

Abstract
Background and Purpose
Selective agonists of the sigma-1 receptor (σ1 protein) are generally reported to protect against neuronal damage and modulate oligodendrocyte differentiation. Human and rodent lymphocytes possess saturable, high-affinity binding sites for compounds binding to the σ1 protein and potential immunomodulatory properties have been described for σ1 protein ligands. Experimental autoimmune encephalomyelitis (EAE) is recognized as a valuable model of the inflammatory aspects of multiple sclerosis (MS). Here, we have assessed the role of a σ1 protein agonist, containing the tetrahydroisoquinoline-hydantoin structure, in EAE.
Experimental Approach
EAE was induced in SJL/J female mice by active immunization with myelin proteolipid protein (PLP)139–151 peptide. The σ1 protein agonist was injected i.p. at the time of immunization (day 0). Disease severity was assessed clinically and by histopathological evaluation of the CNS. Phenotyping of B-cell subsets and regulatory T-cells were performed by flow cytometry in spleen and cervical lymph nodes.
Key Results
Prophylactic treatment of EAE mice with the σ1 protein agonist prevented mononuclear cell accumulation and demyelination in brain and spinal cord and increased T2 B-cells and regulatory T-cells, resulting in an overall reduction in the clinical progression of EAE.
Conclusions and Implications
This σ1 protein agonist, containing the tetrahydroisoquinoline-hydantoin structure, decreased the magnitude of inflammation in EAE. This effect was associated with increased proportions of B-cell subsets and regulatory T-cells with potential immunoregulatory functions. Targeting of the σ1 protein might thus provide new therapeutic opportunities in MS.
Tables of Links
| LIGANDS |
|---|
| BD-1047 |
| IFN-γ |
| IL-17a |
| IL-4 |
| TNF-α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (
Introduction
Experimental autoimmune encephalomyelitis (EAE) is a CNS disease during which an autoimmune inflammatory response causes the destruction of oligodendrocytes, resulting in axonal demyelination. Overall, EAE in animals has proved a highly valuable tool for understanding the pathology of multiple sclerosis (MS) in humans (Batoulis et al., 2011; Ransohoff, 2012). CD4+ Th1 and Th17 cells have been regarded as the main culprits in the pathogenesis. However, recent studies revealed that MS is also mediated by B-cells (DiLillo et al., 2011). B-cells and their products have the capability to promote cellular immune responses. B-cells also present antigens as efficiently as other professional antigen-presenting cells, such as dendritic cells, and function as cellular adjuvants to promote CD4+ T-cell activation, expansion, memory formation and cytokine production in vivo. Thus, B-cells are generally considered as positive regulators of the immune response in an inflammatory context. However, IL10-producing, CD5+CD1dhigh B cells (named B10 cells) have been described as potent negative regulators in both mouse and humans (Matsushita et al., 2008; Iwata et al., 2011). In addition to B10 cells, other B-cell subsets therefore from the B2 lineage, such as transitional 2 (T2) or marginal zone (MZ) B-cells, have been proposed as having suppressive activities. Some regulatory B-cells (Bregs) originate therefore from the B2-cell lineage, although CD5+ B1 cells are also now known to have regulatory functions (DiLillo et al., 2011; Mauri and Bosma, 2012; Kalampokis et al., 2013).
Thus far, cell-specific markers for Bregs, analogous to the expression of the transcription forkhead box protein P3 (FoxP3) by regulatory T-cells (Tregs), have been difficult to establish. At present, the Bregs phenotype is not well defined in EAE (Mauri and Blair, 2010; Kalampokis et al., 2013). However, a comparison of B-cell subsets from susceptible and resistant mice revealed that transitional B-cells are a characteristic feature of EAE (Lee-Chang et al., 2011a). These results were also observed in early phases of MS (Lee-Chang et al., 2011b).
The sigma receptors or binding sites (σ proteins), initially described as a subtype of opiate receptors, are now considered as unique receptors. Pharmacological studies have distinguished two types of σ proteins, termed σ1 and σ2. The σ1 type is an intracellular protein identified as a ligand-regulated molecular chaperone. This 25 kDa protein is present on mitochondrion-associated endoplasmic reticulum (ER) membranes. σ1 agonists potently modulate intracellular Ca2+ mobilizations and extracellular Ca2+ influx, in addition to numerous neurotransmitter responses, and result in activation of signalling pathways (Cobos et al., 2008). In the CNS, this protein is expressed in neurons and oligodendrocytes. The σ1 protein has been shown to affect the action potential process by several modes of action. Indeed, the σ1 protein directly or indirectly modulates voltage-gated ion channels, glutamate and GABA ionotropic receptors, dopamine D1 receptors, muscarinic and nicotinic ACh receptors and neurotrophic tyrosine kinase receptor type 2 (TrkB). The σ1 protein also interacts with intracellular targets such as kinases and inositol trisphosphate receptors (Kourrich et al., 2012). Eliprodil, a high affinity but non-selective σ1 protein ligand with neuroprotective properties also modulated myelination by increasing the amount of myelinated axon segments (Demerens et al., 1999). The σ1 protein is also expressed on lymphocytes, but the significance of this protein in the immune system has been poorly studied. The σ1 protein exhibits a well-established pharmacological profile, with high-affinity synthetic ligands. In vitro, specific σ1 protein ligands inhibit the proliferative responses of mouse CD3+ lymphocytes. In vivo, they inhibit LPS-induced systemic release of IL-1, IL-6, TNF-α and IFN-γ and, interestingly, they can also enhance IL-10 release (Casellas et al., 1994; Bourrie et al., 1995; Derocq et al., 1995).
A synthetic, high-affinity and selective ligand for the σ1 protein, compound 1(S), contains the tetrahydroisoquinoline-hydantoin structure, a limited number of free rotation bonds and a very low cytotoxicity providing a high selectivity index (ratio CC50/IC50), greater than 50 000 (Cazenave Gassiot et al., 2005; Charton et al., 2005; Toussaint et al., 2010). It also has an agonist profile in cocaine-induced hyperlocomotion and locomotor sensitization (Toussaint et al., 2009). In the present study, we have investigated the effect of compound 1(S) on the initiation and clinical development of EAE. Our data have shown that a single injection of compound 1(S) decreased the severity of EAE (clinically and histologically) by a σ1-dependent mechanism, an effect that was associated with an increase of potential immunoregulatory transitional 2 (T2) B-cells as well as Tregs.
Methods
Adsorption, diffusion, metabolism and excretion (ADME) assays
Standardized in vitro ADME experiments were performed by CEREP (Paris, France). The bioavailability-related profile was measured according to Lipinski et al. (2001) for aqueous solubility (in PBS pH 7.4), Sangster (1997) for partition coefficient (logD, n-octanol : PBS, pH 7.4), Banker et al. (2003) for plasma protein binding, Grès et al. (1998) for A-B intestinal permeability (using TC7 cells, pH 6.5/7.4) and Kuhnz and Gieschen (1998) for metabolic stability (human liver microsomes). In parallel, inhibition of cytochrome P450 isoforms was evaluated according to Crespi et al. (1997) for cytochrome 1A2 and cytochrome 2C9, Ono et al. (1996) for cytochrome 2C19 and cytochrome 2D6, and Stresser et al. (2000) for cytochrome 3A4. The LC/MS system for microsomal stability and metabolite identification consisted of an Orbitrap Exactive instrument (Thermo Scientific, Waltham, MA, USA) equipped with an electrospray ionization source used in positive mode (M+H+). The apparatus was managed with Xcalibur software. Inhibition of cloned hERG potassium ion channel repolarization was evaluated in CHO cells, according to Mathes (2006).
Animals
All animal care and experimental procedures complied with the European Communities Council Directives of 24 November 1986 (86/609/EEC) and were approved by the local ethical committee (CEEA 102009R). Efforts were made to minimize the number of animals used and their suffering. Animals that reached severe hind limb paresis (clinical grade 3) were isolated, and hydration and food access were facilitated. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 103 animals were used in the experiments described here. SJL/J mice were purchased from Janvier (Le Genest-St-Isle, France) and bred under conventional barrier protection at the Pasteur Institute (Lille, France).
