

Abstract
Background and Purpose
Hypoxia/reoxygenation induces synthesis of reactive oxygen species (ROS) which can attack macromolecules and cause brain injury. The transcription factor, nuclear factor (erythroid-derived 2)-like 2, (Nrf2), ia potent activator of genes with an antioxidant responsive element and Nrf2 can counteract oxidative injury by increasing expression of several antioxidative genes in response to ROS stress. Here, we show that activation of the δ-opioid receptor (DOR) increasedNrf2 protein expression and translocation, thereby leading to cytoprotection.
Experimental Approach
We used HEK293t cells exposed to 0.5% O2 for 16 h and then reoxygenated for 4 h as a model of hypoxia-reperfusion (H/R) injury. Real time PCR, Western blotting, siRNA and immunohistochemical techniques were used to follow Nrf2 expression and activity. Cell viability and damage (as LDH leakage) were also measured.
Key Results
H/R injury triggered Nrf2 translocation into the nucleus and up-regulated expression of several downstream genes, relevant to antioxidation, such as NAD(P)H:quinone oxidoreductase (NQO1). Incubation with the DOR agonist UFP-512 enhanced Nrf2 protein expression and translocation and up-regulated its downstream genes in normoxia and further increased Nrf2 expression and translocation after H/R, protecting the cells against loss of viability and damage. The effect of UFP-512 on Nrf2 nuclear translocation was blocked by the DOR antagonist, naltrindole. Also, DOR–mediated cytoprotection was strongly inhibited after transfection of HEK293t cells with Nrf2 siRNA.
Conclusions and Implications
The DOR agonist UFP-512 was cytoprotective against H/R injury and this effect was partly dependent on DOR-mediated increase in Nrf2 function.
Tables of Links
| LIGANDS |
|---|
| Calphostin C |
| Keap1 |
| LY294002 |
| Naltrindole |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Introduction
Reactive oxygen species (ROS) are excessively produced during ischaemic/hypoxic stress and they mediate oxidative insult by overcoming the capacity of endogenous intracellular antioxidant systems (Stankowski and Gupta, 2011; Sanderson et al., 2013). Indeed, ROS have been recognized to have a key role in the ischaemic/hypoxic injury (Jung et al., 2010; Eltzschig and Eckle, 2011; Olmez and Ozyurt, 2012). Several lines of evidence suggest that cellular antioxidant activity is critically regulated by the transcription factor, nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (Nguyen et al., 2009; Ungvari et al., 2010). Indeed, Nrf2 is the master transcription factor that controls several antioxidant pathways to counteract the physiological and pathophysiological outcomes of oxidant stress (Kensler et al., 2007; Ma, 2013). Under normal conditions, Nrf2 is retained by the Kelch-like ECH associated protein 1 (Keap1) in the cytoplasm. Once ROS, electrophilic or other insults attack the Nrf2–Keap1 complex, Nrf2 is liberated from the antioxidant response element (ARE) of Keap1 (Wakabayashi et al., 2003) and translocates to the nucleus to regulate the activities of genes involved in maintaining oxidation–reduction equilibrium and to protect cells from oxidative stress (Leonard et al., 2006; Kim et al., 2007). However, the precise mechanisms underlying the cytoprotective action of Nrf2 against ischaemia/hypoxia reperfusion injury have not been fully described.
We and others have demonstrated that the δ-opioid receptor (DOR) mediates responses to stressful stimuli, such as hypoxia and ischaemia, and activation of DOR is cytoprotective against hypoxic/ischaemic injury in various in vitro and in vivo tissue and cell sources, including primary cultured neuron (Zhang et al., 2002), HEK293t cells (Chen et al., 2014), PC12 cells (Wang et al., 2014) and intact animals (Borlongan et al., 2009; Yang et al., 2009; Johnson and Turner, 2010; Staples et al., 2013; Tian et al., 2013). One of the potential mechanisms underlying DOR neuroprotection is related to its ability to attenuate ROS oxidative stress and increase antioxidant capacity in the ischaemic brain (Yang et al., 2009). As DOR signalling is coupled with PKC (Tang et al., 2011; Chao et al., 2012; Higuchi et al., 2012) and PKC is involved in the function of Nrf2 (Huang et al., 2000; Bloom and Jaiswal, 2003), we hypothesized that DOR could act as an upstream regulator of Nrf2 function and that the DOR-mediated protection against hypoxic/ischaemic injury was directly related to its interaction with Nrf2.
Here, we have tested this hypothesis by addressing several fundamental questions regarding the regulation of Nrf2 in hypoxia-reperfusion (H/R) stress and in normoxia. We have also assessed the potential role of DOR in Nrf2-mediated events. Firstly, we determined the effect of DOR activation on the expression, translocation and function of Nrf2 in normoxia and after H/R using several molecular approaches. Furthermore, we investigated whether DOR-induced cytoprotection against H/R injury was dependent upon Nrf2. Finally, we explored the mechanism underlying the DOR-Nrf2 cytoprotection. Our data have elucidated the role of DOR-induced Nrf2 translocation and transactivation in the endogenous capacity of cells to provide cytoprotection against oxidative stress.
Methods
Cell culture and treatment
HEK293t cells were obtained from ATCC (Manassas, VA, USA; Cat. No. CRL-1126). The cells were maintained in DMEM (Sigma-Aldrich; Cat. No. D5796) supplemented with 10% (v/v) FBS (Sigma-Aldrich; Cat. No. F4135). Cells were grown at 37°C in an atmosphere of 5% CO2 and 21% O2, which was referred to as normoxia. To model stress after H/R, the cells were placed in a hypoxic chamber (Galaxy 48R, New Brunswick, Edison, NJ, USA) at 37°C containing 0.5% O2, 5% CO2 for 16 h and then were immediately exposed to normoxic conditions for 4 h. Levels of O2 in the chamber were strictly kept at 0.5% by constantly flushing with nitrogen that was automatically controlled by an Oxygen Sensor. All experiments were carried out at ∼80% confluency with the medium pH being maintained at 7.3–7.5 throughout during the period of normoxia or hypoxia. For treatments under normoxia, UFP-512 (5 μM), naltrindole (1 μM) or UFP-512 (5 μM) plus naltrindole (1 μM) were added to the culture medium and cells incubated for the times shown. In our studies on H/R, UFP-512 and/or naltrindole were added to cultures of HEK293t cells and then immediately exposed to the H/R procedure as described above.
Small interfering RNA (siRNA) transfection
The patented ON-TARGET plus modification pattern SMART siRNA pool targeted to human NFE2L2 gene and negative control pool of four siRNAs were purchased from Dharmacon (Carlsbad, CA, USA). After the cultures of HEK293t cells reached 30–40% confluency, they were transfected with either Nrf2 siRNA (50 nM) or non-target siRNA (50 nM) with the same volume of Lipofectamine RNAi Max (Invitrogen, Grand Island, NY, USA; Cat: 13778075) for 36 h.
