysis by static cytometry (Hoescht fluorescence) in cells treated for 24 h with efavirenz 50 μM, thapsigargin 2 μM or rotenone 25 μM. Statistical analysis was performed by Student's t-test (*P < 0.05, **P < 0.01, ***P < 0.001 vs. the corresponding value of the cell cycle phase in control cells). Thapsigargin and rotenone were analysed separately (#P < 0.05, ##P < 0.01, ###P < 0.001 vs. the corresponding value of the cell cycle phase in control). (F) Cell death analysis. Representative cytograms (bivariate Annexin V/PI analysis) of Hep3B WT and rho° cells treated for 24 h with efavirenz 50 μM, thapsigargin 2 μM, rotenone 25 μM and staurosporine (STS) 1 μM, showing the existence of four cellular populations: AnnV−/PI−, AnnV+/PI−, AnnV−/PI+ and AnnV+/PI+ (upper panel). Histograms expressing quantification of AnnV+/PI−, AnnV−/PI+ and AnnV+/PI+ cellular populations in rho+ and rho° cells treated with increasing concentrations of efavirenz, thapsigargin 2 μM, rotenone 25 μM and STS 1 μM (lower panel). Data (mean ± SEM, n = 3) were calculated as % of total number of nuclei counted and analysed by Student's t-test (**P < 0.01 vs. vehicle). Data for rotenone, thapsigargin and STS were analysed separately (#P < 0.05, ###P < 0.001 vs. untreated cells).
Previous studies by our group have shown that efavirenz exerts a cytotoxic effect on Hep3B cells. When a range of clinically relevant concentrations was evaluated (10–50 μM), the highest concentration (50 μM) was clearly associated with a major drop in cell viability, expressed as a radical decrease in cell number, altered cell cycle and induction of apoptosis. Very importantly, the present study revealed that all these effects are less pronounced in rho° cells; 24 h of treatment of Hep3B WT cells with efavirenz 50 μM led to a marked drop in cell number (a reduction of 60% with respect to vehicle-treated cells), whereas only a slight decrease (approximately 20%) was observed in rho° cells undergoing the same treatment (Figure 5B). A similar phenomenon was observed when chromatin condensation was assessed (namely, mean nuclear fluorescence intensity and nuclear area through Hoechst 33342 fluorescence; Figure 5C and D). WT cells exposed to efavirenz displayed increased Hoechst fluorescence and decreased nuclear area, modifications that were less evident or absent, respectively, in cells lacking functional mitochondria. A similar behaviour regarding both cell number and nuclear size/morphology was recorded with thapsigargin. In the case of exposure to rotenone 25 μM, the increase in Hoechst fluorescence was absent in rho° cells and the reduction in cell number was lower than that of WT cells. Unlike efavirenz and thapsigargin, rotenone produced an increase in the nuclear area, a phenomenon also present in rho° cells (Figure 5C). These results were supported by cell cycle analysis (Figure 5E). Firstly, Hep3B WT and rho° cells displayed slight differences in their cell cycle pattern, expressed as decreased S and G2M populations with an increase in both subG1 and G1. All three stimuli – efavirenz (only 50 μM), thapsigargin and rotenone – induced a change in the cell cycle of WT cells. Efavirenz- and thapsigargin-exposed WT cells manifested an increase in their G2M cellular population and a decrease in the S phase, modifications that were absent in rho° cells. On the other hand, the effect of rotenone was present in both rho° cells and rho+ cells. In order to delve more deeply in the effect of efavirenz on cell viability, we assessed the presence of apoptotic and necrotic cells, in both parental cells and cells lacking functional mitochondria, after 24 h of treatment. As expected, these experiments revealed that under basal conditions, rho° cells have a higher percentage of all three cell death sub-populations: early or typically apoptotic (Ann+/PI−), late apoptotic and/or necrotic (Ann+/PI+) and typically necrotic or damaged (Ann−/PI+). As previously published, in Hep3B WT cells, efavirenz (particularly the highest concentration, efavirenz 50 μM) induced apoptosis and perhaps slightly also necrosis, effects that were shown in the present work to be largely diminished (apoptosis) or absent (necrosis) in respiration-deficient cells (Figure 5F). Interestingly, the effects of rotenone and thapsigargin were somewhat different; they triggered apoptosis in WT cells and this effect seemed to be diminished rho° cells; however, their ability to induce necrosis seemed to be enhanced.