EAE induction and treatment
The method of EAE induction has been described earlier (Lee-Chang et al., 2011a). Randomized 9-week-old female SJL/J mice were inoculated s.c. in the neck with an emulsion containing 100 μg of myelin proteolipid protein (PLP)139–151 peptide and an equal volume of complete Freund's adjuvant (CFA) containing 4 mg·mL−1 of heat-inactivated Mycobacterium tuberculosis H37RA (Difco Laboratories, Detroit, MI, USA) on day 0 (D0). Additionally, mice received 0.3 μg of Bordetella pertussis toxin (BPT; Sigma-Aldrich, Saint Louis, MI, USA) i.p. on D0 and D3. Sham animals only received saline injection. SJL/J mice that only received CFA and BPT were also included in the experiments. Control animals received one administration of saline solution (EAE-vehicle or sham-vehicle mice). Mice showed no apparent toxic side effects of any of the treatment protocols.
Clinical evaluation
Body weight and clinical signs of EAE were monitored daily. Three different treatment groups (EAE-vehicle, EAE-1(S) 1 mg·kg−1 and EAE-1(S) 5 mg·kg−1) were used per experiment, with 5–7 animals per treatment group. Data were compiled from three independent experiments (n = 15–19 per group; Baker and Amor, 2012). The severity of clinical symptoms was scored based on a standard neurological scoring system for EAE, as follows: grade 0, no disease; grade 1, moderate tail hypotonia and/or slightly clumsy gait; grade 2, tail atony and/or clumsy gait; grade 3, severe hind limb paresis; grade 4, paraplegia; grade 5, tetraplegia; and grade 6, dead. Scoring was performed without knowledge of the treatments. Based on the clinical score data, EAE was characterized using the following parameters: incidence, cumulative disease index (CDI), disease peak, score at D14 ± 2, relapse duration and mortality. Incidence of EAE corresponds to the frequency of the new cases reaching grade 2. The CDI was calculated as the sum of the daily clinical scores for each mouse (Bodhankar et al., 2011). Disease peak refers to the first day of maximal clinical score and relapse duration refers to the number of days that mice presented a clinical score ≥2. To ensure a maximum effect of compound 1(S), the next experiments were carried out at a dose of 5 mg·kg−1.
Serum anti-PLP elisa
Mice were deeply anaesthetized with an i.p. injection of pentobarbital. Serum samples were prepared from peripheral blood obtained by cardiac puncture immediately before perfusion. Active immunizations were confirmed by measuring anti-PLP139–151 IgG antibody (Ab), as previously described (El Behi et al., 2007).
Isolation of mononuclear cells
After blood sampling, mice were perfused with an intracardiac injection of chilled PBS. Spleen and cervical lymph nodes (CLNs) were isolated. Cell suspensions were obtained by mechanical dissociation using a nylon mesh of 80 μm (Millipore, Billerica, MA, USA) followed by incubation with erythrocyte lysis reagent (155 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA).
Measurement of cytokines
Serum cytokines were measured using a mouse cytokine-plex assay kit (Bio-Rad, Hercules, CA, USA) with Bio-Plex Manager software version 6.0 in a Bio-Plex TM 200 system (Bio-Rad). This system allows quantitative measurement of IFN-γ, TNF-α, IL-17a and IL-4. Cytokines were evaluated according to the manufacturer's instructions. Four different groups (sham-vehicle; sham-1(S) 5 mg·kg−1; EAE-vehicle and EAE-1(S) 5 mg·kg−1) were used with 5–11 animals per treatment group.
Flow cytometry
Briefly, phenotypic analysis of B1a (CD19+ CD5+ CD43+), MZ (CD19+ CD21high CD23−), transitional 2 (T2) (CD19+ CD93+ CD23+) or follicular (FO) (CD19+ CD21+, CD23high) B-cell subsets and Tregs (CD4+ CD25+ FoxP3+) was performed with the appropriate combination of Abs allowing subset identification, as previously described (Lee-Chang et al., 2011a). Isotype-matched negative controls were used throughout all studies. Cells were analysed using a FACSAria flow cytometer and Cell Quest software (BD Bioscience, Becton Dickinson and Company, Franklin Lakes, NJ, USA). Two independent studies were performed. Four different groups (sham-vehicle; sham-1(S) 5 mg·kg−1; EAE-vehicle and EAE-1(S) 5 mg·kg−1) were used per experiment, with four animals per group (Baker and Amor, 2012).
Histology
After spleen and CLN isolation, mice were intracardially perfused with 4% paraformaldehyde (Carlo Erba Réactifs (SDS), Val-de-Reuil, France) solution in saline. Brain and spinal cord were removed, post-fixed for at least 4 h in the same fixative followed by 12 h in 20% sucrose solution before being embedded in ice-cold OCT (Optimal Cutting Temperature embedding medium, Cell Path, Newtown, UK), frozen in isopentane (−55°C), and stored at −80°C until sectioning.
Using a cryostat, 12 μm serial coronal sections from five levels of the neuroaxis (corpus callosum/striatum, cerebellum/brainstem, cervical, thoracic and lumbar spinal cords) were cut and collected on Superfrost/Plus slides (Thermo Scientific) and kept at −80°C until use. Brain and spinal cord sections were stained by conventional haematoxylin and eosin (H&E) or luxol fast blue (LFB) and examined by light microscopy, without knowledge of the treatments. Inflammation was quantified by counting the mononuclear cell infiltration foci (at least 20 clustered mononuclear cells) in the meninges and parenchyma by a double-blinded investigator. An arbitrary score from 0 to 4 was determined as follows: 0, no mononuclear cell infiltration; 1, few cellular infiltrates in perivascular and/or meninges area; 2, mild cellular infiltrates (≤10); 3, moderate cellular infiltrates (11–20); 4, severe cellular infiltrates (>20). The mean histological score was calculated for each group. Two groups (EAE-vehicle and EAE-1(S) 5 mg·kg−1) were used in this experiment, with three animals per experimentation group. A total of 24 sections per CNS area per animal were analysed.
Data analysis
Data are presented as median and interquartile range (IQR) due to the small size of the animal groups. Clinical parameters were analysed between the three groups (EAE-vehicle, EAE-1(S) 1 mg·kg−1 and 5 mg·kg−1) using a Kruskal–Wallis test, following post hoc pairwise comparisons using a Mann–Whitney U-test for continuous parameters and by Fisher's exact test for qualitative parameters. The clinical score evolution was compared between the three groups with a linear mixed model. This model is an extension of the classical anova taking into account the correlation between measurements of the same subject. The fixed effects were the groups and the time. The random effect was the mice. For each CNS-area H&E analysis, the effect of administration of compound 1(S) on mononuclear cell infiltration was assessed by a linear mixed model in order to take into account that several sections were measured for each animal. The effect of administration of compound 1(S) on cytokine production was studied using a Student's t-test. The effect of compound 1(S) on T2, MZ, B1a, FO B-cells and Tregs in spleen and CLNs was assessed using a Kruskal–Wallis test. Post hoc analyses were performed using a Mann–Whitney U-test. Statistical analyses were performed using SAS software (SAS Institute, Cary, NC, USA; version 9.2). P-values <0.05 were considered statistically significant.
Materials
Compound 1(S) was synthesized according to a previously reported protocol (Toussaint et al., 2010). BD1047 (N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino)ethylamine hydrochloride) was supplied by Tocris (Bristol, UK). All drugs were dissolved in physiological saline and injected i.p. on D0 in a volume of 100 mL per 20 g of body weight. Doses refer to the salt form. For antagonism studies, BD1047 was administered 20 min before compound 1(S).
Results
In vitro ADME properties
A range of physicochemical and biochemical properties of compound 1(S) (Figure 1), including aqueous solubility, logD, cell permeability and effects on CYP isoforms, were measured in vitro using methods already referred to in the Methods. The values obtained are listed in Table 1. Although compound 1(S) was chemically stable, it was rapidly metabolised by human liver microsomes (Table 1) to demethylated and debenzylated compounds (data not shown). However at 10 mM, compound 1(S) did not inhibit of any of the CYP forms tested (Table 1). Cardiotoxicity was estimated using the predictor hERG test. The IC50 for tail current inhibition was 0.55 μM for compound 1(S).