Cell viability assay
Cell viability was measured using CellTiter 96® AQueous Assay (MTS assay; Promega, Madison, WI, USA; Cat. No. G3580). Exponentially growing cells were plated at 5000 cells per well in a 96-well flat bottom plate and allowed to incubate for 24 h before experimental treatments. Wells containing 100 μL of DMEM without cells were set as the control for background readings. Twenty microlitres per well of One Solution Reagent were added and then the cells were incubated at 37°C for 2 h. The absorbance was recorded at 490 nm using a microplate reader (Bio-Rad, Hercules, CA, USA).
Measurements of LDH leakage
Potential cytotoxicity was assessed by measuring LDH leakage into the culture medium using Cytoscan-LDH cytotoxicity assay kit (G-Biosciences, St. Louis, MO, USA; Cat. No. 786-210). The cells were treated with indicated compound(s) and incubated for the desired period. Plates containing 10× lysis buffer and 100 μL of culture medium with or without cells were set as maximum control or spontaneous control respectively. After treatment, the supernatant was transferred to a fresh 96-well plate (50 μL per well) and mixed with the reconstituted substrate mix (50 μL per well). Then, these mixtures were incubated at 37°C for 20 min. After that, 50 μL per well of the Stop Solution was added to terminate the reaction. The absorbance of the solution was recorded by a microplate reader (Bio-Rad) at 490 nm wavelength.
Western blot analysis
The cells were treated with various compounds, as indicated in the Results section. After different treatments, the cells were rinsed with ice-cold Dulbecco's PBS (DPBS) for three times and then lysed in RIPA buffer for total protein extraction or in NE-PER buffer for nuclear and cytoplasmic protein extraction. Protein concentration was determined by the bicinchoninic acid method (Thermo Scientific, Rockford, IL, USA; Cat. No. 23227). Proteins were denatured by heating at 100°C for 8 min. Equal amounts of protein were separated on SDS-PAGE gel and blotted onto PVDF membranes (Millipore, Billerica, MA, USA; Cat. No. IPVH00010) using Trans-Blot semi-dry apparatus (Bio-Rad). The blots were incubated with anti-Nrf2 (1:1,000), anti-β-actin (1:5000) or anti-Lamin B (1:1000) antibody. The membrane was washed with TBST and its composition (Tris-buffered saline, pH 8, with 0.1% Tween 20) for three times with 15 min each time and then incubated with the recommended dilution of labelled secondary antibody in 2% blocking buffer in TBST at room temperature for 1 h. After then, the membrane was washed with TBST for three times with 10 min each time. Blots were visualized with enhanced chemiluminescence plus Western blotting detection reagents (Amersham Biosciences, Uppsala, Sweden; RPN2132).
RNA isolation and reverse transcription real-time PCR analysis
Total cellular RNA was extracted with TRIzol Reagent (Invitrogen; Cat. No. 15596-018) according to the manufacturer's instructions. RNA concentration was determined by measuring UV absorption with Nanodrop 2000c (Thermo Scientific, Waltham, MA, USA), and samples with A260/A280 ratios around 1.85–2.1 were used for reverse transcription-PCR by using Transcriptor First Strand cDNA synthesis kit (Roche, Mannheim, Baden-Wurttemberg, Germany; Cat. No. 04379012001). The sequences of the PCR primer pairs used for the amplification of human Nrf2, Keap1, NQO-1, haem oxygenase 1 (HO-1), glutamate-cysteine ligase-catalytic subunit (GCLC), glutamate-cysteine ligase-modifier subunit (GLCM) and GAPDH are shown in Table 1. Real-time PCR was carried out by using SBYR Green MIX I (Roche; Cat. No. 04707516001) with a real-time system (CFX96; Bio-Rad) and thermal cycler (Bio-Rad). Amplification conditions were 45 cycles of 10 s at 95°C, 20 s at 59°C and 30 s at 72°C.
Table 1.
Primers for real-time PCR
| Gene name | Primers |
|---|---|
| NFE2L2 | F – tcagcgacggaaagagtatga |
| R – ccactggtttctgactggatgt | |
| KEAP1 | F – gtgtccattgagggtatccacc |
| R – gctcagcgaagttggcgat | |
| NQO1 | F –gaagagcactgatcgtactggc |
| R – ggatactgaaagttcgcaggg | |
| HO-1 | F – aagactgcgttcctgctcaac |
| R – aaagccctacagcaactgtcg | |
| GCLM | F – catttacagccttactgggagg |
| R – atgcagtcaaatctggtggca | |
| GCLC | F – ggaggaaaccaagcgccat |
| R – cttgacggcgtggtagatgt | |
| GAPDH | F – acaactttggtatcgtggaagg |
| R – gccatcacgccacagtttc |
Immunocytochemistry
The cells were seeded on chamber slides that were pre-coated with poly-l-lysine and then allowed to grow for 24 h. After treatment, the cells were washed with DPBS, fixed in 4% paraformaldehyde for 30 min at room temperature, permeabilized with 1% Triton X-100 for 10 min and blocked with 1% BSA in PBS for 15 min. The cells were then incubated with anti-Nrf2 (1:100) antibody for 16 h overnight at 4°C, followed by incubation with Alexa Fluor-conjugated anti-rabbit secondary antibody (1:500) for 1 h. The coverslips were mounted on slides using mounting with DAPI medium (Cat. No. P36935; Life Technology). Fluorescence images were visualized with fluorescence microscope.
Data analysis
Data are expressed as mean ± SEM. Statistical significance was examined using one-way anova followed by the post hoc test of least significant difference. P < 0.05 was considered statistically significant.
Materials
UFP-512, a specific and potent DOR agonist (Balboni et al., 2002; Chao et al., 2007), was synthesized by our research team. Naltrindole (a specific and potent DOR antagonist) and mouse monoclonal anti-β-actin antibody were purchased from Sigma-Aldrich (St. Louis, MO, USA). LY294002, a highly selective inhibitor of PI3K, and calphostin C, a potent and selective inhibitor of PKC, were purchased from Tocris Cookson (Bristol, BS, UK; Cat. No. 1130, 1626). Rabbit monoclonal anti-Nrf2 and anti-Lamin B for Western blots were obtained from Cell Signaling Technology (Danvers, MA, USA; Cat. No. 8882, 9087). Mouse or rabbit HRP-conjugated secondary antibodies were purchased from GE Healthcare (Waukesha, WI, USA; Cat. No. NA9310, NA9340). Anti-Nrf2 antibody for immunofluorescence was purchased from Abcam (Cambridge, MA, USA; Cat. No. ab31163), and Alexa Fluor-conjugated donkey anti-rabbit IgG (H + L) was from Life Technology (Grand Island, NY, USA; Cat. No. A31572).
Results
DOR activation increased Nrf2 translocation from cytoplasm to nucleus
To explore potential interactions between DOR and Nrf2, we used UFP-512 (5 μM), a potent and specific agonist of DOR (Balboni et al., 2002; Chao et al., 2007), added to the culture media of HEK293t cells and then extracted the whole cell protein at 0, 2, 8, 16 and 24 h after the administration of UFP-512 to determine if DOR activation altered Nrf2 protein levels in the cells. Our results showed that after 2 h of exposure to UFP-512, the total cellular content of Nrf2 protein tended to increase but this trend did not reach significance over 8h of incubation with UFP-512 (Figure 1A).
Figure 1.