Discussion and conclusions
Recent in vitro studies have attributed a hepatotoxic action to efavirenz that involves (i) interference with mitochondrial function resembling that induced by the pharmacological inhibitor of mitochondrial complex I rotenone and (ii) presence of ER stress with activation of UPR, manifestations triggered by the classic ER stressor thapsigargin (Apostolova et al., 2013). However, several studies have reported differences in the actions of the aforementioned compounds that imply specificity for efavirenz. In this context, thapsigargin not only affects the ER but also undermines mitochondrial function, which is confirmed by the present findings. In contrast, rotenone did not mimic the actions of efavirenz or thapsigargin regarding ER stress/UPR in hepatic cells (data not shown).
Here we have examined the mitochondrial action of efavirenz (24 h of treatment) in cells significantly depleted of functional mitochondria (‘rho°’ throughout the manuscript) and have compared it with the effect of rotenone and thapsigargin in the same model. Rho° cells are respiration-deficient and display an aberrant mitochondrial phenotype with distorted cristae. It is evident that rho° cells are an artificial cellular model of non-respiring mitochondria; however, despite certain limitations, rho° cells are still used as a robust approach to generate a respiration-deficient model in culture, which enables the evaluation of the mitochondrial dependence or independence in the interference of different stimuli including drugs. Although an in vitro cellular model, as the one employed in the present study, cannot fully reflect the hepatic alterations induced by the drug in a living organism and particularly those related to systemic effects, cultured cells can provide relevant knowledge regarding specific drug-induced subcellular responses and provide a starting point for in vivo studies or clinical approximations. Of note, quantitative PCR analysis revealed 30% of the mtDNA remained meaning that the established phenotype was not fully ‘rho°’. Depletion of mtDNA is a characteristic of the so-called mitochondrial diseases, although the amount of remaining mtDNA and its correlation with the severity of these diseases present great variations among patients. A minimum critical proportion of mtDNAs is necessary (a threshold level) before biochemical defects and tissue dysfunction become apparent and it varies in the range of 60–90% mutant to wild-type mtDNA. Although the threshold level can partly explain the disease phenotypes and clinical severity observed in patients, an exact correlation is lacking. Mitochondrial diseases with a major hepatic component (hepatocerebral mtDNA depletion syndromes or isolated hepatic disease) display a significant decrease in liver mtDNA content with most cases usually presenting 20% or less mtDNA compared with age-matched healthy control individuals (Dimmock et al., 2008; Müller-Höcker et al., 2011). It is generally assumed that in mtDNA-depleted cells, the reduced abundance of mtDNA-encoded proteins mirrors the replicative failure of mtDNA. However, liver mitochondrial proteins are soon degraded when they are not properly assembled; therefore, the expression of mtDNA-encoded proteins may be relatively low compared with the depletion in mtDNA, which seems to be the case in our model. Moreover, the number of mtDNA copies relative to nDNA does not always reflect the degree of mitochondrial dysfunction; therefore, functional assays such as measurements of the activity of the specific ETC complexes or evaluation of the overall respiration deficit are needed. The depletion of mtDNA-encoded proteins in the present work was confirmed by studying the protein expression of two mtDNA-encoded proteins, subunit II of cytochrome c oxidase and ND1 of CI; rho° cells, however, maintain an intact nuclear genome, which conferred a comparable protein expression of the β subunit of complex V, β-actin and porin in the parental and mitochondrial DNA-depleted cells. The presence of mitochondria with severely diminished respiration resulted in a differential cellular response to efavirenz in all the parameters studied. When exposed to efavirenz, rho° cells did not display a significant increase in ROS generation, and the increase in the mitochondrial signal (NAO fluorescence) was absent in cells treated with 25 μM efavirenz and only visible in those treated with 50 μM, presumably due to the crossing of a threshold in the stress response. Such a threshold may also account for the effect observed regarding ΔΨm where, interestingly, exposure to efavirenz induced a slightly less evident reduction in rho° cells than in WT, with the exception of treatment with 50 μM. Importantly, the deleterious effect of efavirenz 50 μM on cell number, its triggering of cell cycle arrest and induction of cell death (via apoptosis) was, once again, largely ameliorated in rho° cells. Fluorescence microscopy experiments confirmed that the cell morphology and specifically mitochondrial appearance were less modified in cells lacking functional mitochondria.