Figure 1.
Chemical structure of compound 1(S), the σ1 protein agonist evaluated.
Table 1.
ADME profile of compound 1(S), the σ1 protein agonist used in these studies.
| Values | Experimental conditions | |
|---|---|---|
| Bioavailability | ||
| Aqueous solubility | 278 μM | PBS, pH 7.4 |
| logD | 2.15 | n-octanol/PBS, pH 7.4 |
| P-gp inhibition | 10.3% | 1 μM |
| Plasma protein binding | 93.1% (99.1% recovery) | 8 h, 37°C |
| A-B intestinal permeability (10−6 cm·s−1) | 53.6% (122% recovery) | TC7 cells, pH 6.5/7.4 |
| Metabolism study | ||
| Metabolic stability | 14% | 0 and 1 h, pH 7.4, 37°C human liver microsomes |
| CYP1A2 inhibition | −8% | CEC substrate, 10 μM |
| CYP2C9 inhibition | −1% | MFC substrate, 10 μM |
| CYP2C19 inhibition | 26% | CEC substrate, 10 μM |
| CYP2C6 inhibition | −30% | MFC substrate, 10 μM |
| CYP3A4 inhibition | 40% | BCF substrate, 10 μM |
| Toxicity | ||
| hERG, inhibition of tail current, IC50 | 0.55 μM |
Attenuation of clinical, histological and biological EAE after compound 1 (S) injection
As comparative analysis of B-cell subsets in susceptible and resistant mice at critical time points has demonstrated a homeostatic breakdown of T2 and MZ B-cells in EAE mice at disease peak, a single injection of compound 1(S) was given on D0, at the time of immunization (Lee-Chang et al., 2011a). Thus, mice were actively immunized with PLP139–151 in CFA and immediately injected either with vehicle (saline) or with compound 1(S) i.p. Three different groups, EAE-vehicle, EAE-1(S) 1 mg·kg−1, and EAE-1(S) 5 mg·kg−1, were used per experiment, with 5–7 animals per treatment group. The EAE disease course was followed for 35 days after immunization. Active immunization was confirmed by measuring the serum anti-PLP139–151 IgG antibody at day 35 (D35; data not shown). Data were compiled from three independent experiments (n = 15–19/group).
Clinical observation showed that EAE-vehicle (control) mice experienced onset of symptoms at D11 (Figure 2A). A single injection of compound 1(S) (1 or 5 mg·kg−1) decreased the CDI (P = 0.0246; Table 2). EAE-vehicle mice peaked at D13 and presented a maximal clinical score of 4.0 (Figure 2A; Table 2), as previously described (Tuohy et al., 1989; Magliozzi et al., 2004; Lee-Chang et al., 2011a). However, compound 1(S) (1 or 5 mg·kg−1) did not modify the disease peak (P = 0.1287). In light of this, and in accordance with previous results, disease peak was fixed at D14 ± 2 for the subsequent analysis (Lee-Chang et al., 2011a). The single injection of compound 1(S) (1 or 5 mg·kg−1) reduced the clinical score at this critical point (P < 0.0001). Compound 1(S) injection also significantly changed relapse duration (P = 0.0003), without reducing mortality (P = 0.1371) (Table 2). The effects of compound 1(S) were not dose-related, as there was no difference in the effects on any of the clinical parameters analysed, between 1 mg·kg−1 and 5 mg·kg−1 treatments. Nevertheless, linear mixed model statistical analysis showed that clinical scores were higher in EAE-vehicle mice compared with EAE-1(S) 1 or 5 mg·kg−1 mice (P = 0.007). The clinical scores remained significantly different over the 35 day experimental period (P < 0.0001; Figure 2A).
Figure 2.
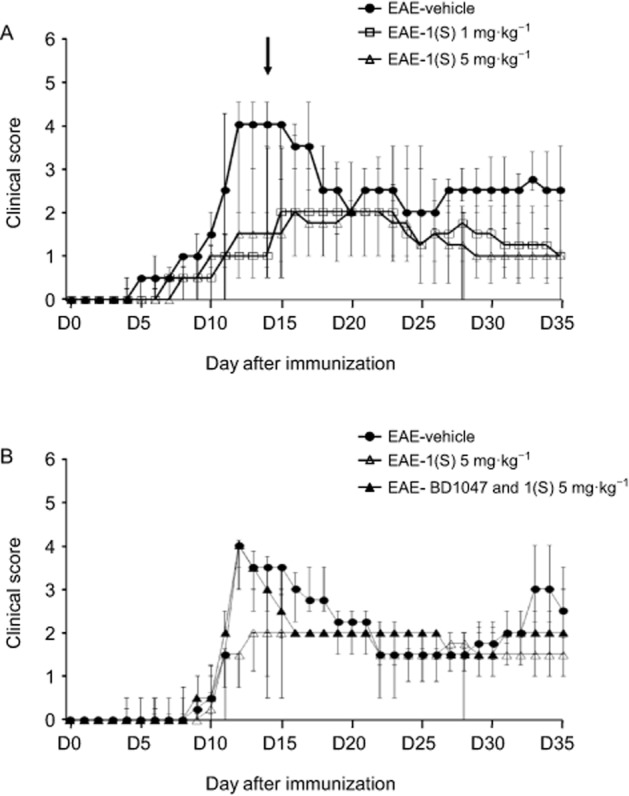
Effect of compound 1(S) on clinical signs of PLP 139–151-induced EAE in SJL/J mice. Mice were immunized on day 0 (D0) and their clinical course was followed for the next 35 days. Data are presented as median ± inter-quartile range IQR. (A) Reduction of EAE clinical score by a single injection of compound 1(S) at D0. Three different groups, EAE-vehicle, EAE-1(S) 1 mg·kg−1 and EAE-1(S) 5 mg·kg−1, were used per experiment with 5–7 animals per treatment group. Data were compiled from three independent experiments (n = 15–19/group). The arrow indicates the time corresponding to the disease peak, when the histological, Tregs and B-cell subsets analyses were carried out. (B) Involvement of the σ1 protein in the beneficial effects of compound 1(S) on the clinical scores. Three different groups, EAE-vehicle, EAE-1(S) 5 mg·kg−1 and EAE-BD1047 and 1(S) 5 mg·kg−1 were used per experiment with nine animals per treatment group. The σ1 protein antagonist BD1047 (10 mg·kg−1) was given i.p., 20 min before compound 1(S) injection. Data are from two separate experiments.
Table 2.
Effect of the σ1 protein agonist, compound 1(S), on EAE developmenta
| Incidence | CDI | Disease peak | Score at D14 ± 2 | Relapse duration | Mortality | |
|---|---|---|---|---|---|---|
| EAE-vehicle | 15/15 | 71.2 (±44.5) | D13 (±4) | 4.0 (±1.0) | 21 (±13.5) | 6/15 |
| EAE-1(S) 1 mg·kg−1 | 15/19 | 45.5 (±35.8)* | D16 (±6) | 1.0 (±0.5)*** | 7.5 (±17.0)* | 2/19 |
| EAE-1(S) 5 mg·kg−1 | 14/19 | 40.0 (±34.3)* | D16 (±5) | 1.5 (±0.5)*** | 7.5 (±11.0)* | 3/19 |
To evaluate the effect of compound 1(S) on EAE development, we used three different groups, EAE-vehicle, EAE-1(S) 1 mg·kg−1 and EAE-1(S) 5 mg·kg−1. Data were compiled from three independent experiments with 5–7 animals per group (n = 15–19/group). Incidence corresponds to the frequency of the new cases reaching clinical grade 2. CDI was calculated as the sum of the daily clinical scores for each mouse (Bodhankar et al., 2011). Disease peak refers to the first day of maximal clinical score. Relapse duration refers to the number of days that mice showed a clinical score ≥2. Data are presented as median ± IQR. No significant differences were observed between any of the effects of 1 mg·kg−1 and 5 mg·kg−1 compound 1(S).