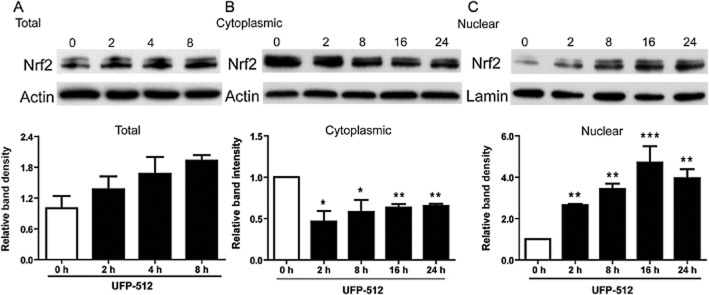
Effect of UFP-512 on Nrf2 expression in different fractions of HEK293t cells. Representative Western blots (upper) and relative quantitation (lower) of Nrf2 in the total protein of HEK293 cells (A); in the cytoplasmic fraction (B); in the nuclear fraction (C). After treatment with UFP-512 for 0, 2, 4, 8, 16 and 24 h in normoxia, the total, cytoplasmic and nuclear fractions of HEK293t cells were extracted for Western blots. Levels of Nrf2 were determined with anti-Nrf2 antibody. Lamin B was chosen as an internal standard for Western blot analysis of the nuclear protein. At least three independent experiments were carried out. *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from 0 h. Note that there are no significant changes in the Nrf2 protein after UFP-512 treatment in whole cell protein, although UFP-512 tended to increase it. UFP-512 decreased Nrf2 density in the cytoplasmic fraction but increased it in the nuclear fraction, suggesting translocation of Nrf2 from cytoplasm to nucleus.
Nrf2 exerts its transcriptional activity after liberation from its cytosolic anchor protein Keap1. The free Nrf2 is then rapidly translocated from the cytosol to the nucleus where it induces gene expression (Itoh et al., 1999, 2010; Tong et al., 2006). To explore whether activation of DOR affected Nrf2 translocation, we measured Nrf2 protein levels in the cytoplasmic and nuclear extracts, separately, in response to DOR activation. In sharp contrast to the lack of significant change in the whole cell extracts, cytoplasmic Nrf2 protein clearly decreased by 40–50% after 2–24 h of exposure to UFP-512 (Figure 1B), whereas nuclear Nrf2 protein significantly increased with increasing exposure duration and reached its peak level at 16–24 h of UFP-512 exposure (Figure 1C). These results indicated that activation of DOR increased the translocation of Nrf2 protein from cytoplasm to nucleus.
To examine this novel finding further, we determined the effects of a DOR antagonist on the UFP-512-induced translocation of Nrf2. We treated HEK293t cells with naltrindole (2 μM), a specific antagonist of DOR (Filizola and Devi, 2012; Granier et al., 2012), added at the same time as the UFP, and then repeated the assays described above. The results showed that the Nrf2 translocation induced by UFP-512 was abolished by naltrindole, whereas naltrindole itself had no appreciable effect on Nrf2 translocation in HEK293t cells in normoxia (Figure 2A and B).
Figure 2.
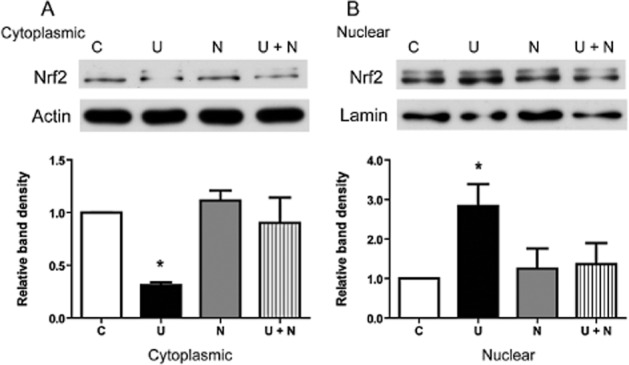
Reversal of UFP-512 induced alteration in Nrf2 protein level by the DOR antagonist naltrindole. Representative Western blots (upper) and relative quantitation (lower) of cytoplasmic Nrf2 protein (A) and nuclear Nrf2 protein (B). The cells were treated with UFP-512 (5 μM), naltrindole (2 μM) or control vehicle for 2 h. Then, cytoplasmic and nuclear proteins were extracted for Western blots and Nrf2 determined with anti-Nrf2 antibody. C, normoxic control. U, UFP-512. N, naltrindole. U + N, simultaneous administration of UFP-512 and naltrindole. At least three independent experiments were conducted in each group. *P < 0.05, significantly different from control. Note that the DOR agonist UFP-512 increased Nrf2 translocation from the cytoplasm to nucleus, whereas addition of the DOR antagonist naltrindole abolished such translocation.
DOR activation enhanced Nrf2-regulated transactivation
As DOR activation increased Nrf2 translocation from the cytoplasm to the nucleus, we next asked if such activation would lead to enhanced transcriptional function of Nrf2. Therefore, we investigated the effects of UFP-512 on transcriptional changes in some Nrf2-regulated genes such as NAD(P)H dehydrogenase, quinone 1 (NQO1) (Jaiswal, 1994; Venugopal and Jaiswal, 1996), haem oxygenase 1 (HO-1) (Alam et al., 2000), glutamate-cysteine ligase-modifier subunit (GCLM) and glutamate-cysteine ligase-catalytic subunit (GCLC) (Solis et al., 2002) with quantitative real-time PCR analysis. In our sample size, the expression of GCLC did not show any significant change in response to incubation with UFP-512. However, all other genes studied showed a significant increase in their mRNA expression. As shown in Figure 3, NQO1, HO-1 and GCLM were increased by ∼40, ∼85 and ∼45% (P < 0.01, P < 0.001 and P < 0.005), respectively, after 2 h of exposure to UFP-512 (Figure 3). Consistent with the changes in Nrf2 proteins, the increase in the expression of these genes in response to UFP-512 exposure was markedly attenuated or abolished by co-exposure to the DOR antagonist naltrindole (2 μM), suggesting that it was the activation of DOR that up-regulated the expression of Nrf2-regulated genes.
Figure 3.
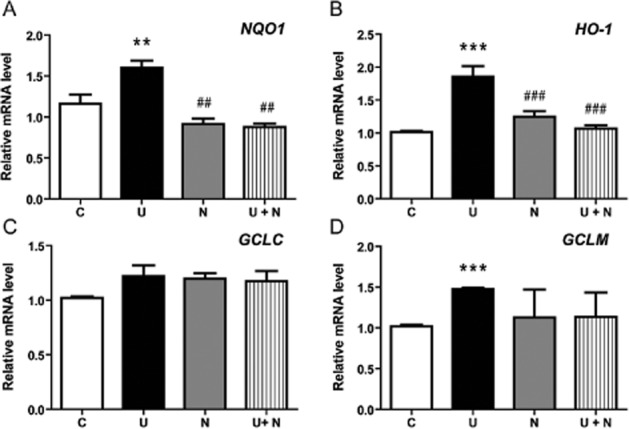
Effect of UFP-512 on Nrf2-targeted gene expression in normoxia. The mRNA expression of Nrf2-dependent genes NQO1, HO-1, GCLC and GCLM was assessed by quantitative real-time PCR in the HEK293t cells after treatment with UFP-512 (U), naltrindole (N) or UFP-512 plus naltrindole (U + N). *P < 0.05, **P < 0 0.01, ***P < 0.005, significantly different from control (C). ##P < 0.01, ###P < 0.005, significantly different from UFP-512 alone. Note that all the genes studied significantly increased or tended to increase their mRNA expression after UFP-512 treatment and that addition of the DOR antagonist naltrindole fully blocked the effects of UFP-512.