Thapsigargin induced similar changes as efavirenz in WT cells with the exception of ΔΨm. We found not a decrease but an increase in ΔΨm, which is in keeping with the results of other studies showing a lack of ΔΨm dissipation during ER stress triggered by thapsigargin. However, to our knowledge ours is the first study to address the mitochondrial action of thapsigargin in cells with greatly diminished respiration. As with efavirenz, the overall effect of thapsigargin on mitochondria and cell viability was largely reduced in rho° cells. However, the effect of thapsigargin on cell survival in respiration-depleted cells is more complex as these cells exhibit a higher percentage of necrotic/damaged (but not apoptotic) cells compared with WT.
The effects of rotenone on WT cells were similar to those of efavirenz, except for that concerning the nuclear area (Hoechst fluorescence), which in the case of rotenone was found to increase. Diminished nuclear area (chromatin condensation and nuclear fragmentation) is a hallmark of apoptosis, and the precise relevance of this slight increment in rotenone-treated cells is unknown. Such an increase in nuclear area after treatment with rotenone was not only present in rho° cells but actually enhanced as well. Another singularity of rotenone treatment was observed in the cell cycle analysis. Unlike efavirenz and thapsigargin, which altered the cell cycle in rho+ but not rho° cells, rotenone produced a similar alteration in both populations. These effects point to the presence of a mitochondria-independent action of rotenone in this cell line used which may also be concentration related. Rotenone at concentrations similar to those employed in the present work has been shown to arrest mammalian cells in metaphase by binding directly to tubulin and preventing microtubule assembly (Meisner and Sorensen, 1966). Alternatively, the interference with the cell cycle may be due to the cancerous nature of the cell line in question, as completion of the cell division in cells with a high metabolic drive and high proliferation rate, such as cancer cells, is particularly sensitive to inhibition of mitochondrial function. This result is related to the fact that in a similar manner to thapsigargin, the percentage of Ann−/PI+ cells in the rotenone- treated samples is enhanced in rho° cells. In summary, the stress response triggered by clinical concentrations of efavirenz is diminished in cells lacking functional mitochondria and this effect shows certain differences when compared with that elicited by other cytotoxic compounds that compromise mitochondria.
Very importantly, the fact that efavirenz exerts an effect on both mitochondria and ER creates a new scenario for understanding liver toxicity. The induction of ER stress and UPR in efavirenz-treated hepatic cells is dependent on mitochondria as several markers of this stress response (increased expression of protein markers such as GRP78 and CHOP, and higher content of ER) were found to be diminished in Hep3B cells lacking functional mitochondria (Apostolova et al., 2013). In order to further link the two effects of efavirenz (mitochondria and ER), we analysed the expression of LONP, whose activation is thought to be an adaptive mechanism in both oxidative and ER stress. LONP has been shown to both regulate proteostasis and participate in the control of mtDNA transcription and replication (Venkatesh et al., 2012). The present work is the first to describe the up-regulation of this protein by efavirenz. This is relevant, as an up-regulation of LONP has been associated with HIV treatment, in particular with the development of lipodystrophy in patients receiving cART, but has not been related to efavirenz until now (Pinti et al., 2010). As expected, in line with that observed for the rest of the mitochondrial parameters evaluated, efavirenz-triggered LONP activation was absent in respiration-deficient cells, which supports the involvement of mitochondria in the onset of the ER stress associated with efavirenz. Although LONP is a promising target for the assessment of the combined mitochondria + ER effect of efavirenz, additional studies are needed to elucidate its effects on this complex and multi-level drug-induced interference.