P < 0.05
P < 0.0005; significantly different from the EAE-vehicle group.
To confirm that the biological activities of compound 1(S) required the presence of the σ1 protein, we blocked its binding sites with the σ1 protein antagonist BD1047, given i.p. (10 mg·kg−1), 20 min before compound 1(S) (1 or 5 mg·kg−1) (Meunier et al., 2006). As shown in Figure 2B, BD1047 blocked the effects of compound 1(S) (5 mg·kg−1) on EAE initiation and development. No significant difference was observed when we compared the median clinical score of EAE-vehicle and EAE-BD1047 and 1(S) 5 mg·kg−1 mice at disease peak, that is, D14 ± 2 (P = 0.0917), even when a significant difference was observed after compound 1(S) injection (P = 0.0012). Similar results were obtained with compound 1(S) injected at 1 mg·kg−1. BD1047 alone displayed no significant effects on EAE (data not shown).
Analysis of cellular infiltration and demyelination were also performed at D14 ± 2 in EAE-vehicle mice (clinical grade 4.0) and EAE-1(S) 5 mg·kg−1 mice (clinical grade 1.5; Figure 2A) (Table 2). An extensive analysis of the CNS was performed using smples of the corpus callosum/striatum, cerebellum/brainstem, and the cervical, thoracic and lumbar spinal cord (Figure 3). Figure 3A shows that EAE-vehicle mice were characterized by infiltration of mononuclear cells in the corpus callosum/striatum (mean scores are shown in Figure 3A), mainly meningeal (Figure 3B, panels A and B). The cell infiltration was higher in the cerebellum and brainstem (Figure 3A); it was characterized by meningeal and perivascular parenchymal infiltrates (data not shown). No differences were observed between the histological scores of cervical, thoracic and lumbar spinal cord. Lesions were typically located within the CNS white matter (Figure 3B, panels D and E). In EAE-1(S) 5 mg·kg−1 mice, histological analyses revealed that the infiltrating mononuclear cell foci score was not significantly changed in the corpus callosum/striatum, but was reduced in the cerebellum and brainstem (P < 0.005). Inflamed vessels were never found in the cervical, thoracic and lumbar spinal cord of EAE-1(S) 5 mg·kg−1 mice (P < 0.005). Panels C and F of Figure 3B show significantly attenuated cellular infiltration in the brain parenchyma and spinal cord of EAE-1(S) 5 mg·kg−1 animals. Analysis of myelin content by LFB staining was also performed at the same time as H&E staining, using serial sections. Demyelination was confirmed in EAE-vehicle mice, mainly in the spinal cord (Figure 3B, panels J and K). Meningeal cuffs observed in the corpus callosum/striatum and cerebellum/brainstem did not lead to demyelination in EAE-vehicle mice (Figure 3B, panels G and H). EAE-1(S) 5 mg·kg−1 mice showed no demyelination in the corpus callosum/striatum, cerebellum/brainstem or spinal cord (Figure 3B, panels I and L).
Figure 3.
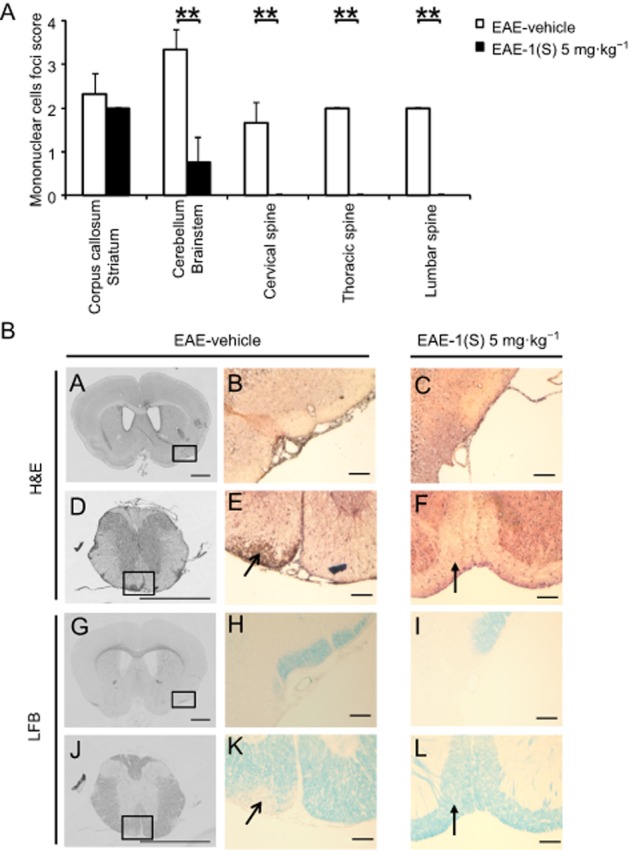
A single injection of compound 1(S) at D0 decreased the infiltration of mononuclear cells and the demyelination in the corpus callosum and the spinal cord in EAE mice. Two different groups, EAE-vehicle and EAE-1(S) 5 mg·kg−1 were used. Mice were immunized to induce EAE and compound 1(S) (5 mg·kg−1) given i.p. Animals were killed at the peak of disease (i.e. on day 14 ± 2), when the clinical grade for the EAE-vehicle group was 4.0 and for the EAE-1(S) 5 mg·kg−1 group was 1.5. This experiment utilized three animals per experimentation group. A total of 24 sections per CNS area per animal were analysed. (A) Quantification of mononuclear cell infiltration in corpus callosum/striatum, cerebellum/brainstem, and cervical, thoracic and lumbar spine. Graph bars indicate the inflammatory score (means ± SD). **P < 0.005, significantly different as indicated. (B) Representative light microscopy of mouse corpus callosum/striatum (panels A–C, G–I) or spinal cord (panels D–F, J–L) sections stained with H&E or LFB. Only EAE-vehicle mice show moderate and extensive inflammatory lesions, in corpus callosum (panels A and B) and spinal cord (panels D and E open arrowhead) respectively. Sections from the same level stained for myelin show loss of myelin in the spinal cord (panels J and K open arrowhead), while white matter was not disrupted in the corpus callosum/striatum area (panels G and H). Minimal infiltration (panels C and F filled arrowhead) and no demyelination (panels I and L filled arrowhead) was observed in sections from EAE-1(S) mice. Scale bars = 100 μm, original magnification: 10×.
To analyse whether the clinical and histological modulation of EAE observed after the single injection of compound 1(S) could be correlated with the modulation of the peripheral inflammatory state, five serum cytokines were also assayed in sham-vehicle, sham-1(S) 5 mg·kg−1, EAE-vehicle and EAE-1(S) 5 mg·kg−1 mice at D14 ± 2 after immunization. The results were inconclusive as to whether a single injection of compound 1(S) at D0 did modulate serum cytokine production (Supporting Information Table S1).
Modulation of Foxp3+ Tregs and B-cell subsets after compound 1(S) injection
CD4+ CD25+ Foxp3+ Tregs depress the autoimmune response causing EAE (O'Connor and Anderton, 2008). Breakdown of T2 and MZ B-cells was observed in susceptible EAE mice at disease peak (D14 ± 2), compared with resistant mice (Lee-Chang et al., 2011a). Tregs and B-cell subsets were analysed in sham-vehicle, sham-1(S) 5 mg·kg−1, EAE-vehicle and EAE-1(S) 5 mg·kg−1 mice, in spleen and brain-draining CLNs at D14 ± 2 after immunization.
As illustrated in Figure 4, in the spleen of sham-vehicle and EAE-vehicle mice, immunization did not modify the amount of Foxp3+ Tregs 14 days after EAE development (Figure 4A). A slight increase in the number of Tregs was observed at the draining site after PLP immunization (i.e. CLNs; Figure 4B). Immunization with CFA-TBP alone had no effect on the number of Tregs (data not shown). On the contrary, a comparison of sham-vehicle vs. sham-1(S) 5 mg·kg−1 and EAE-vehicle vs. EAE-1(S) 5 mg·kg−1 mice demonstrated that a single injection of compound 1(S) at D0 increased Treg numbers in the spleen 14 days after EAE development in both sham and EAE mice (Figure 4A; sham, P = 0.0198 and EAE P = 0.004). Similar results were observed in CLNs [Figure 4B; sham P = 0.001 and EAE, P = 0.039).