DOR activation induced cytoprotection against H/R injury via Nrf2 translocation and up-regulation of its downstream antioxidant-related genes
We have recently demonstrated that DOR activation is protective against hypoxic insults (Chao and Xia, 2010; He et al., 2013) and we therefore tested if this protection might be mediated by the regulation of Nrf2 activity. We exposed HEK293t cells to the H/R procedure (incubation in hypoxia – 0.5% O2 – for 16 h immediately after adding UFP-512 and/or naltrindole to the medium, followed by 4 h in normoxia -21% O2), and then evaluated cell damage and viability with LDH and MTS assays respectively.
As shown in Figure 4A, cell viability, as evaluated by MTS assay, was significantly decreased after H/R, compared with the viability of the cells in normoxia. Incubation with UFP-512 (5 μM) clearly increased cell viability after H/R and addition of the DOR antagonist naltrindole (2 μM) abolished UFP-512-induced cytoprotection. Naltrindole alone did not modify the loss of cell viability after H/R (Figure 4A). To confirm this observation, we also conducted LDH assays to evaluate cell damage under the same conditions (Figure 4B). In these assays, LDH leakage was increased after H/R, compared with cells in normoxia and exposure to UFP-512 decreased this LDH leakage, suggesting less cell damage. Naltrindole (2 μM) abolished the effect of UFP-512 on LDH leakage, although naltrindole alone induced a slight increase in LDH leakage after H/R (Figure 4B). Altogether, these data are consistent with our earlier observations of DOR-mediated cytoprotection in neurons (Zhang et al., 2000; 2002; 2006).
Figure 4.
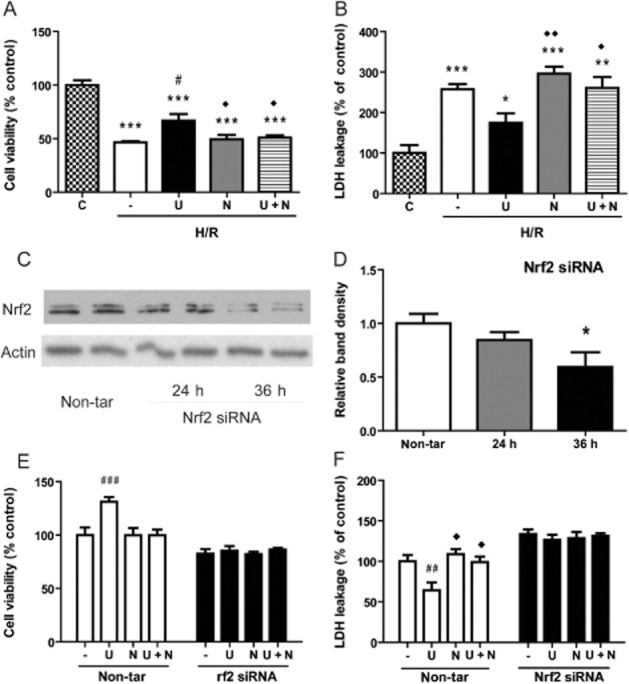
Role of Nrf2 in DOR-mediated protection against H/R injury. In (A), cell viability (with MTS) and, in (B), cell damage (LDH leakage) were measured under different conditions: vehicle treatment in normoxia (C) and after H/R (0.5% O2 for 16h followed by 21% O2 for 4h) vehicle (−), UFP-512 (U), naltrindole (N), or UFP-512 plus naltrindole (U + N) exposure. In (C) and (D), HEK293t cells were transfected with Nrf2 siRNA and assayed for Nrf2 content 24 or 36h later, in normoxia. In (E ) and (F), cells were transfected with Nrf2 or non-target (non-tar) siRNA and exposed to H/R, without further treatment or incubation with UFP-512 (U), naltrindole (N), or UFP-512 plus naltrindole (U + N). In (E), results from assays of cell viability and, in (F), of LDH leakage are compared with those of the control cells transfected with non-target siRNA. Data shown are means ± SEM of three independent experiments. —, H/R alone. *P < 0.05, **P < 0.01, ***P < 0.005, significantly different from control (the non-treated) cells in normoxia; # P < 0.05, ##P < 0.01, ###P < 0.005, significantly different from non-treated cells after H/R stress. ◊P < 0.05, significantly different from UFP-512 after H/R stress. n = 4 per group. Note that H/R decreased cell viability and increased LDH leakage, effects attenuated by UFP-512 (A and B). Co-treatment with naltrindole abolished the UFP-512-induced protection. After Nrf2 siRNA transfection, Nrf2 protein was knocked down (C and D) and DOR activation with UFP-512 no longer induced any protective effect on the cells. In the control cells with non-target siRNA transfection, however, treatment with UFP-512 was still cytoprotective (E and F).
To explore whether DOR-mediated cytoprotection was critically dependent on the presence of Nrf2, we transfected HEK293t cells with Nrf2 siRNA to knock down Nrf2 expression and then tested the effect of UFP-512 on cell viability and injury after H/R. As shown in Figure 4C, Nrf2 protein expression in cells treated with the appropriate siRNA was not significantly changed at 24 h after the transfection but more clearly reduced at 36 h; these experiments were carried out in normoxia. Transfected cells were then exposed to the H/R procedure after 16h, giving a total incubation time of 36h, as in the experiments in normoxia (see Figure 4C and 4D). The transfected+H/R cells were also treated with DOR ligands and both MTS and LDH assays carried out. As shown in Figure 4E and 4F, DOR activation with UFP-512 was still cytoprotective against H/R induced injury in HEK293t cells transfected with the non-target siRNA but no longer induced any cytoprotective effect in the cells with Nrf2 siRNA transfection, suggesting that Nrf2 was critical to DOR-mediated cytoprotection following H/R.
To further elucidate how Nrf2 could mediate DOR cytoprotection, we examined the effect of UFP-512 on Nrf2 translocation after H/R. We found that treatment with the DOR agonist significantly increased Nrf2 accumulation in the nucleus, whereas cytosolic Nrf2 was reduced. after UPF-512 treatment. These changes were reversed by co-administration of naltrindole (Figure 5A–C). Moreover, our fluorescence imaging studies indicated high levels of Nrf2 in the nucleus after DOR activation with UFP-512 after H/R (Figure 6A and B). No such accumulation was found in the cells without DOR activation (Figure 6A). Furthermore, we measured NQO1, HO-1, GCLM and GCLC expressions (Figure 7A–D) using relative quantitative real-time PCR and found that UFP-512 increased the mRNA levels of these Nrf2-regulated genes after H/R, except for GCLC which showed no change, results comparable to those obtained under normoxic conditions, as shown in Figure 3.