Mitochondria play a pivotal role in the development of drug-induced toxicity. The fact that the deleterious effects of drugs can be alleviated in cells with diminished mitochondrial respiration opens a new horizon for understanding mitochondria's involvement in cellular survival. It is tempting to speculate on the concept of a mtDNA threshold and the level of drug-induced or mediated injury. This idea is endorsed by recently published data obtained in a Rho subline of human hepatoma SK-Hep-1 cells showing resistance to bile acid-induced concentration-dependent activation of apoptosis (Marin et al., 2013). Similar phenomena have been described, such as a protective effect of membrane depolarization of isolated rat liver mitochondria, which was shown to attenuate permeability transition pore opening and oxidant production induced by tert-butyl hydroperoxide (Aronis et al., 2002). Effects such as these, which involve alterations in basal mitochondrial function, may account for the idiosyncratic hepatic reactions triggered by anti-HIV drugs and may also explain the different degrees of susceptibility to liver damage seen in patients undergoing antiretroviral therapy. In the case of efavirenz, its mitochondrial effects in the clinic may be directly affected by several factors. Firstly, the concomitant administration of other mitochondriotropic drugs may have a crucial role in the effects of efavirenz on mitochondria. Indeed, efavirenz is never administered individually but as an element within cART together with two NRTI; importantly, these drugs are known to possess mitotoxic potential due to their ability to inhibit mitochondrial DNA polymerase γ (Pol-γ hypothesis) and therefore their administration may generate a different type of mitochondria with a distinct susceptibility to efavirenz. Secondly, the intrinsic genetic variability such as that induced by the mtDNA haplogroup may account for the drug-induced mitotoxicity both under basal or stress conditions. Recently, mitochondrial haplogroups and subhaplogroups have been associated with certain toxicities of NRTI drugs (Kampira et al., 2013); however, to the best of our knowledge, no such correlations have been made with efavirenz. In all, a very intriguing picture is emerging of patient-specific mitochondrial function as a factor that influences the mitotoxic potential of anti-HIV drugs.
Acknowledgments
The authors would like to thank Brian Normanly for editing of the manuscript. Grants PI11/00327 and CIBER CB06/04/0071 (both from Instituto de Salud Carlos III, Ministerio de Economia y Competitividad), PROMETEOII/2014/035, ACOMP/2013/236 and GV/2014/118 (all from Generalitat Valenciana), and UV-INV-PRECOMP12-80613 (Universitat de València). M. P., H. A. F. and F. A. are recipients of Predoctoral Trainee Research Grants (ACIF/2013/136 from Generalitat Valenciana; Fundación Dr Esplugues and FI12/00198 from Instituto de Salud Carlos III, Ministerio de Economía y Competitividad respectively). A. B.-G. is recipient of a Juan de la Cierva contract (JCI-2012-15124, Ministerio de Economía y Competitividad).
Glossary
- 2-DG
2-deoxyglucose
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- ER
endoplasmic reticulum
- HIV
human immunodeficiency virus
- ROS
reactive oxygen species
- UPR
unfolded protein response
- WB
Western blotting
- ΔΨm
mitochondrial membrane potential
Author contributions
M. P., F. A. and H. A. F. performed experiments. A. B.-G., V. M. V., N. A. and J. V. E. designed experiments and supervised research. N. A. and J. V. E. wrote the manuscript.