Figure 4.
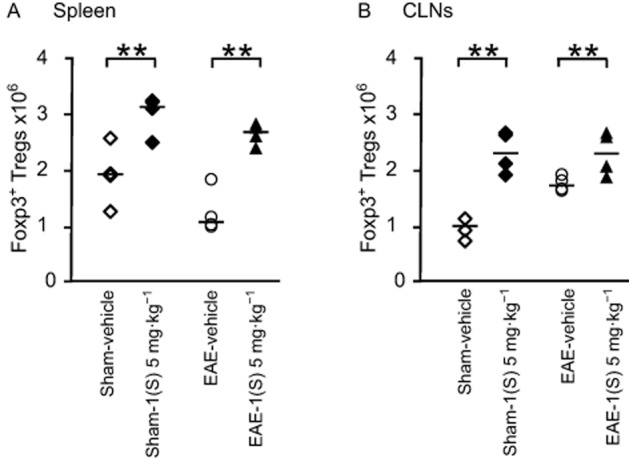
Foxp3+ regulatory T-cells are increased in spleen and CLNs by a single injection of the σ1 protein agonist on D0. Foxp3+ Tregs (A and B) counts were analysed in spleen and CLNs in sham and EAE mice. Compound 1(S) (5 mg·kg−1) was given i.p. Animals were killed at the peak of disease (i.e. on day 14 ± 2) after immunization (clinical grades: sham-vehicle and sham-1(S) 5 mg·kg−1 group, 0; EAE-vehicle, 4.0; EAE-1(S) 5 mg·kg−1 group, 1.5). Horizontal bar indicates median values. **P < 0.005, significantly different as indicated. This experiment utilized four animals per experimentation group and is representative of two independent studies.
EAE-vehicle mice showed significantly reduced MZ and FO B-cell counts in their spleen 14 days after EAE development (Figure 5B and D; MZ, P = 0.042 and FO, P = 0.021). A less marked decrease was noted for B1a B-cells in EAE-vehicle mice (Figure 5C; P = 0.093). An accumulation of B1a and FO B-cells was observed in the CLNs of EAE-vehicle mice 14 days after EAE development (Figure 6A and B; B1a,, P = 0.015 and FO, P = 0.018). Treatment with compound 1(S) (5 mg·kg−1) of sham or EAE mice did not affect MZ, B1a or FO B-cell numbers in the spleen (Figure 5B–D) but induced a specific increase in T2 B-cells (Figure 5A; sham, P = 0.003 and EAE, P = 0.0003). However, in the CLNs, compound 1(S) injection did not modify B1a or FO B-cell counts in sham and EAE mice (Figure 6A and B).
Figure 5.
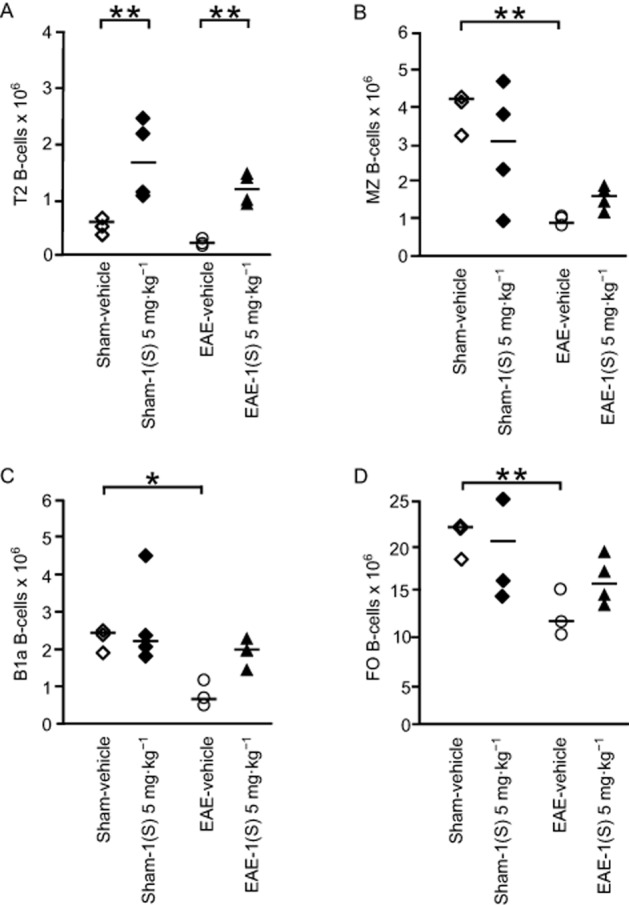
Transitional 2 (T2) B-cell subset is increased in spleen by a single injection of the σ1 protein agonist on D0. T2 (A), MZ (B), B1a (C) and follicular, FO (D) B-cell sub-population counts were analysed in spleen in sham and EAE mice. Compound 1(S) (5 mg·kg−1) was given i.p. Animals were killed at the peak of disease (i.e. on day 14 ± 2) after immunization (clinical grades: sham-vehicle and sham-1(S) 5 mg·kg−1 group, 0; EAE-vehicle, 4.0; EAE-1(S) 5 mg·kg−1 group, 1.5). Horizontal bar indicates median values. Significant differences are indicated as follows: *P < 0.05, **P < 0.005; significantly different as indicated. This experiment utilized four animals per experimentation group and is representative of two independent studies.
Figure 6.
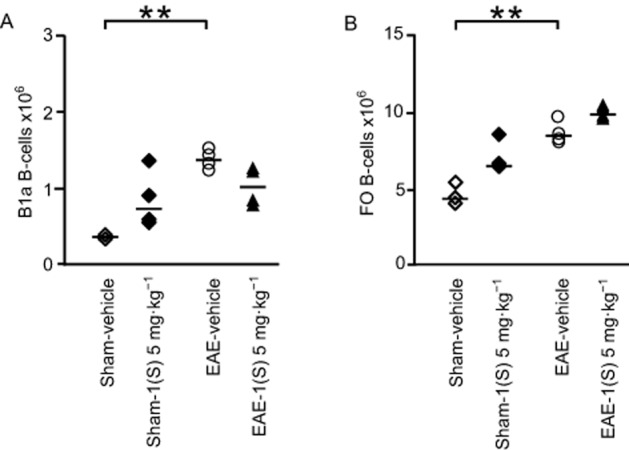
No modulation of B-cell subsets in CLNs by a single injection of σ1 protein agonist on D0. B1a (A) and FO (B) B-cell sub-population counts were analysed in CLNs in sham and EAE mice. Compound 1(S) (5 mg·kg−1) was given i.p. Animals were killed at the peak of disease (i.e. on day 14 ± 2) after immunization (clinical grades: sham-vehicle and sham-1(S) 5 mg·kg−1 group, 0; EAE-vehicle, 4.0; EAE-1(S) 5 mg·kg−1 group, 1.5). Horizontal bar indicates median values. **P < 0.005, significantly different as indicated. This experiment utilized four animals per experimentation group and is representative of two independent studies.