Figure 5.
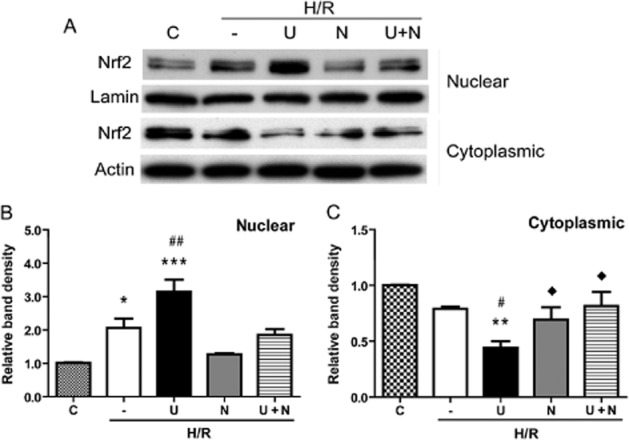
UFP-512-induced Nrf2 translocation from the cytoplasm to nucleus after H/R. (A) Representative Western blots of nuclear Nrf2 protein after DOR activation and/or inhibition in normoxia and H/R. (B) Relative quantitation of Nrf2 protein in the nucleus. (C) Relative quantitation of Nrf2 protein in the cytoplasm. After treatment with UFP-512 and/or naltrindole during the H/R procedure, nuclear and cytoplasmic fractions of the HEK293t cells were extracted for Western blotting and Nrf2 determined with anti-Nrf2 antibody. C, normoxic control. —, H/R alone. U, DOR activation with UFP-512. N, DOR inhibition with naltrindole. U + N, UFP-512 and naltrindole co-administration.*P < 0.05, **P < 0.01, ***P < 0.005, significantly different from control in normoxia, #P < 0.05, ##P < 0.01, significantly different from H/R alone. ◊P < 0.05, significantly different from UFP-512 after H/R stress. At least three independent experiments were performed. Note that H/R stress decreased cytoplasmic Nrf2 protein and increased nuclear Nrf2 protein, suggesting a translocation from the cytoplasm to nucleus. DOR activation with UFP-512 further enhanced such translocation, whereas DOR antagonism with naltrindole abolished the UFP-512-enhanced Nrf2 translocation.
Figure 6.

DOR-induced Nrf2 translocation from the cytoplasm to nucleus after H/R, as assayed by immunofluorescence. Panel (A) shows that after DOR activation with UFP-512 during H/R, Nrf2 staining (red) was merged with the (blue) nucleus stained by DAPI, suggesting high levels of Nrf2 in the nucleus. This accumulation was attenuated by adding the DOR antagonist naltrindole during H/R, as shown by Nrf2 staining outside the nucleus. Panel (B) displays a larger viewing area to further demonstrate the accumulation of Nrf2 in the nucleus induced by UFP-512. Scale bars = 100 μm in (A) and 200 μm in (B).
Figure 7.
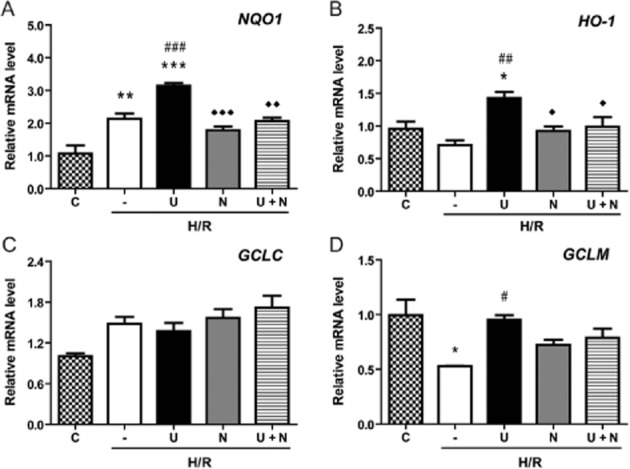
DOR-induced expression of Nrf2-regulated genes after H/R. The expression of Nrf2-targeted gene NQO1, HO-1, GCLC and GCLM (D) mRNAs in the HEK293t cells exposed to UFP-512 and/or naltrindole in H/R condition was assessed by quantitative real-time PCR. C, normoxic control. —, no treatment after H/R. U, UFP-512 during H/R. N, naltrindole during H/R. U + N, UFP-512 and naltrindole co-treatment during H/R. *P < 0.05, **P < 0.01, ***P < 0.005, significantly different from the normoxic control (C). #P < 0.05, ##P < 0.01, ###P < 0.005 significantly different from H/R alone. ◊P < 0.05, ◊◊P < 0.01, ◊◊◊P < 0.005, significantly different from UFP-512 and H/R stress. At least three independent experiments were carried out per group. Note that except for GCLC, all other genes increased their mRNA expression after DOR activation during H/R, an effect blocked by DOR antagonism with naltrindole.
DOR-mediated regulation of Nrf2 differentially involved PKC and PI3K signalling
The Keap1–Nrf2 complex plays a central role in the protection of cells against oxidative injury (Kensler et al., 2007; Vergura et al., 2008; Baird and Dinkova-Kostova, 2011). Keap1 interacts with Nrf2 and the actin cytoskeleton to retain Nrf2 in the cytoplasm to maintain a basal level of gene expression of the cytoprotective molecules and proteins in quiescent cells under normal physiological conditions. Oxidative and/or electrophilic stimuli liberate Nrf2 from its Keap1–Nrf2 complex and allow Nrf2 activity to be expressed. To determine whether UFP-512 regulated both Keap1 and Nrf2, we examined both Keap1 and Nrf2 expression simultaneously, under normoxia or after H/R, using quantitative real-time PCR analysis. Unexpectedly, we found that activation of DOR by UFP-512 had no direct effect on Nrf2 mRNA expression either in normoxia or after H/R. However, mRNA for the anchor protein Keap1 was down-regulated by UFP-512 in normoxia and after H/R but this down-regulation was not reversed by the DOR antagonist naltrindole (Figure 8A–D), suggesting a DOR-independent mechanism.
Figure 8.
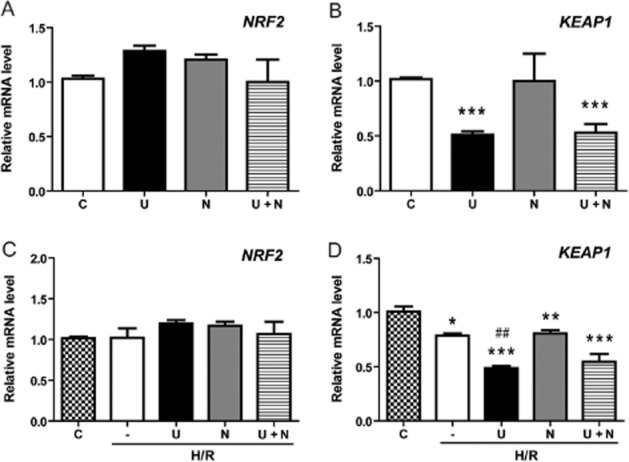
Effects of UFP-512 on Nrf2 and KEAP1 mRNA expression after H/R. In HEK293t cells treated with DOR activation and/or inhibition, Nrf2 and KEAP1 mRNAs were measured by relative quantitative real-time PCR in both normoxia and after H/R. C, normoxic control. —, H/R alone. U, DOR activation with UFP-512 during H/R condition. N, DOR inhibition with naltrindole during H/R condition. U + N, UFP-512 and naltrindole co-administration during H/R condition. *P < 0.05, **P < 0.01, ***P < 0.005, significantly different from control cells in normoxia. ##P < 0.01, significantly different from H/R alone. At least three independent experiments were carried out for each group. Note that UFP-512 had no direct effect on Nrf2 expression.