Conflict of interest
None.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthérieu S, Chesné C, Li R, Camus S, Lahoz A, Picazo L, et al. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab Dispos. 2010;38:516–525. doi: 10.1124/dmd.109.030197. [DOI] [PubMed] [Google Scholar]
- Apostolova N, Gomez-Sucerquia LJ, Moran A, Alvarez A, Blas-Garcia A, Esplugues JV. Enhanced oxidative stress and increased mitochondrial mass during efavirenz-induced apoptosis in human hepatic cells. Br J Pharmacol. 2010;160:2069–2084. doi: 10.1111/j.1476-5381.2010.00866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova N, Blas-García A, Esplugues JV. Mitochondrial interference by anti-HIV drugs: mechanisms beyond Pol-γ inhibition. Trends Pharmacol Sci. 2011a;32:715–725. doi: 10.1016/j.tips.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Apostolova N, Gomez-Sucerquia LJ, Gortat A, Blas-Garcia A, Esplugues JV. Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival-promoting autophagy. Hepatology. 2011b;54:1009–1019. doi: 10.1002/hep.24459. [DOI] [PubMed] [Google Scholar]
- Apostolova N, Gomez-Sucerquia LJ, Alegre F, Funes HA, Victor VM, Barrachina MD, et al. ER stress in human hepatic cells treated with Efavirenz: mitochondria again. J Hepatol. 2013;59:780–789. doi: 10.1016/j.jhep.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Aronis A, Komarnitsky R, Shilo S, Tirosh O. Membrane depolarization of isolated rat liver mitochondria attenuates permeability transition pore opening and oxidant production. Antioxid Redox Signal. 2002;4:647–654. doi: 10.1089/15230860260220157. [DOI] [PubMed] [Google Scholar]
- Blas-García A, Apostolova N, Ballesteros D, Monleón D, Morales JM, Rocha M, et al. Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology. 2010;52:115–125. doi: 10.1002/hep.23647. [DOI] [PubMed] [Google Scholar]
- Burger D, van der Heiden I, la Porte C, van der Ende M, Groeneveld P, Richter C, et al. Interpatient variability in the pharmacokinetics of the HIV non-nucleoside reverse transcriptase inhibitor efavirenz: the effect of gender, race, and CYP2B6 polymorphism. Br J Clin Pharmacol. 2006;61:148–154. doi: 10.1111/j.1365-2125.2005.02536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DF, la Porte CJ, Pirmohamed M, Owen A, Cortes CP. Haplotype structure of CYP2B6 and association with plasma efavirenz concentrations in a Chilean HIV cohort. J Antimicrob Chemother. 2010;65:1889–1893. doi: 10.1093/jac/dkq260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock DP, Zhang Q, Dionisi-Vici C, Carrozzo R, Shieh J, Tang LY, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat. 2008;29:330–331. doi: 10.1002/humu.9519. [DOI] [PubMed] [Google Scholar]
- Faccenda D, Campanella M. Molecular regulation of the mitochondrial F(1)F(o)-ATPsynthase: physiological and pathological significance of the inhibitory factor 1 (IF(1)) Int J Cell Biol. 2012;2012:367934. doi: 10.1155/2012/367934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounden V, van Niekerk C, Snyman T, George JA. Presence of the CYP2B6 516G> T polymorphism, increased plasma Efavirenz concentrations and early neuropsychiatric side effects in South African HIV-infected patients. AIDS Res Ther. 2010;7:32. doi: 10.1186/1742-6405-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez F, Navarro A, Padilla S, Antón R, Masiá M, Borrás J, et al. Prediction of neuropsychiatric adverse events associated with long-term efavirenz therapy, using plasma drug level monitoring. Clin Infect Dis. 2005;41:1648–1653. doi: 10.1086/497835. [DOI] [PubMed] [Google Scholar]
- Hom JR, Gewandter JS, Michael L, Sheu SS, Yoon Y. Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J Cell Physiol. 2007;212:498–508. doi: 10.1002/jcp.21051. [DOI] [PubMed] [Google Scholar]
- Hori O, Ichinoda F, Tamatani T, Yamaguchi A, Sato N, Ozawa K, et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J Cell Biol. 2002;157:1151–1160. doi: 10.1083/jcb.200108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Yasuda K, Ishii N, Ihara K, Ohkubo T, Hiyoshi M, et al. Enhancement of oxidative damage to cultured cells and Caenorhabditis elegans by mitochondrial electron transport inhibitors. IUBMB Life. 2001;51:263–268. doi: 10.1080/152165401753311816. [DOI] [PubMed] [Google Scholar]