Discussion
In mice, EAE exhibits histopathological changes characterized by infiltration of the CNS by peripheral leukocytes, including autoimmune deleterious T- and B-cells, reactive gliosis, demyelination and substantial axonal loss (Brown et al., 1982). The damaged areas observed are similar to those found in active plaques of MS patients. However, there are different gradients in the severity of inflammation and demyelination depending on the mouse strain used. In this study, SJL/J mice were used as severe inflammation is observed in this strain (Dal Canto et al., 1995). Furthermore, this experimental model is a chronic disease with a spontaneous pattern of relapse and remissions (R-EAE), comparable with the most common form of MS (Webb et al., 2004; Ransohoff, 2012). It has already been demonstrated that specific σ1 protein ligands inhibit CD3+ lymphocytes proliferation and LPS-induced release of IL-1, IL-6, TNF-α and IFN-γ, and can enhance LPS-induced release of IL-10 (Casellas et al., 1994; Bourrie et al., 1995; Derocq et al., 1995). The aim of this study was thus to observe the potential role of a σ1 protein agonist on inflammatory processes, particularly in EAE. As B-cells are also known as immunocompetent cells with deleterious and/or beneficial effects in EAE and MS pathogenesis, and as homeostatic breakdown of B-cell subsets has been associated with the induction of EAE (Mann et al., 2012), we decided to inject compound 1(S) at immunization time (D0). Compound 1(S) was injected at 1 and 5 mg·kg−1, two concentrations within a range of σ protein ligands previously tested (Casellas et al., 1994; Derocq et al., 1995; Demerens et al., 1999; Fujino et al., 2003; Toussaint et al., 2009).
No time lag was observed for EAE disease peak in vehicle or in treated mice, but injection of compound 1(S) significantly reduced EAE intensity. Consequently, histological analysis of the CNS was performed 14 ± 2 days after immunization, which corresponds to the disease peak and the time that major mononuclear cell infiltrations were observed in EAE mice (Tuohy et al., 1989; Lee-Chang et al., 2011a). Immunostaining for σ1 protein has been observed throughout the rostrocaudal regions of the CNS, extending from the olfactory bulb to the spinal cord in adult rodents (Alonso et al., 2000). However, the cerebellum presents more specific σ1 protein binding sites than the corpus callosum (Bouchard and Quirion, 1997). Spinal cord, cerebellum and forebrain correspond to areas affected early after the increase of blood–brain-barrier (BBB) permeability (Cross et al., 1993). Consequently, an analysis of the CNS was performed from the corpus callosum/striatum to the lumbar spinal cord in EAE-vehicle mice (clinical grade 4.0) and EAE-1(S) 5 mg·kg−1 mice (clinical grade 1.5). Mononuclear cell infiltrations were attenuated in the cerebellum/brainstem area and absent in the spinal cord of treated mice. It has been suggested that classical EAE models might not be the most suited for studies of myelin repair (Baker and Amor, 2012). However, the most severe lesions are observed in SJL/J mice and primary demyelination is localized around some of the perivascular cuffs (Tuohy et al., 1989; Dal Canto et al., 1995). Therefore, we analysed myelination and mononuclear cell infiltration at the same time (Kozlowski et al., 1987). As expected, little or no demyelination was observed in EAE-1(S) mice, in any of the areas tested.
EAE is characterized by inflammation and demyelination of the CNS. Priming of the myelin-specific response occurs in the secondary lymphoid organs, after which the pathogenic cells migrate to the CNS via the bloodstream. The vast majority of effector cells reside in the periphery and not in the target tissue, even at the EAE disease peak (Targoni et al., 2001). Th1 and Th17 cells are pathogenic in EAE and possibly in MS. Th2 cells are thought to be protective. Cytokines such as IL-4, IL-10 and IL-5 have been associated with inflammation reduction and improvement of symptoms in EAE (El Behi et al., 2010). No modulation of serum levels of cytokines was observed in this study, compatible with the restriction of the major effects of cytokines to the target organ, during autoimmune pathogenesis. As Foxp3+ Tregs represent a well-defined regulatory cell subset, their homeostasis was analysed at disease peak (D14 ± 2) after the single injection of compound 1(S). A significant increase of this subset was observed in the spleen and CLNs of sham-1(S) and EAE-1(S) animals. These results are in agreement with the involvement of Treg cells in autoreactive T-cell effector function and autoimmune disease progression suppression early in the induction of the auto-aggressive response, namely in the lymph nodes. Furthermore, in the PLP-induced model, the susceptibility of different mouse strains correlates inversely with the frequency of PLP-specific Tregs (Fletcher et al., 2010). B-cell depletion could also modulate EAE initiation and disease progression (Matsushita et al., 2008). Until now, identification of the binding sites on σ1 protein in lymphocytes has been performed using [3H] high-affinity ligands (Coccini et al., 1991). On the basis of previous data from our laboratory, B-cell subsets were also analysed in mice spleen and CLNs, after we had confirmed expression of σ1 protein in B lymphocytes (data not shown). Indeed, spleen performs an important immunological function but CLNs may be an important crossroad for processes taking place in the CNS during EAE. B1a, MZ, T2 and FO B-cells were also evaluated in spleen and CLNs to better understand their involvement in the immunological regulation that followed compound 1(S) injection. As expected, EAE led to a significant decrease of MZ and FO B-cells in the spleen. The reduction was less marked for the B1a B-cell subset. Interestingly, a significant increase in T2 B-cell numbers was observed in the spleen of sham-1(S) and EAE-1(S) animals. Indeed, the T2 and MZ B-cell sub-populations are exclusively located in this organ in rodents (Carsetti et al., 2004). Concurrently, EAE significantly increased B1a and FO B-cell subsets in CLNs but no modulation was observed after compound 1(S) injection. T2-MZ B-cells could potentially belong to the Bregs family as B10 cells and transitional 2 marginal-zone precursors (T2-MZP) Breg cells share common surface markers. Indeed, functional differences should exist between B10 cell progenitors (B10pro), B10 and T2-MZP, T2 and MZ B-cell subsets (Mauri and Blair, 2010; Kalampokis et al., 2013). Today, relatively few data are available to explain the precise role of transitional B-cells during EAE initiation and development. In mice, it appears that a slight homeostatic breakdown of T2 might be associated with the induction of and susceptibility to EAE and, as a consequence, with the emergence of pathogenic events (Lee-Chang et al., 2011a). In humans, transitional or ‘late-immature’ B-cells, were considerably reduced in blood samples from MS patients (Lee-Chang et al., 2011b).
The beneficial effect of σ1 protein agonists on the central neurodegenerative process is well documented. Eliprodil has the potential to increase myelination of CNS neurons in vitro (Demerens et al., 1999). Dextrometorphan, a high-affinity σ1 protein agonist, inhibited, in vitro, the cytotoxic effects on oligodendroglia and oligodendroglial progenitors of several molecules that have been shown to be important in the pathogenesis of EAE (Lisak et al., 2014). In the CNS, σ1 protein is also involved in the modulation of astrocytes, macrophages/microglia as well as in the increase of BBB permeability (Yao et al., 2011). Nevertheless, neuroprotection without immunomodulation is not sufficient to reduce first relapse severity in EAE (Hasseldam and Johansen, 2010). The results obtained in this study confirm previous data highlighting the involvement of the σ1 protein in the immunological response. The mechanisms need to be examined in more detail, but the frequency of T2 B-cells and Tregs was clearly augmented by compound 1(S) regardless of the presence of inflammation.
It has been reported that specific ion channels or receptors are not affected by σ1 protein or ligands under normal physiological conditions. The assistance of σ1 chaperones seems to be solely required under pathological conditions (Su et al., 2010). It would be interesting to analyse σ1 protein expression and distribution in EAE and sham animals, with or with out compound 1(S), as the accumulation of σ1 protein is a common finding in various neurodegenerative diseases and its localization at the ER and/or mitochondrion-associated ER membrane is affected by the pharmacological activation of this protein (Su et al., 2010; Hedskog et al., 2013; Miki et al., 2014). Mutations of the σ1 protein gene have also been associated with amyotrophic lateral sclerosis and frontotemporal lobar degeneration (Peviani et al., 2014).
The σ1 protein is involved in various acute and chronic pathologies, in cell survival as well as in cell proliferation. This ligand-regulated molecular chaperone has been described as an intracellular amplifier, creating a supersensitized state for signal transduction. The σ1 protein also affects mitochondrial Ca2+ influx by stabilizing IP3Rs and acting as an inter-organelle signalling modulator of Ca2+-homeostasis, ER stress reaction and apoptosis. Under prolonged stimulation, the σ1 protein interacts with several plasma membrane proteins, including voltage-dependent (Na+, K+, Ca2+) and ligand-operated (NMDA) receptors, ion channels and kinases (Su et al., 2010). All these partners play critical roles in regulating proliferation, cytokine production, cytotoxic function and in vivo migration of lymphocytes. The significant increase in Tregs and T2 B-cell numbers observed after compound 1(S) injection might also be partly attributable to their differential expression on B- and T-cell subsets (Chhabra et al., 2014). Furthermore, apart from the modulation of the level of expression, cell subsets could also express a different profile of σ1 protein partners, depending on their activation states.