Lastly, we identified a role for PKC in this DOR-mediated regulation of Nrf2 and compared it with that for PI3K. Calphostin C (a PKC inhibitor, 100 mM) or LY294002 (a PI3K inhibitor, 10 μM) was co-applied with UFP-512 to the HEK293t cells in normoxia. Then, the levels of cytosolic and nuclear Nrf2 were examined in these cells 16 h later. After blocking the PKC pathway, DOR activation did not increase nuclear Nrf2. In sharp contrast, however, blocking the PI3K pathway further enhanced the increase in nuclear Nrf2 induced by UFP-512. In terms of cytosolic Nrf2, adding calphostin C significantly reduced cytosolic Nrf2, whereas LY294002 had no significant effect (Figure 9A–C).
Figure 9.
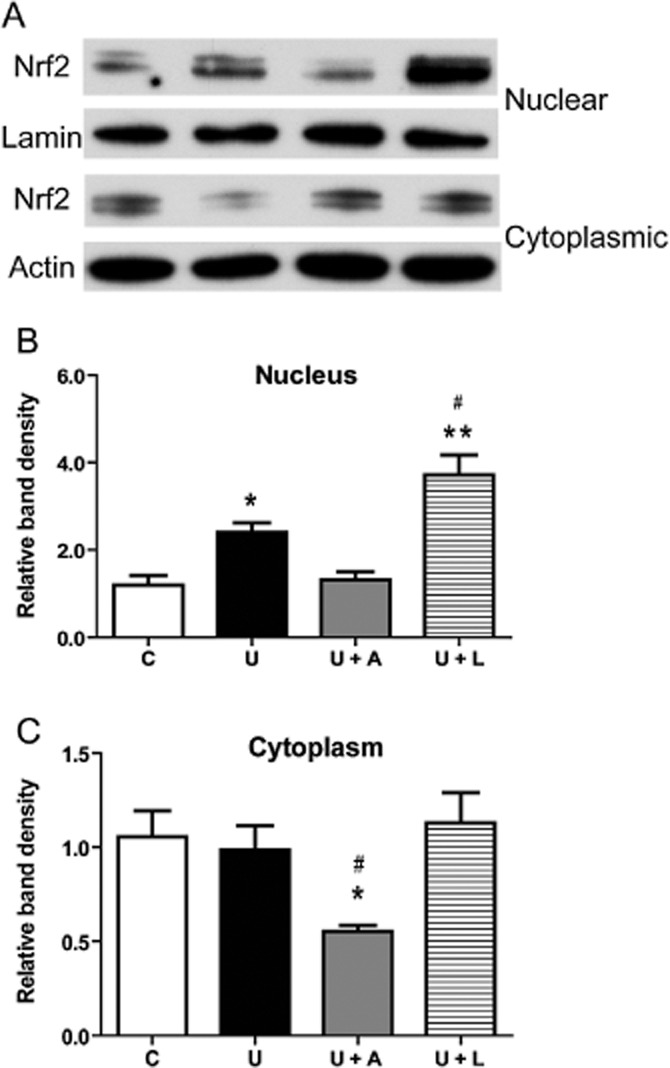
Differential roles of PKC and PI3K pathways in DOR-mediated Nrf2 regulation. (A) Representative Western blots of nuclear and cytoplasmic Nrf2 proteins in the HEK293t cells treated with PKC and PI3K inhibitors. (B) Relative quantitation of Nrf2 protein in the nucleus. (C) Relative quantitation of Nrf2 protein in the cytoplasm. The HEK293t cells were treated with UFP-512 plus calphostin C (a PKC inhibitor, 100 nM) or LY294002 (a PI3K inhibitor, 10 μM) for 16 h in normoxia and then their cytoplasmic and nuclear levels of Nrf2 protein were assayed. C, vehicle (DMSO) control. U, DOR activation with UFP-512. U + CC, co-administration of UFP-512 and calphostin C (A). U + L, co-administration of UFP-512 and LY294002 (L). *P < 0.05, **P < 0.0,1 significantly different from control in normoxia. #P < 0.05, significantly different from UFP-512. At least three independent experiments were carried out in each group. Note that the PKC inhibitor calphostin C lowered the Nrf2 protein in cytoplasm but had no effect on Nrf2 in the nucleus, whereas the PI3K inhibitor LY294002 increased Nrf2 expression in the nucleus, without affecting cytoplasmic levels.
Taken together, these data suggest that although DOR activation may not have direct effects on Nrf2 transcription, it may increase Nrf2 translation to change its protein level via a specific signalling pathway. More importantly, DOR-mediated signalling enhanced Nrf2 translocation from the cytoplasm to the nucleus.
Discussion and conclusions
Our study demonstrated that (i) DOR-mediated cytoprotection against H/R injury relied, at least partly, on the function of Nrf2; (ii) DOR activation increased Nrf2 protein expression and translocation, at least under normoxia, via a PKC-dependent pathway; and (iii) DOR up-regulated, through Nrf2 translocation, the expression of Nrf2-targeted genes relevant to antioxidative action. These novel findings suggest a causal connection between activation of DOR, PKC action and the transcription factor Nrf2 (the ‘DOR-PKC-Nrf2 axis’) in providing cytoprotection against H/R injury.
In this study, we used the HEK293t cell line for several reasons. Firstly, although derived by transformation of primary cultures of HEK cells, they express the neurofilament (NF) subunits NF-L, NF-M, NF-H and α-internexin as well as many other proteins typically found in neurons (Shaw et al., 2002). Secondly, these cells normally express a significant amount of Nrf2. Thirdly, a recent study using absolute quantitative real-time RT-PCR showed that the kidney was, in terms of the opioid receptors, predominantly a ‘DOR tissue’ because DOR expression was almost 45-fold higher than that of KOR, with no detectable expression of MORs (Peng et al., 2012). Indeed, the present study and another independent study in our laboratory (Chen et al., 2014) have detected DOR expression at both mRNA and protein levels. Therefore, this cell line is ideal for exploring the role of DOR in the regulation of Nrf2, as a first step. In fact, our recent results on neurons and glia are consistent with the findings made in the present study.
The transcription factor Nrf2 is a well-known regulator of cellular resistance to oxidants and it controls the basal and induced expression of a group of ARE-dependent genes to combat oxidative stress (Ma, 2013). In the present work, we found that knock-down of Nrf2 expression by Nrf2 siRNA abolished the DOR-mediated protection against H/R injury, suggesting the involvement of Nrf2 in DOR protection against H/R. Indeed, DOR activation induced Nrf2 accumulation in the nucleus and increased transcriptional effect, not only after H/R but also in normoxia. Therefore, we are confident that DOR-mediated cytoprotection against H/R injury is partly mediated through the regulation of Nrf2.