- Jones M, Núñez M. Liver toxicity of antiretroviral drugs. Semin Liver Dis. 2012;32:167–176. doi: 10.1055/s-0032-1316472. [DOI] [PubMed] [Google Scholar]
- Kampira E, Kumwenda J, van Oosterhout JJ, Dandara C. Mitochondrial DNA subhaplogroups L0a2 and L2a modify susceptibility to peripheral neuropathy in Malawian adults on stavudine containing highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2013;63:647–652. doi: 10.1097/QAI.0b013e3182968ea5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Korge P, Weiss JN. Thapsigargin directly induces the mitochondrial permeability transition. Eur J Biochem. 1999;265:273–280. doi: 10.1046/j.1432-1327.1999.00724.x. [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Lin J, Schyschka L, Mühl-Benninghaus R, Neumann J, Hao L, Nussler N, et al. Comparative analysis of phase I and II enzyme activities in 5 hepatic cell lines identifies Huh-7 and HCC-T cells with the highest potential to study drug metabolism. Arch Toxicol. 2012;86:87–95. doi: 10.1007/s00204-011-0733-y. [DOI] [PubMed] [Google Scholar]
- Loko MA, Bani-Sadr F, Winnock M, Lacombe K, Carrieri P, Neau D, et al. Impact of HAART exposure and associated lipodystrophy on advanced liver fibrosis in HIV/HCV-coinfected patients. J Viral Hepat. 2011;18:e307–e314. doi: 10.1111/j.1365-2893.2010.01417.x. [DOI] [PubMed] [Google Scholar]
- Lou PH, Hansen BS, Olsen PH, Tullin S, Murphy MP, Brand MD. Mitochondrial uncouplers with an extraordinary dynamic range. Biochem J. 2007;407:129–140. doi: 10.1042/BJ20070606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggiolo F. Efavirenz: a decade of clinical experience in the treatment of HIV. J Antimicrob Chemother. 2009;64:910–928. doi: 10.1093/jac/dkp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin JJ, Hernandez A, Revuelta IE, Gonzalez-Sanchez E, Gonzalez-Buitrago JM, Perez MJ. Mitochondrial genome depletion in human liver cells abolishes bile acid-induced apoptosis: role of the Akt/mTOR survival pathway and Bcl-2 family proteins. Free Radic Biol Med. 2013;61C:218–228. doi: 10.1016/j.freeradbiomed.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Meisner HM, Sorensen L. Metaphase arrest of Chinese hamster cells with rotenone. Exp Cell Res. 1966;42:291–295. doi: 10.1016/0014-4827(66)90292-8. [DOI] [PubMed] [Google Scholar]
- Müller-Höcker J, Horvath R, Schäfer S, Hessel H, Müller-Felber W, Kühr J, et al. Mitochondrial DNA depletion and fatal infantile hepatic failure due to mutations in the mitochondrial polymerase γ (POLG) gene: a combined morphological/enzyme histochemical and immunocytochemical/biochemical and molecular genetic study. J Cell Mol Med. 2011;15:445–456. doi: 10.1111/j.1582-4934.2009.00819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- Pinti M, Gibellini L, Guaraldi G, Orlando G, Gant TW, Morselli E, et al. Upregulation of nuclear-encoded mitochondrial LON protease in HAART-treated HIV-positive patients with lipodystrophy: implications for the pathogenesis of the disease. AIDS. 2010;24:841–850. doi: 10.1097/QAD.0b013e32833779a3. [DOI] [PubMed] [Google Scholar]
- Tashima KT, Bausserman L, Alt EN, Aznar E, Flanigan TP. Lipid changes in patients initiating efavirenz- and indinavir-based antiretroviral regimens. HIV Clin Trials. 2003;4:29–36. doi: 10.1310/f2v7-3r46-vx6j-241r. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Lee J, Singh K, Lee I, Suzuki CK. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim Biophys Acta. 2012;1823:56–66. doi: 10.1016/j.bbamcr.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XH, Wang CH, Tong YW. Growing tissue-like constructs with Hep3B/HepG2 liver cells on PHBV microspheres of different sizes. J Biomed Mater Res B Appl Biomater. 2007;82:7–16. doi: 10.1002/jbm.b.30698. [DOI] [PubMed] [Google Scholar]