In summary, ADME analysis in vitro demonstrated that compound 1(S) was soluble, bioavailable, able to cross biological barriers and presented little toxicity (Toussaint et al., 2009). A single injection in EAE-susceptible mice at immunization time prevented mononuclear cell accumulation and demyelination in brain and spinal cord and increased T2 B-cells in the spleen and Tregs in spleen and CLNs, resulting in an overall reduction in the clinical progression of disease and confirming the involvement of σ1 protein in this immunological response. Nevertheless, compound 1(S) has only moderate metabolic stability and this has to be taken into account for the further development of new compounds sharing similar profiles.
Acknowledgments
We thank M. Bellard for technical assistance, J. P. Decavel and T. Chassat for invaluable advice on animal manipulations and Dr C. Allet for histology knowledge and experience. We also thank S. Loiseaux, M. Delbeke and Dr A. Sarazin for multiplex assay knowledge and experience, G. Hochart for metabolite identification and Dr LT. Mars for fruitful discussions about the manuscript. We thank Université de Lille and particularly Lille 2 University for their financial support. This work was supported by BiogenIdec. The funding sources had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Glossary
- Bregs
regulatory B cells
- CFA
complete Freund's adjuvant
- CLNs
cervical lymph nodes
- EAE
experimental autoimmune encephalomyelitis
- H&E
haematoxylin and eosin
- LFB
luxol fast blue
- MS
multiple sclerosis
- MZ
marginal zone
- PLP
myelin proteolipid protein
- T2
transitional 2
- Tregs
regulatory T cells
Author contributions
Conceived and designed the experiments: B. O., D. L., P. M., P. V. Performed the experiments: B. O., C. L. C., M. G., M. T., M. D. M. Analysed the data: B. O., A. D. Wrote the paper: B. O.
Critical revision of the manuscript for important intellectual content: C. L. C., L. P., P. M., P. V., P. C., H. Z.
Conflict of interest
The authors declare no competing financial interests.
Supporting Information
Table S1 Effect of σ1 agonist, compound 1(S), on peripheral inflammatory state.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013a;170:1449–1458. [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013c;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-Gated Ion Channels. Br J Pharmacol. 2013d;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso G, Phan V, Guillemain I, Saunier M, Legrand A, Anoal M, et al. Immunocytochemical localization of the sigma-1 receptor in the adult rat central nervous system. Neuroscience. 2000;97:155–170. doi: 10.1016/s0306-4522(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Baker D, Amor S. Publication guidelines for refereeing and reporting on animal use in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2012;242:78–83. doi: 10.1016/j.jneuroim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Banker MJ, Clark TH, Williams JA. Development and validation of a 96-well equilibrium dialysis apparatus for measuring plasma protein binding. J Pharm Sci. 2003;92:967–974. doi: 10.1002/jps.10332. [DOI] [PubMed] [Google Scholar]
- Batoulis H, Recks MS, Addicks K, Kuerten S. Experimental autoimmune encephalomyelitis – achievements and prospective advances. APMIS. 2011;119:819–830. doi: 10.1111/j.1600-0463.2011.02794.x. [DOI] [PubMed] [Google Scholar]
- Bodhankar S, Wang C, Vandenbark AA, Offner H. Estrogen-induced protection against experimental autoimmune encephalomyelitis is abrogated in the absence of B cells. Eur J Immunol. 2011;41:1165–1175. doi: 10.1002/eji.201040992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard P, Quirion R. [3H]1,3-di(2-tolyl)guanidine and [3H](+)pentazocine binding sites in the rat brain: autoradiographic visualization of the putative sigma1 and sigma2 receptor subtypes. Neuroscience. 1997;76:467–477. doi: 10.1016/s0306-4522(96)00221-7. [DOI] [PubMed] [Google Scholar]
- Bourrie B, Bouaboula M, Benoit JM, Derocq JM, Esclangon M, Le Fur G, et al. Enhancement of endotoxin-induced interleukin-10 production by SR 31747A, a sigma ligand. Eur J Immunol. 1995;25:2882–2887. doi: 10.1002/eji.1830251026. [DOI] [PubMed] [Google Scholar]
- Brown A, McFarlin DE, Raine CS. Chronologic neuropathology of relapsing experimental allergic encephalomyelitis in the mouse. Lab Invest. 1982;46:171–185. [PubMed] [Google Scholar]
- Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol Rev. 2004;197:179–191. doi: 10.1111/j.0105-2896.2004.0109.x. [DOI] [PubMed] [Google Scholar]
- Casellas P, Bourrie B, Canat X, Carayon P, Buisson I, Paul R, et al. Immunopharmacological profile of SR 31747: in vitro and in vivo studies on humoral and cellular responses. J Neuroimmunol. 1994;52:193–203. doi: 10.1016/0165-5728(94)90113-9. [DOI] [PubMed] [Google Scholar]
- Cazenave Gassiot A, Charton J, Girault-Mizzi S, Gilleron P, Debreu-Fontaine MA, Sergheraert C, et al. Synthesis and pharmacological evaluation of Tic-hydantoin derivatives as selective sigma1 ligands. Part 2. Bioorg Med Chem Lett. 2005;15:4828–4832. doi: 10.1016/j.bmcl.2005.07.039. [DOI] [PubMed] [Google Scholar]
- Charton J, Cazenave Gassiot A, Girault-Mizzi S, Debreu-Fontaine MA, Melnyk P, Sergheraert C. Synthesis and pharmacological evaluation of Tic-hydantoin derivatives as selective sigma1 ligands. Part 1. Bioorg Med Chem Lett. 2005;15:4833–4837. doi: 10.1016/j.bmcl.2005.07.040. [DOI] [PubMed] [Google Scholar]
- Chhabra S, Chang SC, Nguyen HM, Huq R, Tanner MR, Londono LM, et al. Kv1.3 channel-blocking immunomodulatory peptides from parasitic worms: implications for autoimmune diseases. FASEB J. 2014;28:3952–3964. doi: 10.1096/fj.14-251967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos EJ, Entrena JM, Nieto FR, Cendán CM, Del Pozo E. Pharmacology and therapeutic potential of sigma(1) receptor ligands. Curr Neuropharmacol. 2008;6:344–366. doi: 10.2174/157015908787386113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccini T, Manzo L, Costa LG. 3H-spiperone labels sigma receptors, not dopamine D2 receptors, in rat and human lymphocytes. Immunopharmacology. 1991;22:93–105. doi: 10.1016/0162-3109(91)90034-v. [DOI] [PubMed] [Google Scholar]
- Crespi CL, Miller VP, Penman BW. Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes P450. Anal Biochem. 1997;248:188–190. doi: 10.1006/abio.1997.2145. [DOI] [PubMed] [Google Scholar]
- Cross AH, O'Mara T, Raine CS. Chronologic localization of myelin-reactive cells in the lesions of relapsing EAE: implications for the study of multiple sclerosis. Neurology. 1993;43:1028–1033. doi: 10.1212/wnl.43.5.1028. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Melvold RW, Kim BS, Miller SD. Two models of multiple sclerosis: experimental allergic encephalomyelitis (EAE) and Theiler's murine encephalomyelitis virus (TMEV) infection. A pathological and immunological comparison. Microsc Res Tech. 1995;32:215–229. doi: 10.1002/jemt.1070320305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demerens C, Stankoff B, Zalc B, Lubetzki C. Eliprodil stimulates CNS myelination: new prospects for multiple sclerosis? Neurology. 1999;52:346–350. doi: 10.1212/wnl.52.2.346. [DOI] [PubMed] [Google Scholar]
- Derocq JM, Bourrié B, Ségui M, Le Fur G, Casellas P. In vivo inhibition of endotoxin-induced pro-inflammatory cytokines production by the sigma ligand SR 31747. J Pharmacol Exp Ther. 1995;272:224–230. [PubMed] [Google Scholar]
- DiLillo DJ, Horikawa M, Tedder TF. B-lymphocyte effector functions in health and disease. Immunol Res. 2011;49:281–292. doi: 10.1007/s12026-010-8189-3. [DOI] [PubMed] [Google Scholar]