Our previous studies have established the importance of PKC in DOR-mediated cytoprotection (Ma et al., 2005) and PKC is also involved in Nrf2 regulation (Huang et al., 2000; Bloom and Jaiswal, 2003). Several lines of evidence have suggested that PKC regulates phosphorylation of Nrf2 at Ser40, which is required for Nrf2 translocation (Huang et al., 2000; Bloom and Jaiswal, 2003). We observed that a non-selective PKC inhibitor abolished the Nrf2 translocation induced by UFP-512 and reduced the total Nrf2 protein expression. This finding suggests that PKC plays a critical role in the regulation of Nrf2, and that DOR agonists regulate Nrf2 activity via a PKC-dependent pathway. However, more evidence is needed to fully validate the role of this pathway in the DOR-mediated regulation of Nrf2.
In sharp contrast, inhibition of PI3K markedly increased the accumulation of Nrf2 protein in the nucleus without changing the level of Nrf2 protein in the cytoplasm. This interesting finding further demonstrates the specificity of the PKC effect discussed above and lends further support for the possibility that PI3K inhibition leads to an increase in the synthesis of Nrf2 protein and thus increases its translocation to the nucleus. It is noteworthy that a previous study showed that the inhibition of PI3K/Akt by LY294002, for a relatively short time (from 20 min to 2 h) prevented redistribution of Nrf2 towards the nucleus triggered by oxidative stress. In this case, PI3K/Akt inhibition may activate GSK-3β that phosphorylates Fyn at an unknown threonine residue (Niture et al., 2014). The phosphorylated Fyn may be translocated to the nucleus and phosphorylate Nrf2 (Jain and Jaiswal, 2006; Kaspar and Jaiswal, 2011), which leads to nuclear export and degradation of Nrf2. In the present study, we treated HEK293t cells with LY294002 for a much longer period (16 h). Such a long-term inhibition of the PI3K axis may exhaust the function of GSK-3β and eventually lead to an increased accumulation of Nrf2 in the nucleus. It seems that PI3K signalling has a negative impact on Nrf2 activity and needs to be further investigated.
Keap1 is an anchor protein that negatively regulates Nrf2 function. Keap1 interacts with both Nrf2 and the actin cytoskeleton to retain Nrf2 in the cytoplasm and to maintain it at a low level by promoting its degradation by the ubiquitin-proteasome pathway, in quiescent cells under normal physiological conditions (Kensler et al., 2007). During oxidative stress, ROS stimuli liberate Nrf2 from its Keap1-mediated cytoplasmic binding. The free Nrf2 is rapidly translocated to the nucleus with the assistance of a nuclear localization sequence, where it functions as a strong transcriptional activator of ARE-responsive genes in partnership with other transcription factors (Kensler et al., 2007). In sharp contrast to the increase in Nrf2 translation after DOR activation, KEAP1 mRNA expression was reduced by DOR activation at both normoxic and H/R conditions. The reduction of Keap1 expression may increase the liberation of Nrf2 in the cytoplasm and enhance Nrf2 function in the nucleus. However, the UPF-512 induced reduction of Keap1 mRNA expression was not reversed by naltrindole, suggesting that UFP-512 suppressed Keap1 expression via mechanisms other than DOR activation. More studies are required to clarify this issue.
As Nrf2 regulates several downstream genes, we assessed the expression of some Nrf2-regulated genes after DOR activation to further confirm the existence of the DOR-Nrf2 axis. In the representative genes we studied, NQO1 [NAD(P)H:quinone oxidoreductase] codes for a two-electron reductase that can either bioactivate or detoxify ROS-containing quinones and is transcriptionally activated exclusively by Nrf2 (Venugopal and Jaiswal, 1996). We found that its expression correlated well with the accumulation of Nrf2 in the nucleus. Interestingly, GCLC is less sensitive to DOR activation than other Nrf2-targeted genes. There are several possible explanations for this difference. GCLC is regulated not only by Nrf2, but also by Nrf1. The up-regulation of Nrf2 achieved in our system, by itself may not be enough to increase GCLC expression. Moreover, different Nrf2-regulated genes may respond to Nrf2 with different latencies of action and over a range of times. Therefore, they can display different expression profiles at a fixed time point, as shown in this study. All these findings raise new and interesting questions that should be addressed in further studies.
In summary, we have established a DOR-PKC-Nrf2 axis as a part of the mechanism underlying cytoprotection against H/R injury. Our data may provide clues to the development of new therapeutic strategies for the prevention and/or treatment of H/R injury in vivo, as may occur in a range of clinically important conditions including stroke, myocardial infarction, traumatic injury and transplantation.
Acknowledgments
This work was supported by the NIH (HD-034852 and AT-004422) and the Vivian L Smith Neurologic Foundation.
Glossary
- ARE
antioxidant response element;
- DOR
δ-opioid receptor
- GCLC
glutamate-cysteine ligase-catalytic subunit
- GCLM
glutamate-cysteine ligase-modifier subunit;
- H/R
hypoxia-reperfusion
- HO-1
haem oxygenase 1
- Keap1
Kelch-like ECH associating protein 1
- NQO1
NAD(P)H dehydrogenase [quinone] 1
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- ROS
reactive oxygen species
- siRNA
small interfering RNA
Author contributions
Y. X. and S. C. designed the research; S. C. performed the research; G. B. and H. Z. contributed analytical tools; S. C., D. C. and Y. X. analysed the data; and Y. X., S. C. and D. C. wrote the paper.
Conflict of interest
None.