- El Behi M, Zéphir H, Lefranc D, Dutoit V, Dussart P, Devos P, et al. Changes in self-reactive IgG antibody repertoire after treatment of experimental autoimmune encephalomyelitis with anti-allergic drugs. J Neuroimmunol. 2007;182:80–88. doi: 10.1016/j.jneuroim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- El Behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:189–197. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino M, Funeshima N, Kitazawa Y, Kimura H, Amemiya H, Suzuki S, et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305:70–77. doi: 10.1124/jpet.102.045658. [DOI] [PubMed] [Google Scholar]
- Grès MC, Julian B, Bourrié M, Meunier V, Roques C, Berger M, et al. Correlation between oral drug absorption in humans, and apparent drug permeability in TC-7 cells, a human epithelial intestinal cell line: comparison with the parental Caco-2 cell line. Pharm Res. 1998;15:726–733. doi: 10.1023/a:1011919003030. [DOI] [PubMed] [Google Scholar]
- Hasseldam H, Johansen FF. Neuroprotection without immunomodulation is not sufficient to reduce first relapse severity in experimental autoimmune encephalomyelitis. Neuroimmunomodulation. 2010;17:252–264. doi: 10.1159/000290041. [DOI] [PubMed] [Google Scholar]
- Hedskog L, Pinho CM, Filadi R, Rönnbäck A, Hertwig L, Wiehager B, et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer's disease and related models. Proc Natl Acad Sci U S A. 2013;110:7916–7921. doi: 10.1073/pnas.1300677110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011;117:530–541. doi: 10.1182/blood-2010-07-294249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalampokis I, Yoshizaki A, Tedder TF. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res Ther. 2013;15:S1. doi: 10.1186/ar3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Su TP, Fujimoto M, Bonci A. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012;35:762–771. doi: 10.1016/j.tins.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski PB, Schuller-Lewis GB, Wisniewski HM. Induction of synchronized relapses in SJL/J mice with chronic relapsing experimental allergic encephalomyelitis. Acta Neuropathol. 1987;74:163–168. doi: 10.1007/BF00692847. [DOI] [PubMed] [Google Scholar]
- Kuhnz W, Gieschen H. Predicting the oral bioavailability of 19-nortestosterone progestins in vivo from their metabolic stability in human liver microsomal preparations in vitro. Drug Metab Dispos. 1998;26:1120–1127. [PubMed] [Google Scholar]
- Lee-Chang C, Lefranc D, Salleron J, Faveeuw C, Allet C, Vermersch P, et al. Susceptibility to experimental autoimmune encephalomyelitis is associated with altered B-cell subsets distribution and decreased serum BAFF levels. Immunol Lett. 2011a;135:108–117. doi: 10.1016/j.imlet.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Lee-Chang C, Top I, Zéphir H, Dubucquoi S, Trauet J, Dussart P, et al. Primed status of transitional B cells associated with their presence in the cerebrospinal fluid in early phases of multiple sclerosis. Clin Immunol. 2011b;139:12–20. doi: 10.1016/j.clim.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lisak RP, Nedelkoska L, Benjamins JA. Effects of dextromethorphan on glial cell function: proliferation, maturation, and protection from cytotoxic molecules. Glia. 2014;62:751–762. doi: 10.1002/glia.22639. [DOI] [PubMed] [Google Scholar]
- Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148:11–123. doi: 10.1016/j.jneuroim.2003.10.056. [DOI] [PubMed] [Google Scholar]
- Mann MK, Ray A, Basu S, Karp CL, Dittel BN. Pathogenic and regulatory roles for B cells in experimental autoimmune encephalomyelitis. Autoimmunity. 2012;45:388–399. doi: 10.3109/08916934.2012.665523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathes C. QPatch: the past, present and future of automated patch clamp. Expert Opin Ther Targets. 2006;10:319–327. doi: 10.1517/14728222.10.2.319. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri C, Blair PA. Regulatory B cells in autoimmunity: developments and controversies. Nat Rev Rheumatol. 2010;6:636–643. doi: 10.1038/nrrheum.2010.140. [DOI] [PubMed] [Google Scholar]
- Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Ieni J, Maurice T. Antiamnesic and neuroprotective effects of donepezil against learning impairments induced in mice by exposure to carbon monoxide gas. J Pharmacol Exp Ther. 2006;317:1307–1319. doi: 10.1124/jpet.106.101527. [DOI] [PubMed] [Google Scholar]
- Miki Y, Mori F, Kon T, Tanji K, Toyoshima Y, Yoshida M, et al. Accumulation of the sigma-1 receptor is common to neuronal nuclear inclusions in various neurodegenerative diseases. Neuropathology. 2014;34:148–158. doi: 10.1111/neup.12080. [DOI] [PubMed] [Google Scholar]
- O'Connor RA, Anderton SM. Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol. 2008;193:1–11. doi: 10.1016/j.jneuroim.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Ono S, Hatanaka T, Hotta H, Satoh T, Gonzalez FJ, Tsutsui M. Specificity of substrate and inhibitor probes for cytochrome P450s: evaluation of in vitro metabolism using cDNA-expressed human P450s and human liver microsomes. Xenobiotica. 1996;26:681–693. doi: 10.3109/00498259609046742. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peviani M, Salvaneschi E, Bontempi L, Petese A, Manzo A, Rossi D, et al. Neuroprotective effects of the sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol Dis. 2014;62:218–232. doi: 10.1016/j.nbd.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci. 2012;15:1074–1077. doi: 10.1038/nn.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangster J. Octanol-Water Partition Coefficients: Fundamentals and Physical Chemistry, Vol. 2 of Wiley Series in Solution Chemistry. Chichester: John Wiley & Sons; 1997. [Google Scholar]
- Stresser DM, Blanchard AP, Turner SD, Erve JC, Dandeneau AA, Miller VP, et al. Substrate-dependent modulation of CYP3A4 catalytic activity: analysis of 27 test compounds with four fluorometric substrates. Drug Metab Dispos. 2000;28:1440–1448. [PubMed] [Google Scholar]
- Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci. 2010;31:557–566. doi: 10.1016/j.tips.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targoni OS, Baus J, Hofstetter HH, Hesse MD, Karulin AY, Boehm BO, et al. Frequencies of neuroantigen-specific T cells in the central nervous system versus the immune periphery during the course of experimental allergic encephalomyelitis. J Immunol. 2001;166:4757–4764. doi: 10.4049/jimmunol.166.7.4757. [DOI] [PubMed] [Google Scholar]
- Toussaint M, Delair B, Foulon C, Lempereur N, Vaccher C, Maurice T, et al. Tic hydantoin sigma-1 agonist: pharmacological characterization on cocaine-induced stimulant and appetitive effects. Eur Neuropsychopharmacol. 2009;19:504–515. doi: 10.1016/j.euroneuro.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Toussaint M, Mousset D, Foulon C, Jacquemard U, Vaccher C, Melnyk P. Sigma-1 ligands: Tic-hydantoin as a key pharmacophore. Eur J Med Chem. 2010;45:256–263. doi: 10.1016/j.ejmech.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol. 1989;142:1523–1527. [PubMed] [Google Scholar]
- Webb M, Tham CS, Lin FF, Lariosa-Willingham K, Yu N, Hale J, et al. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J Neuroimmunol. 2004;153:108–121. doi: 10.1016/j.jneuroim.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Yao H, Duan M, Buch S. Cocaine-mediated induction of platelet-derived growth factor: implication for increased vascular permeability. Blood. 2011;117:2538–2547. doi: 10.1182/blood-2010-10-313593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Effect of σ1 agonist, compound 1(S), on peripheral inflammatory state.