References
- Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, et al. Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem. 2000;275:27694–27702. doi: 10.1074/jbc.M004729200. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- Balboni G, Salvadori S, Guerrini R, Negri L, Giannini E, Jinsmaa Y, et al. Potent delta-opioid receptor agonists containing the Dmt-Tic pharmacophore. J Med Chem. 2002;45:5556–5563. doi: 10.1021/jm020336e. [DOI] [PubMed] [Google Scholar]
- Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H: quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- Borlongan CV1, Hayashi T, Oeltgen PR, Su TP, Wang Y. Hibernation-like state induced by an opioid peptide protects against experimental stroke. BMC Biol. 2009;7:1–10. doi: 10.1186/1741-7007-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao D, Xia Y. Ionic storm in hypoxic/ischemic stress: can opioid receptors subside it? Prog Neurobiol. 2010;90:439–470. doi: 10.1016/j.pneurobio.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao D, Bazzy-Asaad A, Balboni G, Xia Y. delta-, but not mu-, opioid receptor stabilizes K(+) homeostasis by reducing Ca(2+) influx in the cortex during acute hypoxia. J Cell Physiol. 2007;212:60–67. doi: 10.1002/jcp.21000. [DOI] [PubMed] [Google Scholar]
- Chao D, He X, Yang Y, Bazzy-Asaad A, Lazarus LH, Balboni G, et al. DOR activation inhibits anoxic/ischemic Na+ influx through Na+ channels via PKC mechanisms in the cortex. Exp Neurol. 2012;236:228–239. doi: 10.1016/j.expneurol.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Li J, Chao D, Sandhu HK, Liao X, Zhao J, et al. Delta-opioid receptor activation reduces alpha-synuclein overexpression and oligomer formation induced by MPP and/or hypoxia. Exp Neurol. 2014;255:127–136. doi: 10.1016/j.expneurol.2014.02.022. [DOI] [PubMed] [Google Scholar]
- Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filizola M, Devi LA. Structural biology: how opioid drugs bind to receptors. Nature. 2012;485:314–317. doi: 10.1038/485314a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, et al. Structure of the δ-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Sandhu HK, Yang Y, Hua F, Belser N, Kim DH, et al. Neuroprotection against hypoxia/ischemia: δ-opioid receptor-mediated cellular/molecular events. Cell Mol Life Sci. 2013;70:2291–2303. doi: 10.1007/s00018-012-1167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi S, Ii M, Zhu P, Ashraf M. Delta-opioid receptor activation promotes mesenchymal stem cell survival via PKC/STAT3 signaling pathway. Circ J. 2012;76:204–212. doi: 10.1253/circj.cj-11-0309. [DOI] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–12480. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Ishii T, Wakabayashi N, Yamamoto M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic Res. 1999;31:319–324. doi: 10.1080/10715769900300881. [DOI] [PubMed] [Google Scholar]
- Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13:1665–16678. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- Jain AK, Jaiswal AK. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J Biol Chem. 2006;281:12132–12142. doi: 10.1074/jbc.M511198200. [DOI] [PubMed] [Google Scholar]
- Jaiswal AK. Antioxidant response element. Biochem Pharmacol. 1994;48:439–444. doi: 10.1016/0006-2952(94)90272-0. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Turner SM. Protecting motor networks during perinatal ischemia: the case for delta-opioid receptors. Ann N Y Acad Sci. 2010;1198:260–270. doi: 10.1111/j.1749-6632.2010.05434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JE, Kim GS, Chen H, Maier CM, Narasimhan P, Song YS, et al. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol. 2010;41:172–179. doi: 10.1007/s12035-010-8102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar JW, Jaiswal AK. Tyrosine phosphorylation controls nuclear export of Fyn, allowing Nrf2 activation of cytoprotective gene expression. FASEB J. 2011;25:1076–1087. doi: 10.1096/fj.10-171553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1–Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Ahn JY, Liang P, Ip C, Zhang Y, Park YM. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 2007;67:546–554. doi: 10.1158/0008-5472.CAN-06-2401. [DOI] [PubMed] [Google Scholar]
- Leonard MO, Kieran NE, Howell K, Burne MJ, Varadarajan R, Dhakshinamoorthy S, et al. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. FASEB J. 2006;20:2624–2626. doi: 10.1096/fj.06-5097fje. [DOI] [PubMed] [Google Scholar]
- Ma MC, Qian H, Ghassemi F, Zhao P, Xia Y. Oxygen-sensitive {delta}-opioid receptor-regulated survival and death signals: novel insights into neuronal preconditioning and protection. J Biol Chem. 2005;280:16208–16218. doi: 10.1074/jbc.M408055200. [DOI] [PubMed] [Google Scholar]
- Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2-an update. Free Radic Biol Med. 2014;66:36–44. doi: 10.1016/j.freeradbiomed.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmez I, Ozyurt H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem Int. 2012;60:208–212. doi: 10.1016/j.neuint.2011.11.009. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Sarkara S, Chang S. Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real-time RT-PCR. Drug Alcohol Depend. 2012;124:223–228. doi: 10.1016/j.drugalcdep.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47:9–23. doi: 10.1007/s12035-012-8344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G, Morse S, Ararat M, Graham F. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- Solis WA, Dalton TP, Dieter MZ, Freshwater S, Harrer JM, He L, et al. Glutamate-cysteine ligase modifier subunit: mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem Pharmacol. 2002;63:1739–17354. doi: 10.1016/s0006-2952(02)00897-3. [DOI] [PubMed] [Google Scholar]
- Stankowski JN, Gupta R. Therapeutic targets for neuroprotection in acute ischemic stroke: lost in translation? Antioxid Redox Signal. 2011;14:1841–1851. doi: 10.1089/ars.2010.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples M, Acosta S, Tajiri N, Pabon M, Kaneko Y, Borlongan CV. Delta opioid receptor and its peptide: a receptor-ligand neuroprotection. Int J Mol Sci. 2013;14:17410–17419. doi: 10.3390/ijms140917410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Zhang Y, Liang R, Yuan P, Du J, Wang H, et al. Activation of the delta-opioid receptor inhibits serum deprivation-induced apoptosis of human liver cells via the activation of PKC and the mitochondrial pathway. Int J Mol Med. 2011;28:1077–1085. doi: 10.3892/ijmm.2011.784. [DOI] [PubMed] [Google Scholar]
- Tian X, Guo J, Zhu M, Li M, Wu G, Xia Y. δ-Opioid receptor activation rescues the functional TrkB receptor and protects the brain from ischemia-reperfusion injury in the rat. PLoS ONE. 2013;8:e69252. doi: 10.1371/journal.pone.0069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Bagi Z, Feher A, Recchia FA, Sonntag WE, Pearson K, et al. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. 2010;299:H18–H24. doi: 10.1152/ajpheart.00260.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H: quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergura R, Balboni G, Spagnolo B, Gavioli E, Lambert DG, McDonald J, et al. Anxiolytic- and antidepressant-like activities of H-Dmt-Tic-NH-CH(CH2-COOH)-Bid (UFP-512), a novel selective delta opioid receptor agonist. Peptides. 2008;29:93–103. doi: 10.1016/j.peptides.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- Wang Q, Chao D, Chen T, Sandhu H, Xia Y. δ-Opioid receptors and inflammatory cytokins in hypoxia: differential regulation between glial and neuron-like cells. Transl Stroke Res. 2014;5:476–483. doi: 10.1007/s12975-014-0342-1. [DOI] [PubMed] [Google Scholar]
- Yang Y, Xia X, Zhang Y, Wang Q, Li L, Luo G, et al. Delta-opioid receptor activation attenuates oxidative injury in the ischemic rat brain. BMC Biol. 2009;7:55. doi: 10.1186/1741-7007-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Haddad GG, Xia Y. Delta-, but not mu- and kappa-, opioid receptor activation protects neocortical neurons from glutamate-induced excitotoxic injury. Brain Res. 2000;885:143–153. doi: 10.1016/s0006-8993(00)02906-1. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gibney GT, Zhao P, Xia Y. Neuroprotective role of delta-opioid receptors in cortical neurons. Am J Physiol Cell Physiol. 2002;282:C1225–C1234. doi: 10.1152/ajpcell.00226.2001. [DOI] [PubMed] [Google Scholar]
- Zhang J, Qian H, Zhao P, Hong SS, Xia Y. Rapid hypoxia preconditioning protects cortical neurons from glutamate toxicity through delta-opioid receptor. Stroke. 2006;37:1094–1099. doi: 10.1161/01.STR.0000206444.29930.18. [DOI] [PubMed] [Google Scholar]